The Challenge of Synthesizing Oligomers for Molecular Wires

Risø National Laboratory for Sustainable Energy, Technical University of Denmark, Frederiksborgvej 399, DK-4000 Roskilde, Denmark
*
Author to whom correspondence should be addressed.
Polymers 2011, 3(1), 545-557; https://doi.org/10.3390/polym3010545
Submission received: 17 January 2011
/
Revised: 6 February 2011
/
Accepted: 23 February 2011
/
Published: 28 February 2011
(This article belongs to the Special Issue Single Molecular Wire)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Controlling the size of the oligomer and introducing functional groups at the ends of the oligomer that allow it to react with separate electrodes are critical issues when preparing materials for molecular wires. We demonstrate a general synthetic approach to oligophenylenevinylene (OPV) derivative molecules with a molecular length up to 9–10 nm which allow for the introduction of aromatic thioacetate functionality in fully conjugated oligomer systems. Oligomers containing 3–15 phenyl units were synthesized by step wise Horner-Wadsworth-Emmons (HWE) reactions of a bifunctional OPV-monomer, which demonstrated good control of the size of the OPVs. Workup after each reaction step ensures a high purity of the final products. End group functionalization was introduced as a last step.
1. Introduction
In recent years, many efforts have been made in order to produce molecular wires, with the prospect of creating nanoelectronics and of better understanding what happens at the molecular level in conductive processes [1-16]. Most of these previous studies have focused on preparation and examination of relatively small molecules (<3–4 nm), but such molecules have limited applications because of their relatively small size. Others have prepared really large molecules from a solid support by step wise elongation, but as very few chemical reactions proceed in quantitative yield the use of a solid support is a problematic pathway for achieving a narrow size distribution, as workup between the steps is not possible. As an illustration, a reaction running at 90% yield (which is normally considered a good yield for a reaction) over 10 steps potentially only contains 0.910 = 35% of the right compound.
Three of the major challenges in the preparation of large monodisperse wires are: First, complete control of the size of the oligomer in order to obtain a pure monodisperse size distribution. In order to obtain this, one must either operate with chemical reactions that proceed in quantitative yields with no byproducts, or a purification step must be included after each step. Unfortunately none of the known quantitative reactions such as ‘Click’-reactions or ‘Living Polymerization’ reactions such as atom transfer radical polymerization (ATRP) or Anionic Polymerization leads to the conjugated system needed in a molecular wire, leaving workup as the only option for obtaining monodispersity. Second, choosing a design of the side chains that will make it possible to both prepare and later dissolve the oligomer. The side chains are essential for keeping the molecules in solution, and thus enabling chemical reactions. It should be emphasized that large molecules generally always requires high dilution which in some cases can render otherwise standard reactions impossible. Third, the introduction of functional groups at the ends of the wire that allows for connecting the molecules to a circuit. Depending on the desired functionality, one has to carefully consider whether this group should be introduced from the start, which requires that the functionality can sustain the reaction conditions of the oligomerization, or whether the introduction should instead be performed as a last step, associated with the potential difficulties in performing chemistry on very large molecules. Due to the excellent binding of sulfur to gold, thiol linkers in conjugation with gold have been used almost exclusively in previous reports, but there should in principle be many other possible substrate materials such as semiconducting oxides (i.e., TiO2, ZnO) where carboxylic acids, alcohols and phenols have a high affinity.
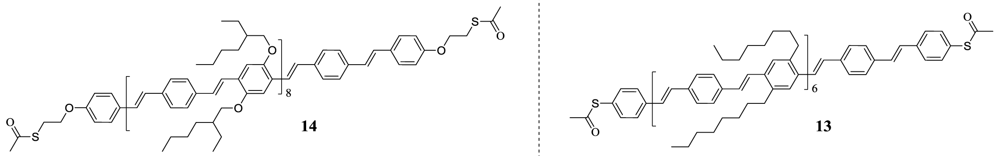
We have previously reported the synthetic approach of up to 12 nm long oligophenylenevinylene (OPV) derivatives (19 phenyls) using step wise Horner-Wadsworth-Emmons (HWE) condensation [16]. Others have reported the synthesis of OPV's of varying size (4–11 phenyls) using the HWE condensation—both unidirectional [17-23] and bidirectional [24,25]—but none as large as these. Final group functionalities were introduced as a last step and successful attachment of thioacetate functionalized oligomers to separate gold electrodes over a 9 nm nanogap was achieved. The thioacetate moiety was introduced to short non-conjugated linkers attached to each end of the molecule (compound 14 in Figure 1). One of the reasons for introducing the gold attaching thioacetate functionality on a non-conjugated linker was the wish to prepare very long wires. The use of ethylhexyloxy (EHO) side chains had previously been shown to provide excellent results, as the solubility was largely improved compared to other side chains [17], but this choice of side chain rendered the only known procedure of introduction of an aromatic thioacetate as a last step impossible, as it involves the cleavage of aromatic tert-butyl thio ethers (Ar–S–C(CH3)3) using BBr3 as a deprotecting reagent [26], i.e., a procedure which will cleave the ether bond of the EHO side chains. Other options for thiol precursors that could potentially be inserted as an initial step, such as thiol itself (–SH) or methyl protected thiol (–SMe), were discarded: the former because of the tendency of thiols to dimerize under formation of a –S–S– bond and the latter because tests showed a much lower reactivity with gold than the thioacetate. Here we report the synthesis of large OPV's up to ∼10 nm (15 benzene rings) that allow for introduction of an aromatic thioacetate functionalization with the potential to create a fully conjugated wire if attached to separate electrodes (Figure 1).
