Internal Dynamics of Dendritic Molecules Probed by Pyrene Excimer Formation

Institute for Polymer Research, Waterloo Institute of Nanotechnology, Department of Chemistry, University of Waterloo, Waterloo, ON N2L 3G1, Canada
Polymers 2012, 4(1), 211-239; https://doi.org/10.3390/polym4010211
Submission received: 5 December 2011
/
Revised: 2 January 2012
/
Accepted: 12 January 2012
/
Published: 17 January 2012
(This article belongs to the Special Issue Dendrimers and Hyperbranched Polymers)
Abstract
:This review exposes the current poor understanding of the internal segmental chain dynamics of dendrimers in solution probed by monitoring the process of excimer formation between pyrene labels covalently attached to the chain ends of dendrimers. The review begins by covering the bases of fluorescence and the kinetics of pyrene excimer formation before describing a procedure based on the Model Free (MF) analysis that is used to analyze quantitatively the fluorescence decays acquired for dendrimers, the ends of which have been fully and covalently labeled with pyrene. Comparison of the various trends obtained by different research groups describing the efficiency of pyrene excimer formation with the generation number of dendrimers illustrates the lack of consensus between the few studies devoted to the topic. One possible reason for this disagreement might reside in the presence of minute amounts of unattached pyrene labels which act as potent fluorescent impurities and affect the analysis of the fluorescence spectra and decays in an uncontrolled manner. The review points out that the MF analysis of the fluorescence decays acquired with pyrene-labeled dendrimers enables one to account for the presence of unattached pyrene and to retrieve information about the internal segmental dynamics of the dendrimer. It provides guidelines that should enable future studies on pyrene-labeled dendrimers to yield results that are more straightforward to interpret.
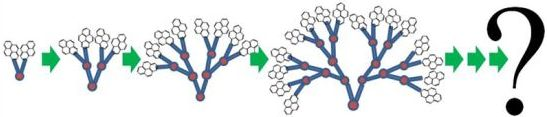
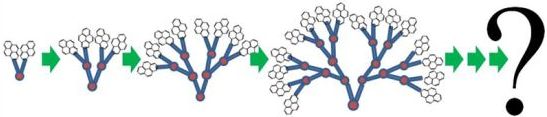
1. Introduction
The exquisite level of synthetic control achieved over the molecular architecture of dendrimers, which was demonstrated more than 25 years ago by the preparation of polyamide and poly(amido amine) (PAMAM) dendrimers by Newkome [1] and Tomalia [2], respectively, has made these macromolecules key elements in a broad range of applications such as for light harvesting devices [3,4], in the biomedical field [5,6], for catalysis [7,8], or as sensors [9,10]. One essential feature afforded by the cascade synthesis of dendrimers [11] is the exponential increase of both the number of reactive end groups and the dendrimer mass with increasing generation number as ~f G where f and G are the branch-juncture multiplicity (f > 1) and G is the generation number. Since the dendrimer volume increases more slowly with G as ~Gα with 1.5 < α < 3.3 according to a number of studies [12,13,14,15,16,17,18,19,20,21], the local density of reactive end groups inside the dendrimer and the overall dendrimer density increase with G. From a practical standpoint, the increase of the reactive group density with G enables the synthetic chemist to use covalent attachment to concentrate molecules of interest like drugs, dyes, or imaging contrast agents in the nanometer-sized volume defined by the dendrimer. From a theoretical standpoint however, the continuous increase in dendrimer density with generation number leads to the prediction that a crossover generation exists where the available free volume inside the dendrimer vanishes. Considering the importance of the end groups for applications, this decrease in free volume with increasing G led numerous theoreticians [12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35] and experimentalists [4,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54] to question what became of the end groups in higher generation dendrimers as they might be trapped in the congested dendrimer interior.
The first theory dealing with this issue was proposed by de Gennes and Hervet (DGH) [22]. It predicted that dendrimers grow outwardly in a manner that concentrates the end groups in a concentric shell located at the surface of the dendrimer. The prediction of a shell-dense location for the end groups is not entropically favorable however as it leads to a decrease in disorder. A computational study carried out by Lescanec and Muthukumar [23] 12 years later contradicted the DGH prediction by finding that many terminal ends of a dendrimer do not remain at the dendrimer periphery but rather fold back into the dendrimer interior. Since then, numerous theoretical [12,13,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35] and experimental [4,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54] studies have been conducted which led to the current consensus [55] that the end groups of dendrimers made of a flexible backbone are distributed throughout the dendrimer interior in a core-dense manner. Only in a few rare studies were the terminal groups found to lay at the dendrimer periphery, when the ends were subject to some repulsive electrostatic forces [35,56,57] or attractive hydrogen-bond interactions [58] or the dendrimer was made of stiff repeating units [50].
