Extractable Free Polymer Chains Enhance Actuation Performance of Crystallizable Poly(ε-caprolactone) Networks and Enable Self-Healing


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
3. Results
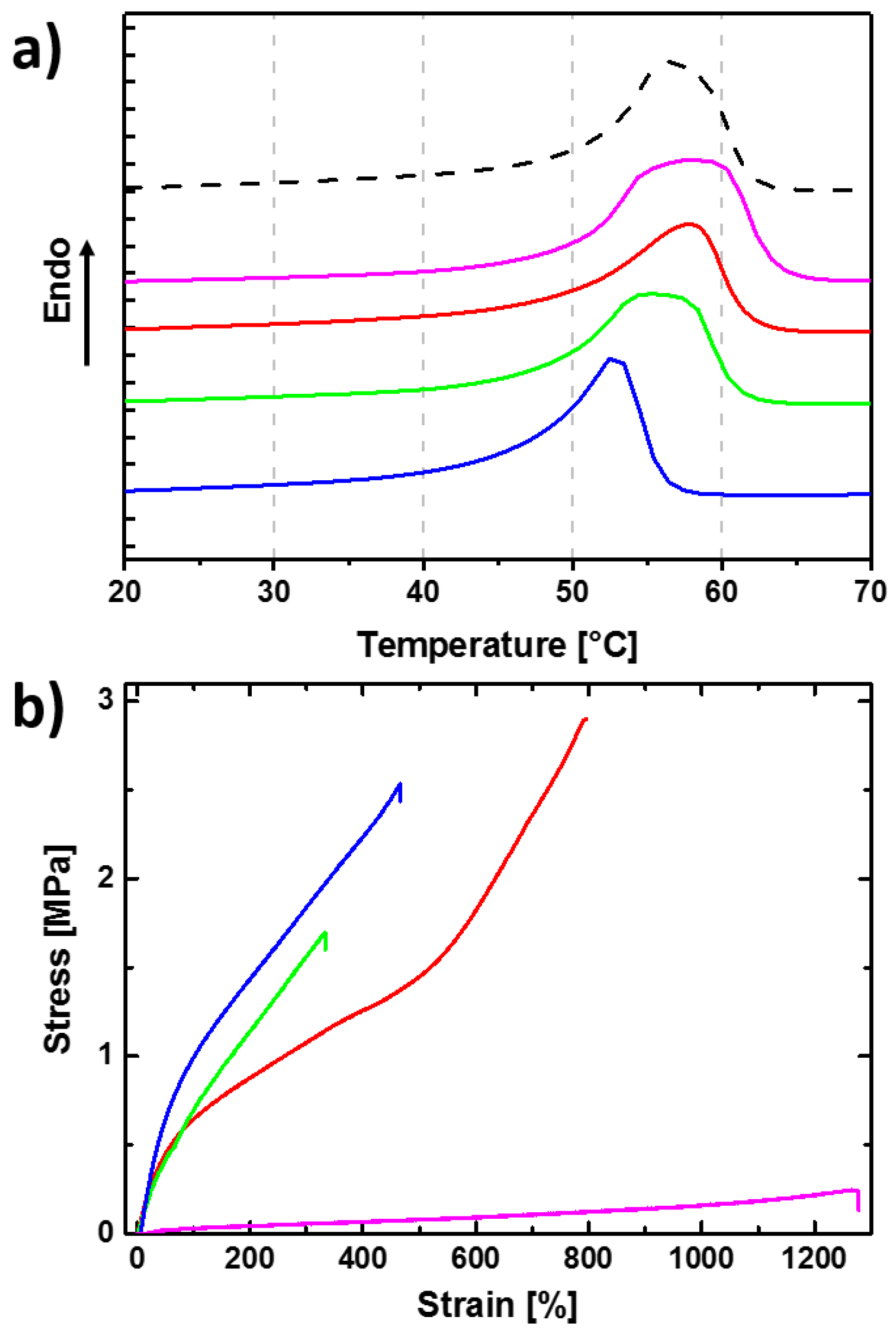
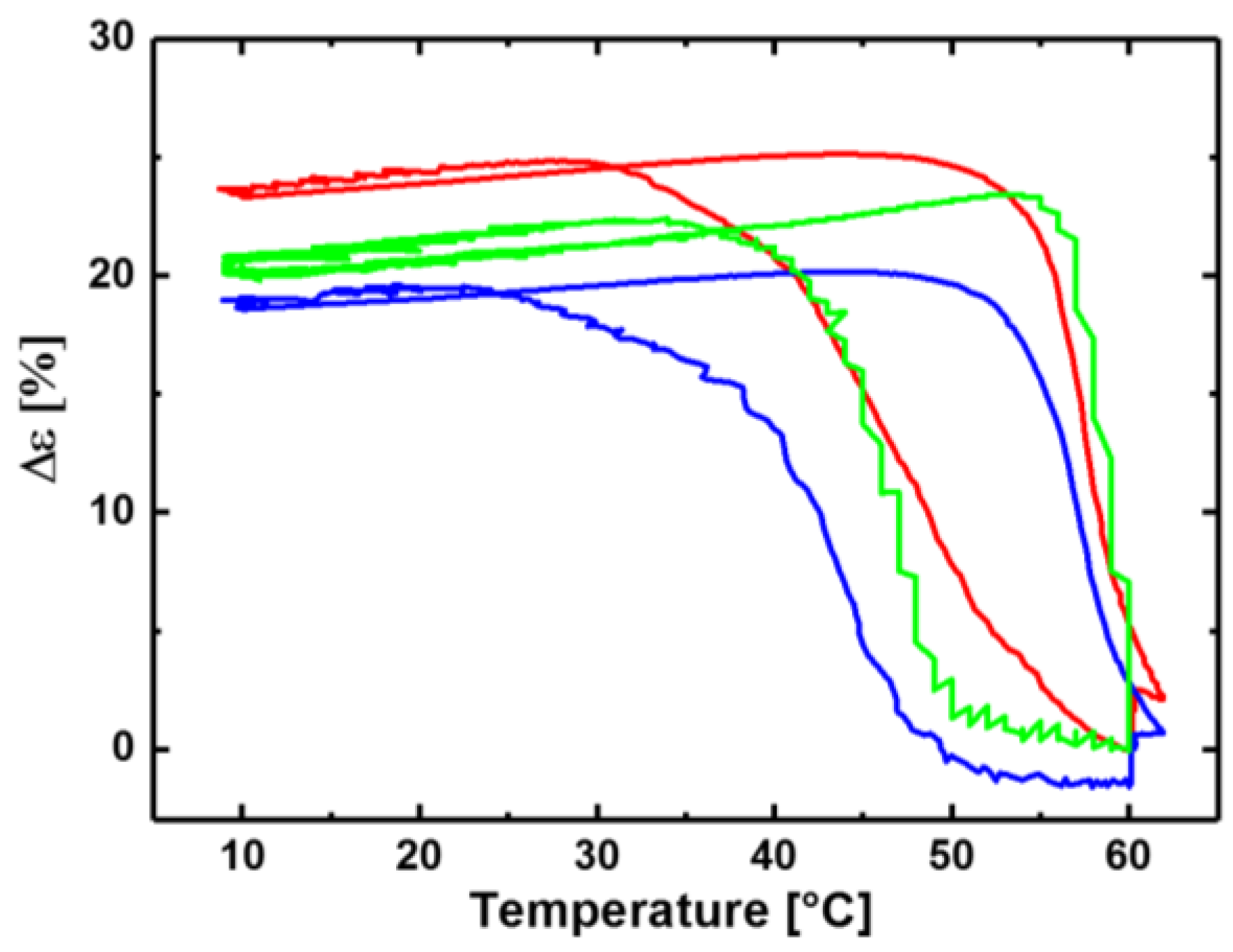
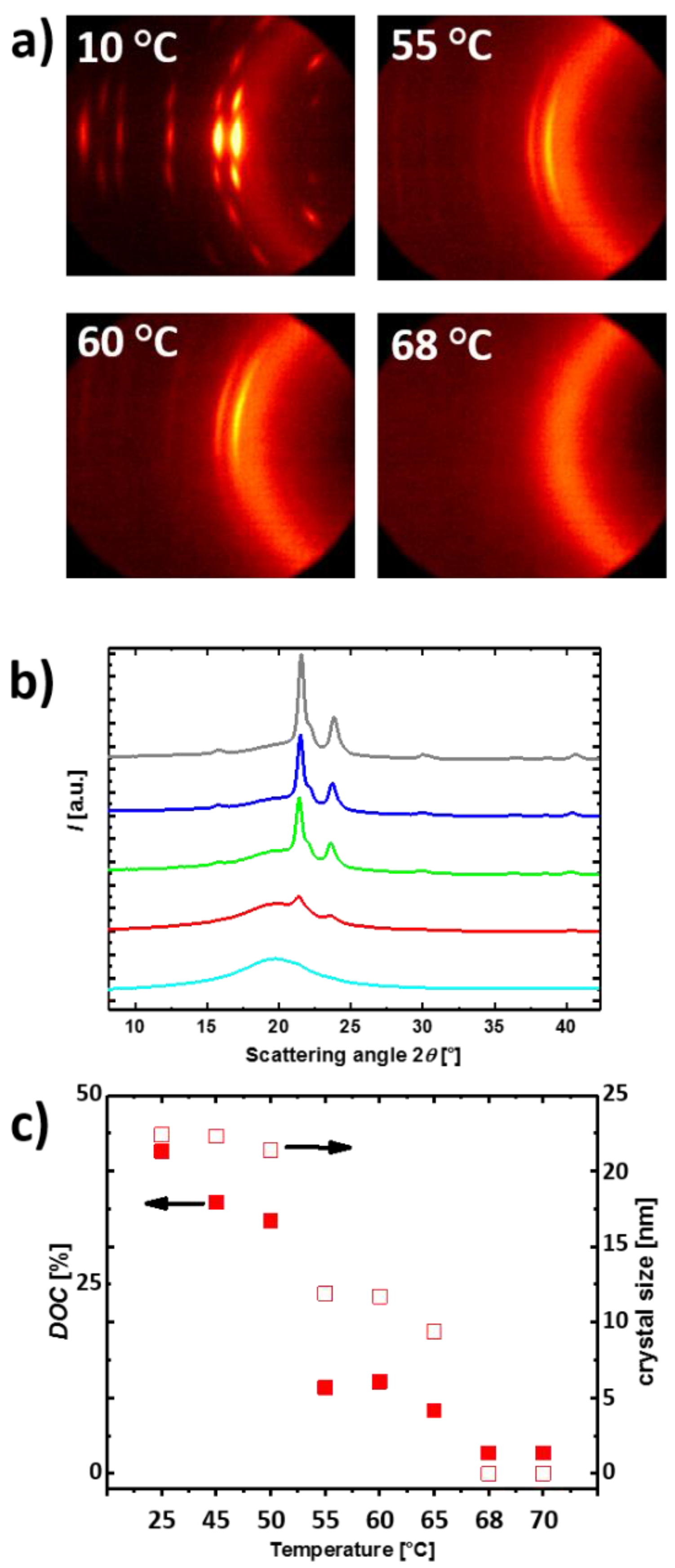
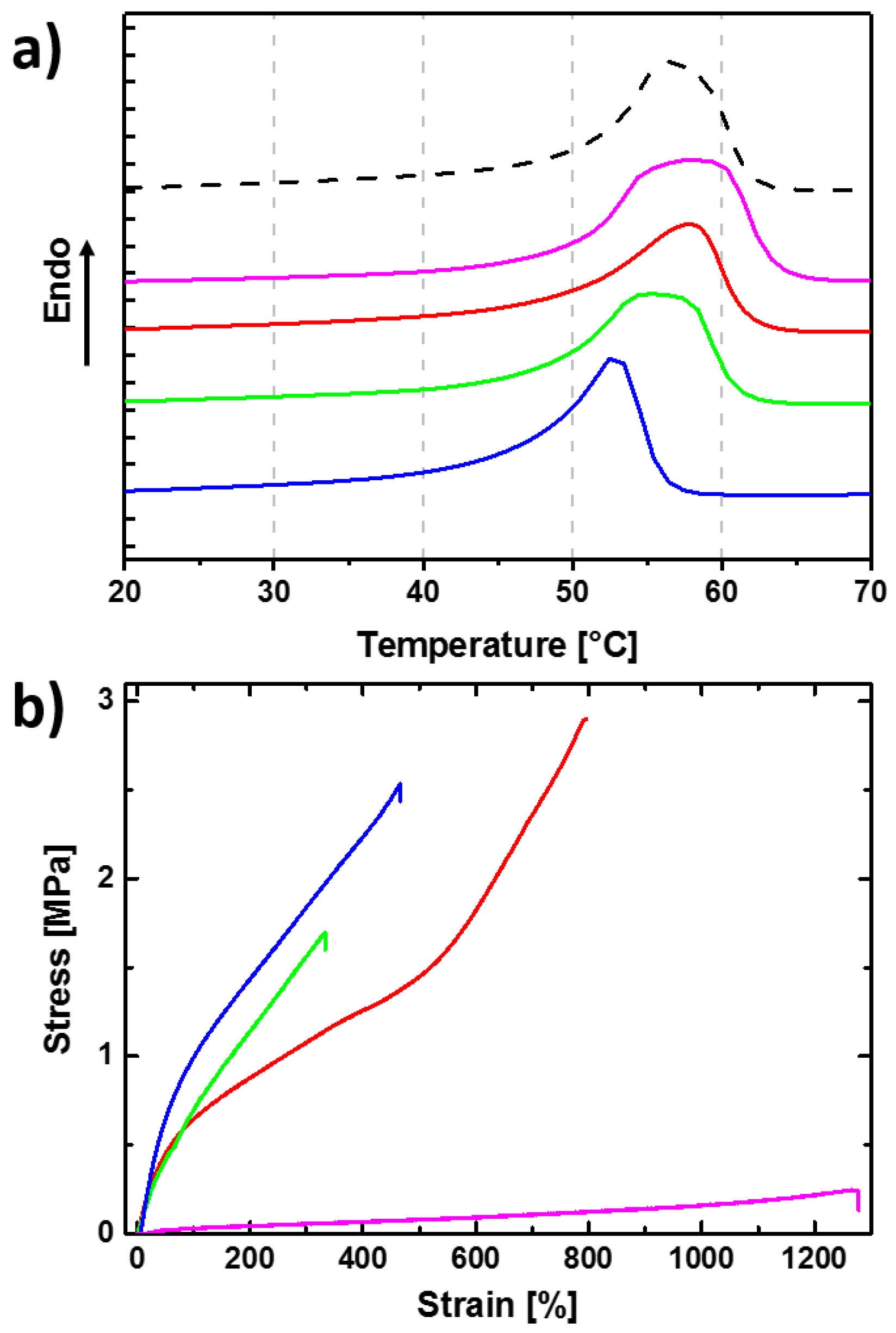
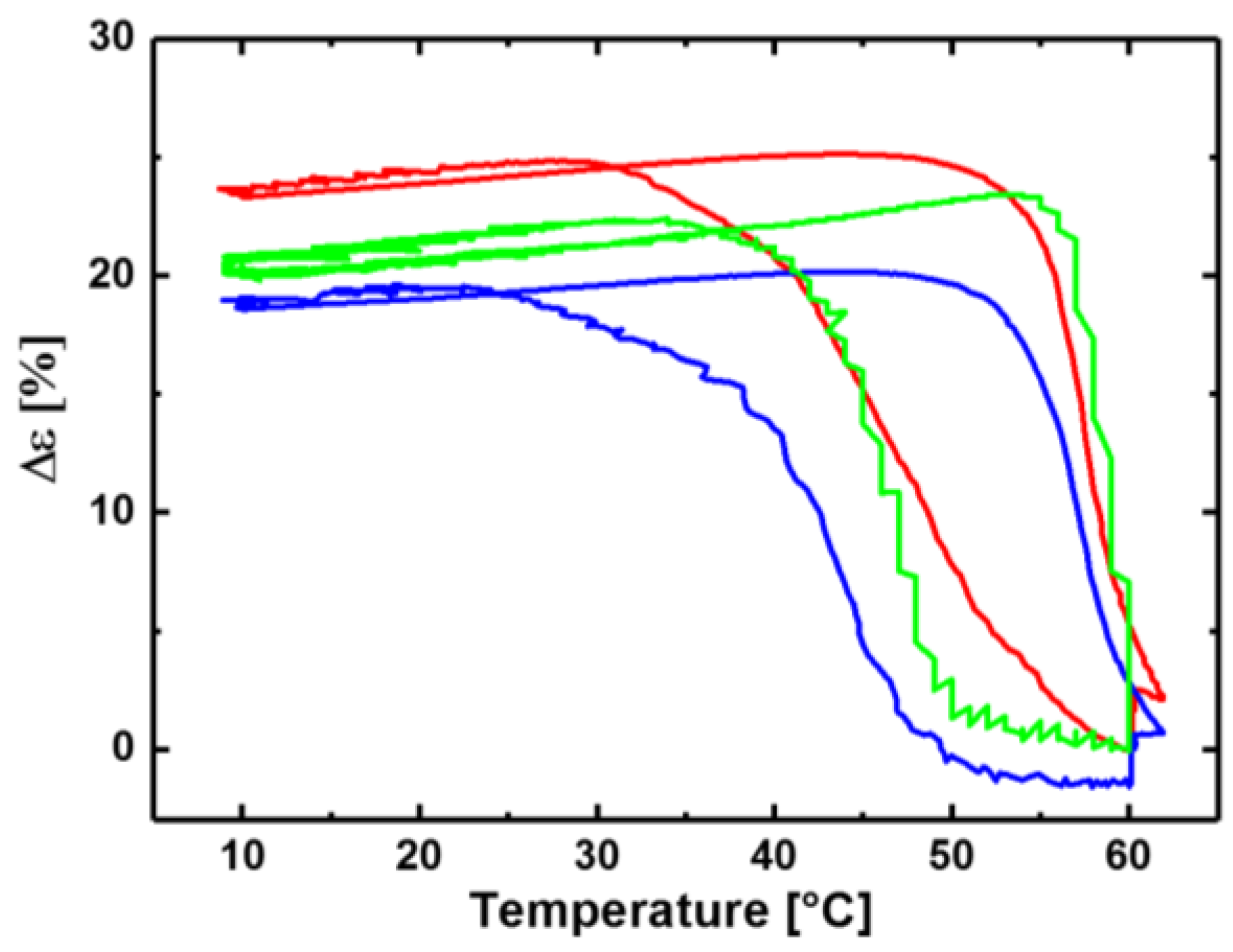
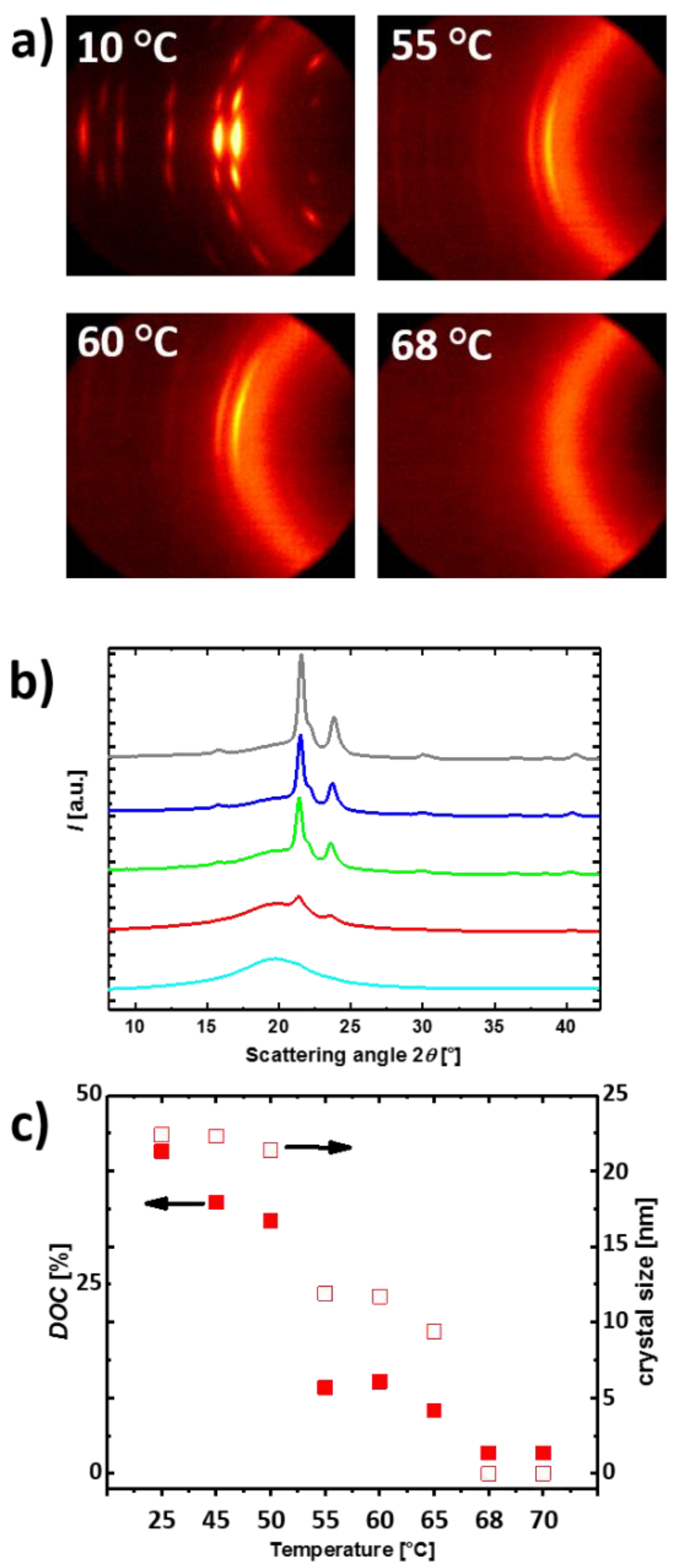
3.1. Reversible Actuation Capabilities
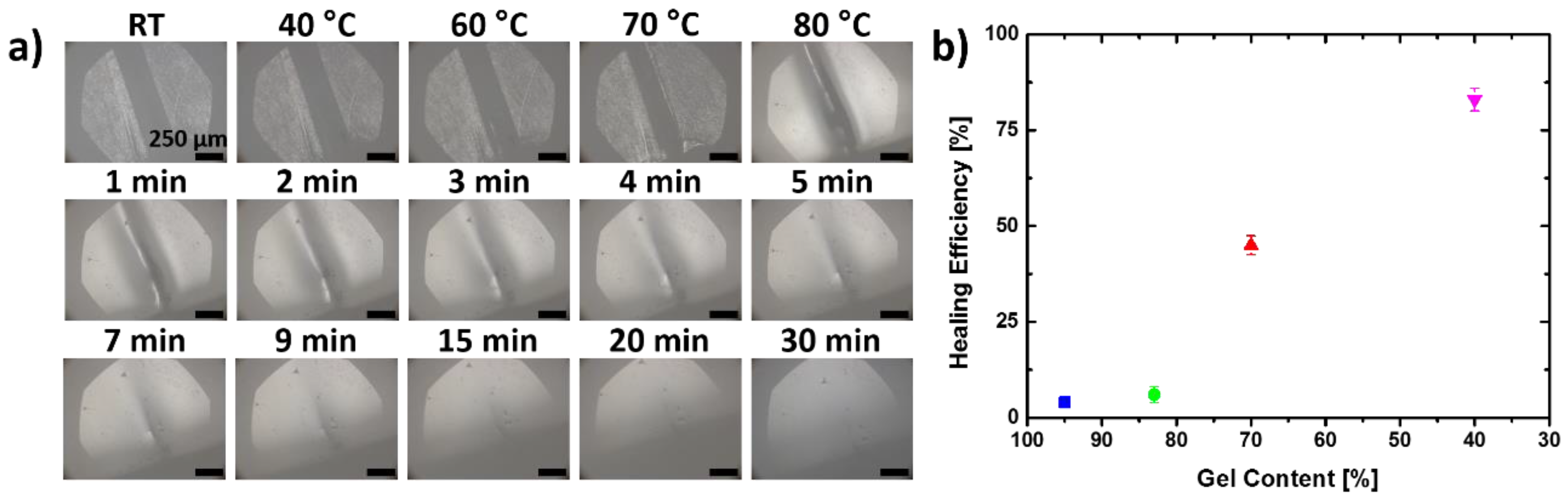
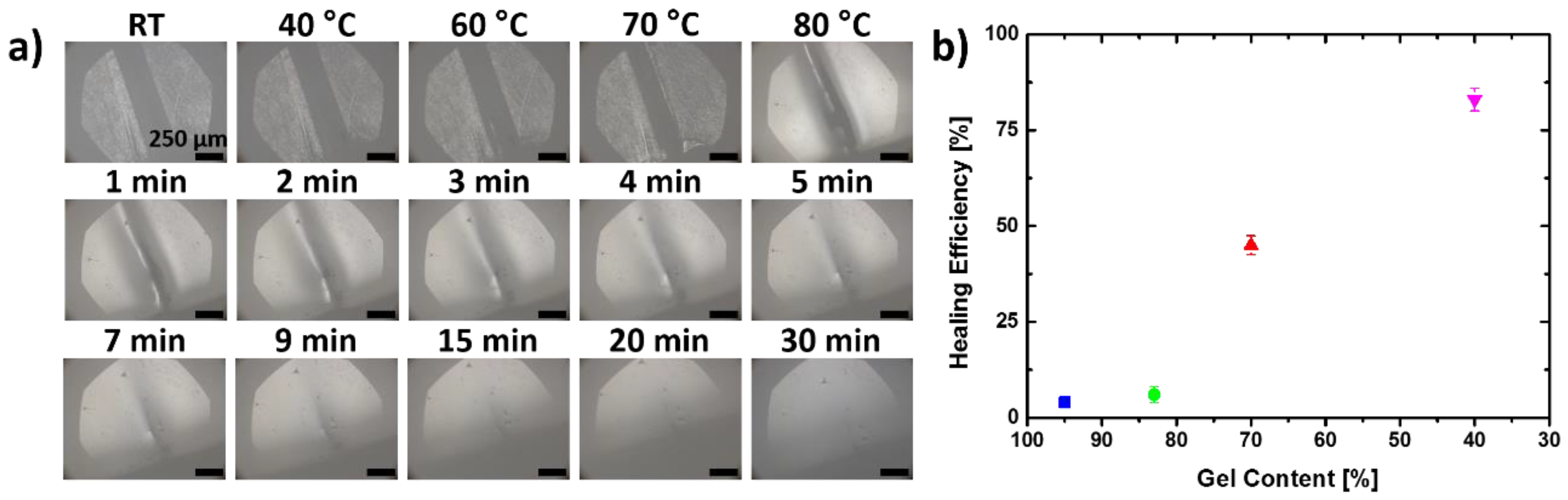
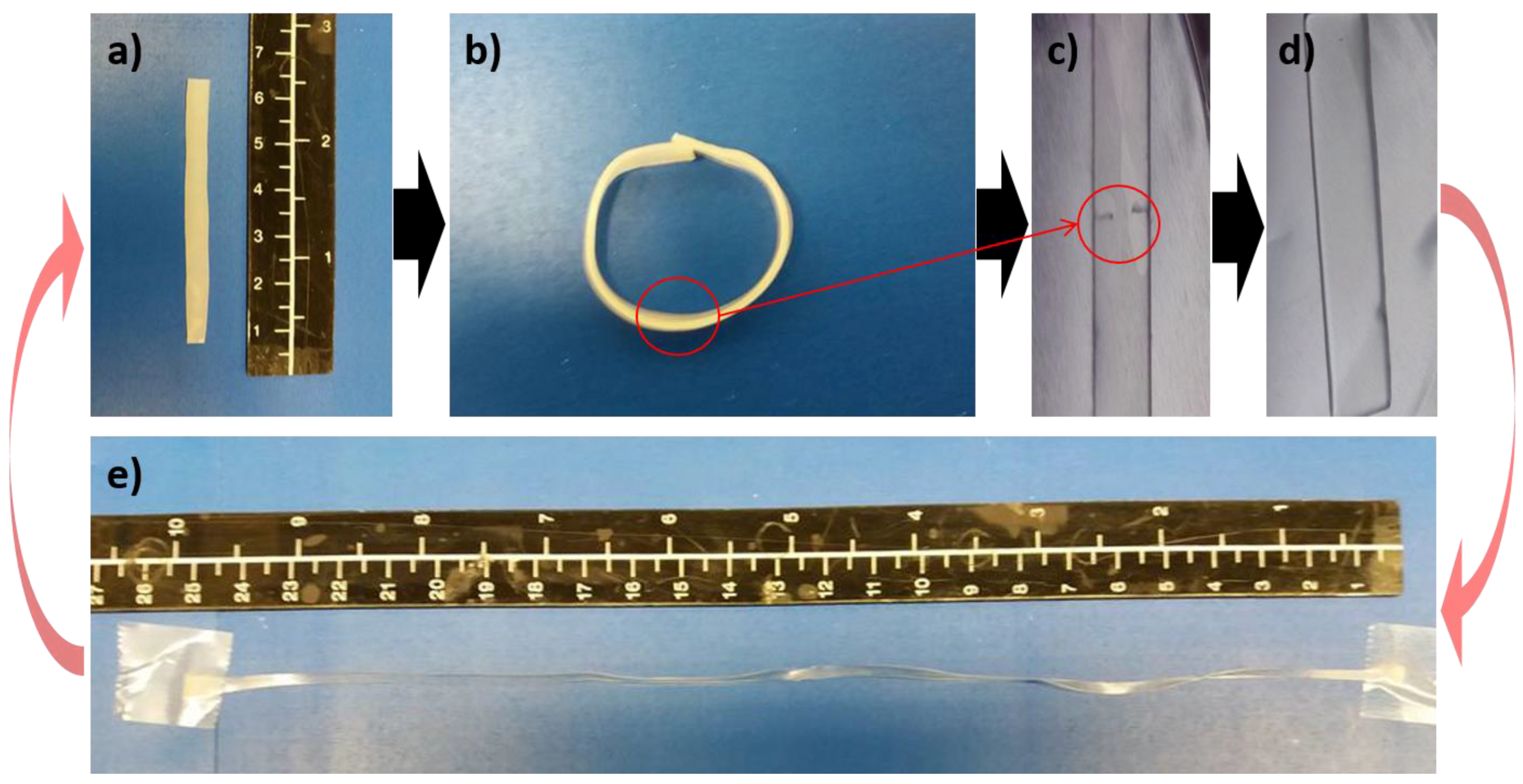
3.2. Self-Healing Capabilities
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tillet, G.; Boutevin, B.; Ameduri, B. Chemical reactions of polymer crosslinking and post-crosslinking at room and medium temperature. Prog. Polym. Sci. 2011, 36, 191–217. [Google Scholar] [CrossRef]
- Rudolph, T.; Schacher, F.H. Selective crosslinking or addressing of individual domains within block copolymer nanostructures. Eur. Polym. J. 2016, 80, 317–331. [Google Scholar] [CrossRef]
- Akiba, M.; Hashim, A.S. Vulcanization and crosslinking in elastomers. Prog. Polym. Sci. 1997, 22, 475–521. [Google Scholar] [CrossRef]
