Efficient Shielding of Polyplexes Using Heterotelechelic Polysarcosines
by
 and
and
Philipp Michael Klein
1,*, 
Kristina Klinker
2,3,
Wei Zhang
1,
Sarah Kern
1,
Eva Kessel
1,
Ernst Wagner
1,4 and
Matthias Barz
2,* 1
Department of Pharmacy, Ludwig-Maximilians-Universität (LMU) Munich, Pharmaceutical Biotechnology, Butenandtstrasse 5-13, D-81377 Munich, Germany
2
Institute of Organic Chemistry, Johannes Gutenberg University, Duesbergweg 10-14, D-55128 Mainz, Germany
3
Graduate School Materials Science in Mainz, Staudinger Weg 9, 55128 Mainz, Germany
4
Nanosystems Initiative Munich, Schellingstraße 4, D-80799 Munich, Germany
*
Authors to whom correspondence should be addressed.
Polymers 2018, 10(6), 689; https://doi.org/10.3390/polym10060689
Submission received: 28 May 2018
/
Revised: 13 June 2018
/
Accepted: 17 June 2018
/
Published: 20 June 2018
(This article belongs to the Special Issue Hydrophilic Polymers)
Abstract
:Shielding agents are commonly used to shield polyelectrolyte complexes, e.g., polyplexes, from agglomeration and precipitation in complex media like blood, and thus enhance their in vivo circulation times. Since up to now primarily poly(ethylene glycol) (PEG) has been investigated to shield non-viral carriers for systemic delivery, we report on the use of polysarcosine (pSar) as a potential alternative for steric stabilization. A redox-sensitive, cationizable lipo-oligomer structure (containing two cholanic acids attached via a bioreducible disulfide linker to an oligoaminoamide backbone in T-shape configuration) was equipped with azide-functionality by solid phase supported synthesis. After mixing with small interfering RNA (siRNA), lipopolyplexes formed spontaneously and were further surface-functionalized with polysarcosines. Polysarcosine was synthesized by living controlled ring-opening polymerization using an azide-reactive dibenzo-aza-cyclooctyne-amine as an initiator. The shielding ability of the resulting formulations was investigated with biophysical assays and by near-infrared fluorescence bioimaging in mice. The modification of ~100 nm lipopolyplexes was only slightly increased upon functionalization. Cellular uptake into cells was strongly reduced by the pSar shielding. Moreover, polysarcosine-shielded polyplexes showed enhanced blood circulation times in bioimaging studies compared to unshielded polyplexes and similar to PEG-shielded polyplexes. Therefore, polysarcosine is a promising alternative for the shielding of non-viral, lipo-cationic polyplexes.
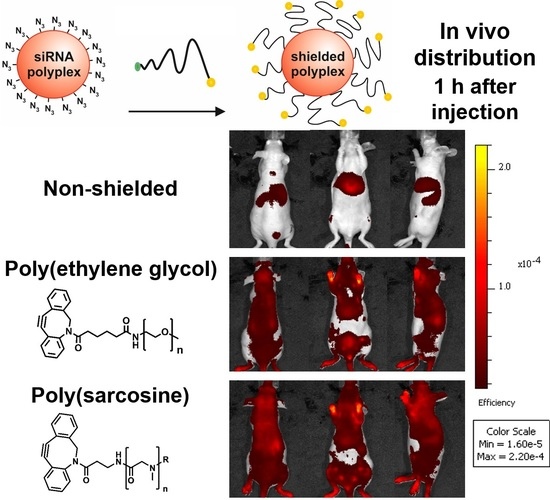
1. Introduction
Therapeutic nucleic acids are powerful tools, which can be used to specifically control gene expression inside cells [1,2,3,4,5]. For several diseases, including severe metastatic tumors, systemic delivery is required to achieve therapeutic effects. Naked oligonucleotides have limited stability in biological fluids because they are actively targeted and consequently, degraded by nucleases. Although this issue might be addressed by chemical modifications [6,7], the renal clearance of small oligonucleotides or siRNA usually occurs within a few minutes, which limits the time to reach their desired site of action [8,9]. Lipopolymer micelles, liposome-based formulations, and polymer-based complexes increase the size usually beyond the renal cut-off and thus enhance circulation times, whenever a stealth-like corona protects the systems from unspecific aggregation [6,10,11,12,13,14,15,16,17,18,19,20,21]. Various cationic lipids, polycations and combinations with helper lipids [4,14,16,17,22,23,24] were used to form lipoplexes or lipid nanoparticles (LNPs) [1,11,25,26,27,28,29,30]. Precise editing of the compounds’ chemical structure enables the fine-tuning of a carrier’s stability and size, but also other properties, which are important for the delivery process like cellular uptake, endosomal escape ability, and cell tolerability [31,32,33,34].
Solid phase-supported synthesis (SPS) is a very convenient and precise way to optimize a delivery system in a step-wise manner [18,35]. Recently, we developed our own customized amino acids, such as succinoyl tetraethylene pentamine (Stp), which contains of short defined repeats of the ethylenediamine motif prepared in boc/fmoc protected form. With artificial building blocks, natural α-amino acids and fatty acids, we sequentially synthesized monodisperse cationic oligomers via SPS, which are highly adaptable to different demands in the field of gene delivery [8,12,18,31,32,33,36,37,38,39]. By precise incorporation of a bioreducible cleavage site between the cationic and a lipophilic block, for instance, it was possible to destabilize polyplexes only after reaching the cytoplasm of the cell [39]. Thereby the carrier system remained stable in serum and transfection efficiency as well as cell viability could be increased in certain cell lines.
Besides size and stability, the surface character of a nanoparticle is of utmost importance for its systemic delivery. Shielding agents attached to the surface prevent interactions with neighboring particles and/or blood components, which usually leads to extended circulation in the body’s bloodstream [40,41,42]. Already in 1990, it could be demonstrated that polyethylene glycol (PEG) could extend the blood circulation half-life of systemically injected liposomes from <30 min to several hours [40]. Its hydrophilic character enables PEG to generate a hydrated shell covering the nanoparticles and thereby sterically reduce unwanted interactions with biomolecules or other poly- or lipoplexes [43]. PEG is the most prominent shielding agent and has often been used to shield cationic polyplexes in numerous applications [44,45,46,47,48,49]. A major drawback, however, is that more and more researchers in academia or industry observe immune responses towards PEGylated nanoparticles [43,50,51,52,53,54,55]. For this reason, several new alternatives were evaluated for shielding, such as natural proteins [56], oligosaccharides [57,58], poly(N-(2-hydroxypropyl)methacrylamide) (pHPMA) [58,59,60], hydroxyethyl starch (HES) [61] or polypeptides (poly(glutamic acid) [62], poly(hydroxyethyl-l-asparagine) [63], poly(hydroxyethyl-l-glutamine) [63], proline-alanine-serine motif (PAS) [64,65]). Nevertheless, according to the Whitesides’ rules for protein resistant surfaces an ideal alternative to PEG should mimic its chemical properties, being a hydrophilic, non-charged polymer and a weak hydrogen acceptor without donor properties, which is not the case for all above-mentioned polymers. In contrast, polysarcosine fulfills all the described criteria and has already demonstrated protein resistant properties on various surfaces [66,67,68]. In addition, it can be also synthesized by living controlled ring-opening polymerization of the corresponding N-carboxyanhydrides (NCAs) [69,70]. However, in vivo data on polysarcosine is rarely reported in literature [71]. In contrast to polypeptides, the side chain of polypeptoids is situated at the nitrogen rather than the α-carbon, in the case of pSar the nitrogen is methylated. As a result, polysarcosine adopts a random coil conformation in aqueous solution and possesses a comparable second virial coefficient and molecular weight dependency like PEG [72]. All these properties mediate high resistance against protein adsorption [73] and make it in theory an ideal material for shielding electrostatic complexes in vivo [74]. Importantly, it has been reported that polysarcosine has so far demonstrated neglectable complement activation or immunogenicity in mouse, rat and rabbit animal models [75,76]. pSar-shielded polyplexes, micelles, colloids and nanohydrogels demonstrated the absence of aggregation in human serum [77,78,79,80].
In the current work, we have incorporated azide domains into a previously described redox-sensitive T-shaped bis-(cholanic acid amido) oligoaminoamide siRNA carrier system [39] and used strain-promoted azide-alkyne cycloaddition (SPAAC) reaction to equip the lipopolyplexes’ surface with ~8 kDa polysarcosine (DP = 119) chains. We report on the ability of polysarcosine to shield siRNA lipoplexes and analyzed the in vivo stability and biodistribution after intravenous administration into mice. In a second approach, we modified the system with a folate ligand to target the folate receptor overexpressed on certain cancer cells [81,82,83,84,85,86].
2. Materials and Methods
2.1. Materials
Protected Fmoc-α-amino acids, N,N-dimethylformamide (DMF), N,N-diisopropylethylamine (DIPEA), trifluoroacetic acid (TFA) and 2-chlorotrityl chloride resin for solid-phase syntheses were purchased from Iris Biotech (Marktredewitz, Germany). 1-hydroxybenzotriazole (HOBt), triisopropylsilane (TIS), 5β cholanic acid. Dimethylformamide (DMF, 99.8% Extra) for Dibenzoazacyclooctyneamine polysarcosine (DBCO-pSar) syntheses was purchased from Acros Organics (Nidderau, Germany), further dried over CaH2 and fractionally distilled in vacuo. Tetrahydrofuran (THF) and folic acid (FolA, 96–102% pure) was purchased from Acros Organics. Triethylamine (TEA) and N,N-diisopropylethylamine (DIPEA) were dried over NaOH and fractionally distilled in vacuo. (Benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluoro-phosphate (PyBOP), 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorphosphate (HBTU) and microreactors were purchased from MultiSynTech (Witten, Germany). Cell culture media, antibiotics and fetal bovine serum (FBS) were obtained from Invitrogen (Karlsruhe, Germany), HEPES from Biomol GmbH (Hamburg, Germany), agarose (NEEO Ultra-quality) from Carl Roth GmbH (Karlsruhe, Germany), glucose from Merck (Darmstadt, Germany), and GelRed™ from VWR (Darmstadt, Germany). d-Luciferin sodium salt and cell culture 5 × lysis buffer were purchased from Promega (Mannheim, Germany). Ready-to-use siRNA duplexes were purchased from Axolabs GmbH (Kulmbach, Germany): eGFP-targeting siRNA (siGFP) (sense: 5′-AuAucAuGGccGAcAA GcAdTsdT-3′; antisense: 5′-UGCUUGUCGGCcAUGAuAUdTsdT-3′) for silencing of eGFPLuc; control siRNA (siCtrl) (sense: 5′-AuGuAuuGGccuGuAuuAGdTsdT-3′; antisense: 5′-CuAAuAc AGGCcAAuAcAUdTsdT-3′); Cy5-labled siRNA (Cy5-siAHA1) (sense: 5′-(Cy5)(NHC6)GGAu GAAGuGGAGAuuAGudTsdT-3′; antisense: 5′-ACuAAUCUCcACUUcA- UCCdTsdT-3′); Cy7-labled siRNA (Cy7-siAHA1) (sense: 5′-(Cy7)(NHC6)GGAuGAAGuGGAG- AuuAGudTsdT-3′; antisense: 5′-ACuAAUCUCcACUUcAUCCdTsdT-3′) small letters: 2’methoxylated; s: phosphorothioate. All other chemicals were obtained from Sigma (Munich, Germany), Merck, Iris Biotech (Marktredwitz, Germany) or AppliChem (Darmstadt, Germany), Acros Organics, Fluka (Munich, Germany) or Alfa Aesar (Karlsruhe, Germany).
2.2. Synthesis of Oligomers and DBCO Shielding Agents
2.2.1. Synthesis of Oligomers
See Supporting Information for detailed descriptions of the syntheses of oligomers.
2.2.2. Synthesis of Sarcosine-N-Carboxyanhydride
The synthesis was performed as described in Klinker et al. [71] Sarcosine (15.16 g, 170.2 mmol, 1 eq) was weighed into a pre-dried three-necked flask and dried under vacuum for one hour. Absolute (abs) tetrahydro furane (THF) (300 mL) was added under a steady nitrogen flow. Two gas washing bottles with aqueous sodium hydroxide solution were connected to the apparatus. Diphosgene (16.26 mL, 134 mmol, 0.8 eq) was added dropwise via syringe. The colorless suspension was heated to 70 °C resulting in a clear solution after three hours of stirring. The solvent was evaporated under reduced pressure resulting in a brown oil as crude reaction product. The obtained oil was heated to 50 °C and dried under reduced pressure yielding an amorphous solid. The solid was redissolved in 60 mL THF and precipitated by adding 300 mL abs n-hexane. The precipitate was filtered off under nitrogen-atmosphere and dried with a stream of dry N2 for 60–90 min to remove remaining traces of solvents. The next day, the solid was dried in high vacuum for 2 h in the sublimation apparatus followed by sublimation at 80–85 °C and <1 × 10−2 mbar. The product was collected from the sublimation apparatus in a glove box the same day. Colorless crystals were obtained (50–67%). mp = 104.3 °C; 1H-NMR (300 MHz, CDCl3) δ [ppm] = 2.86 (s, 3H, N−CH3), 4.22 (s, 2H, N−CH2CO).