While an agreement has been reached about the location of the dendrimer terminal groups, much fewer reports have characterized their large scale motions. Yet, since accessibility of the end groups to the dendrimer surface is a key element in numerous applications with sensing, delivery, or associative purposes, it might actually matter more to determine not whether the end groups are buried in the dendrimer interior but rather, how quickly they can diffuse out of the congested interior to reach the dendrimer surface and achieve their purpose. In short, the long range motions of the dendrimer ends should be characterized. Interestingly, such information is not forthcoming in the scientific literature despite the more than 30,000 papers already published to date and devoted to the study of dendrimers.
The internal dynamics of dendrimers have been the topic of a number of studies. NMR [46,51,59,60], neutron scattering [61,62,63], dielectric relaxation spectroscopy [64,65], rheology [66], fluorescence anisotropy [67], and molecular and Brownian dynamics computation [68,69,70,71] have provided some information about the mobility of the different segments constituting a dendrimer, generally via the rotational correlation time (τC) of the segments. The overall conclusion of these studies is that the chain ends are more mobile than the internal segments, and that their rotational correlation time is not much affected by increasing G values. However, studies that describe local mobility do not provide information about long range motion. This statement is illustrated by considering a fluorescence resonance energy transfer (FRET) experiment where two dyes are attached at the ends of a DNA helix [72]. The dyes are locally mobile so that FRET measurements can be conducted, yet the two dyes cannot encounter due to the rigidity of the DNA helix. Thus studies that probe τC for different segments of dendrimers demonstrate that the chain ends are locally mobile at their position inside the dendrimer, but provide little information about the range and dynamics of their translational displacement which enables the shuttle of the terminal groups from the dendrimer core to its surface. This information can be obtained with techniques that characterize the long range internal dynamics of a macromolecule. One such technique is fluorescence which can be used to probe the process of excimer formation between pyrene fluorophores covalently attached to the terminal ends of a dendrimer, the topic of the present review. Since excimer formation requires that two pyrene units come into direct contact, the extent and rate of excimer formation reflect the internal dynamics of the macromolecule to which the pyrene moieties are covalently attached [73,74,75]. A measure of the extent of excimer formation can be readily determined by acquiring a steady-state fluorescence spectrum of a solution of the pyrene-labeled macromolecule and taking the ratio of the fluorescence intensity of the excimer over that of the monomer, namely the IE/IM ratio [73,75,76]. The IE/IM ratio is often referred to as the coiling index as a large IE/IM ratio indicates that the macromolecule is flexible enough to enable the encounter of two pyrene moieties [77]. Quantitative analysis of the time-resolved fluorescence decays of the pyrene monomer and excimer yields the rate constant of excimer formation which is a direct measure of the internal dynamics of the macromolecule [73,74,75].
Interestingly, whereas the study of the pyrene excimer formation of pyrene-labeled linear polymers has been astonishingly successful at characterizing the internal dynamics of these macromolecules [73,74,75,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116], much fewer pyrene-labeled dendrimers have been prepared to study intramolecular excimer formation [117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134]. Surprisingly for such a rather small set of studies, many of these studies yield trends that are, in the opinion of the author, both internally and externally inconsistent and which, if taken at their face value, would lead the non-expert in fluorescence to the erroneous conclusion that pyrene excimer formation cannot be successfully applied to characterize the internal dynamics of dendrimers. For indeed, pyrene excimer formation is a powerful tool to characterize the internal dynamics of any pyrene-labeled macromolecule including dendrimers.
This review is laid out in the following manner. First, the basic principles of fluorescence and pyrene excimer formation will be introduced. Second, new developments achieved by this laboratory over the past two years in the analysis of the kinetics of pyrene excimer formation will be presented focusing on the study of pyrene end-labeled dendrimers. Third, the parlous state of the current understanding of the process of excimer formation between pyrene pendants covalently attached to the dendrimer terminals will be exposed. Fourth, clear guidelines will be provided on where the field should be heading and how further studies should be conducted.