- Bhattacharya, A. Radiation and industrial polymers. Prog. Polym. Sci. 2000, 25, 371–401. [Google Scholar] [CrossRef]
- Sperling, L.H. Interpenetrating polymer networks and related materials. J. Polym. Sci. Macromol. Rev. 1977, 12, 141–180. [Google Scholar] [CrossRef]
- Sperling, L.H. Interpenetrating polymer networks: An overview. In Interpenetrating Polymer Networks; American Chemical Society: Washington, DC, USA, 1994; Volume 239, pp. 3–38. [Google Scholar]
- Sperling, L.H.; Mishra, V. The current status of interpenetrating polymer networks. Polym. Adv. Technol. 1996, 7, 197–208. [Google Scholar] [CrossRef]
- Dolynchuk, O.; Kolesov, I.; Jehnichen, D.; Reuter, U.; Radusch, H.-J.; Sommer, J.-U. Reversible shape-memory effect in cross-linked linear poly(ε-caprolactone) under stress and stress-free conditions. Macromolecules 2017, 50, 3841–3854. [Google Scholar] [CrossRef]
- Hearon, K.; Nash, L.D.; Volk, B.L.; Ware, T.; Lewicki, J.P.; Voit, W.E.; Wilson, T.S.; Maitland, D.J. Electron beam crosslinked polyurethane shape memory polymers with tunable mechanical properties. Macromol. Chem. Phys. 2013, 214, 1258–1272. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhu, W.; Zhang, X.; Zhao, C.; Xu, M. Shape memory effect of ethylene–vinyl acetate copolymers. J. Appl. Polym. Sci. 1999, 71, 1063–1070. [Google Scholar] [CrossRef]
