Polyurethane Membranes Modified with Isopropyl Myristate as a Potential Candidate for Encapsulating Electronic Implants: A Study of Biocompatibility and Water Permeability

Abstract
:1. Introduction

2. Materials and Methods
2.1. Materials
2.1.1. Synthesis of polyurethane
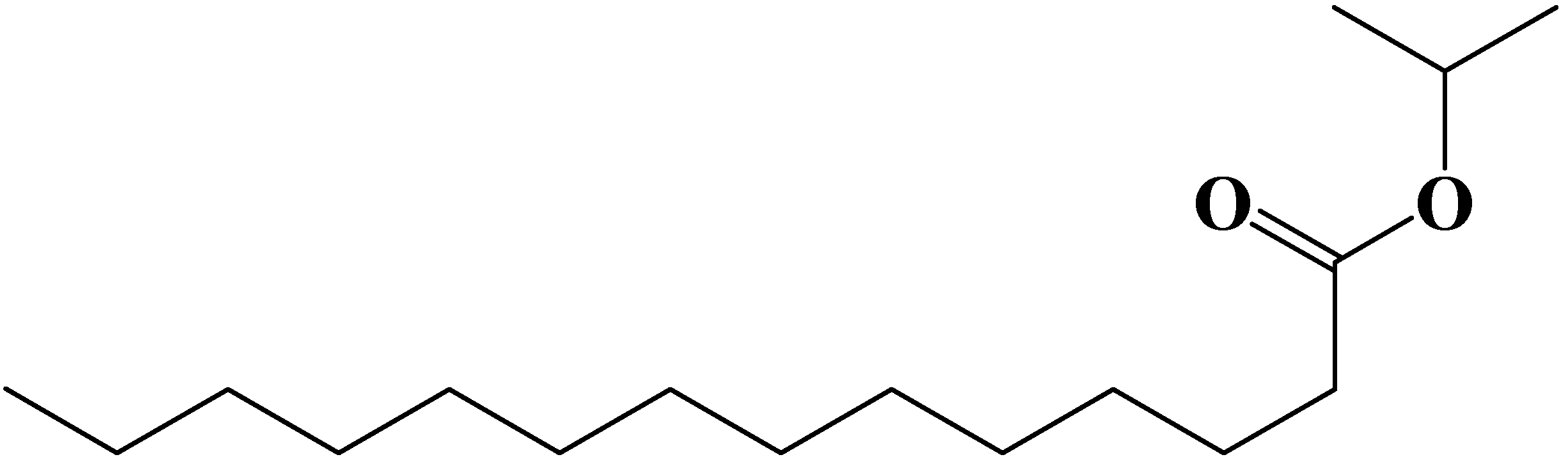
2.1.2. Preparation of membranes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Hard segment in PEU (%) | PTMO (Mw) | IPM loading (wt%) |
|---|---|---|---|
| PEU-1 | 35 | 1,000 | - |
| 0.5IPM-PEU-1 | 35 | 1,000 | 0.5 |
| 1IPM-PEU-1 | 35 | 1,000 | 1 |
| 2IPM-PEU-1 | 35 | 1,000 | 2 |
| 5IPM-PEU-1 | 35 | 1,000 | 5 |
| 10IPM-PEU-1 | 35 | 1,000 | 10 |
| PEU-2 | 25 | 2,000 | - |
| 0.5IPM-PEU-2 | 25 | 2,000 | 0.5 |
| 1IPM-PEU-2 | 25 | 2,000 | 1 |
| 2IPM-PEU-2 | 25 | 2,000 | 2 |
| 5IPM-PEU-2 | 25 | 2,000 | 5 |
| 10IPM-PEU-2 | 25 | 2,000 | 10 |
2.2. Characterization
2.2.1. ATR-FTIR
2.2.2. Contact angle and surface energy
2.2.3. Stability of IPM in PEU
2.2.4. Scanning Electron Microscopy (SEM)
2.2.5. Liquid water transport
2.2.6. Water vapour transport
2.2.7. In vitro cytotoxicity evaluation
2.2.7.1. Cell culture
2.2.7.2. Toxicity test
3. Results and Discussion
3.1. Surface Properties
3.1.1. ATR-FTIR

3.1.2. Contact angle measurement
| Sample | θ° Water* | θ° Bromonaphtalene | (mNm−1) | (mNm−1) | (mNm−1) |
|---|---|---|---|---|---|