2.2.3. Synthesis of DBCO-pSar
DBCO-pSar was synthesized using ring-opening polymerization of SarNCA as described in Klinker et al. [71] In a typical experiment, SarNCA (461.8 mg, 4.012 mmol) were transferred under N2 counter flow into a pre-dried Schlenk-tube, equipped with a stir bar and dried again in vacuum for 30 min. The NCA was dissolved in dry DMF (3.5 mL). A stock solution of DBCO-amine (0.074 mmol, 1/110 eq, M/I = 110) in DMF (2 mL) was freshly prepared and 1 mL of this stock solution was added to the monomer solution with syringe. The solution was stirred at a temperature of 40 °C and kept at a constant pressure of 1.25 bar of dry N2 via the Schlenk-line to prevent impurities from entering into the reaction vessel while allowing CO2 to escape. Completion of the reaction was confirmed by infrared spectroscopy (disappearance of the NCA peaks (1853 and 1786 cm−1)). Directly after completion, the polymer was precipitated in ice-cold diethyl ether and centrifuged (4500 rpm at 4 °C for 15 min). After discarding the liquid fraction, new diethyl ether was added and the polymer was resuspended via sonication. Again, the suspension was centrifuged and the procedure was repeated. The polymer was subsequently dissolved in water and lyophilized to obtain a fluffy powder (279.7 mg, 98%). 1H-NMR: (400 MHz, DMSO−d6): δ [ppm] = 0.88–0.79 (m, ini, 9H, –C(CH3)3), 2.58–3.11 (br, 3nH, N–CH3), 3.73–4.57 (br, 2nH, –CO–CH2–N), = 7.86–7.10 (m, 8H, benzylic protons).
2.2.4. Synthesis of DBCO-pSar-Ac
DBCO-pSar119 (Mn = 8735 g mol−1) (36 mg, 0.004 mmol), acetic anhydride (4.2 mg, 3.9 µL, 0.04 mmol), and triethylamine (10.7 mg, 14.6 µL, 0.08 mmol) were dissolved in absolute DMF (1 mL) and stirred at 25 °C for 24 h under an argon atmosphere. Subsequently, the polymer was precipitated in diethyl ether, extensively dialyzed against water (MWCO = 3500 g mol−1), and lyophilized. Yield after dialysis: 25 mg (69%).
2.2.5. Synthesis of DBCO-pSar-FolA
DBCO-pSar110 (Mn = 8095 g mol−1) (55.8 mg, 0.007 mmol) was separately dissolved in absolute 5mL dimethylsulfoxid (DMSO). Folic acid (30.4 mg, 0.068 mmol), HBTU (26.1 mg, 0.068 mmol), and HOBt (9.31 mg, 0.068 mmol) were dissolved in 5mL DMSO and cooled to 0 °C. DIPEA (17.8 mg, 24.0 µL, 0.138 mmol) was added and the mixture was left to react for 30 min at 0 °C. The in situ formed activated ester was added to the predissolved polymer and the reaction mixture was stirred at 25 °C for 24 h under an argon atmosphere. The crude reaction product was purified by size exclusion chromatography in DMSO using a Sephadex LH-20-packed column. The purified conjugate was lyophilized from water. Yield after SEC: 31 mg (55%).
2.2.6. Gel Permeation Chromatography
Polymer molecular weight and dispersity index were determined using gel permeation chromatography (GPC). GPC in hexafluoro-2-propanol (HFIP) was performed with 3 g L−1 potassium trifluoroacetate (KTFA) at a temperature of 40 °C. The columns were packed with modified silica (PFG columns, particle size: 7 µm; porosity: 100 and 1000 Å). A UV/vis detector (UV-2075 Plus, JASCO, Groß-Umstadt, Germany, λ = 230 nm; λ = 330 nm for folic acid detection) and a refractive index detector (G 1362A RID, Jasco) were used to detect the polymer. Molecular weights were calculated by using calibration performed with PMMA standards (Polymer Standards Services GmbH, Mainz, Germany) Toluene was used as the internal standard.
2.2.7. UV-Vis Spectroscopy
UV-vis absorbance spectra were recorded using a V-630 spectrophotometer (Jasco) with water being the solvent.
2.3. Formation of siRNA Polyplexes
siRNA was diluted with 20 mM HEPES buffered 5% glucose pH 7.4 (HBG) to a concentration of 50 ng/µL for in vitro experiments and 500 ng/µL for in vivo experiments. According to the indicated nitrogen/phosphate (N/P) ratio, the oligomer solution was prepared in a separate tube. Only protonatable nitrogens of an oligomer were considered in the N/P calculation. The same volume of siRNA solution was added to the oligomer. The mixture was rapidly pipetted at least five times and incubated for 40 min at RT yielding a polyplex solution with 25 or 250 ng of siRNA/µL respectively.
2.4. Functionalization of Polyplexes with DBCO Reagents
For functionalization of siRNA polyplexes with DBCO click agents, solutions with DBCO reagents were prepared in ¼ of the volume of the previously prepared polyplex solutions. The concentration of the solution was calculated according to the indicated equivalents (eq) used. Equivalents represent the molar ratio of shielding agent to cationic oligomer in the polyplex solution. The reaction time was 16 h for biophysical and in vitro assays and 4 h for in vivo experiment respectively.
2.5. siRNA Binding Assays
siRNA binding assays were performed analogously as described in Klein et al. [86]. A 1% agarose gel was prepared by dissolving agarose in TBE buffer (10.8 g of Trizma base, 5.5 g of boric acid, 0.75 g of disodium EDTA, and 1 L of water) and subsequent boiling. After cooling down to about 50 °C, GelRedTM was added. Formulations were prepared with 500 ng of siRNA per sample. Samples were placed into the pockets after 4 µL of loading buffer (prepared from 6 mL of glycerine, 1.2 mL of 0.5 M EDTA, 2.8 mL of H2O, 0.02 g of bromophenol blue) was added. Electrophoresis was performed at 70 V for 60 min.
2.6. Particle Size and Zeta Potential Measurements
Polyplexes were formed using 2 µg siRNA in a total volume of 80 µL. Dynamic light scattering (DLS) measurements of polyplex solutions were performed in a folded capillary cell (DTS 1070). A Zetasizer Nano ZS with backscatter detection (Malvern Instruments, Worcestershire, UK) was used for the measurements. For size measurements, the equilibration time was 0 min, the temperature was set to 25 °C and an automatic adjustment of the attenuator was used. The refractive index of the solvent was set to 1.330 and the viscosity was set to 0.8872 mPa·s. Each sample was measured three times. For detection of the zeta potential, the sample was diluted to 800 µL volume with 10 mM NaCl solution. Measurements with at least 6 runs were performed. Zeta potentials were calculated using the Smoluchowski equation. 10 to 15 sub runs each lasting 10 s (n = 3) were measured.
2.7. Cell Culture
The mouse neuroblastoma cells (Neuro2a) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) low glucose medium (Sigma). As FR-expressing cell lines, human cervix carcinoma cells (KB cells), and human cervix carcinoma cells transfected stably with the eGFPLuc (enhanced green fluorescent protein/luciferase) gene (KB/eGFPLuc cells) were cultured in folate-free Roswell Park Memorial Institute medium (RPMI) 1640 (Invitrogen). All media were supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. The cells were cultured in ventilated flasks in the cell incubator at 37 °C and 5% CO2 in a humidified atmosphere. After a confluency of 80–90% was reached, cells were harvested.
2.8. Cell Association and Internalization of siRNA Polyplexes Measured with Flow Cytometry
For untargeted polyplexes, Neuro2a cells were seeded in 24-well plates with 5 × 104 cells/well 24 h prior the experiment, and fresh growth medium was provided immediately before the experiment. Polyplexes containing 1.5 µg of siRNA (20% Cy5-labeled siRNA + 80% unlabeled siRNA) were pipetted into each well and incubated for four hours at 37 °C in 5% CO2. Polyplexes non-specifically associated to the cell surface were then removed by incubating cells with 500 I.U. heparin. For folate-targeted polyplexes, KB cells were seeded in 24-well plates with 5 × 104 cells perwell at 24 h before the experiment, and fresh growth medium was provided before the experiment. Polyplexes containing 1.5 µg of siRNA (20% Cy5-labeled siRNA + 80% unlabeled siRNA) were pipetted into each well, incubated 30 min on ice for cell association or 45 min at 37 °C in 5% CO2 in case of cellular internalization. Cells were then washed with PBS to remove free polyplexes. In case of testing the cellular internalization, polyplexes non-specifically associated to the cell surface were then removed by incubation with 500 I.U. heparin. Next, cells were collected and resuspended in PBS buffer containing 10% of FBS. All samples were analyzed by flow cytometry with CyanTM ADP (Dako, Hamburg, Germany) using an excitation wavelength of 635 nm, and detecting the emission at 665 nm. Dead cells were differentiated by fluorescence of DAPI and removed by gating in order to only analyze cellular uptake of polyplexes in living cells. Flow cytometry data were analyzed using FlowJo 7.6.5 flow cytometric analysis software by FlowJo, LLC (Ashland, OR, USA).
2.9. Confocal Laser Scanning Microscopy (CLSM)
Neuro2a cells were seeded at a density of 3 × 104 cells/well in 300 μL of growth medium into an 8-well Lab-Tek chamber slide (Nunc, Schwerte, Germany) 24 h prior to treatment. Polyplexes were formed as described using a 1.5 µg of a mixture of 80% of siCtrl and 20% Cy5-labeled siRNA and oligomer at N/P 12 in 60 μL of HBG followed by the indicated agent in 20 µL. Cells were incubated with 220 μL of fresh growth medium and the polyplex solution was applied. For the uptake study, the incubation with polyplexes was at 37 °C for 4 h. The growth medium was removed, cells were washed twice with 300 µL of PBS and fixed with 4% PFA solution for 30 min at RT. Cell nuclei were stained with DAPI. For image acquisition, a TCS SP8 confocal microscope (Leica, Mannheim, Germany) was used.
2.10. Mouse Tumor Model
Female six- to seven-week-old nude mice, Rj: NMRI-nu (nu/nu) (Janvier, Le Genest-Saint-Isle, France), were housed in isolated ventilated cages under pathogen-free condition with a 12 h light/dark interval and were acclimated for at least seven days prior to experiments. Water and food were provided ad libitum. Injection of animals was performed subcutaneously with 5 × 106 Neuro2a cells. The tumor volume was measured by caliper and calculated as [0.5 × (longest diameter) × (shortest diameter)2] and the body weight was recorded. All animal experiments were performed according to the terms stated in the proposal “Entwicklung von Sequenz-definierten Oligomeren als Träger für die zielgerichtete Einbringung neuer molekularer Therapeutika in Tumore“ that was approved by the local animal ethics committee and the government of Oberbayern at the 26 May 2014. All animal studies were performed according to guidelines of the German Animal Welfare Act.
2.11. Biodistribution Study
A mixture of 50% unlabeled control siRNA (siCtrl) and 50% of Cy7-labeled siRNA (Cy7-siAHA1) in HBG was used for all near infrared (NIR) in vivo imaging experiments. Tumors of mice were allowed to grow until reaching a size of 500–1000 mm3. The mice (n = 2/per group) were anesthetized with 3% isoflurane in oxygen. siRNA polyplexes containing 50 µg siRNA (50% Cy7-labeled—N/P 10) in 250 µL (prepared from 100 µL of siRNA solution, 100 µL of oligomer solution and 50 µL of agent solution or buffer) or HBG as negative control were injected intravenously (i.v.), and fluorescence intensity was measured with a CCD camera at different time points. For evaluation of images, the intensity of fluorescence signals was analyzed after color bar scales were equalized using the IVIS Lumina system and the Living Image software 3.2 (Caliper Life Sciences, Hopkinton, MA, USA).
3. Results and Discussion
3.1. Design and Synthesis of Lipo-Oligomers for Click Chemistry
Previously, we have established a novel class of redox-sensitive lipo-oligomers that were prepared by solid-phase supported synthesis (SPSS) serving as carriers for siRNA delivery [39]. Beside all beneficial effects of lipid-based delivery systems, such as endosomal escape capability and enhanced nanoparticle stability, the in vivo distribution is often limited to certain tissues, primarily liver, lung and spleen [87,88,89]. In previous work, it has been shown that T-shape oligomers similar to the ones, which were synthesized in this approach demonstrate strongest retention in liver tissue [89,90]. This observation might be related to a high stickiness of cationic particles, but it is also possible that certain serum proteins, which incorporate onto nanoparticle surfaces may impair tissue specificity [88,91,92,93]. An efficient shielding should reduce both types of interactions and should enable a better distribution in the body. Therefore, one of the top candidates from redox-sensitive lipo-oligomers, T-shape structure T-0N3 (published as ID 992 in [39]) was chosen and extended by click-reactive azide functionality. After the formation of siRNA lipopolyplexes, the surface was further modified with the shielding agents.
T-0N3 was chosen as a lead structure and starting point for further modifications, because it forms stable siRNA polyplexes smaller than 200 nm in hydrodynamic diameter, which show high transfection efficiency in mouse neuroblastoma (Neuro2a) cells. This structure combines natural amino acids and artificial building blocks (Figure 1, top). It consists of four repeats of the artificial, cationic polyamino acid succinoyltetraethylenepentamine (Stp) used for complexation of nucleic acid and for endosomal buffering. Two trimeric tyrosine units flanking the cationic domain are able to stabilize the polyplex due to their hydrophobicity and π–π stacking ability. In between the cationic Stp units, two hydrophobic cholanic acids branch off the backbone (T-shape).