- Rodriguez, E.D.; Luo, X.; Mather, P.T. Linear/network poly(ε-caprolactone) blends exhibiting shape memory assisted self-healing (smash). ACS Appl. Mater. Interfaces 2011, 3, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Wang, X.; Fu, C.; Ran, X. Dual-shape memory effect in radiation crosslinked thermoplastic blends: Fabrication, optimization and mechanisms. RSC Adv. 2015, 5, 61601–61611. [Google Scholar] [CrossRef]
- Voit, W.; Ware, T.; Gall, K. Radiation crosslinked shape-memory polymers. Polymer 2010, 51, 3551–3559. [Google Scholar] [CrossRef]
- Xie, F.; Huang, L.; Leng, J.; Liu, Y. Thermoset shape memory polymers and their composites. J. Intell. Mater. Syst. Struct. 2016, 27, 2433–2455. [Google Scholar] [CrossRef]
- Yang, P.; Zhu, G.; Shen, X.; Yan, X.; Nie, J. Poly(ε-caprolactone)-based shape memory polymers crosslinked by polyhedral oligomeric silsesquioxane. RSC Adv. 2016, 6, 90212–90219. [Google Scholar] [CrossRef]
- Zharinova, E.; Heuchel, M.; Weigel, T.; Gerber, D.; Kratz, K.; Lendlein, A. Water-blown polyurethane foams showing a reversible shape-memory effect. Polymers 2016, 8, 412. [Google Scholar] [CrossRef]
