Cellulose Chemistry Meets Click Chemistry: Syntheses and Properties of Cellulose-Based Glycoclusters with High Structural Homogeneity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
: β-1,4-Glucans having oligosaccharide appendages (O-/N -linked β-maltoside and O-/N -linked β -lactoside) at 6C positions of all repeating units can be readily prepared from cellulose through a two step strategy composed of: (1) regio-selective and quantitative bromination/azidation to afford 6-azido-6-deoxycellulose; and (2) the subsequent Cu+-catalyzed coupling with oligosaccharides having terminal alkyne. The resultant cellulose derivatives showed improved water solubility in comparison to native cellulose; they, however, bound to carbohydrate-binding proteins in a rather non-specific manner. Molecular dynamics calculations revealed that these properties are attributable to rigid sheet-like structures of the cellulose derivatives and the subsequent exposure of their hydrophobic moieties to solvents.1. Introduction
Cellulose (linear β-1,4-glucan) is the most promising candidate of eco-materials, since it is the most abundant biopolymer in nature. In addition, cellulose is advantageous as a chiral scaffold, since it has an enormous number of chiral centers. For example, silica gels coated with cellulose derivatives are now widely used as stationary phases for chiral separation [1-4]. Chemically modified celluloses find their application in pharmaceutical/medicinal areas because of their biocompatibility and biodegradability [5,6]. Artificial celluloses carrying multiple copies of carbohydrates are, in this respect, quite attractive bio-materials, since such cellulose derivatives should acquire enhanced affinity towards carbohydrate-binding proteins (lectins), toxins, and viruses, as well as excellent water solubility arising from the clustered carbohydrates [7-11]. A limited example of cellulose-based glycoclusters can be, however, found in the literature [12]. In spite of these great possibilities, chemical modification of cellulose, that is the very first step to access cellulose-based advanced materials, still contain tedious obstacles.
The traditional approach to develop chemically modified celluloses can be divided into two categories. The first one is direct modification of native cellulose, by which various cellulose derivatives have been developed so far [13-18]. This direct approach was adopted to develop cellulose-based glycoclusters [12]. Although this strategy is quite simple, it suffers from one serious problem: that is, random modification and the structural heterogeneity of resultant cellulose derivatives. Native cellulose has no reactive functionality except for hydroxy groups and therefore, modification onto cellulose is mainly based on nucleophilic substitution reactions between these hydroxy groups and electrophiles. The hydroxy groups of cellulose have, however, similar reactivity and therefore, regio-selective and quantitative reactions are hardly accomplished. To the best of our knowledge, no regio-selective/quantitative approach towards cellulose derivatives with perfect structural homogeneity has been established. The cellulose derivatives developed so far, in fact, have functional appendages at random positions along their cellulose main chains. Time and labor consuming characterization processes (1H/13C NMR, HPLC analysis after digestion, etc.) are, therefore, inevitable to reveal their structural details. Furthermore, the structure-function relationship that is essential for tuning or refinement of their properties, is hardly obtained. These difficulties pose a serious barrier for non-specialists of organic chemistry to participate in cellulose chemistry.
The second approach to access cellulose derivatives is enzymatic polymerization of chemically modified glucoside monomers to the corresponding cellulose derivatives (bottom-up approach) [19-21]. The most potential advantage of this approach is perfect structural homogeneity of the resultant cellulose derivatives. Furthermore, if alternatively functionalized (AB-type) cellobiosides (p-1,4-linked diglucosides) are used as monomers, alternatively modified celluloses (…ABABAB…) can be obtained [22-24]. Although this chemo-enzymatic approach is quite powerful, it also suffers from some serious problems. Since preparation of the glucoside/cellobioside monomers requires tedious synthetic routes, research groups having specialists for organic synthesis can pursue this approach. Furthermore, the introduced functional modules are highly limited, since the monomers having large functional appendages cannot be polymerized because of their lowered affinities to the enzymes. In fact, the monomers having substituents larger than O-methyl group were reported to be hardly polymerized to the corresponding cellulose derivatives. Furthermore, hydrolysis (reverse reaction of the enzymes) of the cellulose derivatives simultaneously occurs and usually molecular weight of the obtained cellulose derivatives is quite low. These disadvantages of the bottom-up approach strongly hinder their wide application. Convenient, general, quantitative and regio-selective methods for direct modifications of cellulose are, therefore, strongly desired to accelerate participation of researchers from wide scientific fields in cellulose chemistry.
Recently, one of the authors with Shinkai's research group has developed an easy synthetic approach towards functionalized curdlan (β-1,3-glucan) through a unique two step strategy [25-27]. In this synthetic approach, native curdlan is firstly brominated with triphenylphosphine and carbon tetrabromide and then azidated with sodium azide to afford 6-azido-6-deoxycurdlan (CUR-N3). The structural advantage of 6-azido-6-deoxycurdlan is its perfect homogeneity: that is, all 6-OH groups of curdlan are selectively and quantitatively converted into 6-N3 groups without any side reactions at 2-OH or 3-OH groups. The subsequent coupling with alkyne-terminated functional modules (oligosaccharides, ferrocene, porphyrin, etc.) mediated by Cu+ (click chemistry) afforded curdlan derivatives having the functional modules exclusively at all 6C positions. This success encouraged us to apply this synthetic methodology towards cellulose chemistry, and recently, we reported a short communication in which artificial cellulose having N-linked β-lactosides (Cel-N-Lac) can be synthesized through a similar synthetic strategy [28]. Although Cel-N-Lac can be dissolved into water to some extent to show strong binding affinity towards Recinis communis aggulutinin (RCA120, βGal specific), its water solubility and lectin specificity are not so high. We assumed that hydrophobic phenyl spacers between β-lactoside appendages and cellulose main chain, lower not only the water solubility but also the lectin specificity. In this respect, we newly synthesized three cellulose-based glycoclusters having O-/N -linked β-maltosides and O-linked β-lactosides and carried out detailed investigation on their structure-function relationship that is informative to develop practical cellulose-based glycoclusters. We herein report detailed synthetic protocols of these cellulose-based glycoclusters and their properties including water solubility and lectin affinity.
2. Results and Discussion
2.1. Synthesis of alkyne-terminated oligosaccharides
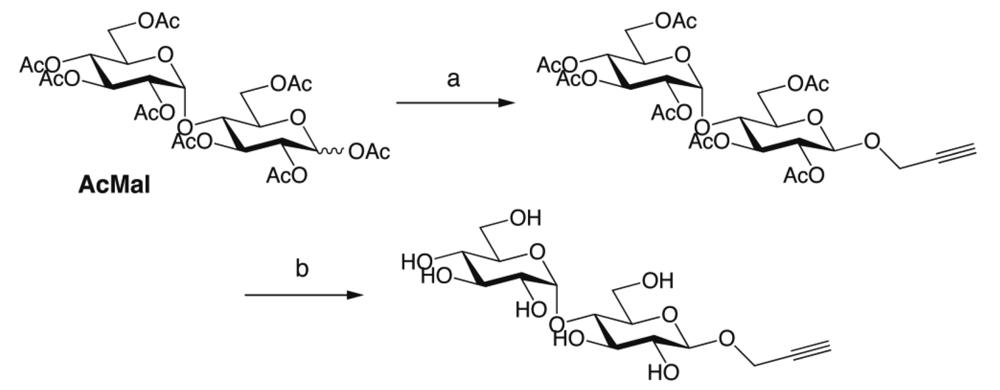
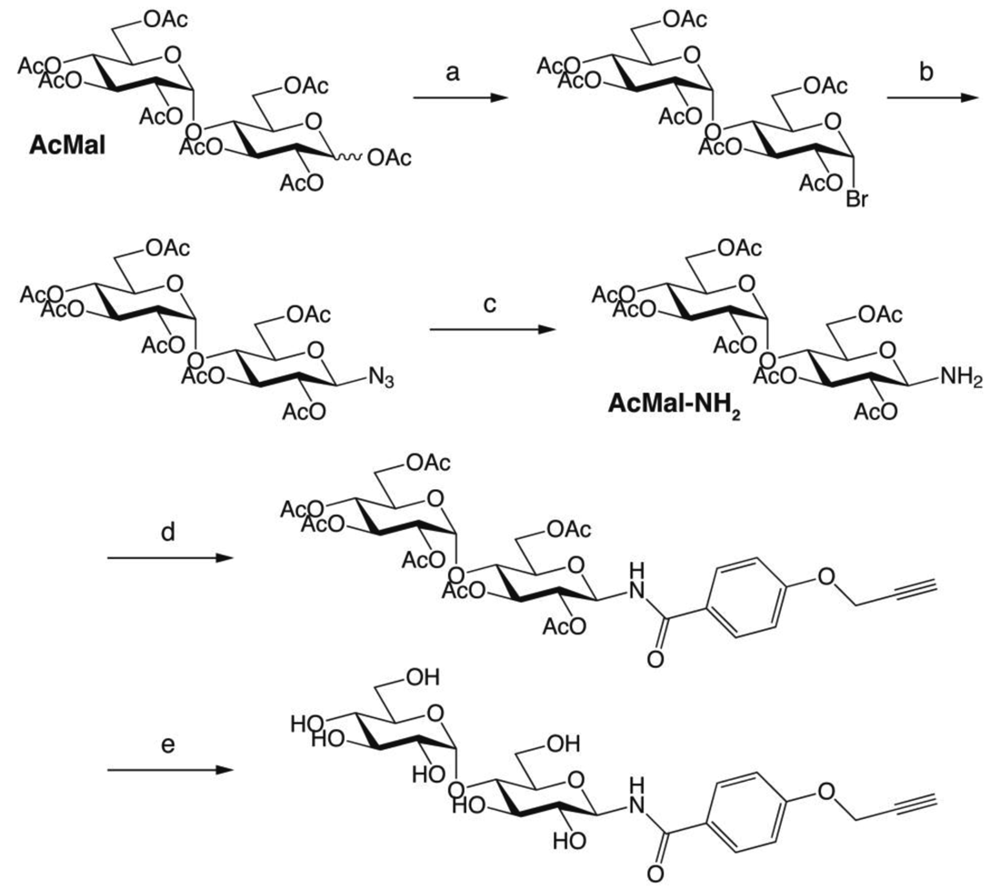
Oligosaccharides carrying terminal alkyne were prepared through two different synthetic routes. In the first one (Scheme 1), per-acetylated maltose (AcMal) and lactose (AcLac) were coupled with propargyl alcohol by treating with boron trifluoride. The following deacetylation attained by sodium methoxide in methanol gave O-linked β-maltoside and β-lactoside having terminal alkyne, respectively. On the other hand, in the second scheme (Scheme 2), AcMal and AcLac were firstly brominated with hydrogen bromide, azidated with sodium azide, and then hydrogenated to give the corresponding oligosaccharide derivatives having amino functionality at their anomeric positions (AcMal-NH2 and AcLac-NH2, respectively). The resultant oligosaccharides were coupled with a benzoic acid derivative having terminal alkyne and then, deacetylated to give N-linked β-maltoside and β-lactoside having terminal alkyne, respectively.