2. Results and Discussion
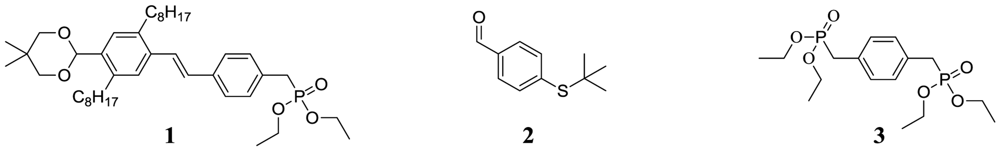
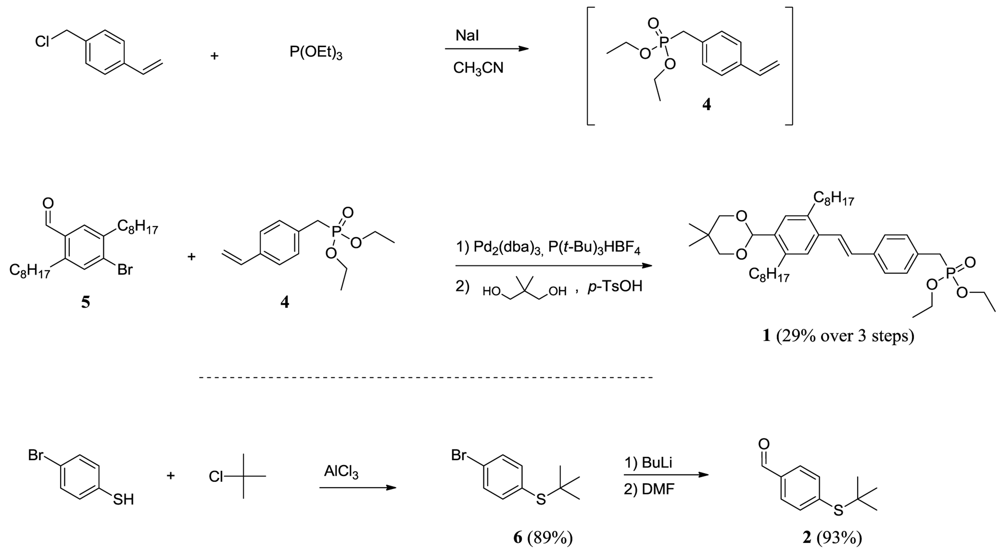
The oligomers were prepared by sequential HWE condensations using three building blocks as shown in Figure 2. The main building block, compound 1, is a modification of the one used for the previously reported oligomer in Figure 1, in which the EHO side chains have been replaced with n-octyl. The prediction was that this would lead to a decrease in solubility compared to the original oligomers, but that it would simultaneously allow for the transformation of aromatic tert-butyl thio ethers into the corresponding thioacetate, by use of BBr3 in the presence of acetyl chloride, without cleavage of the side chains. The synthesis of compound 1 and 2 is outlined in Scheme 1. Initial reaction of 4-(chloromethyl)styrene with triethyl phosphite yielded compound 4, which is very unstable and has to be stored in the freezer if not used immediately. This was then reacted in a Heck coupling with 4-bromo-2,5-dioctylbenzaldehyde (5), which was synthesized as previously described [27], followed by protection of the aldehyde. The instability of compound 4 is reflected in the Heck-reaction, where up to 51% of the bromo starting compound was recollected, which is probably due to polymerization of the vinyl-groups of compound 4.
Compound 2, which carries the initial end-group functionality, was prepared in good yield over two steps by first reacting 4-bromothiophenol and tert-butyl chloride in the presence of AlCl3 to give compound 6 followed by the conversion of the bromide into an aldehyde group by reaction first with n-butyl lithium and then DMF. Compound 3 was prepared as previously described [16].
After coupling compound 1 and 2 in order to functionalize the oligomer, step wise elongation using HWE condensation was then performed, resulting in oligomer ‘arms’ (compound 7, 8, 9 and 11) carrying two different end groups as shown in Scheme 2. The largest of these were then coupled with diphosphonate ester 3, providing a symmetric polymer with a doubling of the length of the molecule in one reaction (Scheme 2). Workup was performed after each step, using gradient silica chromatography and forced precipitation after each step, and the purity was controlled by NMR and size exclusion chromatography (SEC, see supporting information). Compared to previous results using this method of purification, the method seemed somewhat less efficient. This is ascribed to two main properties: higher hydrophobicity and lower solubility of the materials. Due to the more hydrophobic character of the oligomers, separation by gradient column chromatography was shown to be less efficient as the gradient could not be varied to the same extend as for the previous reported oligomers. Furthermore, because of the lower solubility, great care had to be taken in order to prevent precipitation of the larger bifunctional oligomers on the column. The larger symmetric oligomer 10 proved too insoluble for column chromatography, and the largest oligomer 12 was practically insoluble in all solvents. These results really show the difference in solubility for an oligomer carrying EHO side chains compared to n-octyl. Where no problems were encountered in the chromatographic workup of even the largest oligomer (19 phenyls) carrying the EHO, the replacement with n-octyl led to highly reduced solubility at 15 phenyl groups and to insolubility in all ordinary solvents at 19 phenyls.
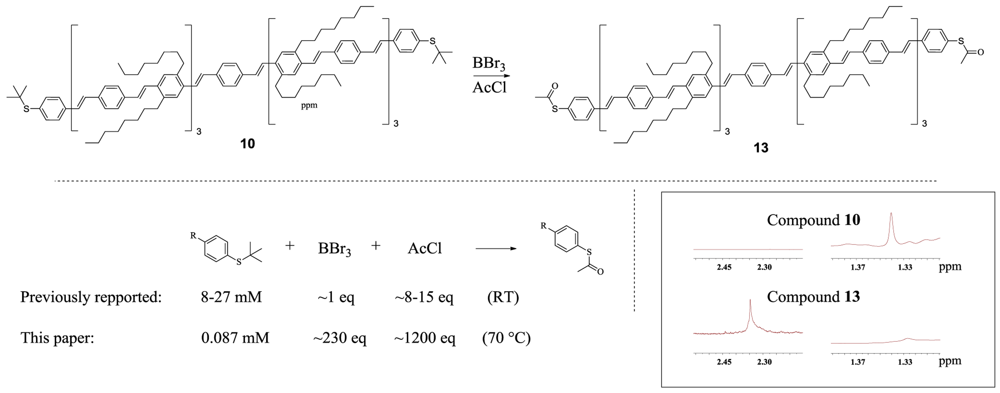
This reduced solubility also played a key role in the preparation of the corresponding aromatic thioacetate of oligomer 10. Due to the heavily reduced solubility, it was impossible to carry out the transformation under the conditions reported for this reaction (see Scheme 3). Dilution of a magnitude of 100–300 times compared to the original description was necessary and a large excess of BBr3 (∼230 times more) and AcOEt (∼80–150 times more) compared to the original report was required in order to obtain the thioacetate. Many experiments were necessary in order to find these realationships, and the final procedure involved running the reaction in deutorated dichlorobenzene with several additions of BBr3 and AcCl in order to follow the reaction by NMR to find the conditions required. These findings are additional proof that reaction conditions, when performed on very large molecules, can sometimes deviate enormously from standard conditions. A final yield of ‘107%’ in the last reaction is ascribed to the difficulty of weighing a very small amount of compound, and is probably due to a change in air humidity or a small amount of residual solvent (C6H4Cl2).