- Zhu, G.; Liang, G.; Xu, Q.; Yu, Q. Shape-memory effects of radiation crosslinked poly(ϵ-caprolactone). J. Appl. Polym. Sci. 2003, 90, 1589–1595. [Google Scholar] [CrossRef]
- Rettler, E.F.J.; Rudolph, T.; Hanisch, A.; Hoeppener, S.; Retsch, M.; Schubert, U.S.; Schacher, F.H. Uv-induced crosslinking of the polybutadiene domains in lamellar polystyrene-block-polybutadiene block copolymer films—An in-depth study. Polymer 2012, 53, 5641–5648. [Google Scholar] [CrossRef]
- Zhao, X. A theory for large deformation and damage of interpenetrating polymer networks. J. Mech. Phys. Solids 2012, 60, 319–332. [Google Scholar] [CrossRef]
- Stachurski, Z.H. Deformation mechanisms and yield strength in amorphous polymers. Prog. Polym. Sci. 1997, 22, 407–474. [Google Scholar] [CrossRef]
- Behl, M.; Kratz, K.; Noechel, U.; Sauter, T.; Lendlein, A. Temperature-memory polymer actuators. Proc. Natl. Acad. Sci. USA 2013, 110, 12555–12559. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.J.; Harrison, K.L. The effect of molecular weight on the crystallization kinetics of polycaprolactone. Polym. Adv. Technol. 2006, 17, 474–478. [Google Scholar] [CrossRef]