2. Fluorescence and Pyrene Excimer Formation
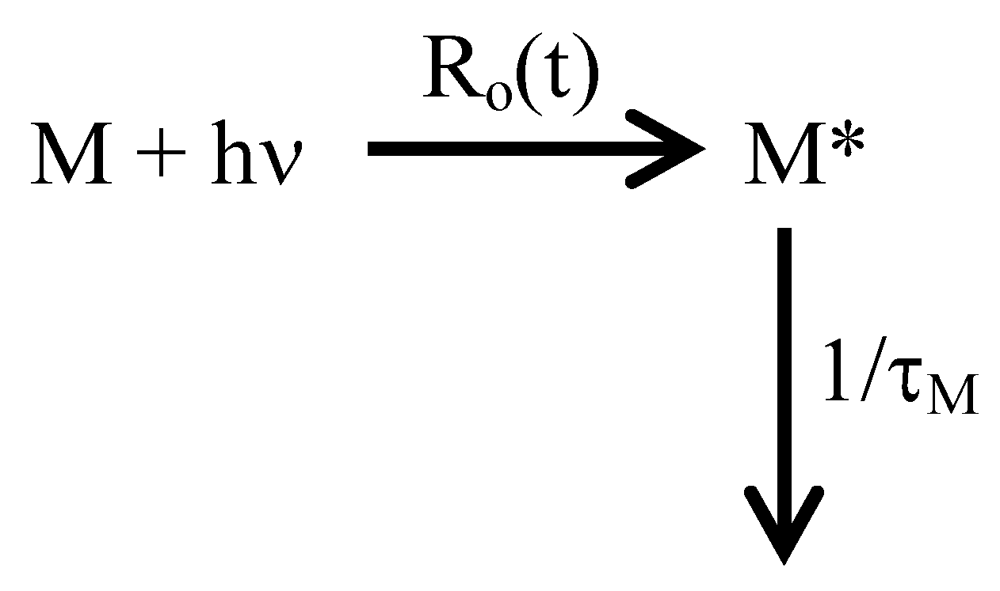
Webster’s unabridged dictionary defines fluorescence as the emission of radiation, especially visible light, by a substance during exposure to external radiation, as light or X-rays. The different photophysical pathways associated with fluorescence have been described in a number of textbooks and they are briefly described hereafter [135,136,137]. Upon absorption of a photon in a process that takes a few femtoseconds, a fluorophore like pyrene gets excited to one of the vibrational levels of its different electronic states. The excited pyrene relaxes then quickly over a few picoseconds to the lowest vibrational level of the first electronic state (S1) via internal conversion. The excited fluorophore relaxes to one of the vibrational levels of the ground-state (S0) by fluorescence in a process that takes several nanoseconds for most fluorophores and more than 100 nanoseconds for many pyrene derivatives. Because fluorescence occurs on a much longer timescale compared to all other photophysical processes involved, excitation of the fluorophore can be considered to be instantaneous with respects to fluorescence. Scheme 1 describes the photophysical processes involved during the fluorescence of a fluorophore (M).
Scheme 1.
Excitation and emission by fluorescence of a fluorophore M.

In Scheme 1, the excited fluorophore M* is generated at a rate Ro(t) and emits fluorescence with its natural lifetime τM. τM−1 represents the rate constant describing the relaxation of the excited fluorophore. It includes a radiative (krad) and a non-radiative (knrad) component such that τM−1 = krad + knrad. According to Scheme 1, the disappearance of M* as a function of time is described by Equation (1).

Solving Equation (1) requires knowledge about whether the fluorescence experiment is conducted with a continuous (steady-state condition, Ro(t) = Ro which is independent of time, and d[M*]/dt = 0) or time resolved (Ro(t) = Ro × δ(t) where δ represent the Dirac function and equals zero for t > 0) excitation. In the case of continuous excitation, the total concentration of M* species ([M*]o) that were generated is given in Equation (2).

Ro in Equation (2) depends on the absorption of the solution, the lamp intensity, and the geometry of the fluorometer used. Multiplying [M*]o by the fluorescence quantum yield of the fluorophore (ϕ = krad × τM) yields the total fluorescence emission IF(M*) given in Equation (3).

For the fluorescence intensity to be strictly linearly proportional to [M*]o, the absorption of the solution needs to be kept below 0.05 to avoid the inner filter effect [137]. In the case of time-resolved excitation, integration of Equation (1) yields Equation (4).

Integration of [M*](t) from time t = 0 to t→ ∞ followed by multiplication by krad yields IF(M*) which is given in Equation (5).

Comparison of Equations (3) and (5) implies that Ro × τM = [M*]o. While this is theoretically true and the absolute values of Ro can be determined by actinometry, in practice actinometry has been shown to be a demanding technique which sometimes results in large variations in quantum yields, for instance, varying from 0.41 to 0.97 for rhodamine B which is often used as a fluorescence standard [138]. Consequently, the determination of Ro is rarely done and the values of Ro and [M*]o remain unknown in the vast majority of fluorescence studies where the fluorescence intensity depends on the fluorometer and the geometry used for fluorescence detection. This comes from the fact that any fluorescence experiment yields a relative value that is made absolute only after proper calibration against a known quantity. However these derivations illustrate the inherent relationship that exists between the results obtained by steady-state and time-resolved fluorescence. This aspect is taken full advantage of when discussing the kinetics of pyrene excimer formation later.
Excimer formation takes place when an excited fluorophore positioned in contact with a ground-state fluorophore results in the formation of an excited complex called an excimer [139]. Many aromatic molecules can form an excimer but among them, it is not excessive to say that pyrene has achieved stardom status. This status is largely due to the fact that the 0–0 transition between the lowest vibrational energy levels of the S0 and S1 electronic states is symmetry forbidden in the case of pyrene [140,141,142,143,144]. As shown in Figure 1, the absence of overlap between the absorption and emission spectra of pyrene demonstrates that little energy transfer can occur between an excited and a ground-state pyrene. Consequently, as an excited pyrene diffuses in solution to encounter a ground-state pyrene, the excitation energy remains localized on the excited pyrene until excimer formation occurs upon contact with a ground-state pyrene. Artefacts due to energy hopping between fluorophores such as with naphthalene [145] or ethidium bromide [146] which both exhibit substantial overlap between their absorption and fluorescence spectra are largely absent in the case of pyrene. This represents an enormous advantage when pyrene excimer formation is used to probe the long range internal dynamics of macromolecules as the large local concentration of fluorophores covalently attached to a macromolecule would otherwise induce a delocalization of the excited state due to energy hopping.