All attempts to do further chemistry on compound 12 proved impossible because of the lack of solubility.
Comparing the synthetic pathway and the resulting properties of the two oligomers in Figure 1 clearly demonstrate the importance of careful consideration when choosing which side chains to use. Although similar in structure, the oligomers have very different solubilities, and they also have different restrictions to what chemistry can be performed in order to carry out end group functionalization. Where the ethylhexyloxy containing OPVs are so soluble that column chromatography can be performed even with very large molecules (19 phenyls), the n-octyl containing OPVs start showing problems of precipitation on the column even in the preparation of the ‘arms’ of (up to 9 phenyls). On the other hand, the latter can sustain chemical conditions that would destroy the former, making it possible to produce an aromatic thioacetate functionalized oligomer.
3. Experimental Section
3.1. General
All solvents used were HPLC grade. Purification by column chromatography was performed by conventional gradient dry column vacuum chromatography [28] or ‘flash’ column chromatography using Merck Silica Gel 60 (15–40 μm) (column height 5–7 cm) and suction to drive the mobile phase.
Size exclusion chromatography (SEC) was performed in chloroform using either of two preparative Knauer systems employing two gel columns in succession with respective pore diameters of 104 Å and 106 Å. The gel columns had dimensions of 25 mmØ × 600 mm.
3.2. Synthesis
p-xylylene-bis-phosphonic acid tetraethyl ester (3)
was prepared as previously described [16].
4-bromo-2,5-dioctylbenzaldehyde (5)
was synthesized according to a previously described procedure [27].
Diethyl 4-vinylbenzylphosphonate (4)
4-(chloromethyl)styrene (115.3 g, 680 mmol) and sodium iodide (102 g, 680 mmol) in acetonitrile (600 mL) under argon was heated to around boiling after which triethyl phosphite (143 mL, 817 mmol) was added in small portions in a manner such that the solution was boiling the whole time. After addition, the mixture was left at reflux for an additional 0.5 h, after which a mixture of ice and water (500 mL) was added, followed by extraction with ether (4 × 100 mL). After removal of the solvent in vacuo the resulting oil was redissolved in ether (400 mL) and washed with water (4 × 60 mL) and brine (60 mL). The organic phase was then dried over MgSO4, followed by evaporation of the solvent. The resulting crude oil (190 g) was distilled at oil pump pressure (134.5 g). The product is unstable and must be kept in a freezer. The product was used in the next step without further purification.
(E)-diethyl4-(4-(5,5-dimethyl-1,3-dioxan-2-yl)-2,5-dioctylstyryl)benzylphosphonate (1)
Compound 4 and 4-bromo-2,5-dioctylbenzaldehyde (42.6 g, 104 mmol) in triethylamine (40 mL) was purged with argon for 5 min. Pd2(dba)3 (180 mg, 0.197 mmol) and P(t-Bu)3HBF4 (260 mg, 0.896 mmol) was added and the reaction mixture was heated to reflux (turns dark) and left for 17 h. The solvent was then removed from the now greenish mixture, followed by dilution in ether (300 mL). The organic phase was washed with aqueous HCl (2 M, 2 × 125 mL), water (2 × 125 mL) and brine (200 mL). After drying over MgSO4, the solvent was removed in vacuo and the crude product was purified by gradient silica column chromatography (heptane/AcOEt, 10% steps). This separation yielded the Bromo-starting compound (21.63 g, 51%) and the initial aldehyde (22.6 g, 37%). This aldehyde was then transformed into the corresponding acetal by reaction with 2,2-dimethylpropane-1,3-diol (4.2 g, 40.3 mmol) in toluene (150 mL) under reflux for 1 hour using a Dean-Stark. The cooled mixture was washed with saturated Na2CO3 (50 mL), water (2 × 50 mL) and brine (50 mL). After drying over MgSO4, removal of the solvent in vacuo yielded the desired protected acetal (26.0 g, 29% over 3 steps from 4-(chloromethyl)styrene).
1H NMR (500 MHz, CDCl3) δ 7.52–6.86 (m, 8H), 5.56–5.45 (m, 1H), 4.12–3.91 (m, 4H), 3.85–3.57 (m, 4H), 3.25–3.06 (m, 2H), 2.78–2.57 (m, 4H), 1.73–1.51 (m, 4H), 1.44–1.20 (m, 29H), 0.91–0.84 (m, 6H), 0.81 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 138.61, 138.20, 136.67, 136.64, 136.15, 135.25, 130.97, 130.89, 130.23, 130.17, 129.41, 129.40, 129.17, 128.36, 127.48, 126.80, 126.78, 126.45, 126.44, 125.43, 100.07, 78.06, 62.33, 62.27, 34.30, 33.38, 33.20, 32.35, 32.06, 32.03, 31.85, 31.52, 30.36, 29.93, 29.87, 29.63, 29.58, 29.42, 23.41, 22.83, 22.80, 22.06, 21.48, 16.55, 16.51, 14.26, 14.24.