- Kristofer, K.W.; Patrick, T.M.; Vikas, P.; Martin, L.D.; Qi, G.; Brendan, M.L.; Qi, H.J. Two-way reversible shape memory effects in a free-standing polymer composite. Smart Mater. Struct. 2011, 20, 065010. [Google Scholar]
- Chung, T.; Romo-Uribe, A.; Mather, P.T. Two-way reversible shape memory in a semicrystalline network. Macromolecules 2008, 41, 184–192. [Google Scholar] [CrossRef]
- Hu, X.; Zhou, J.; Vatankhah-Varnosfaderani, M.; Daniel, W.F.M.; Li, Q.; Zhushma, A.P.; Dobrynin, A.V.; Sheiko, S.S. Programming temporal shapeshifting. Nat. Commun. 2016, 7, 12919. [Google Scholar] [CrossRef] [PubMed]
- Lendlein, A.; Kelch, S. Shape-memory polymers. Angew. Chem. Int. Ed. 2002, 41, 2034–2057. [Google Scholar] [CrossRef]
- Xie, T. Recent advances in polymer shape memory. Polymer 2011, 52, 4985–5000. [Google Scholar] [CrossRef]
- Farhan, M.; Rudolph, T.; Nöchel, U.; Yan, W.; Kratz, K.; Lendlein, A. Noncontinuously responding polymeric actuators. ACS Appl. Mater. Interfaces 2017, 9, 33559–33564. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Kratz, K.; Lendlein, A. Shape-memory properties of degradable electrospun scaffolds based on hollow microfibers. Polym. Adv. Technol. 2015, 26, 1468–1475. [Google Scholar] [CrossRef]