| PEU-1 | 78.43 ± 1.44 | 37.96 ± 0.64 | 4.91 | 35.66 | 40.57 |
| 0.5IPM-PEU-1 | 80.95 ± 1.62 | 38.02 ± 1.02 | 3.99 | 35.63 | 39.62 |
| 1IPM-PEU-1 | 86.94 ± 1.67 | 40.01 ± 0.56 | 2.30 | 34.77 | 37.07 |
| 2IPM-PEU-1 | 85.32 ± 0.81 | 39.65 ± 0.92 | 2.73 | 34.92 | 37.65 |
| 5IPM-PEU-1 | 84.77 ± 1.17 | 39.94 ± 1.42 | 2.92 | 34.79 | 37.71 |
| 10IPM-PEU-1 | 85.43 ± 0.57 | 40.42 ± 0.43 | 2.76 | 34.58 | 37.34 |
| PEU-2 | 89.86 ± 1.04 | 41.95 ± 1.09 | 1.71 | 33.90 | 35.61 |
| 0.5IPM-PEU-2 | 92.56 ± 0.96 | 43.71 ± 1.28 | 1.24 | 33.10 | 34.34 |
| 1IPM-PEU-2 | 94.89 ± 1.00 | 44.54 ± 0.72 | 0.86 | 32.71 | 33.57 |
| 2IPM-PEU-2 | 97.36 ± 0.80 | 47.37 ± 1.36 | 0.62 | 31.36 | 31.98 |
| 5IPM-PEU-2 | 96.56 ± 1.17 | 46.85 ± 1.08 | 0.71 | 31.61 | 32.32 |
| 10IPM-PEU-2 | 95.65 ± 0.57 | 46.05 ± 0.84 | 0.81 | 31.20 | 32.81 |
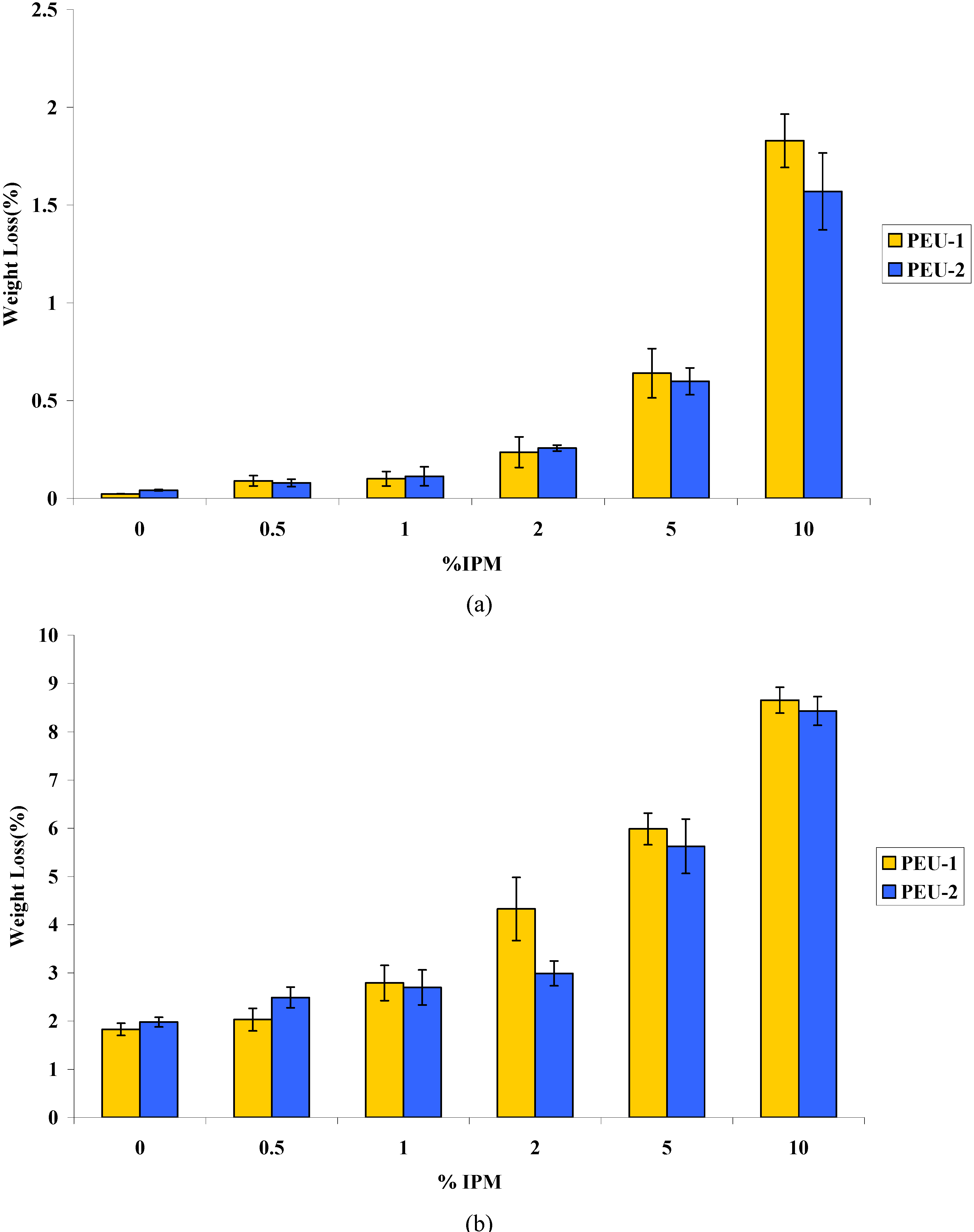
3.2. Aging and Stability


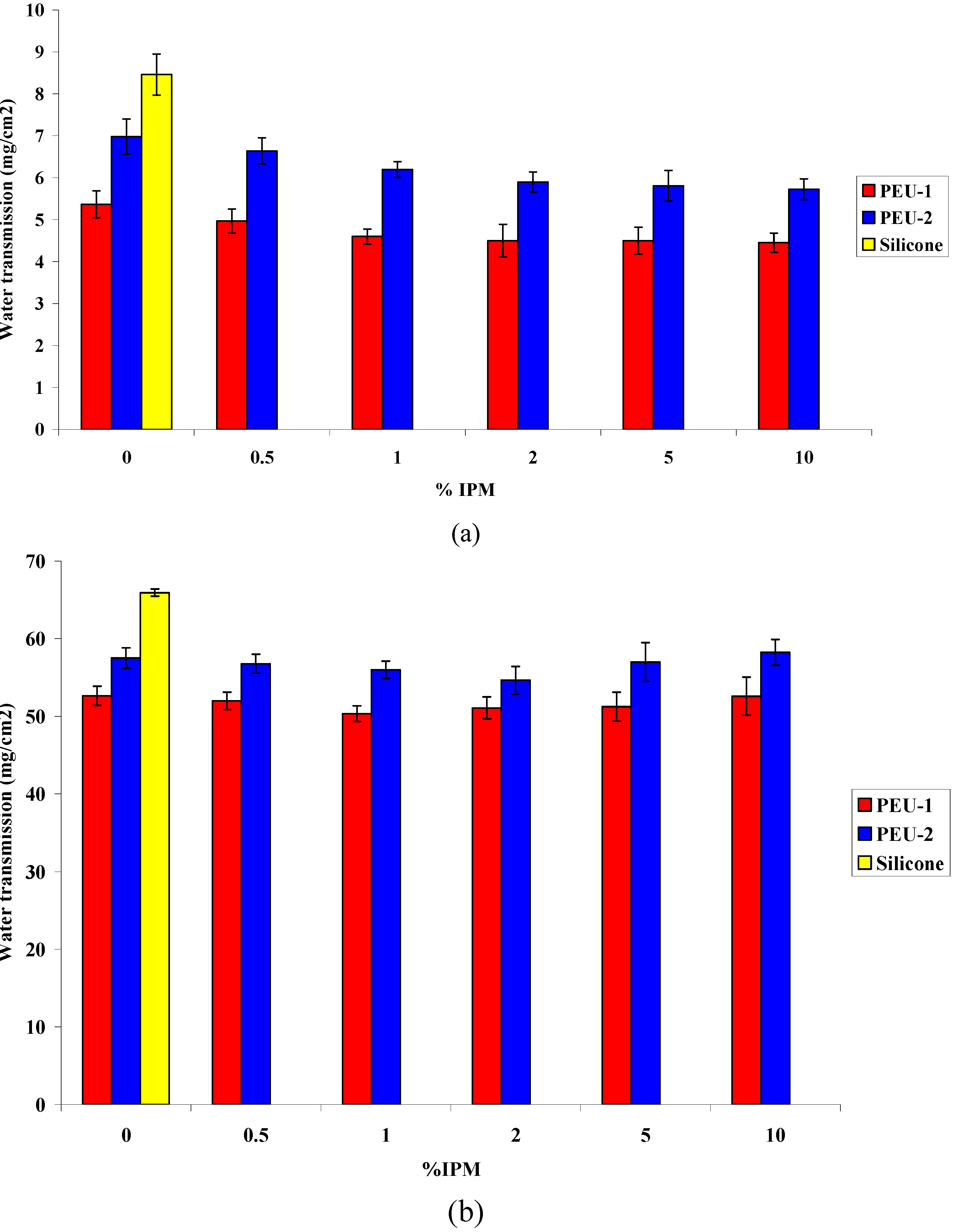
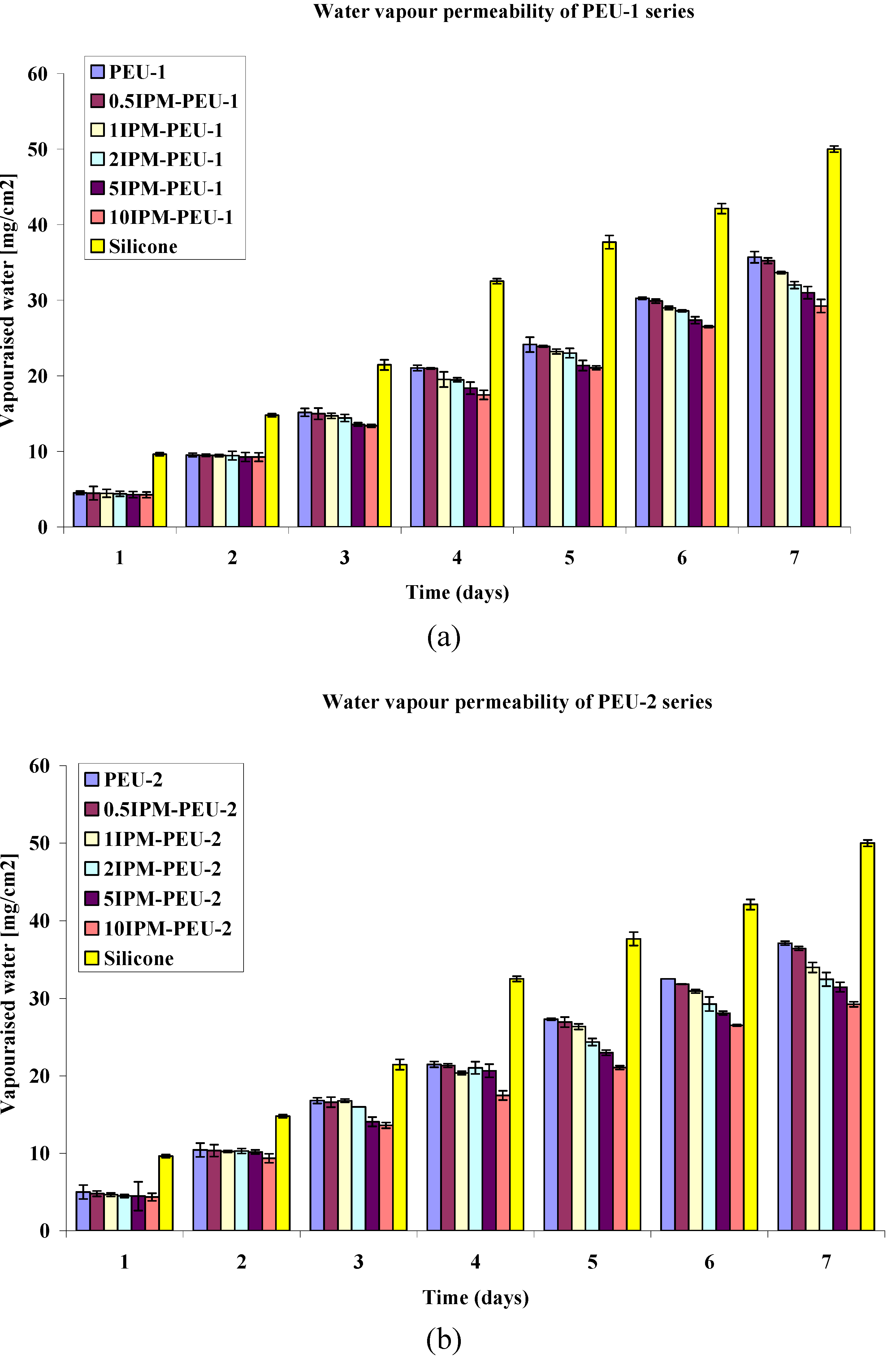
3.3. Water Permeability


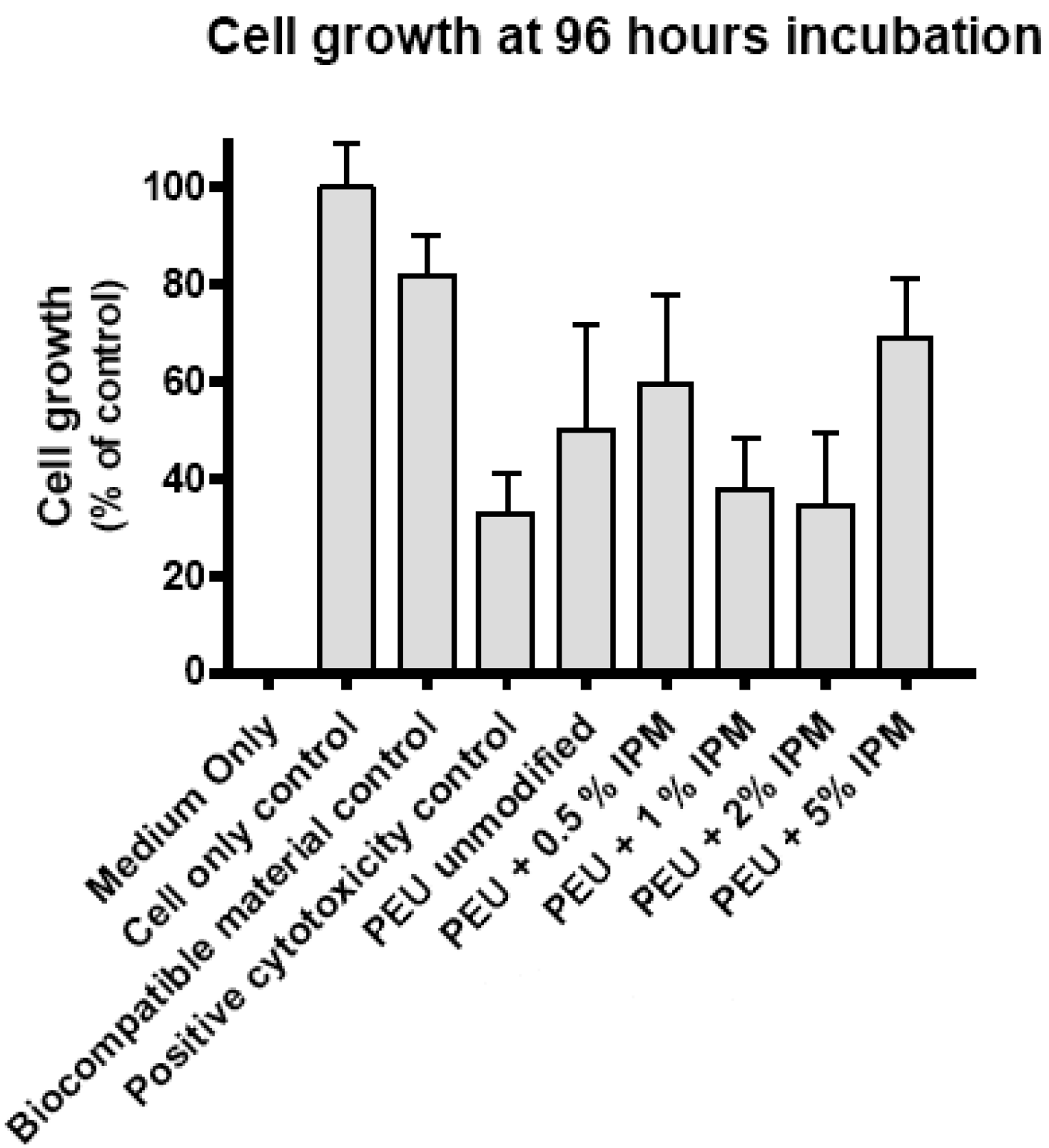
3.4. Cytotoxicity evaluation

4. Conclusion
Acknowledgment
References
- Kotzar, G.; Freas, M.; Abel, P.; Fleischman, A.; Roy, S.; Zorman, C.; Moran, J.M.; Melzak, J. Evaluation of MEMS materials of construction for implantable medical devices. Biomaterials 2002, 23, 2737–2750. [Google Scholar] [CrossRef] [PubMed]