Despite the small molar absorption coefficient of pyrene (εPy) at the wavelength corresponding to the 0–0 transition (372 nm in Figure 1), pyrene exhibits a relatively large εPy value for the S20 transition in the absorption spectrum, usually around 45,000 (±10,000) M−1∙cm−1 depending on the solvent and the type of side group pyrene has been derivatized with. Depending on the pyrene derivative or the solvent being used, the S20 band is usually located between 335 and 345 nm and excitation at these wavelengths away from the pyrene emission minimizes distortions in the fluorescence spectrum of pyrene due to light scattering. This becomes an added benefit when dealing with pyrene-labeled macromolecules which scatter light due to their large size.
Figure 1.
Molar absorption coefficient (solid line) and fluorescence spectrum (dashed line) of pyrene in tetrahydrofuran. For the fluorescence spectrum, [Py] = 2.5 × 10−6 mol∙L−1 and λex = 335 nm (Absorption and fluorescence spectra acquired by Michael A. Fowler).
Figure 1.
Molar absorption coefficient (solid line) and fluorescence spectrum (dashed line) of pyrene in tetrahydrofuran. For the fluorescence spectrum, [Py] = 2.5 × 10−6 mol∙L−1 and λex = 335 nm (Absorption and fluorescence spectra acquired by Michael A. Fowler).

Equation (6) describes how krad is related to εPy [147], which according to Figure 1, takes small values for the S1 transition. In Equation (6), n is the solvent refractive index, ![Polymers 04 00211 i016]() is the wavenumber, and IF is the fluorescence intensity. Based on Equation (6), krad is small for pyrene so that the lifetime of pyrene is large since τM−1 = krad + knrad. Indeed, pyrene and its derivatives have lifetimes of several hundreds of nanoseconds, much longer than any other aromatic fluorophore. The long lifetime of pyrene provides a long time window through which photophysical phenomena occurring over a few tens of picoseconds to several hundreds of nanoseconds can be probed, a dynamic range covering five orders of magnitude.
is the wavenumber, and IF is the fluorescence intensity. Based on Equation (6), krad is small for pyrene so that the lifetime of pyrene is large since τM−1 = krad + knrad. Indeed, pyrene and its derivatives have lifetimes of several hundreds of nanoseconds, much longer than any other aromatic fluorophore. The long lifetime of pyrene provides a long time window through which photophysical phenomena occurring over a few tens of picoseconds to several hundreds of nanoseconds can be probed, a dynamic range covering five orders of magnitude.
 is the wavenumber, and IF is the fluorescence intensity. Based on Equation (6), krad is small for pyrene so that the lifetime of pyrene is large since τM−1 = krad + knrad. Indeed, pyrene and its derivatives have lifetimes of several hundreds of nanoseconds, much longer than any other aromatic fluorophore. The long lifetime of pyrene provides a long time window through which photophysical phenomena occurring over a few tens of picoseconds to several hundreds of nanoseconds can be probed, a dynamic range covering five orders of magnitude.
is the wavenumber, and IF is the fluorescence intensity. Based on Equation (6), krad is small for pyrene so that the lifetime of pyrene is large since τM−1 = krad + knrad. Indeed, pyrene and its derivatives have lifetimes of several hundreds of nanoseconds, much longer than any other aromatic fluorophore. The long lifetime of pyrene provides a long time window through which photophysical phenomena occurring over a few tens of picoseconds to several hundreds of nanoseconds can be probed, a dynamic range covering five orders of magnitude.