- Sisson, A.L.; Ekinci, D.; Lendlein, A. The contemporary role of ε-caprolactone chemistry to create advanced polymer architectures. Polymer 2013, 54, 4333–4350. [Google Scholar] [CrossRef]
- Lendlein, A.; Schmidt, A.M.; Langer, R. Ab-polymer networks based on oligo(epsilon-caprolactone) segments showing shape-memory properties. Proc. Natl. Acad. Sci. USA 2001, 98, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Ujhelyiova, A.; Marcincin, A.; Legen, J. DSC analysis of polypropylene-low density polyethylene blend fibres. Fibres Text. East. Eur. 2005, 13, 129–132. [Google Scholar]
- Kolesov, I.S.; Radusch, H.J. Multiple shape-memory behavior and thermal-mechanical properties of peroxide cross-linked blends of linear and short-chain branched polyethylenes. eXPRESS Polym. Lett. 2008, 2, 461–473. [Google Scholar] [CrossRef]
- Iyer, A.R.; Samuelson, J.J.; Barone, G.F.; Campbell, S.W.; Bhethanabotla, V.R. Sorption of benzene, dichloroethane, dichloromethane, and chloroform by poly(ethylene glycol), polycaprolactone, and their copolymers at 298.15 k using a quartz crystal microbalance. J. Chem. Eng. Data 2017, 62, 2755–2760. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer Network (a) | Tm (b) (°C) | ∆Hm (c) (J·g−1) | χc (d) (%) | E (e) (MPa) | εb (f) (%) | E (g) (MPa) | εb (h) (%) | G (i) (%) |