- Receveur, R.A.M.; Lindemans, F.W.; de Rooij, N.F. Microsystem technologies for implantable applications. J. Micromech. Microeng. 2007, 17, 50–80. [Google Scholar] [CrossRef]
- Rodger, D.C.; Tai, Y.C. Microelectronic packaging for retinal prostheses. IEEE Eng. Med. Biol. Mag. 2005, 24, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, M.; Levesque, S.; Guidion, R.G. Biomedical applications of polyurethanes. In Biomedical Applications of Polyurethanes; Vermette, P., Griesser, H.J., Laroche, G., Guidoin, R., Eds.; Landes Bioscience: Georgetown, TX, USA, 2001; pp. 220–251. [Google Scholar]
- Donaldson, P. Encapsulating microelectronic implants in one-part silicone rubbers. Med. Biol. Eng. Comput. 1989, 27, 93–94. [Google Scholar] [CrossRef] [PubMed]
- Galland, G.; Lam, T.M. Permeability and diffusion of gases in segmented polyurethanes—structure properties relations. J. Appl. Polym. Sci. 1993, 50, 1041–1058. [Google Scholar] [CrossRef]
- Roohpour, N.; Wasikiewicz, J.M.; Paul, D.; Vadgama, P.; Rehman, I.U. Synthesis and characterisation of enhanced barrier polyurethane for encapsulation of implantable medical devices. J. Mater. Sci. Mater. Med. 2009, 20, 1803–1814. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, D.M.; Gordon, B.; Rosenberg, G.; Snyder, A.J.; Benesi, A.; Runt, J. Synthesis and characterization of amphiphilic poly(urethaneurea)-comb-polyisobutylene copolymers. Macromolecules 2000, 33, 4380–4389. [Google Scholar] [CrossRef]