This hydrophobic domain mediates efficient lipopolyplex stabilization. The lipid and cationic domains are connected with a bioreducible linking unit (ssbb) [39]. In this approach, the azide function was incorporated into the lipo-oligomer during standard Fmoc solid phase-supported synthesis via an azidolysine residue at the N- and/or C-terminus of the backbone (structures T-1N3 with one azide and T-2N3 with two azides, Figure 1, top). Consequently, the structure can be subsequently further modified with an alkyne-bearing functional group via click chemistry.
3.2. Synthesis of DBCO-Modified Polysarcosine
Polysarcosine is a hydrophilic, nonionic peptoidic structure with exclusively weak hydrogen bond acceptor properties. As shown empirically by Whitesides and co-workers for protein-resistant surfaces, these properties are essential to achieve “stealth”-like properties in a material [71,74]. Polysarcosine can be functionalized at its N-terminal (via post-polymerization modification) and C-terminal (via functional initiators) end. It is conveniently synthesized by controlled living ring-opening polymerization of α-amino acid N-carboxyanhydrides (NCA) with low dispersity index (ÐGPC ≤ 1.1; see Table S1) [71]. Initiating the reaction with dibenzoazacyclooctyneamine (DBCO-amine) leads to a C-terminal DBCO end group (Figure S1A) [94]. To ensure end group accessibility and steric stabilization at once, we aimed for a degree of polymerization of around 115, which correlates with the PEG5k used as reference material. Therefore, a theoretical degree of polymerization of 115 was set by the monomer to initiator ratio. The polymerization was carried out under the conditions recently reported by Klinker et al. [95], NMR end group and SEC analysis (using pSar standards as described by Weber et al. [72]) revealed a number average degree of polymerization (DP) of 119 and a number average molecular weight of 8735 g mol−1 (Table S1), which is within the experimental error. The experimentally determined molecular weight is perfectly in line with the calculated one. The synthesized polymer displays a symmetrical SEC elugram indicating a Poisson-like molecular weight distribution, as expected for the amine initiated N-substituted glycine N-carboxyanhydride (NNCA) polymerization. The terminal DBCO can be clearly detected in the 1H-NMR spectra and thus can be employed for the strain promoted azide-alkyne cycloadditon (SPAAC) with azides. For SPAAC no catalyst is needed, no toxic by-products are generated, and no side reactions with other functional domains of the oligomer can occur [96]. Mixing azide-modified cationic lipo-oligomers with siRNA leads to spontaneous assembly of lipopolyplexes. Due to oligomer excess, several azide functionalities are accessible on the polyplex surface and can serve as attachment points for functionalization with DBCO-modified pSar. The N-terminal free amine group can further be modified with carboxylic acid-bearing molecules to introduce a second functionality, e.g., targeting ligands such as folic acid or alternatively may be capped by acetylation to remove the terminal amine, which is positively charged in aqueous solution of neutral pH. All synthesized agents are presented in Figure 1.
3.3. Polyplex Formation and pSar-Shielding
For polyplex formation, the structures T-1N3 and T-2N3 were incubated with siRNA for 40 min at a final concentration of 25 ng siRNA/µL. The nucleic acid binding ability was evaluated by measuring the electrophoretic mobility of incorporated siRNA within an agarose gel. Different N/P values depict the ratio of protonatable amines (N) within an oligomer to the phosphates (P) of the siRNA. Like its azide-free analogue T-0N3 [39], the new azide-containing oligomers showed complete retention of the nucleic acid in the pockets of the gel at an N/P ratio of 12 (Figure S2). The binding ability observed for structures containing one or two azides did not differ.
Next, the heterotelechelic polysarcosine with DBCO-end group (DBCO-pSar119; Figure 1, bottom) was used to react with the azides on the preformed polyplexes (N/P ratio 12) to introduce a shielding layer. The SPAAC was allowed to proceed until full conversion for 16 h (see scheme in Figure 1, bottom).
Afterwards, the influence of the pSar shielding agent on electrophoretic mobility was evaluated with respect to the number of azide functions incorporated into the polyplex-forming core structures (N3 = 0, 1, 2). The amount of DBCO-pSar119 added to the polyplexes was kept constant (Figure 2A). In this experiment, only the azide-bearing polyplexes migrated in the agarose gel towards the cathode, whereas the azide-free polyplex remained in the loading pocket. This demonstrates that a covalent bond connecting the shielding agent to the nanoparticle is crucial to provide this migratory effect. The migration of the integrated nucleic acid against its own negative charge shows that it is fully shielded against the force of the electric field. With increasing equivalents of DBCO-pSar119, stronger migration could be observed. This effect can be explained by the degree of polyplex surface modification (Figure 2B). For the oligomer with only one azide functionality (T-1N3), maximum migration was achieved with equimolar amounts of DBCO click agent (1 eq/oligomer). More DBCO-pSar119 (2 eq) did not increase the effect. T-2N3 siRNA polyplexes with two azide functionalities within the carrier also revealed the maximum migration for equimolar ratios of azide to DBCO (2 eq DBCO-pSar119). These findings are in line with covalent modification with PEG5k [86].
A second indirect measure for the efficiency of polyplex shielding is the zeta potential or electrochemical mobility. The latter can be determined by measuring a particle’s mobility in an electric field with light scattering.
In this respect, we observed that the positive zeta potential of an unshielded particle can be strongly reduced from 21 mV to 6 mV in case of T-1N3 polyplexes and from 17 mV to 3 mV in case of T-2N3 polyplexes, when the particle is shielded with an excess of DBCO-pSar119 (Table 1). By using 0.5 eq DBCO-pSar119/oligomer, the zeta potential can already be reduced to 50%. It should be noted here that due to the N-terminal cationic tail group, the zeta potential always remained slightly positive. As determined by single-angle dynamic light scattering (DLS), hydrodynamic diameters of the polyplexes were approximately 100 nm. With increasing amounts of DBCO-pSar119, the nanoparticle size increased by up to 16 nm in diameter. The most plausible explanation for the increase in size seems to be pSar covering the particle surface.
3.4. Evaluation of pSar-Shielding Agents In Vitro
Through the incorporation of pSar the unspecific interaction of polyplexes with cell membranes should be efficiently reduced as already demonstrated for other stealth-like polymers, e.g., PEG. To prove our assumption we performed uptake studies with pSar-shielded polyplexes. Formulations were prepared with Cy5-labled siRNA for this assay to follow the fluorescent cargo, incubated with neuroblastoma Neuro2a cells for 4 h at standard culture conditions and analyzed by flow cytometry. The signal intensity of cells bearing fluorescent dyes correlates with the amount of polyplexes being internalized (Table 2 and Figure S3). Unshielded material and material shielded with low equivalents of DBCO-pSar119 showed significant uptake into cells already after 4 h incubation time for polyplex formulations prepared with one or two azide-bearing backbones (T-1N3 and T-2N3). For T-1N3 formulations, a significant reduction in fluorescence intensity of more than 50% was observed for 1 eq of DBCO-pSar119 per oligomer, whereas 2 eq of DBCO-pSar119 were needed for T-2N3 formulations to reduce cell uptake (Table 2).
The effect on internalization can be visualized by confocal laser scanning microscopy (CLSM). Cells were incubated with T-1N3 siRNA formulations for 4 h and the Cy5-labeled siRNA (red) representing the localization of the polyplex was detected (Figure 3). Compared to the unshielded material that was avidly taken up by cells, 0.5 eq of DBCO-pSar119 showed a slight reduction in cellular internalization. For 1 eq of DBCO-pSar119, only a few polyplexes were taken up by cells, indicating efficient shielding ability. This experiment reconfirmed the observations already made in the flow cytometry studies.
In conclusion, covalent surface modification of polyplexes by SPAAC reduced cell binding and uptake substantially. Interestingly, the increase of azide functionalities in the T-2N3 backbone did not lead to a better surface passivation of the formed polyplex. As depicted in Table 2, both polyplexes behave comparably and differ only slightly at full polysarcosinylation levels. This observation may relate to differences in microstructure between T-1N3 and T-2N3 based polyplexes, which seem to influence the accessibility of azide on the polyplex surface.
3.5. Distribution of pSar-Functionalized Polyplexes In Vivo
After the shielding ability of pSar-functionalized polyplexes could be demonstrated in biophysical and in vitro assays, we aim to explore the in vivo behavior of polysarcosinylated polyplexes. For in vivo biodistribution studies, the unshielded T-1N3 siRNA polyplex, which showed the lowest interaction with cells, was used to prepare a formulation to which either DBCO-PEG5k and a formulation using acetylated polysarcosine (DBCO-pSar119-Ac; Figure 1, bottom) was covalently linked by SPAAC. The acetyl end group of DBCO-pSar119-Ac seems to be better comparable to the commercial methoxylated PEG agent in terms of surface polarity. The cap of the N-terminal sarcosine slightly reduced migration distance of polyplexes in the gel in comparison to the non-acetylated pSar (Table S2, Figure S4). T-1N3 siRNA polyplexes were prepared with 50% Cy7-labeled siRNA followed by 4 h incubation with 1 eq of the respective shielding agent per oligomer. The final concentration of siRNA in the formulation was 200 ng/µL in this experiment. 50 µg of siRNA and oligomers at an N/P ratio of 10 were used.
The formulations were administered to Neuro2a tumor-bearing mice via tail-vain injection and the distribution of the near infrared (NIR) fluorescent dye attached to the siRNA was monitored at various time points over 24 h by bioimaging in mice (Figure 4 and Figure S5). The unshielded polyplexes started accumulating in the liver after 15 min. Such a finding could also be observed for other unshielded siRNA formulations with cationic T-shape structures in previous work [89,90]. In contrast to the unshielded polyplexes, both shielded formulations showed much-extended circulation times and tumor accumulation. 60 min after i.v. injection of the material, the shielded formulations were still detectable in all parts of the body including the tumor site. The intensity of the signals decreased after 4 h, indicating a slow removal of polyplexes from circulation. The strongest signals remained in liver and bladder, which indicates that the stability of polyplexes coated with PEG or pSar is enhanced but not infinite and the labeled siRNA can leave the polyplex ending up in liver and kidney. To further enhance stability bioreversible core cross-linking is required [79,80,97]. In mice injected with shielded polyplexes, a strong signal was detected in the exposed periphery, such as the paws after more than 4 h (Figure S5). In direct comparison, polyplexes with the DBCO-pSar119-Ac and the DBCO-PEG5k displayed negligible differences in biodistribution, circulation time or tumor accumulation. A pronounced accumulation at the tumor site, however, could not be observed and may require further strategies to enhance tumor accessibility and retention.
3.6. Attachment of the Targeting Ligand Folate to Polysarcosine
The inhibition of unspecific cell association is an important requirement for any specific interaction with cell surface receptors or proteins in solution. Thus, attaching a targeting ligand onto shielded polyplexes is expected to enable targeting of specific cell surface receptors. Since pSar can be further functionalized at its free secondary amino function, we chose folic acid (FolA) as a ligand to be conjugated to the N-terminus of DBCO-pSar110 by peptide bond formation. Folic acid is an interesting ligand, because it is a commercially available small molecule with carboxyl groups for conjugation and it is the natural ligand to the folic acid receptor (FR) overexpressed on several tumor types, e.g., prostate cancer [82,83,84,98,99,100,101]. The applied coupling conditions using equimolar amounts of folic acid, DBCO-pSar polymer and coupling reagents (2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), 1-hydroxybenzotriazole (HOBt)) and the steric hindrance of the polymer avoid the formation of divalent folic acid conjugates. Concerning regioselectivity, it has been reported that both isoforms result for N,N-dicyclohexylcarbodiimide (DCC)-mediated amidation in DMSO or DMSO/DMF, but with an observed regioselectivity of 80% for the γ-conjugate (DBCO-pSar110-FolA; Figure 1 and Figure 5, Table S1) [102].
The properties of T-1N3 siRNA polyplexes equipped with this negatively charged ligand changed in an unexpected way. For increasing equivalents, aggregates with high polydispersity were found by DLS (Table S1). An interesting observation was the change in size from around 80 to 25 nm of nanoparticles when folic acid was involved, which indicates strong compactation of the polyion complex. For small degrees of particle surface modification with DBCO-pSar110-FolA, the size of polyplexes only changed marginal compared to unshielded polyplexes.
For higher equivalents, aggregates were found in DLS measurements. Similar findings were observed for folic acid-targeted lipopolyplexes before [103]. In the latter case, aggregation could be prevented by incorporation of tetra-glutamylated folic acid into the agent. We observed that for further increase of DBCO-pSar110-FolA to equimolar ratios, small defined particles of ~25 nm were found. This can be explained with DBCO-pSar110-FolA-induced instability following a complete rearrangement of particles to a uniform population. The folic acid’s chemical properties—its hydrophobic character and negative charge—seem to play a major role in this reassembly process, because it was not observed for untargeted polysarcosine-shielded particles. We need to state clearly that currently the effect of FolA on polyplex formation is not understood and requires more detailed physicochemical studies.