(4-bromophenyl)tert-butylthioether (6)
A mixture of 4-bromothiophenol (10.0 g, 52.9 mmol) in tert-butyl chloride (25 mL, 52.9 mmol) was placed under argon, after which aluminum trichloride (0.35 g, 2.62 mmol) was added in portions followed by heating until HCl-gas started to form (the gas was neutralized by leading it through a NaOH solution). The reaction was followed by TLC until no more starting compound was present. During the reaction, additional aluminum trichloride (0.35 g, 2.62 mmol) was added portion wise in order to keep the reaction running. After reaction end, water (50 mL) was added and the mixture was extracted with pentane (3 × 40 mL). The collected organic phases were washed with water (3 × 25 mL) and brine (25 mL). After drying over MgSO4, the solvent was removed in vacuo and the resulting slightly yellowish oil (crude 12.45 g) was micro-distilled to obtain the pure product as a clear oil (11.5 g, 89%). 1H NMR (500 MHz, CDCl3) δ 7.50–7.33 (m, 4H), 1.28 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 139.05, 132.02, 131.79, 123.59, 46.26, 31.03.
4-(tert-butylthio)benzaldehyde (2)
(4-bromophenyl)(tert-butyl)sulfane (10.5 g, 42.8 mmol) was added drop wise to a solution of n-BuLi (28 mL, 44.8 mmol) in anhydrous THF (45 mL) cooled to −40 °C causing precipitation to occur. The temperature was then lowered to −78 °C for 20 min after which anhydrous DMF (9 mL, 116 mmol) was added. Cooling was removed and the mixture was allowed to reach RT where it was stirred for an additional 0.5 h. Water was added and the mixture was extracted with hexane. The combined organic phases were washed with water and brine before drying over MgSO4 and removal of the solvent. The slightly yellow crude oil (8.37 g) was purified by kugelrohr distillation (approx 120 °C) to yield a clear oil (8.12 g, 93%). 1H NMR (500 MHz, CDCl3) δ 10.03 (s, 1H), 7.83 (d, J = 8.1 Hz, 2H), 7.68 (d, J = 8.1 Hz, 2H), 1.34 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 191.65, 141.35, 137.00, 135.89, 129.42, 47.16, 31.15.
4-((E)-4-((E)-4-(tert-butylthio)styryl)styryl)-2,5-dioctylbenzaldehyde (7)
Compound 1 (5.37 g, 8.03 mmol) and compound 2 (1.80 g, 9.26 mmol) in THF (80 mL) were cooled on an acetone/dry ice bath after which potassium tert-butoxide (2.50 g, 22.3 mmol) was added followed by removal from the cold and stirring at RT for 1 hour. The reaction mixture was then recooled followed by addition of THF (100 mL) and a premixed blend of H2SO4:H2O (100 mL, 9:1) in THF (100 mL). The reaction was then allowed to reach RT followed by heating to 40 °C for 1 hour. The mixture was then extracted with ether (4 × 100 mL), which was washed with water (3 × 100 mL), saturated NaHCO3 (100 mL) and brine (100 mL). Evaporation of the solvent after drying over MgSO4 yielded a crude (5.60 g) which was purified by flash chromatography (heptane:CHCl3, 1:1) yielding the pure compound (4.67 g, 93%) 1H NMR (500 MHz, CDCl3) δ 10.25 (s, 1H), 7.67–7.03 (m, 14H), 3.07–2.97 (m, 4H), 2.83–2.73 (m, 4H), 1.73–1.44 (m, 4H), 1.44–1.13 (m, 29H), 0.88 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 191.91, 143.60, 141.32, 138.92, 137.90, 137.75, 137.27, 136.85, 132.72, 132.68, 132.37, 132.21, 129.17, 128.38, 127.98, 127.37, 127.20, 126.63, 125.33, 46.41, 33.05, 32.78, 32.46, 32.03, 31.14, 31.12, 29.81, 29.68, 29.61, 29.57, 29.42, 29.40, 29.17, 22.84, 22.81, 14.26.
4-((E)-4-((E)-4-((E)-4-((E)-4-(tert-butylthio)styryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylbenzaldehyde (8)
Compound 7 (4,6 g, 7,38 mmol) and compound 1 (5.81 g, 8.69 mmol) in dry THF (100 mL) under argon were cooled on acetone/dry ice. Potassium tert-butoxide (2.54 g, 22.6 mmol) was added and the reaction was removed from the cold (turned dark red) and was left stirring while being monitored by TLC for approximately 1 hour. THF was then added and the mixture cooled on acetone/dry ice followed by slow addition of a premixed solution of H2SO4:H2O (60 mL, 9/1) in THF (60 mL). The mixture was allowed to reach RT followed by heating to 40 ° C for 0.5–1 hour while monitoring by TLC. The mixture was extracted with ether, and the collected organic phases were washed with water, saturated NaHCO3 and brine. Drying over MgSO4 followed by removal of the solvent in vacuo yielded a dark yellow crude (9.49 g) which was purified by flash chromatography (CHCl3:heptane, 60:40) yielding the pure compound as a yellow solid (6.26 g, 81%). 1H NMR (500 MHz, CDCl3) δ 10.24 (s, 1H), 7.74–6.94 (m, 24H), 3.08–2.97 (m, 2H), 2.85–2.70 (m, 6H), 1.74–1.57 (m, 8H), 1.47–1.21 (m, 49H), 0.87 (m, 12H). 13C NMR (126 MHz, CDCl3) δ 191.92, 143.61, 141.40, 138.90, 138.85, 138.08, 137.90, 137.65, 136.53, 136.50, 135.28, 135.09, 132.69, 132.31, 132.18, 129.38, 129.14, 128.96, 127.96, 127.89, 127.37, 127.14, 127.09, 127.03, 126.89, 126.59, 126.24, 125.10, 46.39, 33.47, 33.07, 32.79, 32.47, 32.08, 32.04, 31.63, 31.15, 29.83, 29.69, 29.64, 29.62, 29.58, 29.47, 29.43, 29.40, 29.17, 22.84, 22.82, 14.28, 14.26.