- Ratner, B.D.; Hoffman, A.; Schoen, F.; Lemons, J. Biomaterials science: An introduction to materials in medicine; Elsevier Academic Press: San Diego, CA, USA, 2004. [Google Scholar]
- Xu, R.J.; Manias, E.; Snyder, A.J.; Runt, J. Low penneability biomedical polyurethane nanocomposites. J. Biomed. Mater. Res. A 2003, 64A, 114–119. [Google Scholar] [CrossRef]
- Wang, X.D.; Luo, X.; Wang, X.F. Study on blends of thermoplastic polyurethane and aliphatic polyester: Morphology, rheology, and properties as moisture vapor permeable films. Polym. Test. 2005, 24, 18–24. [Google Scholar] [CrossRef]
- Hsieh, K.H.; Liao, D.C.; Chen, C.Y.; Chiu, W,Y. Interpenetrating polymer networks of polyurethane and maleimide-terminated polyurethane for biomedical applications. Polym. Adv. Technol. 1996, 7, 265–272. [Google Scholar] [CrossRef]
- Wang, D.A.; Ji, J.; Gao, C.Y.; Yu, G.H.; Feng, L.X. Surface coating of stearyl poly(ethylene oxide) coupling-polymer on polyurethane guiding catheters with poly(ether urethane) film-building additive for biomedical applications. Biomaterials 2001, 22, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Dube, N.; Al-Malaika, S.; Laroche, G.; Vermette, P. Additives in biomedical polyurethanes In Biomedical Applications of Polyurethanes; Vermette, P., Griesser, H.J., Laroche, G., Guidoin, R., Eds.; Landes Bioscience: Georgetown, TX, USA, 2001; pp. 55–76. [Google Scholar]