When testing the folate targeted formulation with DBCO-pSar110-FolA on a FR-overexpressing KB/eGFPLuc cell line, we found that targeted polyplexes showed increased binding to the cell surface, which could be blocked by folic acid competition (Figure S6A) ensuring FR mediated binding. Much to our surprise, the internalization of the polyplexes into the cell was extremely low (Figure S6A,B). As a consequence, no gene silencing activity was achieved with such systems (Figure S6C). Limitations in endosomal escape, often reported as being responsible for bad transfection efficiencies [104,105,106,107,108], can be excluded, since co-incubation with the lysosomotropic agent chloroquine did not improve gene silencing activity (Figure S6D). The trafficking of the vitamin folate via FR is reported to occur by a non-caveolar, non-clathrin pathway also known as CLIC/GEEC endocytosis pathway [105,109]. For folate-targeted nanoparticles however, it could be demonstrated that pathways like caveolae- and clathrin-mediated endocytosis occur [106,107,108]. The size and the ligand density on the particle surface were reported to influence the cellular uptake pathway. The well shielded, ~25 nm siRNA polyplexes do not seem to trigger any of the pathways in HeLa-derived KB cells efficiently. Further, the bioreducible carrier T-1N3 within the liopolyplex might be an easy prey for disulfide cleavage, which was reported to occur distinctly in the extracellular environment of HeLa cells [110]. The consequence of insufficient cellular uptake was a lack of gene silencing activity. This effect has been observed for folate-targeted polyplexes with 3.5 kDa PEG chains before [86]. At this point, we cannot provide an explanation for the observed findings and further studies need to be conducted to understand the fact that specific receptor binding was achieved, while receptor mediated endocytosis seems to be inhibited. In light of these in vitro data, a transfer of targeted polyplexes to in vivo studies was not performed based on ethical considerations.
4. Conclusions
To investigate the use of ability of pSar to shield polyplexes and enhance their circulation times and reduce unspecific interactions, we synthesized a polyplex formulation based on sequence defined lipo-oligomers and applied PEG and pSar based polymers for shielding of the preformed polyplexes. In previous work, a novel class of redox-sensitive lipo-oligomers for siRNA delivery was successfully established [39]. One of the best performing candidates from this work was chosen and extended by a click-reactive azide functionality, resulting in carrier T-1N3. After the formation of siRNA polyplexes, the particle surface was functionalized with the shielding agent polysarcosine. The SPAAC could be performed between the DBCO-pSar polymer and the azide-containing lipo-oligomers within the polyplex. In addition, it was demonstrated that the grafting could be controlled stoichiometrically introducing a shielding layer. The shielding of the formed pSar corona has been observed in vitro in gel retardation assays and cell studies. In contrast to unmodified polyplexes, binding and cellular uptake was substantially reduced for all pSar-modified systems.
Furthermore, biodistribution in mice revealed that 8 kDa polysarcosine can clearly expand the circulation of the siRNA lipopolyplexes from several minutes to hours. The difference in non-shielded and shielded formulations is most pronounced at the 60 min time point, where in case of non-shielded polyplexes, most of the polyplexes have accumulated in the liver, but stable circulation is still observed for pSar-shielded polyplexes. While the biodistribution between non-modified polyplexes and polysarcosinylated systems differ substantially, such systems behaved similar to PEGylated polyplexes in vivo. Therefore, we can conclude that in terms of polyplex shielding, pSar and PEG behave identically and can be both applied to reduce unspecific interactions of lipo-oligomer polyplexes and thus enhance blood circulation substantially from minutes to hours. When DBCO-pSar was, however, modified with folic acid to target cell surface receptors, not only the size of polyplexes was reduced from 80 to 25 nm, but also specific binding to FR-positive KB cell membranes did not boost cellular internalization. Therefore, we need to conclude that further investigations are necessary to combine favorable in vivo shielding with efficient receptor-targeted gene silencing for pSar-functionalized lipo-oligomer polyplexes.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4360/10/6/689/s1, Figure S1: Synthesis of heterotelechelic DBCO-pSar polyplex shielding agents by NCA polymerization and subsequent amidation for further introduction of functionalities, Figure S2: siRNA binding ability of T-shape structures analyzed with an agarose gel shift assay, Figure S3: Cellular internalization of siRNA formulations shielded with increasing equivalents of DBCO-pSar119 determined by flow cytometry, Figure S4: Electrophoretic mobility of formulations, Figure S5: Biodistribution of siRNA formulations in NMRI-nude mice bearing Neuro2A tumors after i.v. administration, Figure S6: Cellular binding, cellular internalization and transfection efficiency of folate targeted siRNA polyplexes and untargeted analogues, Table S1: Analytical data of synthesized heterotelechelic DBCO-pSar ligands, Table S2: Particle size and zeta potential of siRNA formulations determined with a DLS zetasizer.
Author Contributions
P.M.K. performed chemistry of oligomers, preparation of the formulations and biophysical and cell-free in vitro experiments. K.K. synthesized and analyzed the DBCO agents. W.Z. performed transfections and FACS studies. S.K. and E.K. performed the in vivo experiment. E.W. and M.B. supervised the experimental work and contributed with scientific discussions. P.M.K. wrote the draft manuscript, K.K., M.B. and E.W. edited the manuscript, all other authors checked and contributed to the finalization of the manuscript.
Funding
This work was supported by German Research Foundation (DFG) CRC1066-1/-2 Projects A6, B5 and B12 as well as the Excellence Cluster Nanosystems Initiative Munich (NIM). K.K. would like to thank “Materials Science in Mainz” (MAINZ) and HaVo-foundation. W.Z. appreciates receiving a Bayerischen Gleichstellungsförderung fellowship as support for her postdoctoral research at LMU Munich.
Acknowledgments
We thank Miriam Höhn for generating the CLSM images.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tabernero, J.; Shapiro, G.I.; LoRusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-humans trial of an rna interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013, 3, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Kacsinta, A.D.; Dowdy, S.F. Current views on inducing synthetic lethal RNAi responses in the treatment of cancer. Expert Opin. Biol. Ther. 2016, 16, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Haussecker, D. Current issues of RNAi therapeutics delivery and development. J. Control. Release 2014, 195, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in humans from systemically administered sirna via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.T.; Mixson, A.J. SiRNA nanoparticles: The future of RNAi therapeutics for oncology? Nanomedicine 2014, 9, 2251–2254. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.K.; Willoughby, J.L.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent n-acetylgalactosamine-conjugated sirna localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef] [PubMed]
- Parmar, R.; Willoughby, J.L.; Liu, J.; Foster, D.J.; Brigham, B.; Theile, C.S.; Charisse, K.; Akinc, A.; Guidry, E.; Pei, Y.; et al. 5′-(e)-vinylphosphonate: A stable phosphate mimic can improve the RNAi activity of sirna-galnac conjugates. Chembiochem 2016, 17, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Dohmen, C.; Edinger, D.; Frohlich, T.; Schreiner, L.; Lachelt, U.; Troiber, C.; Radler, J.; Hadwiger, P.; Vornlocher, H.P.; Wagner, E. Nanosized multifunctional polyplexes for receptor-mediated sirna delivery. ACS Nano 2012, 6, 5198–5208. [Google Scholar] [CrossRef] [PubMed]
- van de Water, F.M.; Boerman, O.C.; Wouterse, A.C.; Peters, J.G.; Russel, F.G.; Masereeuw, R. Intravenously administered short interfering rna accumulates in the kidney and selectively suppresses gene function in renal proximal tubules. Drug Metab. Dispos. 2006, 34, 1393–1397. [Google Scholar] [CrossRef] [PubMed]
- Meade, B.R.; Gogoi, K.; Hamil, A.S.; Palm-Apergi, C.; van den Berg, A.; Hagopian, J.C.; Springer, A.D.; Eguchi, A.; Kacsinta, A.D.; Dowdy, C.F.; et al. Efficient delivery of RNAi prodrugs containing reversible charge-neutralizing phosphotriester backbone modifications. Nat. Biotechnol. 2014, 32, 1256–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, Y.; Hatakeyama, H.; Sato, Y.; Hyodo, M.; Akita, H.; Harashima, H. Gene silencing via RNAi and sirna quantification in tumor tissue using mend, a liposomal sirna delivery system. Mol. Ther. 2013, 21, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Philipp, A.; Oskuee, R.; Schmidt, C.; Wagner, E. Breathing life into polycations: Functionalization with ph-responsive endosomolytic peptides and polyethylene glycol enables sirna delivery. J. Am. Chem. Soc. 2008, 130, 3272–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.; Dohmen, C.; Philipp, A.; Kiener, D.; Maiwald, G.; Scheu, C.; Ogris, M.; Wagner, E. Synthesis and biological evaluation of a bioresponsive and endosomolytic sirna-polymer conjugate. Mol. Pharm. 2009, 6, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E. Polymers for sirna delivery: Inspired by viruses to be targeted, dynamic, and precise. Acc. Chem. Res. 2012, 45, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E. Biomaterials in RNAi therapeutics: Quo vadis? Biomater. Sci. 2013, 1, 804–809. [Google Scholar] [CrossRef]
- Pittella, F.; Cabral, H.; Maeda, Y.; Mi, P.; Watanabe, S.; Takemoto, H.; Kim, H.J.; Nishiyama, N.; Miyata, K.; Kataoka, K. Systemic sirna delivery to a spontaneous pancreatic tumor model in transgenic mice by pegylated calcium phosphate hybrid micelles. J. Control. Release 2014, 178, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Lachelt, U.; Wagner, E. Nucleic acid therapeutics using polyplexes: A journey of 50 years (and beyond). Chem. Rev. 2015, 115, 11043–11078. [Google Scholar] [CrossRef] [PubMed]
- Leng, Q.; Chou, S.T.; Scaria, P.V.; Woodle, M.C.; Mixson, A.J. Increased tumor distribution and expression of histidine-rich plasmid polyplexes. J. Gene Med. 2014, 16, 317–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zintchenko, A.; Philipp, A.; Dehshahri, A.; Wagner, E. Simple modifications of branched pei lead to highly efficient sirna carriers with low toxicity. Bioconjug. Chem. 2008, 19, 1448–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehead, K.A.; Dorkin, J.R.; Vegas, A.J.; Chang, P.H.; Veiseh, O.; Matthews, J.; Fenton, O.S.; Zhang, Y.; Olejnik, K.T.; Yesilyurt, V.; et al. Degradable lipid nanoparticles with predictable in vivo sirna delivery activity. Nat. Commun. 2014, 5, 4277. [Google Scholar] [CrossRef] [PubMed]
- Love, K.T.; Mahon, K.P.; Levins, C.G.; Whitehead, K.A.; Querbes, W.; Dorkin, J.R.; Qin, J.; Cantley, W.; Qin, L.L.; Racie, T.; et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. USA 2010, 107, 1864–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, Q.; Mixson, A.J. Small interfering rna targeting raf-1 inhibits tumor growth in vitro and in vivo. Cancer Gene Ther. 2005, 12, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Miyata, K.; Nishiyama, N.; Kataoka, K. Rational design of smart supramolecular assemblies for gene delivery: Chemical challenges in the creation of artificial viruses. Chem. Soc. Rev. 2012, 41, 2562–2574. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Takemoto, H.; Yi, Y.; Zheng, M.; Maeda, Y.; Chaya, H.; Hayashi, K.; Mi, P.; Pittella, F.; Christie, R.J.; et al. Precise engineering of sirna delivery vehicles to tumors using polyion complexes and gold nanoparticles. ACS Nano 2014, 8, 8979–8991. [Google Scholar] [CrossRef] [PubMed]