4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-(tert-butylthio)styryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylbenzaldehyde (9)
Compound 8 (5.54 g, 5.27 mmol) and compound 3 (4.11 g, 6.14 mmol) in dry THF (160 mL) under argon were cooled until precipitation started. Potassium tert-butoxide (1.85 g, 16.5 mmol) was added and the reaction was left stirring at RT while being monitored by TLC for approximately 1 hour. A premixed solution of H2SO4:H2O (100 mL, 9/1) in THF (200 mL) was added (which caused precipitation) followed by heating to 40 ° C for 0.5–1 hour while monitoring by TLC. The mixture was extracted with ether, and the collected organic phases were washed with water, saturated NaHCO3 and brine. Drying over MgSO4 followed by removal of the solvent in vacuo yielded a crude, which showed the presence of approximately 10% acetal. After a coarse workup by flash chromatography, the mixture was redissolved in THF (200 mL), premixed solution of H2SO4:H2O (50 mL, 9/1) in THF (100 mL) was added and the mixture stirred at 40 ° C for 1 hour. After workup (extraction with ether, wash with water, saturated NaHCO3, brine and drying over MgSO4) the crude product was purified by gradient flash chromatography (CHCl3:heptane, 40:60–60:40) yielding the desired compound (5.87 g, 75%). 1H NMR (500 MHz, CDCl3) δ 10.27 (s, 1H), 7.74–6.98 (m, 34H), 3.07–2.96 (m, 2H), 2.78 (t, J = 6.5 Hz, 10H), 1.74–1.57 (m, 12H), 1.49–1.20 (m, 69H), 0.89 (m, 18H). 13C NMR (126 MHz, CDCl3) δ 191.92, 143.61, 141.40, 138.89, 138.85, 138.82, 138.79, 138.10, 137.90, 137.68, 137.34, 136.49, 135.34, 135.23, 135.13, 135.03, 132.69, 132.32, 132.17, 129.40, 129.24, 129.16, 129.05, 128.92, 127.96, 127.86, 127.37, 127.14, 127.08, 127.03, 126.87, 126.59, 126.29, 126.07, 126.00, 125.09, 46.38, 33.48, 33.07, 32.79, 32.48, 32.08, 32.04, 31.63, 31.15, 29.84, 29.69, 29.65, 29.62, 29.58, 29.47, 29.43, 29.40, 22.84, 22.82, 14.28, 14.27.
1,4-bis((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-(tert-butylthio)styryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)benzene (10)
Compound 9 (300 mg, 0.203 mmol) and compound 3 (35.2 mg, 0.093 mmol) in THF (60 mL) were heated to 50 °C, after which potassium tert-butoxide (100 mg, 0.891 mmol) was added. Stirring was continued for approximately 0.5 h after which the heat was removed and stirring was continued for an additional 0.5 h. The volume of the mixture was then reduced to approximately half, and the resulting mixture was filtered and the solid washed with THF and dried in vacuum (175 mg, 62%).
1H NMR (500 MHz, CDCl3) δ 7.66–6.94 (m, 76H), 2.87–2.70 (m, 24H), 1.71–1.61 (m, 24H), 1.49–1.22 (m, 138H), 0.94–0.82 (m, 36H). 13C NMR (126 MHz, CDCl3) δ 138.83, 138.81, 137.94, 137.90, 137.69, 137.34, 135.25, 135.21, 135.15, 132.19, 129.44, 129.16, 129.07, 127.85, 127.15, 127.03, 126.95, 126.86, 126.60, 126.33, 126.08, 46.39, 33.49, 32.09, 31.64, 31.16, 29.86, 29.66, 29.48, 22.85, 14.29.
4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-(tert-butylthio)styryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylbenzaldehyde (11)
Compound 9 (1.50 g, 1.01 mmol) and compound 1 (0.79 g, 1.18 mmol) were dissolved in THF (120 mL) under argon, after which potassium tert-butoxide (0.346 g, 3.08 mmol) was added and the mixture was left at RT for 1 hour while monitoring by TLC. Deprotection of the acetal was performed by treatment with a cold mixture of H2SO4:H2O (50 mL, 9:1) in THF (100 mL) for 1 hour at 40 °C, followed by extraction with ether, washing of the organic phase with water, saturated NaHCO3 and brine. Evaporation of the solvent after drying over MgSO4 gave a crude that still showed the presence of unhydrolyzed acetal on NMR. The crude was therefore redissolved in THF (100 mL) and re-treated with a cold mixture of H2SO4:H2O (50 mL, 9:1) in THF (100 mL). After extraction, washing and drying, the crude was purified by flash chromatography using pure CHCl3 as eluent as attempts to use a gradient of CHCl3:heptane caused precipitation on the column. (Yield: 1.40 g, 72%). 1H NMR (500 MHz, CDCl3) δ 10.24 (s, 1H), 7.71–6.96 (m, 44H), 3.06–2.98 (m, 2H), 2.82–2.74 (m, 14H), 1.74–1.55 (m, 16H), 1.48–1.19 (m, 89H), 0.96–0.82 (m, 24H).
1,4-bis((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-(tert-butylthio)styryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)benzene (12)
Compound 11 (31.3 mg, 0.016 mmol) and compound 3 (3.04 mg, 8.04 μmol) were dissolved in dry THF (40 mL) and heated to 50 °C. Potassium tert-butoxide (9.0 mg, 0.080 mmol) was then added causing a slight color shift and precipitation. The mixture was stirred overnight. Filtering of the solid and washing with THF resulted in an insoluble solid (21.4 mg).
1,4-bis((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-(acetylthio)styryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)benzene 13
Compound 10 (1.05 mg, 0.346 μmol) in CD4Cl2 (2.0 mL) under argon was heated to 70 °C after which a total amount of tribromoborane (80 μL, 0.080 mmol) and acetyl chloride (30 μL, 0.422 mmol) was added in small portions over the next 24 hours while continuously monitoring by NMR. The reaction mixture was then diluted with C6H4Cl2 (20 mL) and the solvent and excess BBr3 and AcCl was removed in high vacuum. After removal, the crude was redissolved in C6H4Cl2 (20 mL) and filtrated before the solvent was removed once more to obtain the final product (1.11 mg, ‘107%’).