- Krishna, O.D.; Kim, K.; Byun, Y. Covalently grafted phospholipid monolayer on silicone catheter surface for reduction in platelet adhesion. Biomaterials 2005, 26, 7115–7123. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.J.; Kim, H.D. Characteristics of crosslinked blends of Pellethene((R)) and multiblock polyurethanes containing phospholipid. Biomaterials 2005, 26, 2877–2886. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Kundu, P.P. Addition polymers from natural oils—A review. Prog. Polym. Sci. 2006, 31, 983–1008. [Google Scholar] [CrossRef]
- Morimoto, N.; Watanabe, A.; Iwasaki, Y.; Akiyoshi, K.; Ishihara, K. Nano-scale surface modification of a segmented polyurethane with a phospholipid polymer. Biomaterials 2004, 25, 5353–5361. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.Y.; Lee, M.H.; Kim, B.K. Surface modification of waterborne polyurethane. Colloids Surf. A Physicochem. Eng. Asp. 2006, 290, 178–185. [Google Scholar] [CrossRef]
- Abraham, M.H.; Acree, W.E. Characterisation of the water-isopropyl myristate system. Int. J. Pharm. 2005, 294, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Moulik, S.P. Biocompatible microemulsions and their prospective uses in drug delivery. J. Pharm. Sci. 2008, 97, 22–45. [Google Scholar] [CrossRef] [PubMed]