- Li, S.D.; Chen, Y.C.; Hackett, M.J.; Huang, L. Tumor-targeted delivery of sirna by self-assembled nanoparticles. Mol. Ther. 2008, 16, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Ramusovic, S.; Nguyen, T.; Lu, Z.R. Novel polymerizable surfactants with ph-sensitive amphiphilicity and cell membrane disruption for efficient sirna delivery. Bioconjug. Chem. 2007, 18, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Xu, R.; Wu, X.; Gillespie, D.; Jensen, R.; Lu, Z.R. Targeted systemic delivery of a therapeutic sirna with a multifunctional carrier controls tumor proliferation in mice. Mol. Pharm. 2009, 6, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Gujrati, M.; Vaidya, A.; Lu, Z.R. Multifunctional ph-sensitive amino lipids for sirna delivery. Bioconjug. Chem. 2016, 27, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Schafer, J.; Hobel, S.; Bakowsky, U.; Aigner, A. Liposome-polyethylenimine complexes for enhanced DNA and sirna delivery. Biomaterials 2010, 31, 6892–6900. [Google Scholar] [CrossRef] [PubMed]
- Siegwart, D.J.; Whitehead, K.A.; Nuhn, L.; Sahay, G.; Cheng, H.; Jiang, S.; Ma, M.; Lytton-Jean, A.; Vegas, A.; Fenton, P.; et al. Combinatorial synthesis of chemically diverse core-shell nanoparticles for intracellular delivery. Proc. Natl. Acad. Sci. USA 2011, 108, 12996–13001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, J.J.; Langer, R.; Anderson, D.G. A combinatorial polymer library approach yields insight into nonviral gene delivery. Acc. Chem. Res. 2008, 41, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Uchida, H.; Miyata, K.; Oba, M.; Ishii, T.; Suma, T.; Itaka, K.; Nishiyama, N.; Kataoka, K. Odd-even effect of repeating aminoethylene units in the side chain of n-substituted polyaspartamides on gene transfection profiles. J. Am. Chem. Soc. 2011, 133, 15524–15532. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, T.; Edinger, D.; Klager, R.; Troiber, C.; Salcher, E.; Badgujar, N.; Martin, I.; Schaffert, D.; Cengizeroglu, A.; Hadwiger, P.; et al. Structure-activity relationships of sirna carriers based on sequence-defined oligo (ethane amino) amides. J. Control. Release 2012, 160, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Krzyszton, R.; Salem, B.; Lee, D.J.; Schwake, G.; Wagner, E.; Radler, J.O. Microfluidic self-assembly of folate-targeted monomolecular sirna-lipid nanoparticles. Nanoscale 2017, 9, 7442–7453. [Google Scholar] [CrossRef] [PubMed]
- Leng, Q.; Scaria, P.; Zhu, J.; Ambulos, N.; Campbell, P.; Mixson, A.J. Highly branched hk peptides are effective carriers of sirna. J. Gene Med. 2005, 7, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stoter, M.; et al. Image-based analysis of lipid nanoparticle-mediated sirna delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Sahay, G.; Querbes, W.; Alabi, C.; Eltoukhy, A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.; Chen, D.; Zoncu, R.; et al. Efficiency of sirna delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 2013, 31, 653–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittrup, A.; Lieberman, J. Knocking down disease: A progress report on sirna therapeutics. Nat. Rev. Genet. 2015, 16, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Klein, P.M.; Reinhard, S.; Lee, D.J.; Muller, K.; Ponader, D.; Hartmann, L.; Wagner, E. Precise redox-sensitive cleavage sites for improved bioactivity of sirna lipopolyplexes. Nanoscale 2016, 8, 18098–18104. [Google Scholar] [CrossRef] [PubMed]
- Klibanov, A.L.; Maruyama, K.; Torchilin, V.P.; Huang, L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990, 268, 235–237. [Google Scholar] [CrossRef] [Green Version]
- Senior, J.; Delgado, C.; Fisher, D.; Tilcock, C.; Gregoriadis, G. Influence of surface hydrophilicity of liposomes on their interaction with plasma protein and clearance from the circulation: Studies with poly (ethylene glycol)-coated vesicles. Biochim. Biophys. Acta Biomembr. 1991, 1062, 77–82. [Google Scholar] [CrossRef]
- Mori, A.; Klibanov, A.L.; Torchilin, V.P.; Huang, L. Influence of the steric barrier activity of amphipathic poly (ethyleneglycol) and ganglioside gm1 on the circulation time of liposomes and on the target binding of immunoliposomes in vivo. FEBS Lett. 1991, 284, 263–266. [Google Scholar] [CrossRef]
- Yang, Q.; Lai, S.K. Anti-peg immunity: Emergence, characteristics, and unaddressed questions. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 655–677. [Google Scholar] [CrossRef] [PubMed]
- Plank, C.; Mechtler, K.; Szoka, F.C., Jr.; Wagner, E. Activation of the complement system by synthetic DNA complexes: A potential barrier for intravenous gene delivery. Hum. Gene Ther. 1996, 7, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Tockary, T.A.; Osada, K.; Motoda, Y.; Hiki, S.; Chen, Q.; Takeda, K.M.; Dirisala, A.; Osawa, S.; Kataoka, K. Rod-to-globule transition of pdna/peg–poly (l-lysine) polyplex micelles induced by a collapsed balance between DNA rigidity and peg crowdedness. Small 2016, 12, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Merkel, O.M.; Librizzi, D.; Pfestroff, A.; Schurrat, T.; Buyens, K.; Sanders, N.N.; De Smedt, S.C.; Béhé, M.; Kissel, T. Stability of sirna polyplexes from poly (ethylenimine) and poly (ethylenimine)-g-poly (ethylene glycol) under in vivo conditions: Effects on pharmacokinetics and biodistribution measured by fluorescence fluctuation spectroscopy and single photon emission computed tomography (spect) imaging. J. Control. Release 2009, 138, 148–159. [Google Scholar]
- Kursa, M.; Walker, G.F.; Roessler, V.; Ogris, M.; Roedl, W.; Kircheis, R.; Wagner, E. Novel shielded transferrin-polyethylene glycol-polyethylenimine/DNA complexes for systemic tumor-targeted gene transfer. Bioconjug. Chem. 2003, 14, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Fella, C.; Walker, G.F.; Ogris, M.; Wagner, E. Amine-reactive pyridylhydrazone-based peg reagents for ph-reversible pei polyplex shielding. Eur. J. Pharm. Sci. 2008, 34, 309–320. [Google Scholar] [CrossRef] [PubMed]
- DeRouchey, J.; Walker, G.F.; Wagner, E.; Rädler, J.O. Decorated rods: A “bottom-up” self-assembly of monomolecular DNA complexes. J. Phys. Chem. B 2006, 110, 4548–4554. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Hunter, A.C.; Dadswell, C.M.; Savay, S.; Alving, C.R.; Szebeni, J. Causative factors behind poloxamer 188 (pluronic f68, flocor™)-induced complement activation in human sera: A protective role against poloxamer-mediated complement activation by elevated serum lipoprotein levels. Biochim. Biophys. Acta Mol. Basis Dis. 2004, 1689, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, H.; Hennink, W.E.; Brinks, V. The immunogenicity of polyethylene glycol: Facts and fiction. Pharm. Res. 2013, 30, 1729–1734. [Google Scholar] [CrossRef] [PubMed]
- Wenande, E.; Garvey, L. Immediate-type hypersensitivity to polyethylene glycols: A review. Clin. Exp. Allergy 2016, 46, 907–922. [Google Scholar] [CrossRef] [PubMed]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly (ethylene glycol) in drug delivery: Pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. 2010, 49, 6288–6308. [Google Scholar] [CrossRef] [PubMed]
- Hamad, I.; Hunter, A.; Szebeni, J.; Moghimi, S.M. Poly (ethylene glycol) s generate complement activation products in human serum through increased alternative pathway turnover and a masp-2-dependent process. Mol. Immunol. 2008, 46, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Dewachter, P.; Mouton-Faivre, C. Anaphylaxis to macrogol 4000 after a parenteral corticoid injection. Allergy 2005, 60, 705–706. [Google Scholar] [CrossRef] [PubMed]
- Kircheis, R.; Wightman, L.; Schreiber, A.; Robitza, B.; Rössler, V.; Kursa, M.; Wagner, E. Polyethylenimine/DNA complexes shielded by transferrin target gene expression to tumors after systemic application. Gene Ther. 2001, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tetley, L.; Uchegbu, I.F. The level of hydrophobic substitution and the molecular weight of amphiphilic poly-l-lysine-based polymers strongly affects their assembly into polymeric bilayer vesicles. J. Colloid Interf. Sci. 2001, 237, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Toncheva, V.; Wolfert, M.A.; Dash, P.R.; Oupicky, D.; Ulbrich, K.; Seymour, L.W.; Schacht, E.H. Novel vectors for gene delivery formed by self-assembly of DNA with poly (l-lysine) grafted with hydrophilic polymers. Biochim. Biophys. Acta Gen. Subj. 1998, 1380, 354–368. [Google Scholar] [CrossRef]
- Oupický, D.; Howard, K.A.; Koňák, Č.; Dash, P.R.; Ulbrich, K.; Seymour, L.W. Steric stabilization of poly-l-lysine/DNA complexes by the covalent attachment of semitelechelic poly [n-(2-hydroxypropyl) methacrylamide]. Bioconjug. Chem. 2000, 11, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T.; Ulbrich, K. Hpma Copolymers: 30 Years of Advances; Elsevier: New York, NY, USA, 2010. [Google Scholar]
- Noga, M.; Edinger, D.; Kläger, R.; Wegner, S.V.; Spatz, J.P.; Wagner, E.; Winter, G.; Besheer, A. The effect of molar mass and degree of hydroxyethylation on the controlled shielding and deshielding of hydroxyethyl starch-coated polyplexes. Biomaterials 2013, 34, 2530–2538. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wallace, S. Polymer-drug conjugates: Recent development in clinical oncology. Adv. Drug Deliv. Rev. 2008, 60, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Romberg, B.; Metselaar, J.M.; Baranyi, L.; Snel, C.J.; Bunger, R.; Hennink, W.E.; Szebeni, J.; Storm, G. Poly(amino acid)s: Promising enzymatically degradable stealth coatings for liposomes. Int. J. Pharm. 2007, 331, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Schlapschy, M.; Binder, U.; Börger, C.; Theobald, I.; Wachinger, K.; Kisling, S.; Haller, D.; Skerra, A. Pasylation: A biological alternative to pegylation for extending the plasma half-life of pharmaceutically active proteins. Protein Eng. Des. Sel. 2013, 26, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Mendler, C.T.; Friedrich, L.; Laitinen, I.; Schlapschy, M.; Schwaiger, M.; Wester, H.-J.; Skerra, A. High Contrast Tumor Imaging with Radio-Labeled Antibody Fab Fragments Tailored for Optimized Pharmacokinetics via Pasylation. MAbs 2015, 7, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Birke, A.; Kim, J.; Jang, Y.; Song, S.Y.; Ryu, S.; Kim, B.S.; Kim, B.G.; Barz, M.; Char, K. Cooperative catechol-functionalized polypept(o)ide brushes and ag nanoparticles for combination of protein resistance and antimicrobial activity on metal oxide surfaces. Biomacromolecules 2018, 19, 1602–1613. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Fetsch, C.; Amin, I.; Jordan, R.; Luxenhofer, R. Polypeptoid brushes by surface-initiated polymerization of n-substituted glycine n-carboxyanhydrides. Langmuir 2013, 29, 6983–6988. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Tang, Z.; Richter, M.; Marschelke, C.; Forster, P.; Wegener, E.; Amin, I.; Zimmermann, H.; Scharnweber, D.; Braun, H.G.; et al. Patterned polypeptoid brushes. Macromol. Biosci. 2016, 16, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Birke, A.; Ling, J.; Barz, M. Polysarcosine-containing copolymers: Synthesis, characterization, self-assembly, and applications. Prog. Polym. Sci. 2018, 81, 163–208. [Google Scholar] [CrossRef]
- Fetsch, C.; Grossmann, A.; Holz, L.; Nawroth, J.F.; Luxenhofer, R. Polypeptoids from n-substituted glycine n-carboxyanhydrides: Hydrophilic, hydrophobic, and amphiphilic polymers with poisson distribution. Macromolecules 2011, 44, 6746–6758. [Google Scholar] [CrossRef]
- Klinker, K.; Barz, M. Polypept(o)ides: Hybrid systems based on polypeptides and polypeptoids. Macromol. Rapid Commun. 2015, 36, 1943–1957. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Birke, A.; Fischer, K.; Schmidt, M.; Barz, M. Solution properties of polysarcosine: From absolute and relative molar mass determinations to complement activation. Macromolecules 2018, 51, 2653–2661. [Google Scholar] [CrossRef]
- Wei, Q.; Becherer, T.; Angioletti-Uberti, S.; Dzubiella, J.; Wischke, C.; Neffe, A.T.; Lendlein, A.; Ballauff, M.; Haag, R. Protein interactions with polymer coatings and biomaterials. Angew. Chem. 2014, 53, 8004–8031. [Google Scholar] [CrossRef] [PubMed]
- Hörtz, C.; Birke, A.; Kaps, L.; Decker, S.; Wächtersbach, E.; Fischer, K.; Schuppan, D.; Barz, M.; Schmidt, M. Cylindrical brush polymers with polysarcosine side chains: A novel biocompatible carrier for biomedical applications. Macromolecules 2015, 48, 2074–2086. [Google Scholar] [CrossRef]
- Sela, M. Immunological studies with synthetic polypeptides. Adv. Immunol. 1966, 5, 29–129. [Google Scholar] [PubMed]