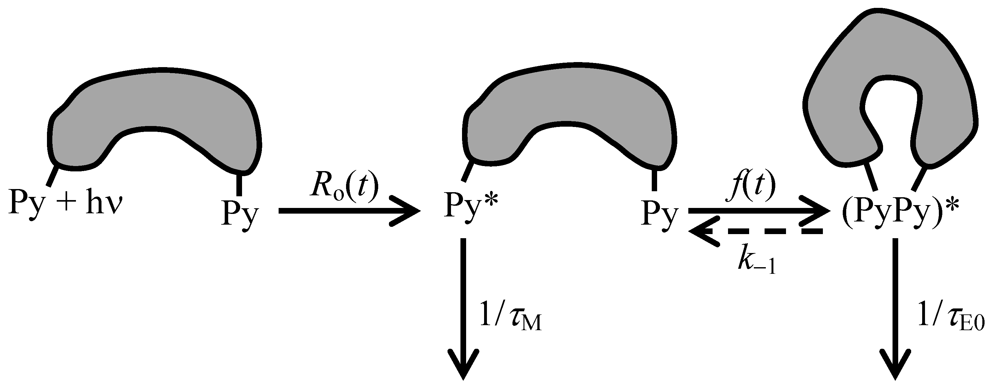
The kinetics of excimer formation between pyrene labels covalently attached to a macromolecule are described by Scheme 2 where the parameters τM and τE0 represent the lifetime of the pyrene monomer and excimer, respectively. Two temperature regimes have been defined for excimer formation depending on whether a Steven-Ban plot [148] of the logarithm Ln(IE/IM) (where IE and IM are the fluorescence intensity of the pyrene excimer and monomer, respectively) increases or decreases linearly with increasing 1/T [149]. In the high temperature regime, Ln(IE/IM) increases with increasing 1/T and k−1 is large compared to 1/τE0. In the low temperature regime where Ln(IE/IM) decreases with increasing 1/T, the excimer dissociation rate constant k−1 is small and can be neglected. In the case of pyrene excimer formation, usually k−1 can be neglected at temperatures lower than 30 °C so that it is often neglected for experiments carried out at room temperature (T≈ 25 °C) [81]. Scheme 2 is referred to as Birks’ scheme in recognition of J.B. Birks who characterized the kinetics of excimer formation for aromatic molecules in solution [139].
Scheme 2.
Excimer formation between pyrene groups covalently attached to a macromolecule.

The rate of excimer formation is given by the function f(t) which depends on the distribution of distances spanning every two pyrenes [75,150]. The time-dependent behavior of the concentration of pyrene monomer and excimer is obtained by integrating Equations (7) and (8) where excimer dissociation has been neglected.

The subscript diff used in Equation (7) emphasizes that these pyrenes form excimer by diffusion. These differential equations are coupled which implies that any decrease of ![Polymers 04 00211 i017]() in Equation (7) due to excimer formation results in an equivalent increase in [E0*] in Equation (7). Integration of Equations (7) and (8) yields the time-dependent concentrations of
in Equation (7) due to excimer formation results in an equivalent increase in [E0*] in Equation (7). Integration of Equations (7) and (8) yields the time-dependent concentrations of ![Polymers 04 00211 i017]() (t) and [E0*](t). How this derivation is mathematically handled with the specific application to pyrene-labeled dendrimers is the topic of the next section.
(t) and [E0*](t). How this derivation is mathematically handled with the specific application to pyrene-labeled dendrimers is the topic of the next section.
 in Equation (7) due to excimer formation results in an equivalent increase in [E0*] in Equation (7). Integration of Equations (7) and (8) yields the time-dependent concentrations of (t) and [E0*](t). How this derivation is mathematically handled with the specific application to pyrene-labeled dendrimers is the topic of the next section.
in Equation (7) due to excimer formation results in an equivalent increase in [E0*] in Equation (7). Integration of Equations (7) and (8) yields the time-dependent concentrations of (t) and [E0*](t). How this derivation is mathematically handled with the specific application to pyrene-labeled dendrimers is the topic of the next section.3. Analysis of the Kinetics of Excimer Formation for Pyrene-Labeled Dendrimers
In homogeneous solutions, pyrene excimer formation takes place with a rate constant k1 (i.e., f(t) = k1 × [Py] × ![Polymers 04 00211 i017]() in Scheme 2 and in Equations (7) and (8)) [139]. In that case, the kinetics of excimer formation are simple enough and the dissociation rate constant can actually be recovered from a more detailed analysis than that proposed in Equations (7) and (8). Whereas
in Scheme 2 and in Equations (7) and (8)) [139]. In that case, the kinetics of excimer formation are simple enough and the dissociation rate constant can actually be recovered from a more detailed analysis than that proposed in Equations (7) and (8). Whereas ![Polymers 04 00211 i017]() (t) would be found to decay monoexponentially if Equation (7) was integrated assuming a function Ro(t) = Ro × δ(t) (i.e., instantaneous excitation of pyrene), the complete treatment of the kinetics of excimer formation that includes the dissociation rate constant k−1 in Scheme 2 is described by J.B. Birks yielding a biexponential decay for
(t) would be found to decay monoexponentially if Equation (7) was integrated assuming a function Ro(t) = Ro × δ(t) (i.e., instantaneous excitation of pyrene), the complete treatment of the kinetics of excimer formation that includes the dissociation rate constant k−1 in Scheme 2 is described by J.B. Birks yielding a biexponential decay for ![Polymers 04 00211 i017]() (t) [139]. The contribution to the overall decay of the exponential having the shorter decay time is usually rather small (less than 15% of the total pre-exponential weight) so that the decay of
(t) [139]. The contribution to the overall decay of the exponential having the shorter decay time is usually rather small (less than 15% of the total pre-exponential weight) so that the decay of ![Polymers 04 00211 i017]() (t) can be considered to be essentially monoexponential as would be expected for a process where k−1 is small compared to τE−1 in Scheme 2 (i.e., the excimer fluoresces more quickly than it can dissociate).