1H NMR (500 MHz, C6D4Cl2) δ 7.96–6.57 (m, 76H), 2.93 (s, 24H), 2.35 (s, 6H), 1.88–1.63 (m, 24H), 1.57–1.16 (m, 120H), 0.93 (t, J = 7.0 Hz, 36H).
4. Conclusions
A chemical approach to prepare large alkyl substituted phenylene vinylene oligomers up to ∼10 nm (15 phenyls), which can be end group functionalized with aromatic thioacetates as a final step, was shown. The introduction of a thioacetate onto the conjugated backbone of the oligomer makes this a potential candidate for the preparation of a fully conjugated OPV molecular wire attached directly to two separate gold electrodes. The oligomers were prepared by step wise HWE condensation, using the three building blocks shown in Figure 2, to create bifunctional ‘arms’ that were then reacted with a symmetrical core, creating a symmetrical oligomer and at the same time doubling the size. The thioacetate functionality was introduced as a last step by cleavage of aromatic tertbutylthio ethers, situated at the end of the symmetrical oligomer, with a large excess of BBr3 in the presence of large excess of acetyl chloride. These reaction conditions differ largely with those previously reported in the respect that high dilution of the oligomer and a large excess of the other reactants was necessary—conditions that are ascribed to the very low solubility of the larger oligomers which makes this otherwise standard reaction very difficult.
The generally low solubility of the oligomers made workup more tedious when compared with previously reported oligomers with more or less identical structures except for the side chains. Where the present oligomers carry n-octyl side chains in order to be resilient to the final aromatic functionalization (treatment with BBr3), the previously reported oligomers carried ethylhexyloxy chains, which are very comparable in size with the n-octyl but involve an ether functionality, which makes treatment with BBr3, and thus the aromatic thioacetalization, impossible. These examples demonstrate that one has to carefully select which side chains to choose when preparing larger oligomers.
Supplementary Material
polymers-03-00545-s001.pdf
Figure 1.
left: Chemical structure of previously reported oligomer 14 containing an aliphatic thioacetate functionality which allows for attachment between two separate gold electrodes, allowing it to function as a molecular wire [16]. As the thioacetate functionality is not in direct contact with the conjugate oligomer, the transport of electrons between the oligomer and the gold electrodes of electrons has to happen through tunneling. right: A newly synthesized oligomer 13 containing an aromatic thioacetate functionality which has the potential to create a fully conjugated organic system between two gold electrodes by creation of a Ar-S-Au bond.
Figure 1.
left: Chemical structure of previously reported oligomer 14 containing an aliphatic thioacetate functionality which allows for attachment between two separate gold electrodes, allowing it to function as a molecular wire [16]. As the thioacetate functionality is not in direct contact with the conjugate oligomer, the transport of electrons between the oligomer and the gold electrodes of electrons has to happen through tunneling. right: A newly synthesized oligomer 13 containing an aromatic thioacetate functionality which has the potential to create a fully conjugated organic system between two gold electrodes by creation of a Ar-S-Au bond.

Figure 2.
Chemical structure of the three building blocks used for in the preparation of the oligomers.
Figure 2.
Chemical structure of the three building blocks used for in the preparation of the oligomers.

Scheme 1.
Synthetic route to Compound 1 and 2.
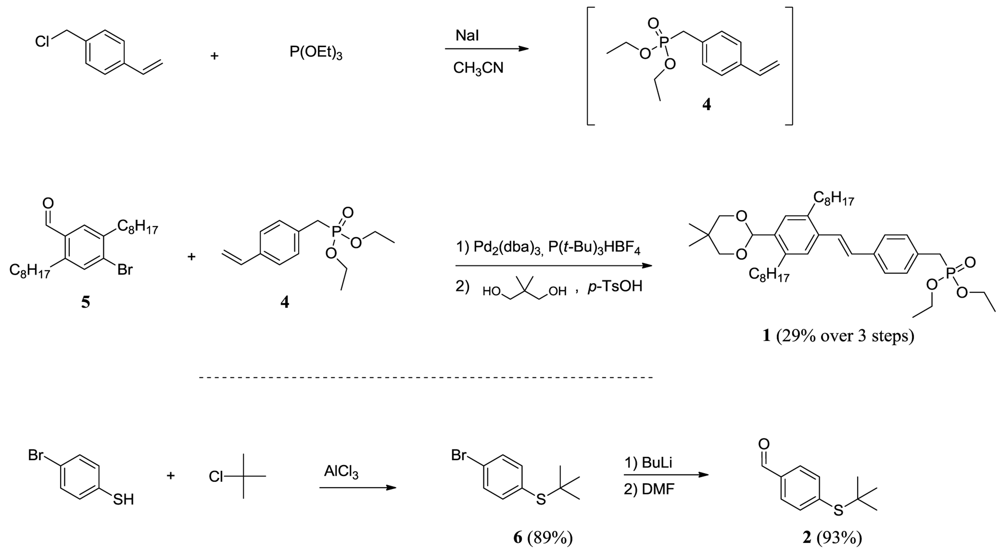
Scheme 2.
Reaction steps in the preparation of the oligomers. The synthesis of the “arms” of the molecule starts with the introduction of the termination group followed by growth of the arm and final doubling of the size of the molecule by attaching two “arms” to a core molecule, which in this work is a simple benzene ring.
Scheme 2.
Reaction steps in the preparation of the oligomers. The synthesis of the “arms” of the molecule starts with the introduction of the termination group followed by growth of the arm and final doubling of the size of the molecule by attaching two “arms” to a core molecule, which in this work is a simple benzene ring.