2.2. Preparation of 6-azido-6-deoxycellulose
Cellulose (DPn = 280) was dissolved into N,N-dimethylacetoamide (DMA) containing LiCl by stirring at 80 °C for 24 h and then, converted into 6-bromo-6-deoxycellulose (Cel-Br) through activation of its primary hydroxy groups with triphenylphosphine, followed by bromination with carbon tetrabromide (Scheme 3). It should be emphasized that the most popular and well studied reagent for the bromination of cellulose includes N-bromosuccinimide (NBS) [13-18]. We, however, used carbon tetrabromide instead of NBS. Although we first carried out 13C NMR measurement to reveal structural details of Cel-Br, its 13C NMR spectrum was too noisy to be assigned, suffering from its low solubility in any deuterated solvents (e.g., DMSO-d6). We therefore next carried out azidation reaction without full characterization of Cel-Br. The subsequent azidation was attained by treating Cel-Br with NaN3 in a DMA/dimethylsulfoxide (DMSO) mixed solvent system at 85 °C for 42 h to afford 6-azide-6-deoxycellulose (Cel-N3). It should be noted that solubility of these cellulose derivatives drastically changes, that is, Cel-Br is relatively soluble in DMA but hardly soluble in DMSO, and Cel-N3 is less soluble in DMA but well-soluble in DMSO. When we carried out this azidation in DMA solution throughout the reaction, partially azidated cellulose was precipitated and perfect conversion from Cel-Br to Cel-N3 was never achieved. We therefore started the azidation in DMA solution and then suitably added DMSO to the reaction mixture to keep the mixture homogeneous.
Regio-selectivity and quantitativity of these reactions were assessed by 13C NMR spectrum of Cel-N3 (Figure 1). The peak assignable to hydroxymethyl groups (–CH2OH, 60.90 ppm) entirely disappears and that of azidomethyl group (–CH2N3, 50.66 ppm) newly appears. Furthermore, the simple monosaccharide-like spectrum is composed of six predominant peaks which implies a highly homogeneous structure of Cel-N3 along its main chain. It should, however, be noted that some small but unneglectable peaks accompany the predominant peaks, indicating that some undesired side reactions occur at the other positions, presumably 2-OH and 3-OH groups. This superfluous azidation was supported by elemental analysis, in which Cel-N3 shows N/C ratio of 0.60567 ± 0.00059. This N/C ratio means that an averaged number of azide group introduced onto the repeating unit, or degree of substitution (DS), is 1.0386 ± 0.0010, slightly higher than that expected for Cel-N3 with perfect homogeneity (DS = 1.0000). Although a limited side reaction was observed as mentioned above, it should be emphasized that the structural homogeneity (C6-selectivity and quantitativity) attained through our protocol is equal with, or higher than, that attained through conventional methodology using NBS as a bromination reagent [13-18].
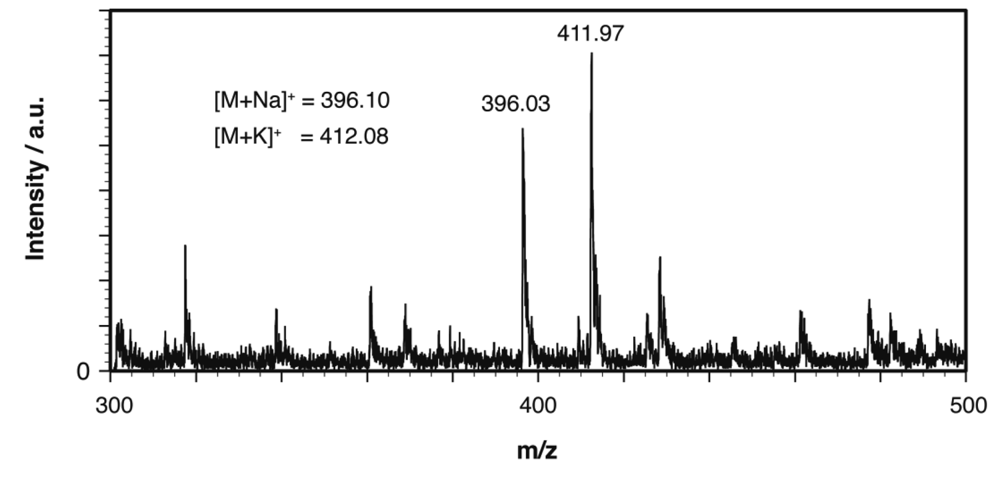
The structural homogeneity of Cel-N3 was also confirmed by acid-catalyzed hydrolysis of Cel-N3 followed by tin layer chromatographic (TLC) and matrix assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectral analysis of the hydrolysate. Briefly, we incubated Cel-N3 in aqueous HCl (35 % v/v) at the ambient temperature for five days and the resultant hydrolysate was acetylated in a mixture of acetic anhydride and pyridine. The TLC analysis of the acetylated product showed only one main spot arising presumably from 1,2,3,4-tetra-O-acetyl-6-azide-6-deoxyglucose (Ac4Glc-6-N3) and no other spot attributable to the byproducts can be detected. The quantitative conversion was also supported by the MALDI-TOF mass spectral analysis showing two main peaks at m/z = 396.03 and 411.97 that can be attributed to [M + Na ]+ (calc. 396.10) and [M + K ]+ (calc. 412.08), respectively (Figure 2). Together with the aforementioned IR spectral, 13C NMR spectral, and elemental analysis data, these two additional data clearly indicate the high structural homogeneity of Cel-N3.
2.3. Cu+-catalyzed [3 + 2]-cycloadditions of the alkyne-terminated oligosaccharides onto Cel-N3
Although Cel-N3 with highly homogeneous repeating structure was obtained as mentioned above, the azide functionality has little function, such as protein recognition and light harvesting functions, and therefore, Cel-N3 itself cannot find any practical applications. On the other hand, Cel-N3 can give full scope to its ability when it is used as a substrate for further modification to access cellulose-based advanced materials. However, if the further modification reaction is accompanied by undesired side reactions and/or incomplete conversions, the resultant cellulose derivatives finally have heterogeneous repeating units. Exclusive chemoselectivity and perfect conversion yield are therefore highly required for the further modification reaction. To satisfy these criteria, we applied click chemistry for the further modification on Cel-N3 [29-33].
A coupling of the alkyne-terminated oligosaccharides and Cel-N3 can be achieved in DMSO containing CuBr2, ascorbic acid, and propylamine. The resultant cellulose derivatives having O-/N -linked &beta-lactoside (Cel-O-Lac and Cel-N-Lac, respectively) and O-/N -linked β-maltoside (Cel-O-Mal and Cel-N-Mal, respectively) can be obtained as white powder after purification through dialysis (water, MWCO8000) and the subsequent lyophilization.