|---|---|---|---|---|---|---|---|---|
| PCL-L100 | 60 ± 1 | 65 ± 1 | 48 ± 1 | - (j) | - (j) | 152 ± 10 | 790 ± 10 | - (j) |
| cPCL40-L60 | 59 ± 1 | 63 ± 1 | 47 ± 1 | 0.06 ± 0.01 | 1210 ± 20 | 175 ± 10 | 910 ± 20 | 40 ± 2 |
| cPCL70-L30 | 59 ± 1 | 63 ± 1 | 47 ± 1 | 1.15 ± 0.1 | 790 ± 30 | 210 ± 6 | 715 ± 35 | 70 ± 3 |
| cPCL83-L17 | 58 ± 1 | 63 ± 1 | 47 ± 1 | 1.61 ± 0.06 | 310 ± 20 | 226 ± 13 | 460 ± 30 | 85 ± 3 |
| cPCL95-L05 | 53 ± 1 | 61 ± 1 | 45 ± 1 | 1.78 ± 0.12 | 460 ± 15 | 257 ± 7 | 550 ± 15 | 95 ± 1 |
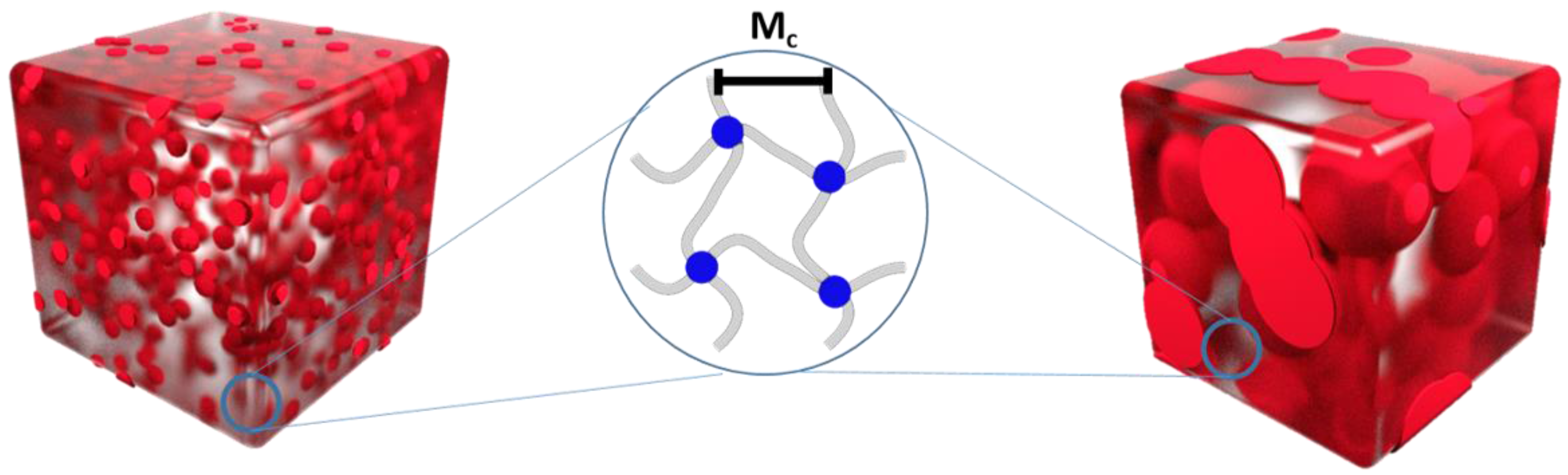
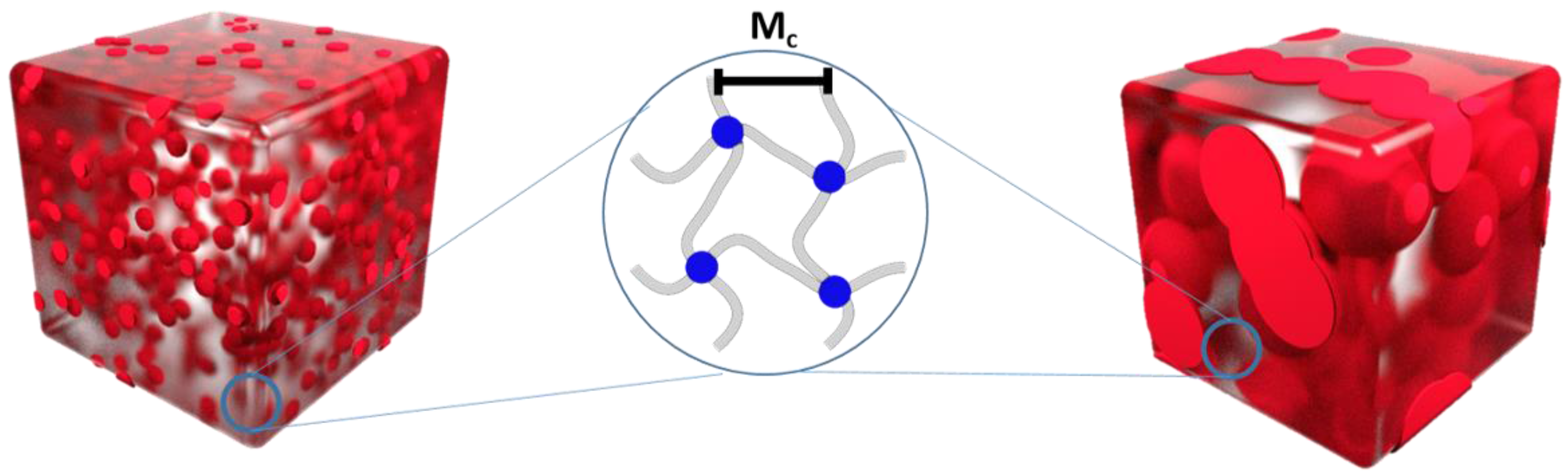
| Polymer Network (a) | υc (b) (mol·cm−3) (Mooney-Rivlin) | Mc (c) (g·mol−1) (Mooney-Rivlin) | υc (d) (mol·cm−3) (Flory-Rehner) | Mc (e) (g·mol−1) (Flory-Rehner) |
|---|---|---|---|---|
| cPCL40-L60 | 1.83·10−5 | 1.71·105 | n.d. | n.d. |
| cPCL70-L30 | 1.09·10−4 | 1.05·104 | n.d. | n.d. |
| cPCL83-L17 | 2.22·10−4 | 5.14·103 | n.d. | n.d. |
| cPCL95-L05 | 2.32·10−4 | 4.92·103 | n.d. | n.d. |
| cPCL70 | 1.03·10−4 | 1.11·104 | 1.68·10−4 | 6.77·103 |