(t) can be considered to be essentially monoexponential as would be expected for a process where k−1 is small compared to τE−1 in Scheme 2 (i.e., the excimer fluoresces more quickly than it can dissociate).
in Scheme 2 and in Equations (7) and (8)) [139]. In that case, the kinetics of excimer formation are simple enough and the dissociation rate constant can actually be recovered from a more detailed analysis than that proposed in Equations (7) and (8). Whereas (t) would be found to decay monoexponentially if Equation (7) was integrated assuming a function Ro(t) = Ro × δ(t) (i.e., instantaneous excitation of pyrene), the complete treatment of the kinetics of excimer formation that includes the dissociation rate constant k−1 in Scheme 2 is described by J.B. Birks yielding a biexponential decay for (t) [139]. The contribution to the overall decay of the exponential having the shorter decay time is usually rather small (less than 15% of the total pre-exponential weight) so that the decay of (t) can be considered to be essentially monoexponential as would be expected for a process where k−1 is small compared to τE−1 in Scheme 2 (i.e., the excimer fluoresces more quickly than it can dissociate).The first study of intramolecular pyrene excimer formation was reported by Zachariasse in 1976 for a series of n-alkanes terminated at both ends with a 1-pyrenemethyl group [151]. It was followed by numerous others that provided information about the ensemble of conformations adopted by the short alkane chain as it folds to bring its two ends in close proximity to form an excimer [152,153,154,155,156,157,158,159,160]. The scope of these studies was then expanded to include the end-to-end cyclization (EEC) of polymer chains by covalently attaching a pyrenyl group at both ends of a series of poly(ethylene oxide) (Py2-PEO) [78]. These early fluorescence experiments carried out with continuous excitation provided relative information about the EEC rate constant (kEEC). In 1980, Winnik applied the Birks scheme to analyze the fluorescence decays of a series of pyrene end-labeled polystyrenes (Py2-PS) and retrieve kEEC quantitatively [79]. The procedure introduced by Winnik to determine kEEC was then expanded to numerous polymeric backbones such as polydimethylsiloxane [81,96,97], poly(bisphenol A-diethylene glycol carbonate) [84], poly(tetramethylene oxide) [86], or poly(ε-caprolactone) [94]. At about the same time, it was also realized that kEEC provided not only information about the EEC process, but maybe more importantly, also a measure of the long range internal dynamics of the polymer chain. The procedure was further applied to more biologically relevant polymers by attaching dyes other than pyrene to the ends of monodisperse polypeptides of well-defined repeating sequences [161,162,163,164,165,166,167,168,169,170,171,172,173]. In so doing, kEEC was obtained for these polypeptides by the analysis of fluorescence decays and it was found to yield information about the polypeptide relative chain flexibility.
The experiments with end-labeled oligomers or polymers dealt with well-defined constructs with a single chain length spanning two pyrenes. In this case, theory predicts that f(t) in Scheme 2 equals kEEC [174,175] which was found experimentally to scale as Nα with α taking values between 0.9 and 1.9 [74,81,84,85,170,171,172,173]. In turn, this result implied that kEEC decreases strongly with increasing distance spanning two pyrenes. This had a detrimental consequence for studying macromolecules with more than two pyrene pendants and where the pyrene pendants are separated by more than one chain length, as is the case for pyrene end-labeled dendrimers. In this case, f(t) is unknown.
Although f(t) is undefined, work from this laboratory has proposed a mathematical procedure that provides a quantitative measure of the intramolecular rate constant of excimer formation for pyrene end-labeled dendrimers without the explicit knowledge of f(t) [150]. The treatment is based on the observation that although f(t) is not known, any fluorescence decay can be fitted with a sum of exponentials. Thus the fluorescence decay of the pyrene monomer for a pyrene end-labeled dendrimer can be fitted with Equation (9).

In Equation (9), the pre-exponential factors ai are normalized so that their sum equals unity ( ![Polymers 04 00211 i018]() ). Since
). Since ![Polymers 04 00211 i017]() (t) is known, its expression can be used in Equation (7) to determine f(t) which for times t > 0 is given by Equation (10).
(t) is known, its expression can be used in Equation (7) to determine f(t) which for times t > 0 is given by Equation (10).
 ). Since (t) is known, its expression can be used in Equation (7) to determine f(t) which for times t > 0 is given by Equation (10).
). Since (t) is known, its expression can be used in Equation (7) to determine f(t) which for times t > 0 is given by Equation (10).
Equation (10) can then be introduced in Equation (7) to yield the expression of [E0*](t) given in Equation (11).