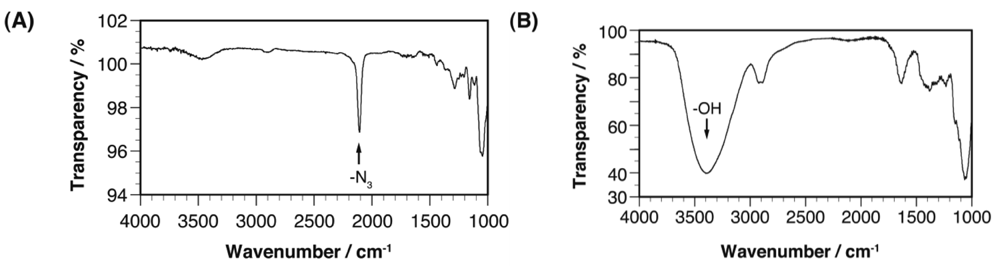
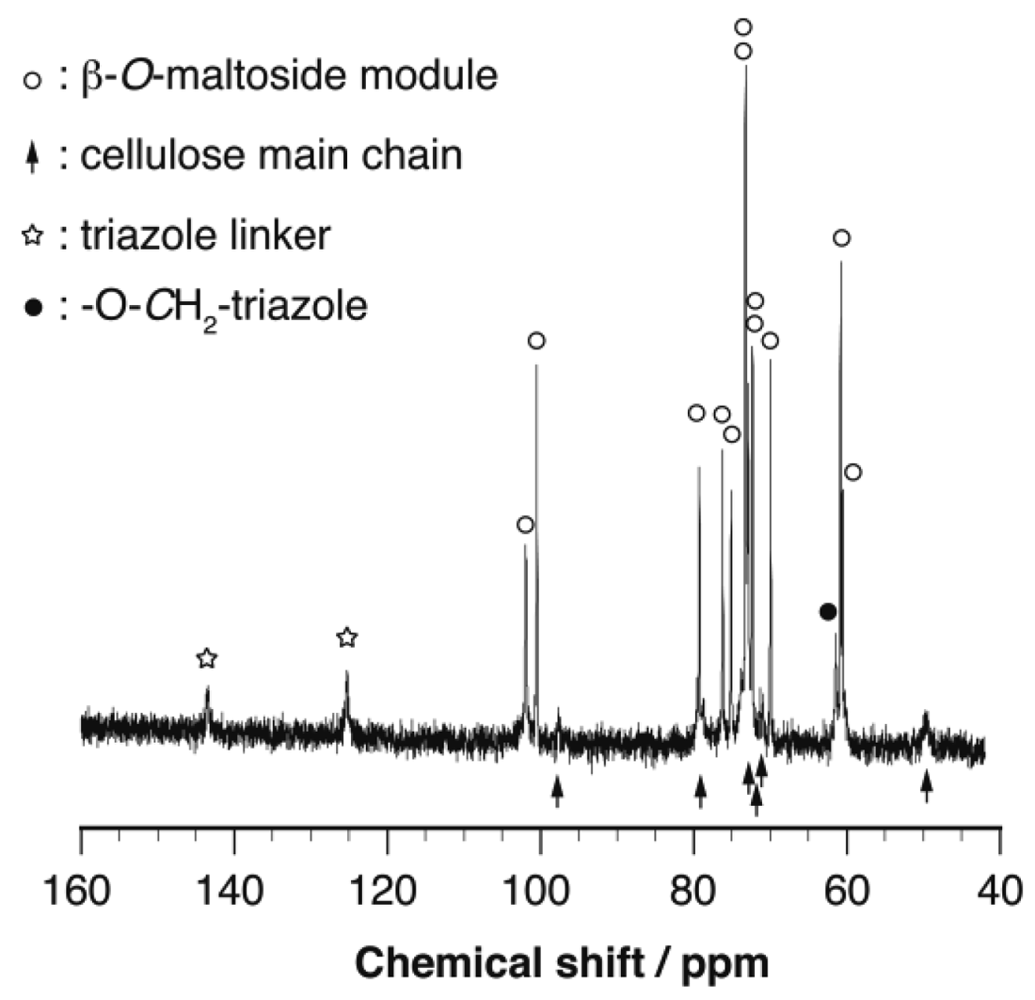
Characterization of these cellulose-based glycoclusters was achieved based on their IR and 13C NMR spectra. In their IR spectra, the strong azide peak (2,101 cm−1) observed in that of Cel-N3 was entirely diminished, suggesting perfect conversion of azidomethyl functionalities into oligosaccharides tethered with 1,4-triazole linkers (Figure 3). The quantitative conversion of Cel-N3 into the corresponding cellulose-based glycoclusters was also supported by their 13C NMR spectra, in which the peak assignable to azidomethyl functionalities (–CH2N3) entirely disappeared and those of 1,4-triazole linkers (ca. 125 and 142 ppm) and oligosaccharide appendages newly appeared (Figure 4). Only a limited number of unassignable peaks were observed in the 13C NMR spectra, indicating that structural homogeneity is maintained through the coupling reaction taking full advantages of click chemistry. This effective conversion of the azidomethyl group to the triazole-tethered oligosaccharides was also supported by elemental analysis. For example, Cel-O-Mal showed N/C ratio of 0.1700 ± 0.0014 indicating that an average number of β-maltoside units attached onto the repeating unit (DS) is 1.0198 ± 0.0084, that is comparable to that of Cel-N3. It should be emphasized here that these extremely bulky carbohydrate modules can never be introduced into polysaccharides through the chemo-enzymatic bottom-up strategy. To the best of our knowledge, our approach is the only one to access β-1,4-glucan-based glycoclusters both with high structural homogeneity of the cellulose main chain and large multivalency of the oligosaccharide appendages.
2.4. Water solubility of the cellulose derivatives
Cellulose itself is hardly soluble in water, mainly because of intramolecular hydrogen bonding networks (e.g., between O5 and 3-OH of the preceding repeating unit). The resultant rigid tape-like conformation induces enhanced packings of cellulose strands stabilized by intermolecular hydrogen bondings and hydrophobic interactions. Chemical modification of cellulose to increase its water solubility is quite important to develop cellulose-based advanced materials, especially those for pharmaceutical and medicinal purposes. To assess water solubility of the cellulose-based glycoclusters, we added the cellulose-based glycoclusters into small amounts of water and then, the resultant turbid mixtures were kept at 60 °C for one day to prepare their saturated aqueous solutions. After cooling to an ambient temperature followed by centrifugations to remove precipitates, carbohydrate concentrations in their supernatants were assayed through a well-known phenol-sulfric acid protocol.
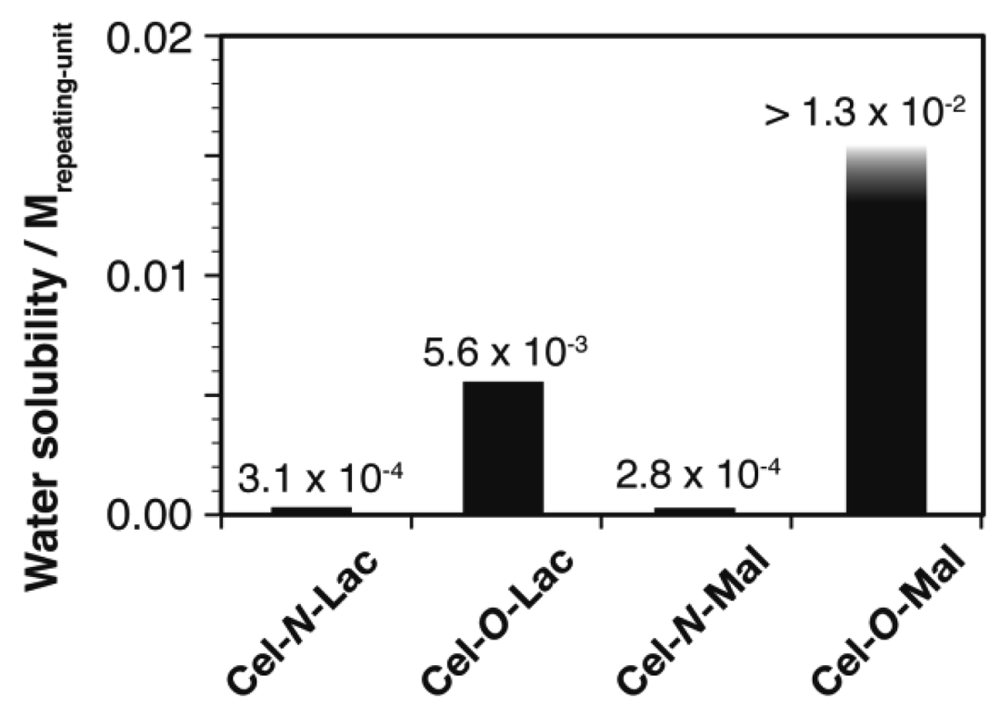
Although cellulose (DPn = 280) is hardly soluble in water, the cellulose-based glycoclusters, especially Cel-O-Lac and Cel-O-Mal, show enhanced water solubility arising from their bulky and water-soluble oligosaccharide appendages (Figure 5). The water solubility of the cellulose-based glycoclusters, however, drastically differ from each other depending on chemical structures of the appendages. Through comparison of these data, two important tendencies can be found in their structure-solubility relationship. First, one is a critical effect of hydrophobic spacers: that is, cellulose-based glycoclusters having N-linked oligosaccharides are less soluble in water than the corresponding O-linked counterparts. This low water solubility of N-linked glycoclusters is clearly attributable to hydrophobic phenyl spacers linking between the cellulose main chain and the oligosaccharide appendages. The other is the importance of its oligosaccharide structure: that is, Cel-O-Mal has much higher water solubility than that of Cel-O-Lac. The low water solubility of Cel-O-Lac probably arises from its hydrophobic α-face of non-reducing β-galactoside units.