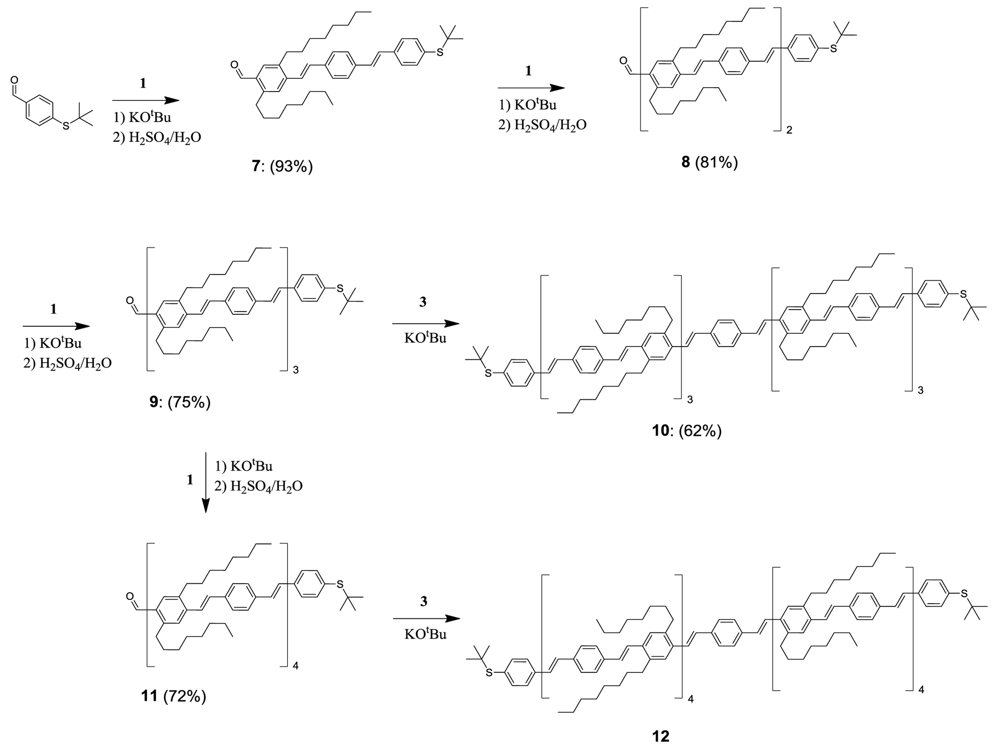
Scheme 3.
top: Thioacetate functionalization of oligomer with 15 phenyls. Bottom left: Schematic representation of the reaction conditions previously reported for the conversion of aromatic tert-butyl thioether into the corresponding aromatic thioacetates (Ref. [26]) compared to the reaction conditions required for the transformation of oligomer 10. This demonstrates that reaction conditions on very large molecules cannot always be directly transferred from standard reactions. Bottom right: 1H-NMR of compound 10 and 13 in C6D4Cl2 showing disappearance of the singlet signal (∼1.33 ppm) from the tert-butyl thioether of compound 10 and formation of the singlet signal (2.35 ppm) from the CH3-group in the formed aromatic thioactetate 13.
Scheme 3.
top: Thioacetate functionalization of oligomer with 15 phenyls. Bottom left: Schematic representation of the reaction conditions previously reported for the conversion of aromatic tert-butyl thioether into the corresponding aromatic thioacetates (Ref. [26]) compared to the reaction conditions required for the transformation of oligomer 10. This demonstrates that reaction conditions on very large molecules cannot always be directly transferred from standard reactions. Bottom right: 1H-NMR of compound 10 and 13 in C6D4Cl2 showing disappearance of the singlet signal (∼1.33 ppm) from the tert-butyl thioether of compound 10 and formation of the singlet signal (2.35 ppm) from the CH3-group in the formed aromatic thioactetate 13.

Acknowledgments
This work was supported by EU STREP project no. 026714 with the acronym VERSATILE.
Reference
- Ashwell, G.J.; Tyrrell, W.D.; Urasinska, B.; Wang, C.S.; Bryce, M.R. Organic rectifying junctions from an electron-accepting molecular wire and an electron-donating phthalocyanine. Chem. Comm. 2006. [Google Scholar] [CrossRef]
- Ashwell, G.J.; Urasinska, B.; Wang, C.S.; Bryce, M.R.; Grace, I.; Lambert, C.J. Single-molecule electrical studies on a 7 nm long molecular wire. Chem. Comm. 2006. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, B.; Frisbie, CD. Electrical resistance of long conjugated molecular wires. Science 2008, 320, 1482–1486. [Google Scholar]
- Choi, S.H.; Frisbie, CD. Enhanced hopping conductivity in low band gap donor-acceptor molecular wires up to 20 nm in length. J. Am. Chem. Soc. 2010, 132, 16191–16201. [Google Scholar]
- Danilov, A.; Kubatkin, S.; Kafanov, S.; Hedegard, P.; Stuhr-Hansen, N.; Moth-Poulsen, K.; Bjørnholm, T. Electronic transport in single molecule junctions: Control of the molecule-electrode coupling through intramolecular tunneling barriers. Nano Lett. 2008, 8, 1–5. [Google Scholar]
- Huang, W.; Masuda, G.; Maeda, S.; Tanaka, H.; Ogawa, T. Molecular junctions composed of oligothiophene dithiol-bridged gold nanoparticles exhibiting photoresponsive properties. Chem. Eur. J. 2006, 12, 607–619. [Google Scholar]
- Kubatkin, S.; Danilov, A.; Hjort, M.; Cornil, J.; Bredas, J.L.; Stuhr-Hansen, N.; Hedegard, P.; Bjørnholm, T. Single-electron transistor of a single organic molecule with access to several redox states. Nature 2003, 425, 698–701. [Google Scholar]
- Li, Z.H.; Pobelov, I.; Han, B.; Wandlowski, T.; Blaszczyk, A.; Mayor, M. Conductance of redox-active single molecular junctions: An electrochemical approach. Nanotechnology 2007, 18, 044018. [Google Scholar]
- Liang, T.T.; Naitoh, Y.; Horikawa, M.; Ishida, T.; Mizutani, W. Fabrication of steady junctions consisting of alpha,omega-bis(thioacetate) oligo(p-phenylene vinylene)s in nanogap electrodes. J. Am. Chem. Soc. 2006, 128, 13720–13726. [Google Scholar]
- Liang, Y.Y.; Wang, H.B.; Yuan, S.W.; Lee, Y.G.; Yu, L.P. Conjugated block copolymers and co-oligomers: From supramolecular assembly to molecular electronics. J. Mater. Chem. 2007, 17, 2183–2194. [Google Scholar]
- Moth-Poulsen, K.; Bjørnholm, T. Molecular electronics with single molecules in solid-state devices. Nat. Nanotechnol. 2009, 4, 551–556. [Google Scholar]