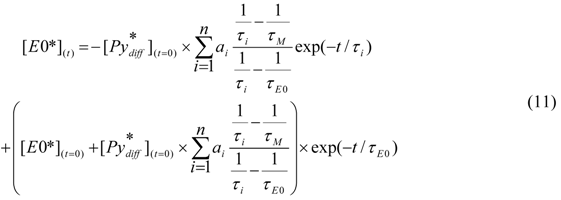
Theoretically, Equations (9) and (11) should be enough to fit the fluorescence decays of, respectively, the pyrene monomer and excimer of any pyrene-labeled macromolecule forming excimer by diffusion. Unfortunately, these equations need to be adjusted to account for two complications that are specific to pyrene-labeled macromolecules in general and dendrimers in particular [126,176]. The first complication is due to the unavoidable presence of traces of unattached pyrene label in any pyrene-labeled dendrimer. As will become clear later, the long lifetime and large fluorescence quantum yield of pyrene monomers unattached to the dendrimer constitute a major challenge to the interpretation of the fluorescence results. Second, the high local pyrene concentration inside the congested dendrimer interior does not always enable two pyrenyl units to encounter and adopt the ideal stacking required for excimer formation. The resulting dimers D* constituted of poorly stacked pyrenes emit with a lifetime τD that is different from τE0. Whereas τE0 takes values between 40 and 70 ns in organic solvents [139], lifetimes τD have been obtained that were as short as 2–4 ns [108,126] or as long as 100–150 ns [103,112,113,114,116].
To account for the unattached pyrene labels that do not form excimer under the dilute solution conditions used to acquire the fluorescence decays, typically 2.5 × 10−6 M in pyrene, and emit as free pyrenes would, the concentration ![Polymers 04 00211 i032]() (t) given in Equation (12) is added to Equation (9) to yield Equation (13).
(t) given in Equation (12) is added to Equation (9) to yield Equation (13).
 (t) given in Equation (12) is added to Equation (9) to yield Equation (13).
(t) given in Equation (12) is added to Equation (9) to yield Equation (13).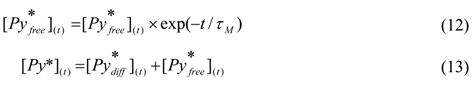
Depending on whether the dimers are pre-stacked or formed by diffusion, two expressions for [D*](t) have been derived [126,150,176,177] from which only the simpler one used to deal with the former type of dimers is given in Equation (14).

The overall excimer fluorescence decay combines the contributions from both the pyrene excimer E* and excited dimers D* by summing both contributions in Equation (15).

Equations (13) and (15) can then be used to fit the fluorescence decays of the pyrene monomer and excimer respectively. Because the expressions of ![Polymers 04 00211 i017]() (t), and [E0*](t) were obtained by making no assumption about the nature of f(t), this type of mathematical treatment used to fit the fuorescence decays has been referred to as the Model Free (MF) analysis [126,176,177].
(t), and [E0*](t) were obtained by making no assumption about the nature of f(t), this type of mathematical treatment used to fit the fuorescence decays has been referred to as the Model Free (MF) analysis [126,176,177].
(t), and [E0*](t) were obtained by making no assumption about the nature of f(t), this type of mathematical treatment used to fit the fuorescence decays has been referred to as the Model Free (MF) analysis [126,176,177].At first glance, Equations (13) and (15) are sums of exponentials similar to those typically found to fit the fluorescence decays of the pyrene monomer and excimer [74,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,159], although the exact form of the sums of exponentials encountered in the literature is different from that of Equations (13) and (15). As for the other analyses, the monomer and excimer decays are fitted gobally to improve the accuracy on the retrieved parameters [178,179,180]. However, our analysis departs in one important manner from the current art in the way the pre-exponential factors ![Polymers 04 00211 i019]() in Equation (9) and
in Equation (9) and ![Polymers 04 00211 i020]() and
and ![Polymers 04 00211 i021]() in Equation (11) are being handled because αi, βi, and γi are not treated as floating parameters in the optimization, but rather are fitted as a function of the decay times τi and τE0 and the pre-exponential factors ai. As it turns out, analyzing the fluorescence decays of the pyrene monomer and excimer in this manner retrieves the parameters ai, the decay times τi and τE0, and the molar fractions fdiff, ffree, fE0, and fD of the pyrene species
in Equation (11) are being handled because αi, βi, and γi are not treated as floating parameters in the optimization, but rather are fitted as a function of the decay times τi and τE0 and the pre-exponential factors ai. As it turns out, analyzing the fluorescence decays of the pyrene monomer and excimer in this manner retrieves the parameters ai, the decay times τi and τE0, and the molar fractions fdiff, ffree, fE0, and fD of the pyrene species ![Polymers 04 00211 i017]() ,
, ![Polymers 04 00211 i032]() , E0* and D* found in solution with improved accuracy [126,150,176,177] compared to more traditional analyses [74,108,160]. Furthermore, it also enables the calculation of the ratio
, E0* and D* found in solution with improved accuracy [126,150,176,177] compared to more traditional analyses [74,108,160]. Furthermore, it also enables the calculation of the ratio ![Polymers 04 00211 i022]() given in Equation (16) which constitutes an absolute measure of the IE/IM ratio obtained by steady-state fluorescence.