- Hara, E.; Ueda, M.; Kim, C.J.; Makino, A.; Hara, I.; Ozeki, E.; Kimura, S. Suppressive immune response of poly-(sarcosine) chains in peptide-nanosheets in contrast to polymeric micelles. J. Pept. Sci. 2014, 20, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Birke, A.; Huesmann, D.; Kelsch, A.; Weilbacher, M.; Xie, J.; Bros, M.; Bopp, T.; Becker, C.; Landfester, K.; Barz, M. Polypeptoid-block-polypeptide copolymers: Synthesis, characterization, and application of amphiphilic block copolypept(o)ides in drug formulations and miniemulsion techniques. Biomacromolecules 2014, 15, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Otter, R.; Klinker, K.; Spitzer, D.; Schinnerer, M.; Barz, M.; Besenius, P. Folding induced supramolecular assembly into ph-responsive nanorods with a protein repellent shell. Chem. Commun. 2018, 54, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Heller, P.; Hobernik, D.; Lachelt, U.; Schinnerer, M.; Weber, B.; Schmidt, M.; Wagner, E.; Bros, M.; Barz, M. Combining reactive triblock copolymers with functional cross-linkers: A versatile pathway to disulfide stabilized-polyplex libraries and their application as pdna vaccines. J. Control. Release 2017, 258, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Klinker, K.; Schafer, O.; Huesmann, D.; Bauer, T.; Capeloa, L.; Braun, L.; Stergiou, N.; Schinnerer, M.; Dirisala, A.; Miyata, K.; et al. Secondary-structure-driven self-assembly of reactive polypept(o)ides: Controlling size, shape, and function of core cross-linked nanostructures. Angew. Chem. 2017, 56, 9608–9613. [Google Scholar] [CrossRef] [PubMed]
- Duro-Castano, A.; Movellan, J.; Vicent, M.J. Smart branched polymer drug conjugates as nano-sized drug delivery systems. Biomater. Sci. 2015, 3, 1321–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.K.; Kim, S.Y.; Lee, S.B.; Lee, Y.M. Folate-conjugated methoxy poly(ethylene glycol)/poly(epsilon-caprolactone) amphiphilic block copolymeric micelles for tumor-targeted drug delivery. J. Control. Release 2005, 109, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Low, P.S. Folate-targeted therapies for cancer. J. Med. Chem. 2010, 53, 6811–6824. [Google Scholar] [CrossRef] [PubMed]
- Sudimack, J.; Lee, R.J. Targeted drug delivery via the folate receptor. Adv. Drug Deliv. Rev. 2000, 41, 147–162. [Google Scholar] [CrossRef]
- Low, P.S.; Henne, W.A.; Doorneweerd, D.D. Discovery and development of folic-acid-based receptor targeting for imaging and therapy of cancer and inflammatory diseases. Acc. Chem. Res. 2008, 41, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Klein, P.M.; Kern, S.; Lee, D.J.; Schmaus, J.; Hohn, M.; Gorges, J.; Kazmaier, U.; Wagner, E. Folate receptor-directed orthogonal click-functionalization of sirna lipopolyplexes for tumor cell killing in vivo. Biomaterials 2018. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Keough, E.; Matter, A.; Leander, K.; Young, S.; Carlini, E.; Sachs, A.B.; Tao, W.; Abrams, M.; Howell, B.; et al. Biodistribution of small interfering rna at the organ and cellular levels after lipid nanoparticle-mediated delivery. J. Histochem. Cytochem. 2011, 59, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Muller, K.; Kessel, E.; Reinhard, S.; He, D.; Klein, P.M.; Hohn, M.; Rodl, W.; Kempter, S.; Wagner, E. Targeted sirna delivery using a lipo-oligoaminoamide nanocore with an influenza peptide and transferrin shell. Adv. Healthc. Mater. 2016, 5, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Troiber, C.; Edinger, D.; Kos, P.; Schreiner, L.; Klager, R.; Herrmann, A.; Wagner, E. Stabilizing effect of tyrosine trimers on pdna and sirna polyplexes. Biomaterials 2013, 34, 1624–1633. [Google Scholar] [CrossRef] [PubMed]
- Schottler, S.; Becker, G.; Winzen, S.; Steinbach, T.; Mohr, K.; Landfester, K.; Mailander, V.; Wurm, F.R. Protein adsorption is required for stealth effect of poly(ethylene glycol)- and poly(phosphoester)-coated nanocarriers. Nat. Nanotechnol. 2016, 11, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, D.; Caracciolo, G.; Marchini, C.; Montani, M.; Amici, A.; Callipo, L.; Capriotti, A.L.; Cavaliere, C.; Lagana, A. Surface adsorption of protein corona controls the cell uptake mechanism in efficient cationic liposome/DNA complexes in serum. J. Control. Release 2010, 148, e94–e95. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, M.; Stigler, J.; Elia, G.; Lynch, I.; Cedervall, T.; Dawson, K.A. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc. Natl. Acad. Sci. USA 2008, 105, 14265–14270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fokina, A.; Klinker, K.; Braun, L.; Jeong, B.G.; Bae, W.K.; Barz, M.; Zentel, R. Multidentate polysarcosine-based ligands for water-soluble quantum dots. Macromolecules 2016, 49, 3663–3671. [Google Scholar] [CrossRef]
- Klinker, K.; Holm, R.; Heller, P.; Barz, M. Evaluating chemical ligation techniques for the synthesis of block copolypeptides, polypeptoids and block copolypept(o)ides: A comparative study. Polym. Chem. 2015, 6, 4612–4623. [Google Scholar] [CrossRef]
- Dommerholt, J.; Rutjes, F.; van Delft, F.L. Strain-promoted 1,3-dipolar cycloaddition of cycloalkynes and organic azides. Top. Curr. Chem. 2016, 374, 16. [Google Scholar] [CrossRef] [PubMed]
- Schafer, O.; Barz, M. Of thiols and disulfides: Methods for chemoselective formation of asymmetric disulfides in synthetic peptides and polymers. Chemistry 2018. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.J.; Kessel, E.; Lehto, T.; Liu, X.; Yoshinaga, N.; Padari, K.; Chen, Y.C.; Kempter, S.; Uchida, S.; Radler, J.O.; et al. Systemic delivery of folate-peg sirna lipopolyplexes with enhanced intracellular stability for in vivo gene silencing in leukemia. Bioconjug. Chem. 2017, 28, 2393–2409. [Google Scholar] [CrossRef] [PubMed]
- Dohmen, C.; Frohlich, T.; Lachelt, U.; Rohl, I.; Vornlocher, H.P.; Hadwiger, P.; Wagner, E. Defined folate-peg-sirna conjugates for receptor-specific gene silencing. Mol. Ther. Nucleic Acids 2012, 1, e7. [Google Scholar] [CrossRef] [PubMed]
- Leamon, C.P.; Low, P.S. Folate-mediated targeting: From diagnostics to drug and gene delivery. Drug Discov. Today 2001, 6, 44–51. [Google Scholar] [CrossRef]
- Leamon, C.P.; DePrince, R.B.; Hendren, R.W. Folate-mediated drug delivery: Effect of alternative conjugation chemistry. J. Drug Target. 1999, 7, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lee, R.J.; Mathias, C.J.; Green, M.A.; Low, P.S. Synthesis, purification, and tumor cell uptake of 67ga-deferoxamine—Folate, a potential radiopharmaceutical for tumor imaging. Bioconjug. Chem. 1996, 7, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.; Kessel, E.; Klein, P.M.; Hohn, M.; Wagner, E. Post-pegylation of sirna lipo-oligoamino amide polyplexes using tetra-glutamylated folic acid as ligand for receptor-targeted delivery. Mol. Pharm. 2016, 13, 2332–2345. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Gunther, M.; Gu, Z.; Wagner, E. Pyridylhydrazone-based pegylation for ph-reversible lipopolyplex shielding. Biomaterials 2011, 32, 858–869. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Baez, L.; Williamson, C.; Donaldson, J.G. Clathrin-independent endocytosis: A cargo-centric view. Exp. Cell Res. 2013, 319, 2759–2769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.L.; Van Cuong, N.; Hsieh, M.F. Endocytosis pathways of the folate tethered star-shaped peg-pcl micelles in cancer cell lines. Polymers 2014, 6, 634–650. [Google Scholar] [CrossRef]
- Langston Suen, W.L.; Chau, Y. Size-dependent internalisation of folate-decorated nanoparticles via the pathways of clathrin and caveolae-mediated endocytosis in arpe-19 cells. J. Pharm. Pharmacol. 2014, 66, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Dalal, C.; Saha, A.; Jana, N.R. Nanoparticle multivalency directed shifting of cellular uptake mechanism. J. Phys. Chem. C 2016, 120, 6778–6786. [Google Scholar] [CrossRef]
- Sabharanjak, S.; Sharma, P.; Parton, R.G.; Mayor, S. Gpi-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev. Cell 2002, 2, 411–423. [Google Scholar] [CrossRef]
- Brulisauer, L.; Kathriner, N.; Prenrecaj, M.; Gauthier, M.A.; Leroux, J.C. Tracking the bioreduction of disulfide-containing cationic dendrimers. Angew. Chem. 2012, 51, 12454–12458. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Overview of chemical compounds. Table, top: structural illustration of sequence-defined oligomers with T-shape topology; T-0N3 (ID: 992 published [39]), T-1N3 (ID: 1073) and T-2N3 (ID: 1086) with no, one or two terminal azidolysines K(N3). Further units of the oligomers: Stp: succinoyl-tetraethylene-pentamine, ssbb: succinoyl-cystamine, CholA: 5β-Cholanic acid, Y: tyrosine, K: lysine, G: glycine. The broken lines represent amide linkages, the triangle (▲) is the starting point of the synthesis. IDs are unique database identification numbers. Table, bottom: chemical structure of the shielding agents DBCO-pSar119, DBCO-pSar119-Ac and DBCO-pSar110-FolA. Scheme of the formulation of a shielded polyplex.
Figure 1.
Overview of chemical compounds. Table, top: structural illustration of sequence-defined oligomers with T-shape topology; T-0N3 (ID: 992 published [39]), T-1N3 (ID: 1073) and T-2N3 (ID: 1086) with no, one or two terminal azidolysines K(N3). Further units of the oligomers: Stp: succinoyl-tetraethylene-pentamine, ssbb: succinoyl-cystamine, CholA: 5β-Cholanic acid, Y: tyrosine, K: lysine, G: glycine. The broken lines represent amide linkages, the triangle (▲) is the starting point of the synthesis. IDs are unique database identification numbers. Table, bottom: chemical structure of the shielding agents DBCO-pSar119, DBCO-pSar119-Ac and DBCO-pSar110-FolA. Scheme of the formulation of a shielded polyplex.

Figure 2.
Electrophoretic mobility of siRNA lipopolyplex formulations analyzed with an agarose gel shift assay (A) siRNA polyplexes formed with lipo-oligomers bearing no (T-0N3), one (T-1N3) and two (T-2N3) azide functions incubated with 0.75 equivalents of DBCO-pSar119 1% agarose gel, 70 V, 80 min runtime; (B) Formulations with increasing equivalents (eq mol/mol) of DBCO-pSar119. 0.75% agarose gel, 100 V, 80 min runtime. Polyplexes were incubated for 40 min at N/P 12, followed by DBCO-pSar119 addition for 16 h at room temperature. The right lane in each gel shows the running distance of free siRNA not complexed by lipo-oligomers.
Figure 2.
Electrophoretic mobility of siRNA lipopolyplex formulations analyzed with an agarose gel shift assay (A) siRNA polyplexes formed with lipo-oligomers bearing no (T-0N3), one (T-1N3) and two (T-2N3) azide functions incubated with 0.75 equivalents of DBCO-pSar119 1% agarose gel, 70 V, 80 min runtime; (B) Formulations with increasing equivalents (eq mol/mol) of DBCO-pSar119. 0.75% agarose gel, 100 V, 80 min runtime. Polyplexes were incubated for 40 min at N/P 12, followed by DBCO-pSar119 addition for 16 h at room temperature. The right lane in each gel shows the running distance of free siRNA not complexed by lipo-oligomers.

Figure 3.
Intracellular distribution of T-1N3 siRNA formulations in Neuro2a-eGFP-Luc cells with increasing equivalents (eq mol/mol) of DBCO-pSar119 acquired with a confocal laser scanning microscop. Cells were incubated with the formulations for 4 h and washed with PBS buffer. siRNA was spiked with 20% Cy5-labeled siRNA (red), nuclei were stained with DAPI (blue). The overlay image shows the light microscope image and the merged DAPI and Cy5 channels. Scale bar: 10 µm.
Figure 3.
Intracellular distribution of T-1N3 siRNA formulations in Neuro2a-eGFP-Luc cells with increasing equivalents (eq mol/mol) of DBCO-pSar119 acquired with a confocal laser scanning microscop. Cells were incubated with the formulations for 4 h and washed with PBS buffer. siRNA was spiked with 20% Cy5-labeled siRNA (red), nuclei were stained with DAPI (blue). The overlay image shows the light microscope image and the merged DAPI and Cy5 channels. Scale bar: 10 µm.

Figure 4.
Biodistribution of T-1N3 siRNA formulations (50 μg siRNA; 50% Cy7-labeled) in NMRI-nude mice bearing Neuro2a tumors after i.v. administration. NIR fluorescence bioimages show formulations with 1 eq DBCO-PEG5k, 1 eq acetylated DBCO-pSar119-Ac or HBG buffer (non-shielded). Experiments were performed with two mice per group for time points until 60 min and one mouse per group for later time points; a representative mouse of each group is shown. Mice are presented in the dorsal, ventral and lateral view.
Figure 4.
Biodistribution of T-1N3 siRNA formulations (50 μg siRNA; 50% Cy7-labeled) in NMRI-nude mice bearing Neuro2a tumors after i.v. administration. NIR fluorescence bioimages show formulations with 1 eq DBCO-PEG5k, 1 eq acetylated DBCO-pSar119-Ac or HBG buffer (non-shielded). Experiments were performed with two mice per group for time points until 60 min and one mouse per group for later time points; a representative mouse of each group is shown. Mice are presented in the dorsal, ventral and lateral view.

Figure 5.
Characterization of DBCO-pSar ligands for post-shielding of polyplexes. (A) GPC elugrams of DBCO-pSar in HFIP with different end groups; (B) UV-vis spectrum of DBCO-pSar and DBCO-pSar-FolA, respectively; (C) 1H-NMR spectrum of DBCO-pSar-FolA in DMSO-d6 (400 MHz).
Figure 5.