- Osorio, E.A.; O'Neill, K.; Wegewijs, M.; Stuhr-Hansen, N.; Paaske, J.; Bjørnholm, T.; van der Zant, H.S.J. Electronic excitations of a single molecule contacted in a three-terminal configuration. Nano Lett. 2007, 7, 3336–3342. [Google Scholar]
- Seferos, D.S.; Blum, A.S.; Kushmerick, J.G.; Bazan, G.C. Single-molecule charge-transport measurements that reveal technique-dependent perturbations. J. Am. Chem. Soc. 2006, 128, 11260–11267. [Google Scholar]
- van der Zant, H.S.J.; Kervennic, Y.V.; Poot, M.; O'Neill, K.; de Groot, Z.; Thijssen, J.M.; Heersche, H.B.; Stuhr-Hansen, N.; Bjørnholm, T.; Vanmaekelbergh, D.; van Walree, C.A.; Jenneskens, L.W. Molecular three-terminal devices: Fabrication and measurements. Faraday Discuss. 2006, 131, 347–356. [Google Scholar]
- Wang, C.S.; Batsanov, A.S.; Bryce, M.R. Nanoscale aryleneethynylene oligomers incorporating fluorenone units as electron-dopable molecular wires. Faraday Discuss. 2006, 131, 221–234. [Google Scholar]
- Søndergaard, R.; Strobel, S.; Bundgaard, E.; Norrman, K.; Hansen, A.G.; Albert, E.; Csaba, G.; Lugli, P.; Tornow, M.; Krebs, F.C. Conjugated 12 nm long oligomers as molecular wires in nanoelectronics. J. Mater. Chem. 2009, 19, 3899–3908. [Google Scholar]
- Alstrup, J.; Norrman, K.; Jørgensen, M.; Krebs, F.C. Lifetimes of organic photovoltaics: Design and synthesis of single oligomer molecules in order to study chemical degradation mechanisms. Sol. Energy Mater. Sol. Cells 2006, 90, 2777–2792. [Google Scholar]
- Hagemann, O.; Jørgensen, M.; Krebs, F.C. Synthesis of an all-in-one molecule (for organic solar cells). J. Org. Chem. 2006, 71, 5546–5559. [Google Scholar]
- Jørgensen, M.; Krebs, F.C. Stepwise and directional synthesis of end-functionalized single-oligomer OPVs and their application in organic solar cells. J. Org. Chem. 2004, 69, 6688–6696. [Google Scholar]
- Jørgensen, M.; Krebs, F.C. Stepwise unidirectional synthesis of oligo phenylene vinylenes with a series of monomers. Use in plastic solar cells. J. Org. Chem. 2005, 70, 6004–6017. [Google Scholar]
- Peeters, E.; van Hal, P.A.; Knol, J.; Brabec, C.J.; Sariciftci, N.S.; Hummelen, J.C.; Janssen, R.A.J. Synthesis, photophysical properties, and photovoltaic devices of oligo(p-phenylene vinylene)-fullerene dyads. J. Phys. Chem. B 2000, 104, 10174–10190. [Google Scholar]
- Sugiono, E.; Metzroth, T.; Detert, H. Practical synthesis of vinyl-substituted p-phenylenevinylene oligomers and their triethoxysilyl derivatives. Adv. Synth. Catal. 2001, 343, 351–359. [Google Scholar]
- Wong, M.S.; Li, Z.H. Multifunctional properties of monodisperse end-functionalized oligophenylenevinylenes. Pure Appl. Chem. 2004, 76, 1409–1419. [Google Scholar]
- Detert, H.; Sadovski, O. Triarylamines connected via phenylenevinylene segments. Synth. Met. 2003, 138, 185–188. [Google Scholar]
- Giacalone, F.; Segura, J.L.; Martin, N.; Ramey, J.; Guldi, D.M. Probing molecular wires: Synthesis, structural, and electronic study of donor-acceptor assemblies exhibiting long-range electron transfer. Chem. Eur. J. 2005, 11, 4819–4834. [Google Scholar]
- Stuhr-Hansen, N.; Christensen, J.B.; Harrit, N.; Bjørnholm, T. Novel synthesis of protected thiol end-capped stilbenes and oligo(phenylenevinylene)s (OPVs). J. Org. Chem. 2003, 68, 1275–1282. [Google Scholar]
- Krebs, F.C.; Jørgensen, M. High carrier mobility in a series of new semiconducting PPV-type polymers. Macromolecules 2003, 36, 4374–4384. [Google Scholar]
- Pedersen, D.S.; Rosenbohm, C. Dry column vacuum chromatography. Synthesis-Stuttgart 2001. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Søndergaard, R.; Krebs, F.C. The Challenge of Synthesizing Oligomers for Molecular Wires. Polymers 2011, 3, 545-557. https://doi.org/10.3390/polym3010545
AMA Style
Søndergaard R, Krebs FC. The Challenge of Synthesizing Oligomers for Molecular Wires. Polymers. 2011; 3(1):545-557. https://doi.org/10.3390/polym3010545
Chicago/Turabian StyleSøndergaard, Roar, and Frederik C. Krebs. 2011. "The Challenge of Synthesizing Oligomers for Molecular Wires" Polymers 3, no. 1: 545-557. https://doi.org/10.3390/polym3010545