The observed negative effects arising from the hydrophobic spacers and the α-face on their water solubility are much more drastic in comparison with those observed for glycoclusters, based on flexible artificial polymers such as polystyrene [34-40] and polyacrylamide [41-46]. For example, polystyrenes having β-lactoside appendages still have excellent water solubility. In these glycoclusters, the flexible nature of the polymer main chain permits its structural rearrangement and therefore, these glycoclusters take unique cylindrical structures composed of hydrophobic cores and hydrophilic surfaces [47-49]. As a result, hydrophobic moieties interact with each other in an intrapolymer fashion, and intermolecular interactions to induce polymer aggregates are highly limited. Whereas, in the case of the cellulose-based glycoclusters, the rigid nature of the cellulose main chain should hinder their structural rearrangements and then, their hydrophobic moieties should still remain exposed to the solvent. Especially, in the case of Cel-N-Mal and Cel-N-Lac, the aforementioned structural rigidity, and the resultant exposure of both phenyl and triazole spacers, critically lower the water solubility.
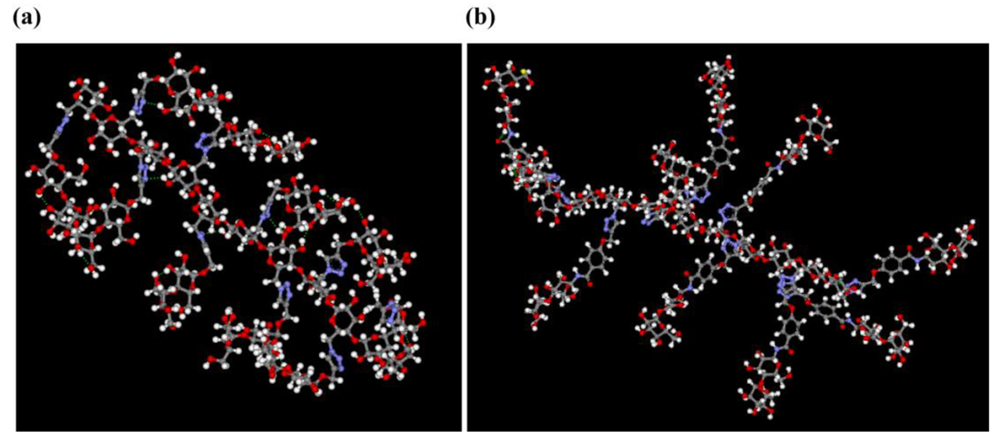
Our hypothesis was supported by molecular dynamics (MD) calculation, in which 10-mers of Cel-O/N-Mal were constructed and then, dynamics calculations (1,000,000 steps, CHARMm, 300 K, NVT, GBSW solvent model) were carried out after minimization, heating, and equilibration processes. The most stable conformations for these cellulose-based glycopolymers during the dynamics processes are shown in Figure 6, in which they both take sheet-like structures with their hydrophobic phenyl/triazole spacers exposed to the solvent. These sheet-like structures should result in enhanced intermolecular networks composed of hydrophobic interactions and hydrogen bondings to reduce water solubility. Since the most stable conformations of Cel-O-Mal and Cel-N-Mal take sheet-like forms and are not different from each other, the observed much lower water solubility of Cel-N-Mal in comparison to that of Cel-O-Mal should arise from the additional hydrophobic phenyl spacers in their structures.
2.5. Affinity between the cellulose-based glycoclusters and lectins
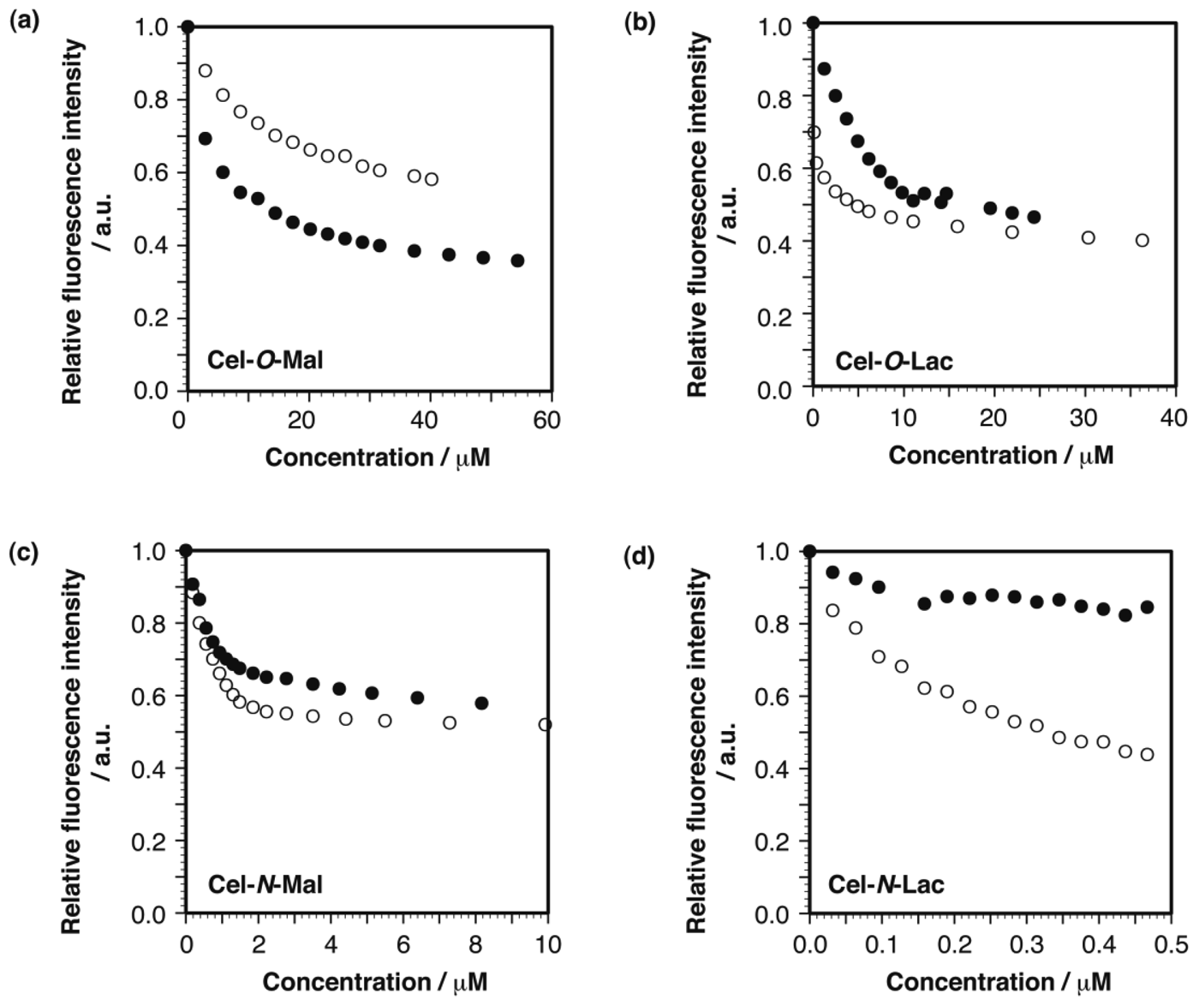
Affinity between the cellulose-based glycoclusters and lectins was assessed through fluorescence titration assay using fluorescein isothiocyanate (FITC) -labeled lectins [50,51]. In this assay, we used concanavalin A (ConA) and ricinis communis agglutinin (RCA120) as lectins that specifically bind to αMan/αGlc and βGal, respectively [52]. We, therefore, expected that Cel-O/N-Mals specifically bound to FITC-ConA and Cel-O/N-Lacs to FITC-RCA120. Results of the binding assays were, however, much different from our expectations: that is, little lectin specificity was observed for the cellulose-based glycoclusters. For example, as shown in Figure 7(a), an injection of Cel-O-Mal induced a drastic decrement in fluorescent intensity not only for FITC-ConA but also for FITC-RCA120, indicating a non-specific binding. Such non-specific lectin bindings were also observed for the other cellulose derivatives, as shown in Figure 7(b–d). We assume that the non-specific lectin bindings arose from hydrophobic interactions between the cellulose-based glycoclusters and lectins. The aforementioned sheet-like structure and the resultant exposure of hydrophobic moieties should play negative roles not only for their water solubility but also for lectin specificity.
3. Experimental Section
3.1. General
1H and 13C NMR spectra were acquired on a JEOL AL300 (Jeol Datum, Ltd.) in CDCl3, CD3OD, D2O, or DMSO-d6 at 300 MHz. The chemical shifts were reported in ppm (δ) relative to Me4Si. IR spectra were recorded on a JASCO FT/IR-4100 fourier transform infrared spectrometer (Jasco Co., Ltd.). Matrix assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectra were recorded on SHIMAZU AXIMA-CFR+ (Shimazu, Ltd.). Fluorescence titration assay was carried out using JASCO FP-6200 spectrofluorometer (Jasco Co., Ltd.). Molecular dynamics calculation was carried out by using Discovery Studio 2.0 program (Accelrys Co., Ltd.). Elemental analysis was conducted by Chemical Analysis Center in Chiba University. Silica gel 60 N (particle size 63–210 mm) for column chromatography was purchased from Kanto Chemical Co. Inc. Thin layer chromatography (TLC) was carried out with Merck TLC aluminum sheets pre-coated with silica gel 60 F254. Cellulose (Avicel microcrystalline cellulose, DPn = 280) was kindly supplied from FMC Co. All other chemicals were purchased from Wako Pure Chemical Industries Ltd., Kishida Chemicals Co. Ltd., Kanto Chemical Co. Ltd. and Funakoshi Co. Ltd. and used without purification.