Characterization of DBCO-pSar ligands for post-shielding of polyplexes. (A) GPC elugrams of DBCO-pSar in HFIP with different end groups; (B) UV-vis spectrum of DBCO-pSar and DBCO-pSar-FolA, respectively; (C) 1H-NMR spectrum of DBCO-pSar-FolA in DMSO-d6 (400 MHz).


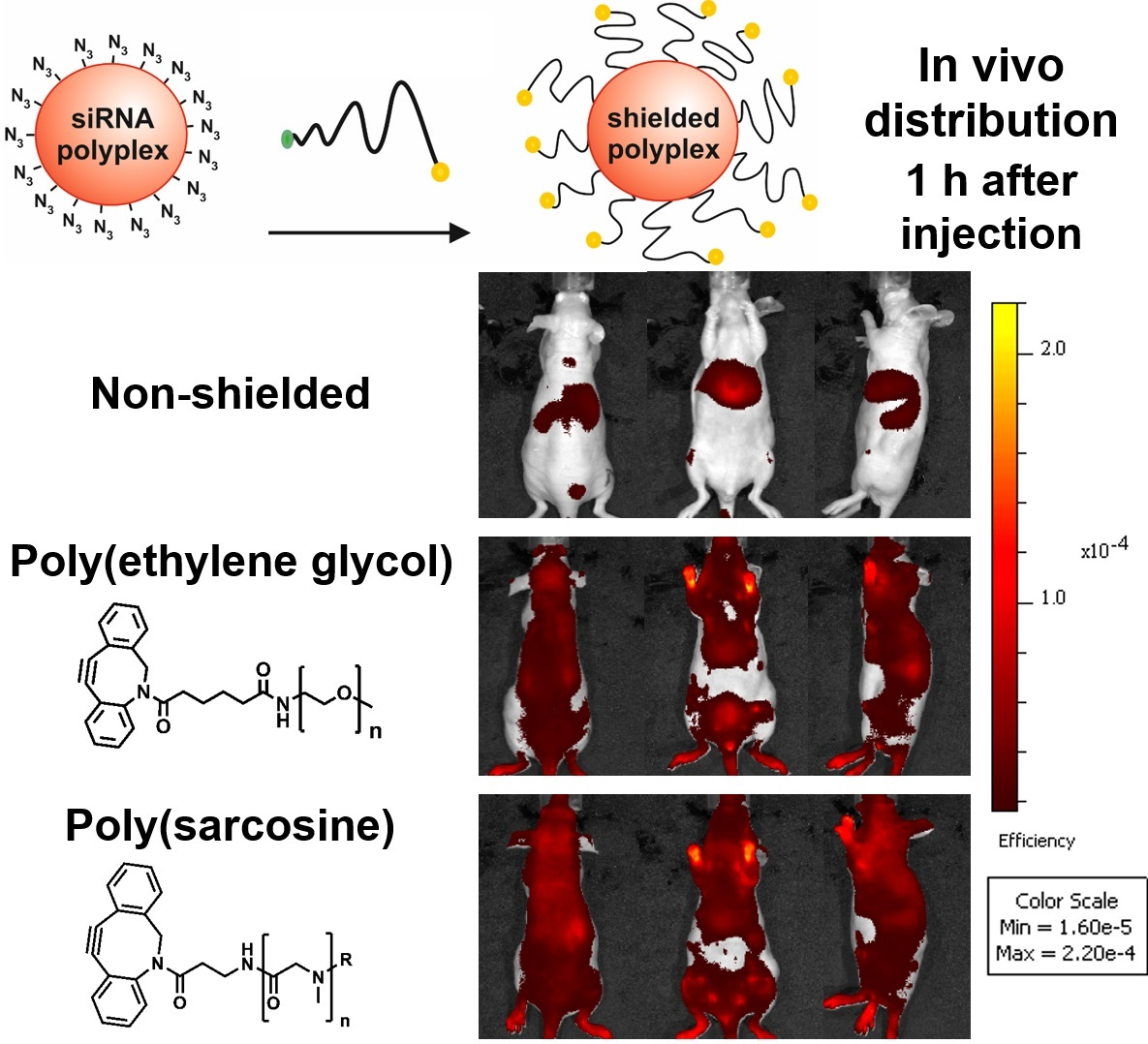{kind=link}
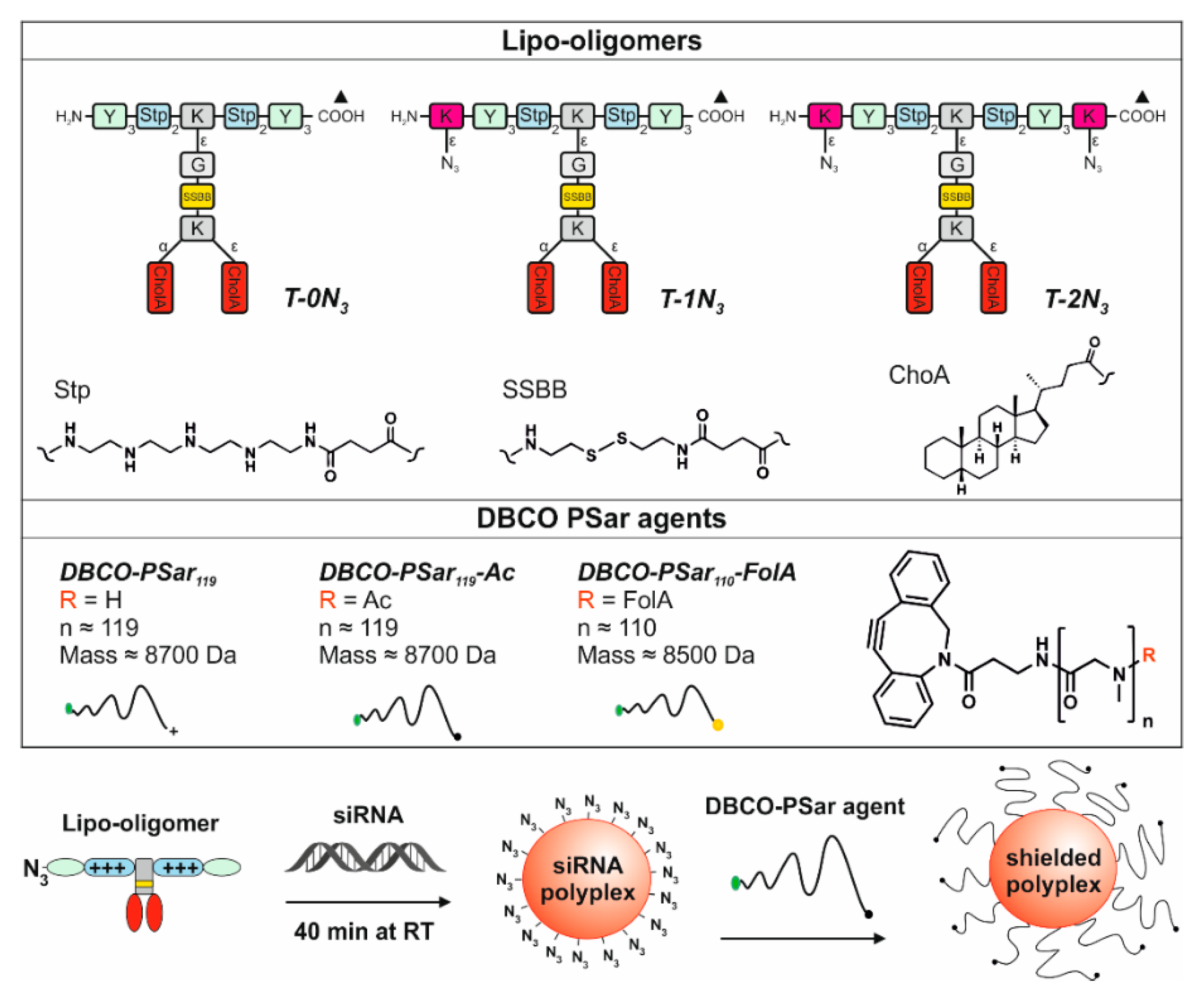{kind=link}
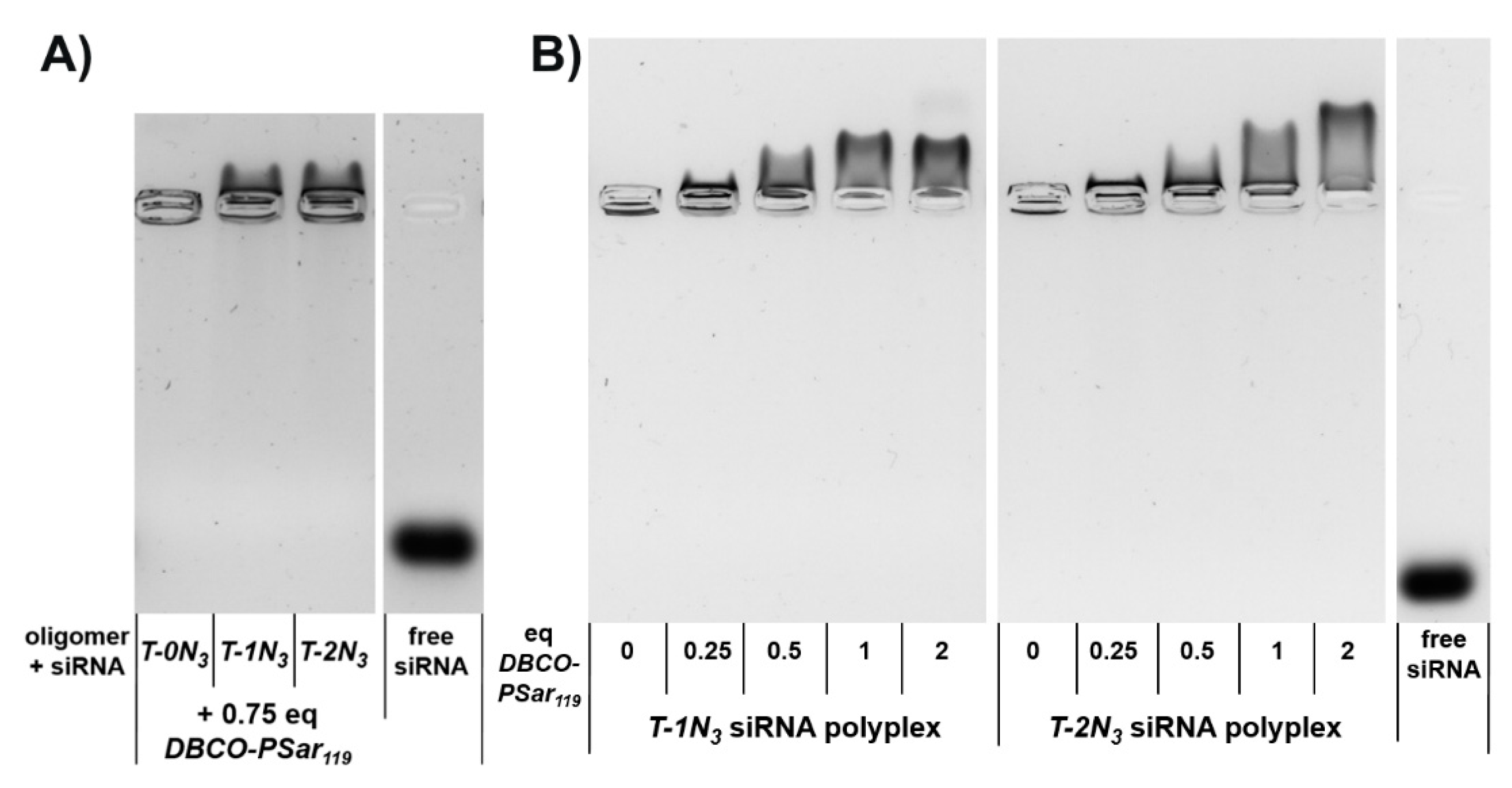{kind=link}
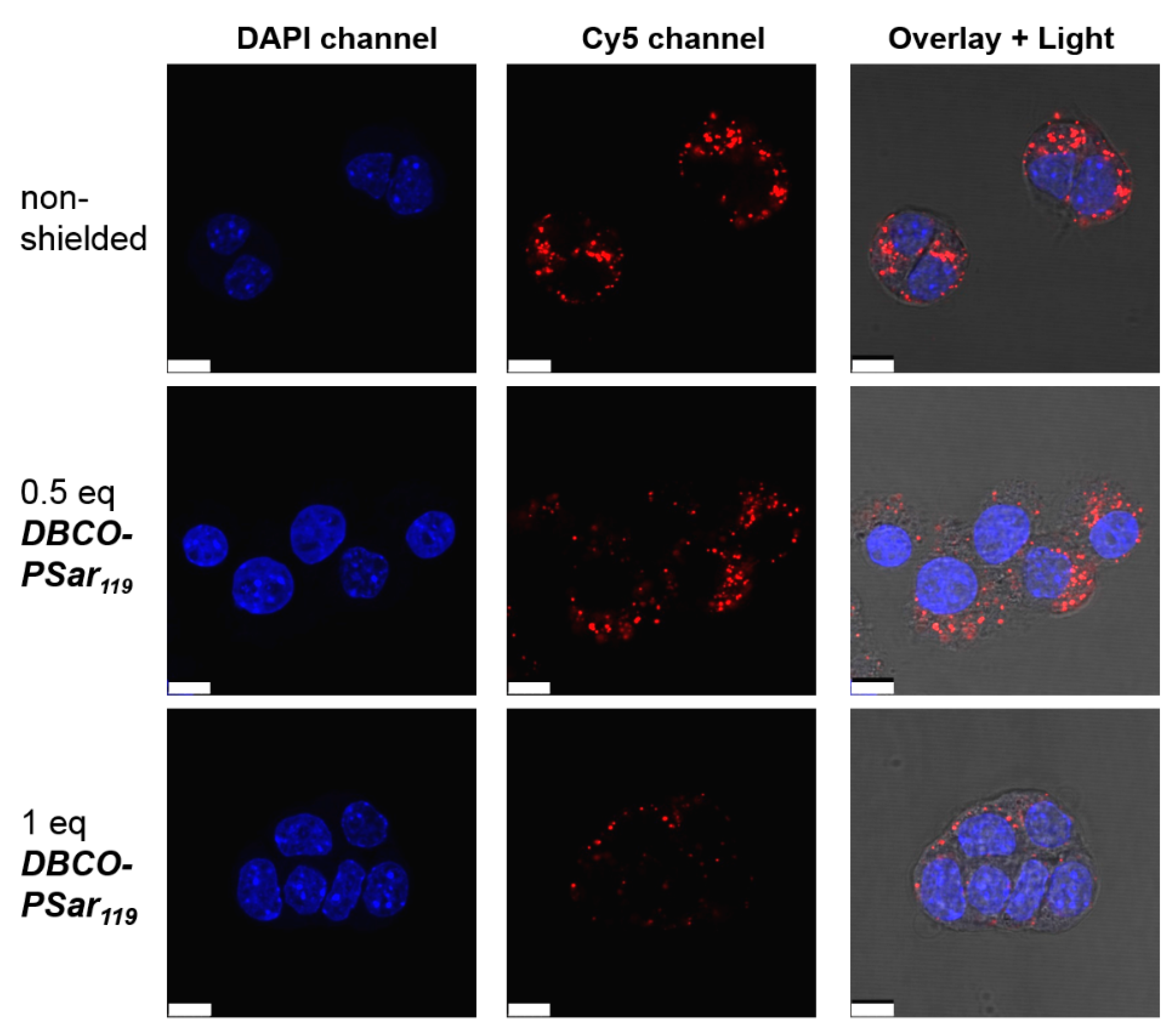{kind=link}
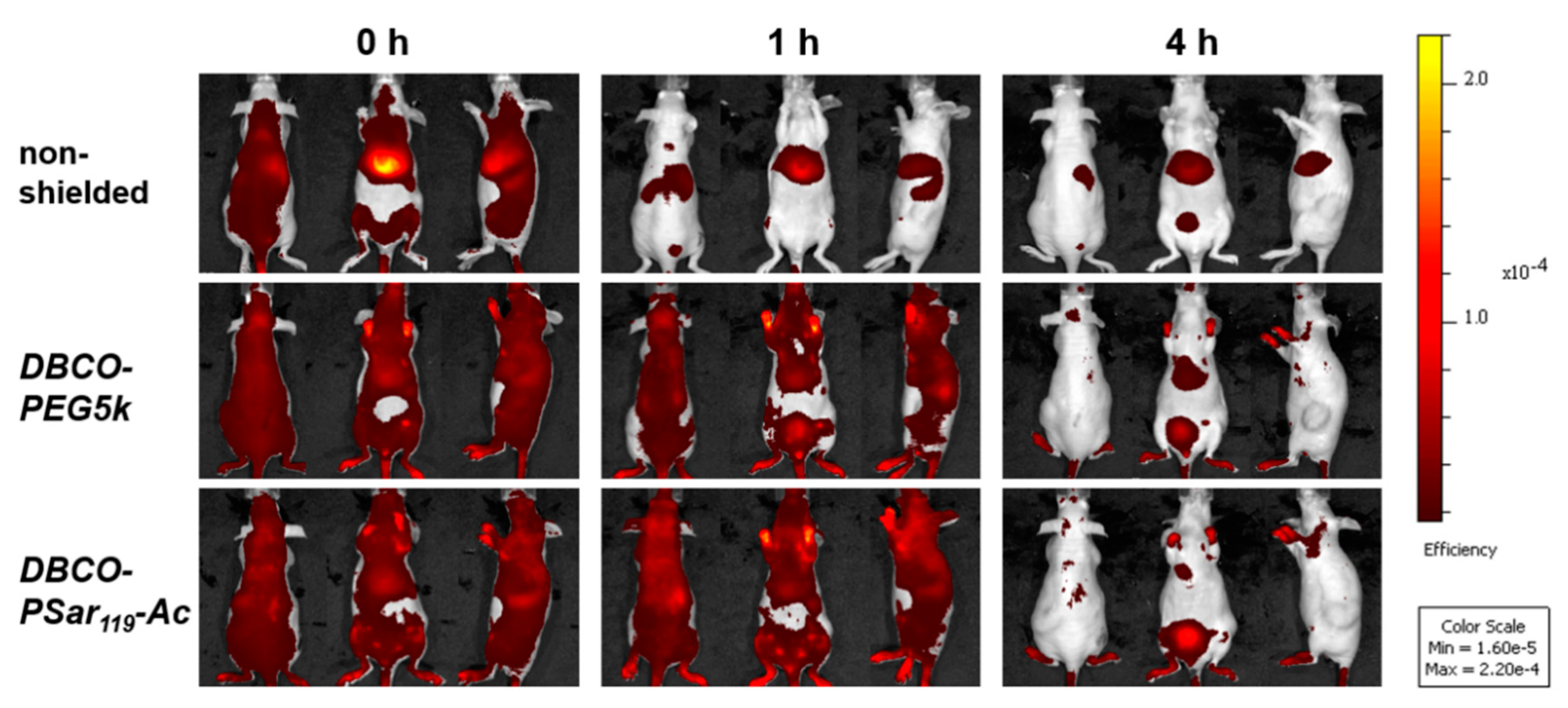{kind=link}
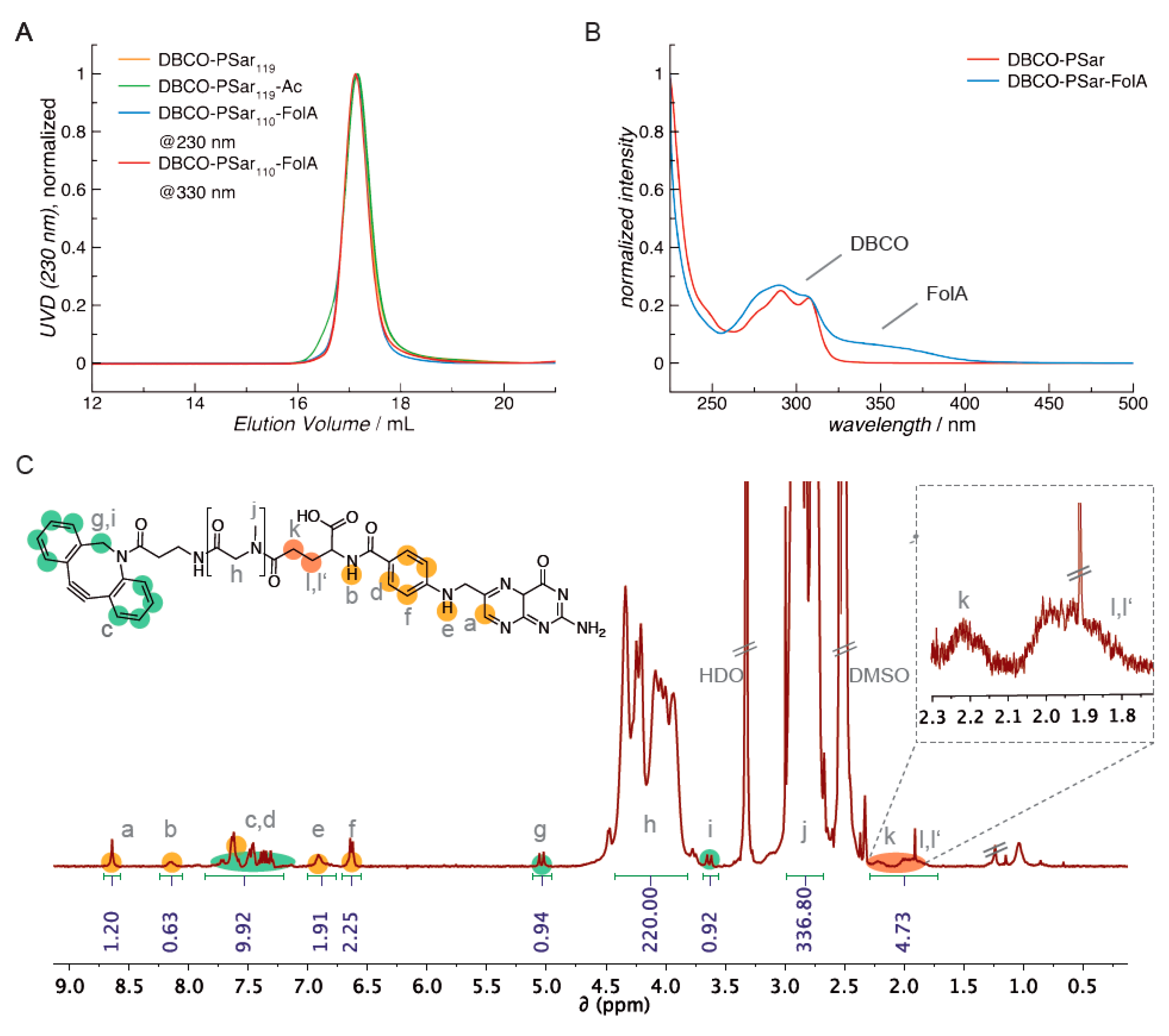{kind=link}
Table 1.
Particle size (z-average) and zeta potential of pSar-shielded siRNA formulations determined by a dynamic light scattering (DLS) zetasizer. siRNA polyplexes were prepared at N/P 12.
Table 1.
Particle size (z-average) and zeta potential of pSar-shielded siRNA formulations determined by a dynamic light scattering (DLS) zetasizer. siRNA polyplexes were prepared at N/P 12.
| siRNA Formulation | eq DBCO-pSar119 | z-Average (nm) | PDI | Mean Zeta Potential (mV) |
|---|---|---|---|---|
| T-1N3 | 0 | 81.0 ± 5.0 | 0.26 ± 0.02 | 20.9 ± 0.9 |
| 0.5 | 86.7 ± 2.8 | 0.24 ± 0.02 | 9.4 ± 0.5 | |
| 1 | 91.8 ± 2.9 | 0.26 ± 0.02 | 8.5 ± 0.6 | |
| 2 | 96.8 ± 4.0 | 0.27 ± 0.02 | 6.0 ± 1.1 | |
| T-2N3 | 0 | 90.6 ± 0.9 | 0.15 ± 0.03 | 17.2 ± 0.8 |
| 0.5 | 98.7 ± 1.3 | 0.15 ± 0.01 | 7.7 ± 0.6 | |
| 1 | 102.3 ± 2.1 | 0.19 ± 0.01 | 6.3 ± 1.0 | |
| 2 | 105.1 ± 1.9 | 0.17 ± 0.01 | 2.5 ± 0.3 |
Table 2.