| cPCL83 | 1.60·10−4 | 7.11·103 | 2.39·10−4 | 4.76·103 |
| cPCL95 | 2.14·10−4 | 5.33·103 | 2.72·10−4 | 4.19·103 |
| Polymer Network (a) | Tsw,act (b) (°C) | Tsw,rec (c) (°C) | Δε (d) (%) |
|---|---|---|---|
| cPCL40-L60 | - | - | 3 ± 1 |
| cPCL70-L30 | 49 ± 1 | 58 ± 1 | 24 ± 1 |
| cPCL83-L17 | 47 ± 1 | 59 ± 1 | 21 ± 2 |
| cPCL95-L05 | 43 ± 1 | 57 ± 1 | 20 ± 1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farhan, M.; Rudolph, T.; Nöchel, U.; Kratz, K.; Lendlein, A. Extractable Free Polymer Chains Enhance Actuation Performance of Crystallizable Poly(ε-caprolactone) Networks and Enable Self-Healing. Polymers 2018, 10, 255. https://doi.org/10.3390/polym10030255
Farhan M, Rudolph T, Nöchel U, Kratz K, Lendlein A. Extractable Free Polymer Chains Enhance Actuation Performance of Crystallizable Poly(ε-caprolactone) Networks and Enable Self-Healing. Polymers. 2018; 10(3):255. https://doi.org/10.3390/polym10030255
Chicago/Turabian StyleFarhan, Muhammad, Tobias Rudolph, Ulrich Nöchel, Karl Kratz, and Andreas Lendlein. 2018. "Extractable Free Polymer Chains Enhance Actuation Performance of Crystallizable Poly(ε-caprolactone) Networks and Enable Self-Healing" Polymers 10, no. 3: 255. https://doi.org/10.3390/polym10030255