3.2. Synthesis of methyl p-propargyloxybenzoate
To methyl p-hydroxybenzoate (1.05 g) in DMF (50 mL), K2CO3 (3.41 g) was added and the resultant mixture was stirred at 60 °C for 1 h. After cooling down to an ambient temperature, propargyl bromide (1.28 mL) was added, and then stirring was continued at room temperature overnight. The reaction mixture was then diluted with ethyl acetate and washed with NaCl-saturated aqueous solution and deionized water repeatedly. The resultant organic layer was dried over anhydrous MgSO4, and evaporated to give methyl p-propargyloxybenzoate as a white powder in 87% yield: [M + Na ]+ = 213.14 (calc. 213.10); 1H NMR (300 MHz, CDCl3, TMS): 8.01 (d, J = 9.0 Hz, 2H), 7.00 (d, J = 9.0 Hz, 2H), 4.75 (d, J = 2.4 Hz, 2H), 3.89 (s, 3H), 2.56 (t, J = 2.4 Hz, 1H); 13C NMR (300 MHz, CDCl3, TMS): 166.72, 161.14, 131.56, 123.43, 114.47, 77.81, 76.10, 55.81, 51.96; IR (KBr, cm −1): 1703.
3.3. Synthesis of p-propargyloxybenzoic acid
To methyl p-propargyloxybenzoate (0.42 g) in THF (500 mL), KOH (1.20 g) in water (10 mL) was added and then, the mixture was refluxed for 14 h. After acidifying the resultant reaction mixture (ca. pH 2) by an addition of a proper amount of aqueous HCl, the mixture was evaporated to dryness and purified by chromatography on a silica gel column (30 cm long; 3 cm i.d.; chloroform-methanol = 9:1 in v/v) to give p-propargyloxybenzoic acid as a white powder (31%): [M + Na ]+ = 169.98 (calc. 169.06); 1H NMR (300 MHz, CD3OD, TMS): 7.97 (d, J = 9.0 Hz, 2H), 7.03 (d, J = 9.0 Hz, 2H), 4.80 (d, J = 2.4 Hz, 2H), 2.98 (t, J = 2.4 Hz, 1H); 13C NMR (300 MHz, CDCl3 containing a small amount of CD3OD, TMS): 169.2, 161.8, 131.98, 124.31, 114.52, 77.38, 76.20, 55.90; IR (KBr, cm−1): 1,680.
3.4. General procedure for the synthesis of p-propargyloxybenzamido-2,3,6,2′,3′,4′,6′-hepta-O-acetyl-β-maltoside/lactoside
A thionyl chloride solution (10 mL) containing p-propargyloxybenzoic acid (0.14 g) was refluxed for 5 h and evaporated to dryness. The resultant yellow solid was dissolved into THF and then, mixed with 2,3,6,2′,3′,4′,6′-hepta-O-acetyl-β-maltosylamine or -lactosylamine (0.53 g) in THF. Triethylamine was added to the reaction mixture immediately and the resultant mixture was stirred for 5 min, diluted with ethyl acetate, and washed with 0.5 N HCl aq and NaHCO3 aq, repeatedly. The resultant organic layer was dried over anhydrous MgSO4, evaporated, and purified by chromatography on a silica gel column (30 cm long; 3 cm i.d.; chloroform-methanol = 25:1 in v/v) to give p-propargyloxybenzamido-2,3,6,2′,3′,4′,6′-hepta-O-acetyl-β-maltoside or -lactoside as a white powder.
3.4.1. p-propargyloxybenzamido-2,3,6,2′,3′,4′,6′-hepta-O-acetyl-β-maltoside
Yield: 42%; [M + Na ]+ = 817.32 (calc. 816.24); 1H NMR (300 MHz, CDCl3 containing a small amount of CD3OD, TMS) d 7.74 (d, J = 9.0 Hz, 2H), 7.01 (d, J = 9.0 Hz, 2H), 5.46 (t, J = 9.0 Hz, 1H), 5.47–5.40 (m, 2H), 5.38 (dd, J = 8.4 and 9.0 Hz, 1H), 5.07 (t, J = 9.9 Hz, 1H), 4.91 (d, J = 9.9 Hz, 1H), 4.87 (d, J = 5.4 and 6.3 Hz, 1H), 4.74 (d, J = 2.4 Hz, 2H), 4.46 (dd, J = 2.7 and 12.3 Hz, 1H), 4.30–4.20 (m, 2H), 4.08–3.87 (m, 4H), 2.56 (t, J = 2.4 Hz, 1H), 2.10 (s, 3H), 2.09 (s, 3H), 2.05 (s, 3H), 2.04 (s, 3H), 2.02 (s, 6H), and 2.01 (s, 3H).
3.4.2. p-propargyloxybenzamido-2,3,6,2′,3′,4′,6′-hepta-O-acetyl-β-lactoside
Yield: 48%; [M + Na ]+ = 817.29 (calc. 816.24); 1H NMR (300 MHz, CDCl3, TMS) d 8.11 (d, J = 9.0 Hz, 2H), 7.33 (d, J = 9.0 Hz, 2H), 6.92 (d, J = 8.4 Hz, 1H), 5.42 (t, J = 9.0 Hz, 1H), 5.37 (m, 2H), 5.12 (dd, J = 8.1 and 10.5 Hz, 1H), 5.00 (t, J = 9.6 Hz, 1H), 4.96 (dd, J = 3.6 and 10.5 Hz, 1H), 4.82 (d, J = 2.4 Hz, 1H), 4.50–4.45 (m, 1H), 4.19–4.15 (m, 2H), 4.10–4.06 (m, 1H), 3.92 (s, 3H), 3.89 (t, J = 7.5 and 7.5 Hz, 1H), 3.86-3.81 (m, 2H), 2.58 (t, J = 2.4 Hz, 1H), 2.16 (s, 3H), 2.12 (s, 3H), 2.08 (s, 3H), 2.07 (s, 3H), 2.05 (s, 3H), 2.04 (s, 3H), and 1.96 (s, 3H); 13C NMR (300 MHz, CDCl3, TMS) d 171.60, 170.35, 170.32, 170.12, 170.06, 169.35, 168.98, 165.31, 160.03, 130.68, 121.44, 113.99, 100.86, 79.13, 78.58, 77.93, 76.00, 74.57, 72.24, 71.10, 70.92, 70.67, 68.93, 66.56, 61.85, 60.81, 55.80, 20.83, 20.74, 20.70, 20.61, 20.60, 20.58, and 20.46; IR (KBr, cm−1): 1,734, 1,639, and 1,577.
3.5. General procedure for the synthesis of p-propargyloxybenzamido-β-maltoside/lactoside
To p-propargyloxybenzamido-2,3,6,2′,3′,4′,6′-hepta-O-acetyl-β-maltoside or -lactoside (0.01 g) in MeOH/THF mixture (1:1 v/v, 60 mL), aqueous ammonia (10 mL) was added. After stirring at an ambient temperature overnight, the resultant mixture was evaporated to dryness and the residue was re-dissolved into water and the resultant solution was lyophilized to give p-propargyloxybenzamido-β-maltoside or -lactoside as white powder.
3.5.1. p-propargyloxybenzamido-β-maltoside
Yield: 99%; [M + Na ]+ = 523.05 (calc. 522.47); 1H NMR (300 MHz, D2O): 7.71 (d, J = 9.0, 2H), 7.01 (d, J = 9.0, 2H), 5.30 (d, J = 3.9, 1H), 5.05 (d, J = 9.0, 1H), 4.73 (d, J = 2.4, 2H), 3.75–3.19 (m, 12H), 2.82 (t, J = 2.4, 1H); IR (KBr, cm−1): 3,343, 1,641, and 1,569.
3.5.2. p-propargyloxybenzamido-2,3,6,2′,3′,4′,6′-hepta-O-acetyl-β-lactoside
Yield: 97%; [M + Na ]+ = 522.89 (calc. 522.47); 1H NMR (300 MHz, DMSO-d6, TMS): 8.76 (d, J = 8.7, 1H), 7.89 (d, J = 9.0, 2H), 7.04 (d, J = 9.0, 2H), 5.14 (d, J = 4.2, 1H), 5.07 (d, J = 5.1, 1H), 4.97 (t, J = 8.7, 1H), 4.87 (d, J = 2.4, 2H), 4.81 (d, J = 4.8, 1H), 4.74 (s, 1H), 4.68 (t, J = 4.8, 1H), 4.57 (t, J = 6.3, 1H), 4.54 (d, J = 4.5, 1H), 4.23 (d, J = 7.2, 1H), 3.76-3.28 (br m, 12H), 2.49 (t, J = 2.4 Hz, 1H); 13C NMR (300 MHz, DMSO-d6, TMS): 165.88, 159.51, 129.14, 126.97, 114.13, 103.13, 80.46, 80.03, 78.64, 78.02, 76.45, 75.61, 75.37, 73.13, 71.62, 70.51, 68.03, 60.46, 60.28, 55.50; IR (KBr, cm −1): 3,361, 1,639, and 1,577.
3.6. General procedure for the synthesis of 2,3,6,2′,3′,4′,6′-hepta-O-acetyl-2″-propargyloxy-β-maltoside/lactoside
BF3OEt2 (5.0 mL) was added to 1,2,3,6,2′3′4′6′-octa-O-acetyl-maltose or -lactose (3.2 g) and propargyl alcohol (5.0 mL) in anhydrous CH2Cl2 (10 mL) at room temperature and stirring was continued for 45 h under nitrogen atmosphere. The resultant mixture was diluted with ethyl acetate and washed with NaHCO3 saturated aqueous solution. The organic layer was dried over anhydrous MgSO4, filtered and concentrated to dryness. In the case of the maltoside, MALDI-TOF-MS and TLC analysis of the residue showed partially deacetylated product. The residue was, therefore, subjected to full deacetylation (next step) without purification and characterization at this step.