Mean fluorescence intensity (MFI) of Cy5-labeled siRNA formulations (top: T-1N3; bottom: T-2N3) shielded with increasing equivalents (eq mol/mol) of DBCO-pSar119 for cellular internalization determined by flow cytometry.
Table 2.
Mean fluorescence intensity (MFI) of Cy5-labeled siRNA formulations (top: T-1N3; bottom: T-2N3) shielded with increasing equivalents (eq mol/mol) of DBCO-pSar119 for cellular internalization determined by flow cytometry.
| siRNA Formulation | eq DBCO-pSar119 | MFI |
|---|---|---|
| T-1N3 | 0 | 881.5 ± 25.5 |
| 0.25 | 780.5 ± 2.5 | |
| 0.5 | 715.0 ± 24.0 | |
| 1 | 359.0 ± 14.0 | |
| 2 | 245.5 ± 8.0 | |
| T-2N3 | 0 | 883.0 ± 86.0 |
| 0.25 | 870.5 ± 62.5 | |
| 0.5 | 785.5 ± 38.5 | |
| 1 | 602.5 ± 37.5 | |
| 2 | 263.5 ± 4.5 | |
| untreated cells | 2.4 ± 0.2 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Klein, P.M.; Klinker, K.; Zhang, W.; Kern, S.; Kessel, E.; Wagner, E.; Barz, M. Efficient Shielding of Polyplexes Using Heterotelechelic Polysarcosines. Polymers 2018, 10, 689. https://doi.org/10.3390/polym10060689
AMA Style
Klein PM, Klinker K, Zhang W, Kern S, Kessel E, Wagner E, Barz M. Efficient Shielding of Polyplexes Using Heterotelechelic Polysarcosines. Polymers. 2018; 10(6):689. https://doi.org/10.3390/polym10060689
Chicago/Turabian StyleKlein, Philipp Michael, Kristina Klinker, Wei Zhang, Sarah Kern, Eva Kessel, Ernst Wagner, and Matthias Barz. 2018. "Efficient Shielding of Polyplexes Using Heterotelechelic Polysarcosines" Polymers 10, no. 6: 689. https://doi.org/10.3390/polym10060689
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.