- Pauliukaite, R.; Schoenleber, M.; Vadgama, P.; Brett, C.M.A. Development of electrochemical biosensors based on sol-gel enzyme encapsulation and protective polymer membranes. Anal. Bioanal. Chem. 2008, 390, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Christie, I.M.; Treloar, P.H.; Vadgama, P. Plasticized poly(vinyl chloride) as a permselective barrier membrane for high-selectivity amperometric sensors and biosensors. Anal. Chim. Acta 1992, 269, 65–73. [Google Scholar] [CrossRef]
- Reddy, S.M.; Vadgama, P.M. Surfactant-modified poly(vinyl chloride) membranes as biocompatible interfaces for amperometric enzyme electrodes. Anal. Chim. Acta 1997, 350, 77–89. [Google Scholar] [CrossRef]
- Bremner, T.; Hill, D.J.T.; Killeen, M.I.; Odonnell, J.H.; Pomery, P.J.; StJohn, D.; Whittaker, A.K. Development of wear-resistant thermoplastic polyurethanes by blending with poly(dimethyl siloxane). II. A packing model. J. Appl. Polym. Sci. 1997, 65, 939–950. [Google Scholar] [CrossRef]
- Roohpour, N.; Wasikiewicz, J.; Moshaverinia, A.; Paul, D.; Rehman, I.; Vadgama, P. Isopropyl myristate-modified polyether-urethane coatings as protective barriers for implantable medical devices. Materials 2009, 2, 719–733. [Google Scholar] [CrossRef]