3.6.1. 2,3,6,2′,3′,4′,6′-hepta-O-acetyl-2″-propargyloxy-β-maltoside
[M+Na ]+ = 696.85 (calc. 697.21).
3.6.2. 2,3,6,2′,3′,4′,6′-hepta-O-acetyl-2″-propargyloxy-β-lactoside
Yield: 41%; 1H NMR (300 MHz, CDCl3, TMS): 5.35 (d, J = 2.7 Hz, 1H), 5.23 (t, J = 9.3 Hz, 1H), 5.11 (dd, J = 8.1 and 10.2 Hz, 1H), 4.96 (dd, J = 3.3 and 7.8 Hz, 1H), 4.75(d, J = 7.8 Hz, 1H), 4.34 (d, J = 2.4 Hz, 2H), 4.14–4.05 (m, 2H), 3.90–3.79 (m, 2H), 3.66–3.62 (m, 1H), 2.47 (t, J = 2.1 Hz, 1H), 2.18 (s, 3H), 2.13 (s, 3H), 2.07–2.05 (m, 12H), 1.97 (s, 3H); [M+Na ]+ = 697.26 (calc. 697.21); IR (KBr, cm −1) 1,753 (acetyl).
3.7. General procedure for the synthesis of 2″-propargyloxy-β-maltoside/lactoside
A catalytic amount of sodium methoxide was added to crude Ac-Mal-yn in a MeOH (50 mL)/THF (50 mL) mixed solvent system and the resultant mixture was stirred for 6 h. The resultant mixture was neutralized on DOWEX and evaporated to dryness. The resultant residue was subjected to purification by silica-gel column chromatography (CHCl3/MeOH (2/1)) to obtain 2″-propargyloxy-β-maltoside (βMal-yn) as white powder after lyophilization. In the case of 2″-propargyloxy-β-lactoside (βLac-yn), pure Ac-Lac-yn was used as a substrate in this reaction. We, therefore, obtained pure βLac-yn without any column chromatographic purification process.
3.7.1. 2″-propargyloxy-β-maltoside
Yield: 38% (2 steps); [M+Na ]+ = 402.66 (calc. 403.12); 1H NMR (300 MHz, D2O, HOD): 5.26 (d, J = 3.6 Hz, 1H), 4.52 (d, J = 8.1 Hz, 1H), 4.33 (s, 2H), 3.78 (t, J = 12.9 Hz, 1H), 3.70–3.48 (m, 8H), 3.43 (dd, J = 3.6 and 9.9 Hz, 1H), 3.27 (t, J = 9.3 Hz, 1H), 3.19 (t, J = 8.7 Hz, 1H), 2.78–2.75 (m, 1H); 13C NMR (300 MHz, D2O): 101.016 (αGlc1), 100.200 (βGlc1), 79.390 (–CH2–CCH), 77.231, 77.000, 76.843 (–CH2–CCH), 76.643, 75.294, 73.448, 73.357, 72.294, 69.978, 61.308 (Glc6), 61.127 (Glc6), 57.204 (–CH2–CCH); IR (KBr, cm−1) 3,381.
3.7.2. 2″-propargyloxy-β-lactoside
Yield: 98%; [M+Na ]+ = 403.09 (calc. 403.12); 1H NMR (300 MHz, D2O, HOD): 4.53 (d, J = 7.93 Hz 1H), 4.35 (dd, J = 1.90 and 8.66 Hz, 1H), 4.32 (d, J = 7.72 Hz, 1H), 3.85 (d, J = 11.5 Hz, 1H), 3.80 (d, J = 3.03 Hz, 1H), 3.70–3.58 (m, 5H), 3.55–3.52 (m, 3H), 3.49–3.48 (m, 1H), 3.42 (t, J = 8.11 Hz, 1H), 3.32 (t, J = 8.28 Hz, 2H), 2.82 (s, 1H); 13C NMR (300 MHz, D2O): 103.282, 100.739, 79.136, 78.602, 76.769, 75.707, 75.206, 74.711, 72.959, 72.880, 71.311, 68.911, 61.403, 60.359, 56.958; IR (KBr, cm−1) 3,397, 2,919.
3.8. Synthesis of 6-bromo-6-deoxycellulose (Cel-Br)
Cellulose (DPn = 280, 2.05 g) and LiCl (5.65 g) was added into toluene (10 mL) and then, evaporated to dryness. To the resultant white powder, DMA (150 ml) was added under N2 atmosphere and the resultant mixture was stirred at 80 °C for 24 h. After cooling down to an ambient temperature, triphenylphosphine (12.2 g) in DMA (40 mL) was added and the stirring was continued at room temperature for 4 h. Carbon tetrabromide (10.5 g) in DMA (10 mL) was added, and then, the resultant reaction mixture was stirred at 60 °C overnight. The resultant reaction mixture was poured into water and then, the precipitate was washed with methanol and chloroform repeatedly to obtain Cel-Br as white powder in 71% yield. As mentioned in the manuscript, Cel-Br was hardly soluble in DMSO-d6 and its 13C NMR spectrum was too noisy to be assigned. We, therefore, carried out next azidation reaction without full characterization.
3.9. Synthesis of 6-azide-6-deoxycellulose (Cel-N3)
Cel-Br (2.00 g) was dissolved into DMA (150 mL) containing sodium azide (5.05 g) and then, the resultant mixture was stirred at 85 °C for 42 h. During the reaction, appropriate amounts of DMSO were suitably added to the reaction mixture to keep it homogeneous (the total amount of added DMSO was 150 mL). The resultant reaction mixture was poured into water and then, the precipitate was washed with cold water and methanol repeatedly to obtain Cel-N3 as white powder in 80% yield: 13C NMR (DMSO-d6, TMS): 101.76, 79.00, 73.92, 73.23, 73.20, 50.66; IR (ATR, cm-1): 3,479 and 2,101.
3.10. General procedure for the chemo-selective coupling between Cel-N3 and alkyne-terminatedoligosaccharides
CuBr2 (1.8 mg), ascorbic acid (7.5 mg), the alkyne-terminated oligosaccharides (ca. 400 mg) and propylamine (400 mL) were added to 6-azido-6-deoxycellulose (50 mg) in DMSO (0.80 mL) and the mixture was incubated at room temperature for 12 h. The mixture was dialyzed (water, MWCO8000) followed by lyophilization to give the cellulose-based glycoclusters (Cel-N-Mal, Cel-N-Lac, Cel-O-Mal and Cel-O-Lac).
3.10.1. Cel-N-Mal
13C NMR (300 MHz, DMSO-d6, TMS): 166.00, 160.43, 142.13, 129.26, 126.44, 125.87, 114.05, 100.55, 80.15, 79.39, 78.66, 77.10, 76.76, 73.75, 73.20, 73.20, 73.20, 73.01, 72.71, 72.34, 71.55, 69.97, 61.12, 60.81, 60.43; IR (ATR, cm−1): 3,371, 1,645, and 1,542.
3.10.2. Cel-N-Lac
13C NMR (300 MHz, DMSO-d6, TMS): 166.08, 160.45, 142.04, 129.17, 126.48, 125.73, 114.08, 103.55, 101.78, 80.47, 80.05, 79.88, 76.46, 75.63, 75.38, 75.37, 74.71, 73.15, 72.11, 71.64, 71.28, 70.55, 70.54, 68.07, 62.05, 60.50; IR (ATR, cm−1): 3,402, 1,643, and 1,567.
3.10.3. Cel-O-Mal
13C NMR (300 MHz, DMSO-d6, TMS): 143.45, 125.24, 101.91, 100.47, 97.66, 79.25, 78.75, 76.22, 75.07, 73.80, 73.21, 73.18, 72.90, 72.86, 72.64, 72.30, 69.95, 61.43, 60.81, 60.57, 49.74; IR (KBr, cm−1): 3,389, 2,920, 1,632, and 1,064.
3.10.4. Cel-O-Lac
13C NMR (300 MHz, DMSO-d6, TMS): 143.51, 125.44, 103.59, 101.79, 97.76, 79.81, 78.75, 75.22, 75.07, 73.80, 73.33, 72.90, 72.64, 71.30, 67.95, 61.43, 60.81, 60.57, 60.31, 59.61, 49.74; IR (ATR, cm−1): 3,391, 2,930, 1,633, and 1,045.
4. Conclusions
We established a simple synthetic strategy to access cellulose-based glycoclusters with excellent structural homogeneity: that is, the carbohydrate modules are attached exclusively onto the C6 position of the each repeating unit. Such structural homogeneity could never be achieved through the conventional glycosylation on native polysaccharides (polymer-modification approach). The resultant cellulose-based glycoclusters, however, showed low lectin specificity, possibly arising from the hydrophobic interactions between their hydrophobic moieties and hydrophobic residues of lectins. The enhanced hydrophobic interactions are attributable to the rigid sheet-like structure of cellulose-based glycoclusters and the subsequent exposure of the hydrophobic moieties to the solvent. Whereas, cellulose derivatives obtained by the conventional random modification have carbohydrate units not only at the C6 position but also at C2 and C3. Comparison of these data suggests an importance of C3 glycosylation to develop cellulose-based glycoclusters with excellent water solubility and lectin specificity. Since it is widely recognized that the extended tape-like conformation of cellulose is stabilized mainly by intrastranded O5-HO3 hydrogen bondings [53], the introduction of large carbohydrate units, especially onto the C3 position, should break the intrapolymer hydrogen bonding network, change the original tape-like conformation to a random coiled one, and increase water solubility of the cellulose derivatives. Such structure-function relationships, obtained by using cellulose derivatives with high structural homogeneity, should help in developing practical cellulose-based fine materials.
The facts reported in this paper strongly underline the usefulness of our synthetic strategy to develop cellulose-based advanced materials maintaining the intrinsic tape-like conformations. We are now directing our research efforts to develop such tape-like cellulose derivatives with unique properties. One such example includes nucleotide-appended cellulose to solubilize single-walled carbon nanotubes. The result will soon be reported elsewhere.










Acknowledgments
This work was partially supported by Grant-in-Aid for Scientific Research (No. 18750103) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. Financial support was also given by Special Research Fund of Toyo University. The cellulose used in this work was kindly supplied from FMC Co. Ltd.
References
- Li, J.-Q.; Ikai, T.; Okamoto, Y. Preparation and HPLC Application of Chiral Stationary Phase from 4-tert-Butylphenylcarbamates of Cellulose and Amylose Immobilized onto Silica Gel. J. Sep. Sci. 2009, 32, 2885–2891. [Google Scholar]
- Ikai, T.; Yamamoto, C.; Kamigaito, M.; Okamoto, Y. Organic-Inorganic Hybrid Materials for Efficient Enantioseparation Using Cellulose 3,5-Dimethylphenylcarbamate and Tetraethyl Orthosilicate. Chem. Asian J. 2008, 3, 1494–1499. [Google Scholar]
- Tang, S.; Ikai, T.; Tsuji, M.; Okamoto, Y. Immobilization and Chiral Recognition of 3,5-Dimethylphenylcarbamates of Cellulose and Amylose Bearing 4-(Trimethoxysilyl)phenylcarbamate Groups. Chirality 2010, 22, 165–172. [Google Scholar]
- Ikai, T.; Okamoto, Y. Structure Control of Polysaccharide Derivatives for Efficient Separation of Enantiomers by Chromatography. Chem. Rev. 2009, 109, 6077–6101. [Google Scholar]
- Tarng, D.-C.; Huang, T.-P.; Liu, T.-Y.; Chen, H.-W.; Sung, Y.-J.; Wei, Y.-H. Effect of Vitamin E-Bonded Membrane on the 8-Hydroxy 2′-Deoxyguanosine Level in Leukocyte DNA of Hemodialysis Patients. Kidney Int. 2000, 58, 790–799. [Google Scholar]
- Kanda, H.; Kubo, K.; Hamasaki, K.; Kanda, Y.; Nakao, A.; Kitamura, T.; Fujita, T.; Yamamoto, K.; Mimura, T. Influence of Various Hemodialysis Membranes on the Plasma (1-3)-beta-D-Glucan Level. Kidney Int. 2001, 60, 319–323. [Google Scholar]
- Miura, Y.; Ikeda, T.; Wada, N.; Sato, H.; Kobayashi, K. Chemoenzymatic Synthesis of Glycoconjugate Polymers: Greening the Synthesis of Biomaterials. Green Chem. 2003, 5, 610–614. [Google Scholar]
- Sato, H.; Miura, Y.; Saito, N.; Kobayashi, K.; Takai, O. A Micropatterned Carbohydrate Display for Tissue Engineering by Self-Assembly of Heparin. Surf. Sci. 2007, 601, 3871–3875. [Google Scholar]
- Mahon, E.; Aastrup, T.; Barboiu, M. Dynamic Glycovesicle Systems for Amplified QCM Detection of Carbohydrate-Lectin Multivalent Biorecognition. Chem. Commun. 2010, 46, 2441–2443. [Google Scholar]
- Hayashida, O.; Mizuki, K.; Akagi, K.; Matsuo, A.; Kanamori, T.; Nakai, T.; Sando, S.; Aoyama, Y. Macrocyclic Glycoclusters. Self-Aggregation and Phosphate-Induced Agglutination Behaviors of Calix[4]resorcarene-Based Quadruple-Chain Amphiphiles with a Huge Oligosaccharide Pool. J. Am. Chem. Soc. 2003, 125, 594–601. [Google Scholar]
- Aoyama, Y.; Matsuda, Y.; Chuleeraruk, J.; Nishiyama, K.; Fujimoto, K.; Fujimoto, T.; Shimizu, T.; Hayashida, O. Molecular Delivery Systems Using Macrocyclic Sugar Clusters. Pure Appl. Chem. 1998, 70, 2379–2384. [Google Scholar]
- Matsuzaki, K.; Sato, T.; Enomoto, K.; Yamamoto, I.; Oshima, R.; Hatanaka, K.; Uryu, T.; Kaku, H.; Sone, Y.; Misaki, A. Synthesis of Water-Soluble, Branched Polysaccharides Having D-Mannopyranose, D-Arabinofuranose, or Oligo-D-Arabinofuranose Side-Chains and Their Antitumor Activity. Carbohydr. Res. 1986, 157, 171–182. [Google Scholar]
- Wei, Y.; Cheng, F. Synthesis and Aggregates of Cellulose-Based Hydrophobically Associating Polymer. Carbohydr. Polym. 2007, 68, 734–739. [Google Scholar]
- Furuhata, K.; Aoki, N.; Suzuki, S.; Sakamoto, M.; Saegusa, Y.; Nakamura, S. Bromination of Cellulose with Tribromoimidazole, Triphenylphosphine and Imidazole under Homogeneous Conditions in LiBr-Dimethylacetamide. Carbohydr. Polym. 1995, 26, 25–29. [Google Scholar]
- Vigo, T.L.; Sachinvala, N. Deoxycelluloses and Related Structures. Polym. Adv. Technol. 1999, 10, 311–320. [Google Scholar]
- Heinze, T.; Liebert, T. Unconventional Methods in Cellulose Functionalization. Prog. Polym. Sci. 2001, 26, 1689–1762. [Google Scholar]
- Furuhata, K.; Koganei, K.; Chang, H.-S.; Aoki, N.; Sakamoto, M. Dissolution of Cellulose in Lithium Bromide-Organic Solvent Systems and Homogeneous Bromination of Cellulose with N-Bromosuccinimide-Triphenylphosphine in Lithium Bromide-N,N-Dimethylacetamide. Carbohydr. Res. 1992, 230, 165–177. [Google Scholar]
- Tseng, H.; Furuhata, K.; Sakamoto, M. Bromination of Regenerated Chitin with N-Bromosuccinimide and Triphenylphosphine under Homogeneous Conditions in Lithium Bromide-N,N-Dimethylacetamide. Carbohydr. Res. 1995, 270, 149–161. [Google Scholar]
- Kobayashi, S.; Uyama, H.; Kimura, S. Enzymatic Polymerization. Chem. Rev. 2001, 101, 3793–3818. [Google Scholar]
- Jahn, M.; Stoll, D.; Warren, R.A.J.; Szabó, L.; Singh, P.; Gilbert, H.J.; Ducros, V.M.-A.; Davies, G.J.; Withers, S.G. Expansion of the Glycosynthase Repertoire to Produce Defined Manno-Oligosaccharides. Chem. Commun. 2003, 1327–1329. [Google Scholar]
- Good, F.; Schuerch, C. Improved Synthesis of Substituted 2,6-Dioxabicyclo[3.1.1]heptanes: 1,3-Anhydro-2,4,6-tri-O-Benzyl-2,4,6-tri-O-p-Bromobenzyl- and -2,4,6-tri-O-p-Methylbenzyl-β-d-Glucopyranose. Carbohydr. Res. 1984, 125, 165–171. [Google Scholar]
- Okamoto, E.; Kiyosada, T.; Shoda, S.; Kobayashi, S. Synthesis of Alternatingly 6-O-Methylated Cellulose via Enzymatic Polymerization of a Substituted Cellobiosyl Fluoride Monomer Catalyzed by Cellulase. Cellulose 1997, 4, 161–172. [Google Scholar]
- Kobayashi, S.; Shoda, S. Chemical Synthesis of Cellulose and Cello-Oligomers Using a Hydrolysis Enzyme as a Catalyst. Int. J. Biol. Macromol. 1995, 17, 373–379. [Google Scholar]
- Shoda, S.; Okamoto, E.; Kiyosada, T.; Kobayashi, S. Synthesis of 6- and/or 6′-O-Methylated Cellobiosyl Fluorides: New Monomers for Enzymatic Polymerization. Macromol. Rapid Comm. 1994, 15, 751–756. [Google Scholar]
- Hasegawa, T.; Umeda, M.; Numata, M.; Li, C.; Bae, A.-H.; Fujisawa, T.; Haraguchi, S.; Sakurai, K.; Shinkai, S. ‘Click Chemistry’ on Polysaccharides: A Convenient, General, and Monitorable Approach to Develop (1→3)-β-D-Glucans with Various Functional Appendages. Carbohydr. Res. 2006, 341, 35–40. [Google Scholar]
- Hasegawa, T.; Umeda, M.; Numata, M.; Fujisawa, T.; Haraguchi, S.; Sakurai, K.; Shinkai, S. Click Chemistry on Curdlan: A Regioselective and Quantitative Approach to Develop Artificial β-1,3-Glucans with Various Functional Appendages. Chem. Lett. 2006, 35, 82–83. [Google Scholar]