- Kwok, D.Y.; Neumann, A.W. Contact angle measurement and contact angle interpretation. Adv. Colloid Interface Sci. 1999, 81, 167–249. [Google Scholar] [CrossRef]
- Lee, J.H.; Ju, Y,M,; Kim, D.M. Platelet adhesion onto segmented polyurethane film surfaces modified by addition and crosslinking of PEO-containing block copolymers. Biomaterials 2000, 21, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Standard Test Methods for Water Vapor Transmission of Materials (ASTM E 96); American Society for Testing and Materials: Washington, DC, USA, 1980.
- Castner, D.G.; Ratner, B.D. Biomedical surface science: Foundations to frontiers. Surf. Sci. 2002, 500, 28–60. [Google Scholar] [CrossRef]
- Lin, D.T.; Young, T.H.; Fang, Y. Studies on the effect of surface properties on the biocompatibility of polyurethane membranes. Biomaterials 2001, 22, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Freij-Larsson, C.; Jannasch, P.; Wesslen, B. Polyurethane surfaces modified by amphiphilic polymers: effects on protein adsorption. Biomaterials 2000, 21, 307–315. [Google Scholar] [CrossRef]
- Khayet, M.; Suk, D.E.; Narbaitz, R.M.; Santerre, J.P.; Matsuura, T. Study on surface modification by surface-modifying macromolecules and its applications in membrane-separation processes. J. Appl. Polym. Sci. 2003, 89, 2902–2916. [Google Scholar] [CrossRef]