- Hasegawa, T.; Numata, M.; Okumura, S.; Kimura, T.; Sakurai, K.; Shinkai, S. Carbohydrate-Appended Curdlans as a New Family of Glycoclusters with Binding Properties both for a Polynucleotide and Lectins. Org. Biomol. Chem. 2007, 5, 2404–2412. [Google Scholar]
- Yamashita, E.; Okubo, K.; Negishi, K.; Hasegawa, T. Regioselective and Quantitative Modification of Cellulose to Access Cellulose-based Advanced Materials: Cellulose-Based Glycoclusters. Chem. Lett. 2009, 38, 122–123. [Google Scholar]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar]
- Lee, L.V.; Mitchell, M.L.; Huang, S.-J.; Fokin, V.V.; Sharpless, K.B.; Wong, C.-H. A Potent and Highly Selective Inhibitor of Human α-1,3-Fucosyltransferase via Click Chemistry. J. Am. Chem. Soc. 2003, 125, 9588–9589. [Google Scholar]
- Devaraj, N.K.; Miller, G.P.; Ebina, W.; Kakaradov, B.; Collman, J.P.; Kool, E.T.; Chidsey, C.E.D. Chemoselective Covalent Coupling of Oligonucleotide Probes to Self-Assembled Monolayers. J. Am. Chem. Soc. 2005, 127, 8600–8601. [Google Scholar]
- Fazio, F.; Bryan, M.C.; Blixt, O.; Paulson, J.C.; Wong, C.-H. Synthesis of Sugar Arrays in Microtiter Plate. J. Am. Chem. Soc. 2002, 124, 14397–14402. [Google Scholar]
- Opsteen, J.A.; van Hest, J.C.M. Modular Synthesis of Block Copolymers via Cycloaddition of Terminal Azide and Alkyne Functionalized Polymers. Chem. Commun. 2005, 57–59. [Google Scholar]
- Muñoz-Bonilla, A.; Ibarboure, E.; Bordegé, V.; Fernández-Garćia, M.; Rodrígues-Hernández, J. Fabrication of Honeycomb-Structured Porous Surfaces Decorated with Glycopolymers. Langmuir 2010, 26, 8552–8558. [Google Scholar]
- Ting, S.R.S.; Min, E.H.; Escalé, P.; Save, M.; Billon, L.; Stenzel, M.H. Lectin Recognizable Biomaterials Synthesized via Nitroxide-Mediated Polymerization of a Methacryloyl Galactose Monomer. Macromolecules 2009, 42, 9422–9434. [Google Scholar]
- Park, K.-H.; Na, K.; Lee, Y.S.; Chang, W.-K.; Park, J.-K.; Akaike, T.; Kim, D. Effects of Mannosylated Glycopolymers on Specific Interaction to Bone Marrow Hematopoietic and Progenitor Cells Derived from Murine Species. J. Biomed. Mater. Res. A 2007, 82A, 281–287. [Google Scholar]
- You, L.; Schlaad, H. An Easy Way to Sugar-Containing Polymer Vesicles or Glycosomes. J. Am. Chem. Soc. 2006, 128, 13336–13337. [Google Scholar]
- Park, K.-H.; Sung, W.J.; Kim, S.; Kim, D.H.; Akaike, T.; Chung, H.-M. Specific Interaction of Mannosylated Glycopolymers with Macrophage Cells Mediated by Mannose Receptor. J. Biosci. Bioeng. 2005, 99, 285–289. [Google Scholar]
- Tsuchida, A.; Matsuura, K.; Kobayashi, K. A Quartz-Crystal Microbalance Study of Adsorption Behaviors of Artificial Glycoconjugate Polymers with Different Saccharide Chain Lengths and with Different Backbone Structures. Macromol. Chem. Phys. 2000, 201, 2245–2250. [Google Scholar]
- Kobayashi, K.; Kamiya, S.; Matsuyama, M.; Murata, T.; Usui, T. Synthesis and Functions of a Glycopolymer Carrying Ga1β1→4(GlcNAc)3 Tetrasaccharide. Polym. J. 1998, 30, 653–658. [Google Scholar]
- Toyoshima, M.; Miura, Y. Preparation of Glycopolymer-Substituted Gold Nanoparticles and Their Molecular Recognition. J. Polym. Sci. A1 2009, 45, 1412–1421. [Google Scholar]
- Teng, D.; Yin, W.; Zhang, X.; Wang, Z.; Li, C. New Glycoconjugate Polyacrylamide with Water-Solubility and Additional Activated Groups: Synthesis and Characterization. J. Polym. Res. 2009, 16, 311–316. [Google Scholar]
- Oubihi, M.; Oshima, K.; Aoki, N.; Kobayashi, K.; Kitajima, K.; Matsuda, T. An ELISA-Based Assay for Detergent-Solubilized Cellular beta 1,4-Galactosyltransferase Activity. Use of a Polyacrylamide Derivative with GlcNAc-beta Side Chains as a Solid Phase Acceptor Substrate. Biosci. Biotech. Biochem. 2000, 64, 785–792. [Google Scholar]
- Hashimoto, K.; Ohsawa, R.; Saito, H. Glycopolymeric Inhibitors of β-Glucuronidase. II. Synthesis of Glycopolymers Containing Pendant l-Gulonic Moieties and Effects of the Carboxyl Group in the Glycopolymers upon the Activity of β-Glucuronidase. J. Polym. Sci. A1 1999, 37, 2773–2779. [Google Scholar]
- Tsuchida, A.; Akimoto, S.; Usui, T.; Kobayashi, K. Synthesis of Artificial Glycoconjugate Polymers Starting from Enzymically Synthesized Oligosaccharides and Their Interactions with Lectins. J. Biochem. 1998, 123, 715–721. [Google Scholar]
- Nishimura, S.-I.; Yamada, K. Transfer of Ganglioside GM3 Oligosaccharide from a Water Soluble Polymer to Ceramide by Ceramide Glycanase. A Novel Approach for the Chemical-Enzymatic Synthesis of Glycosphingolipids. J. Am. Chem. Soc. 1997, 119, 10555–10556. [Google Scholar]
- Wataoka, I.; Urakawa, H.; Kobayashi, K.; Akaike, T.; Schmidt, M.; Kajiwara, K. Structural Characterization of Glycoconjugate Polystyrene in Aqueous Solution. Macromolecules 1999, 32, 1816–1821. [Google Scholar]
- Wataoka, I.; Urakawa, H.; Kobayashi, K.; Ohno, K.; Fukuda, T.; Akaike, T.; Kajiwara, K. Molecular Specification of Homopolymer of Vinylbenzyl-Lactose-Amide in Aqueous Solution. Polym. J. 1999, 31, 590–594. [Google Scholar]
- Wataoka, I.; Kobayashi, K.; Kajiwara, K. Effect of the Carbohydrate Side-Chain on the Conformation of a Glycoconjugate Polystyrene in Aqueous Solution. Carbohydr. Res. 2005, 340, 989–995. [Google Scholar]
- Hasegawa, T.; Umeda, M.; Matsumoto, T.; Numata, M.; Mizu, M.; Koumoto, K.; Sakurai, K.; Shinkai, S. Lactose-Appended Schizophyllan is a Potential Candidate as a Hepatocyte-Targeted Antisense Carrier. Chem. Commun. 2004, 382–383. [Google Scholar]
- Hasegawa, T.; Yonemura, T.; Matsuura, K.; Kobayashi, K. Artificial Metalloglycoclusters: Compact Saccharide Shell to Induce High Lectin Affinity as Well as Strong Luminescence. Bioconj. Chem. 2003, 14, 728–737. [Google Scholar]
- Mahon, E.; Aastrup, T.; Barboiu, M. Multivalent Recognition of Lectins by Glyconanoparticle Systems. Chem. Commun. 2010, 46, 5491–5493. [Google Scholar]
- Qian, X. The Effect of Cooperativity on Hydrogen Bonding Interactions in Native Cellulose Iβ from ab Initio Molecular Dynamics Simulations. Mol. Simulat. 2008, 34, 183–191. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Negishi, K.; Mashiko, Y.; Yamashita, E.; Otsuka, A.; Hasegawa, T. Cellulose Chemistry Meets Click Chemistry: Syntheses and Properties of Cellulose-Based Glycoclusters with High Structural Homogeneity. Polymers 2011, 3, 489-508. https://doi.org/10.3390/polym3010489
Negishi K, Mashiko Y, Yamashita E, Otsuka A, Hasegawa T. Cellulose Chemistry Meets Click Chemistry: Syntheses and Properties of Cellulose-Based Glycoclusters with High Structural Homogeneity. Polymers. 2011; 3(1):489-508. https://doi.org/10.3390/polym3010489
Chicago/Turabian StyleNegishi, Kaori, Yoichi Mashiko, Erika Yamashita, Atsushi Otsuka, and Teruaki Hasegawa. 2011. "Cellulose Chemistry Meets Click Chemistry: Syntheses and Properties of Cellulose-Based Glycoclusters with High Structural Homogeneity" Polymers 3, no. 1: 489-508. https://doi.org/10.3390/polym3010489