- Szelest-Lewandowska, A.; Masiulanis, B.; Szymonowicz, M.; Pielka, S.; Paluch, D. Modified polycarbonate urethane: Synthesis, properties and biological investigation in vitro. J. Biomed. Mater. Res. A 2007, 82A, 509–520. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Roohpour, N.; Wasikiewicz, J.M.; Moshaverinia, A.; Paul, D.; Grahn, M.F.; Rehman, I.U.; Vadgama, P. Polyurethane Membranes Modified with Isopropyl Myristate as a Potential Candidate for Encapsulating Electronic Implants: A Study of Biocompatibility and Water Permeability. Polymers 2010, 2, 102-119. https://doi.org/10.3390/polym2030102
Roohpour N, Wasikiewicz JM, Moshaverinia A, Paul D, Grahn MF, Rehman IU, Vadgama P. Polyurethane Membranes Modified with Isopropyl Myristate as a Potential Candidate for Encapsulating Electronic Implants: A Study of Biocompatibility and Water Permeability. Polymers. 2010; 2(3):102-119. https://doi.org/10.3390/polym2030102
Chicago/Turabian StyleRoohpour, Nima, Jaroslaw M. Wasikiewicz, Alireza Moshaverinia, Deepen Paul, Mike F. Grahn, Ihtesham U. Rehman, and Pankaj Vadgama. 2010. "Polyurethane Membranes Modified with Isopropyl Myristate as a Potential Candidate for Encapsulating Electronic Implants: A Study of Biocompatibility and Water Permeability" Polymers 2, no. 3: 102-119. https://doi.org/10.3390/polym2030102