given in Equation (16) which constitutes an absolute measure of the IE/IM ratio obtained by steady-state fluorescence.
 in Equation (9) and
in Equation (9) and  and
and  in Equation (11) are being handled because αi, βi, and γi are not treated as floating parameters in the optimization, but rather are fitted as a function of the decay times τi and τE0 and the pre-exponential factors ai. As it turns out, analyzing the fluorescence decays of the pyrene monomer and excimer in this manner retrieves the parameters ai, the decay times τi and τE0, and the molar fractions fdiff, ffree, fE0, and fD of the pyrene species , , E0* and D* found in solution with improved accuracy [126,150,176,177] compared to more traditional analyses [74,108,160]. Furthermore, it also enables the calculation of the ratio
in Equation (11) are being handled because αi, βi, and γi are not treated as floating parameters in the optimization, but rather are fitted as a function of the decay times τi and τE0 and the pre-exponential factors ai. As it turns out, analyzing the fluorescence decays of the pyrene monomer and excimer in this manner retrieves the parameters ai, the decay times τi and τE0, and the molar fractions fdiff, ffree, fE0, and fD of the pyrene species , , E0* and D* found in solution with improved accuracy [126,150,176,177] compared to more traditional analyses [74,108,160]. Furthermore, it also enables the calculation of the ratio  given in Equation (16) which constitutes an absolute measure of the IE/IM ratio obtained by steady-state fluorescence.
given in Equation (16) which constitutes an absolute measure of the IE/IM ratio obtained by steady-state fluorescence. (16)
(16)The SPC superscript in Equation (16) reminds the reader that (IE/IM)SPC is obtained by time-correlated single photon counting experiments whereas the (IE/IM)SS ratio corresponds to the IE/IM ratio obtained from the steady-state fluorescence spectra. In effect, the ratios (IE/IM)SPC and (IE/IM)SS both represent the extent of excimer formation, the only difference being that the former and latter ratios represent absolute and relative values, respectively. One major advantage of Equation (16) is that the emission due to free pyrene labels can be isolated so that the true IE/IM ratio of a pure pyrene-labeled dendrimer is obtained simply by setting ffree in Equation (16) to equal zero to yield ![Polymers 04 00211 i023]() . In turn,
. In turn, ![Polymers 04 00211 i023]() is expected to be proportional to the average rate constant of pyrene excimer formation <k> defined hereafter, as has been found in a number of studies [73,74,78,125,126,176]. A discrepancy between
is expected to be proportional to the average rate constant of pyrene excimer formation <k> defined hereafter, as has been found in a number of studies [73,74,78,125,126,176]. A discrepancy between ![Polymers 04 00211 i023]() and <k> would imply that something is amiss in the analysis. Discrepancies between the ratios (IE/IM)SPC and (IE/IM)SS and <k> typically indicate that some free pyrene is present in solution that has not been accounted for.
and <k> would imply that something is amiss in the analysis. Discrepancies between the ratios (IE/IM)SPC and (IE/IM)SS and <k> typically indicate that some free pyrene is present in solution that has not been accounted for.
 . In turn, is expected to be proportional to the average rate constant of pyrene excimer formation <k> defined hereafter, as has been found in a number of studies [73,74,78,125,126,176]. A discrepancy between and <k> would imply that something is amiss in the analysis. Discrepancies between the ratios (IE/IM)SPC and (IE/IM)SS and <k> typically indicate that some free pyrene is present in solution that has not been accounted for.
. In turn, is expected to be proportional to the average rate constant of pyrene excimer formation <k> defined hereafter, as has been found in a number of studies [73,74,78,125,126,176]. A discrepancy between and <k> would imply that something is amiss in the analysis. Discrepancies between the ratios (IE/IM)SPC and (IE/IM)SS and <k> typically indicate that some free pyrene is present in solution that has not been accounted for.Finally, a measure of the time scale over which pyrene excimer formation takes place can be obtained from the average excimer formation rate constant <k> which can be approximated by two expressions given in Equations (17) and (18).

Both equations provide a measure of the rate constant of excimer formation but Equation (18) has been shown to provide more reliable results in at least one instance [176].
4. Internal Segmental Dynamics of Pyrene-Labeled Dendrimers
The ability of dendrimers to bring reactants into close proximity with absolute control over their spacing has made them essential building blocks in the toolbox of photochemists. Fluorescence resonance energy transfer (FRET), energy hopping or migration, and electron transfer between a donor and an acceptor are all photophysical processes whose efficiency depends strongly on the distance between the donor and acceptor [135,136,137]. The design of dendrimers used for this type of applications consists in placing many donors at the dendrimer periphery and a single acceptor molecule at the dendrimer focal point [3,36,39,40,41,42,43,44,120,121,122,123,125]. As the dendrimer generation increases, the efficiency of the photophysical process of interest decreases according to the increased distance separating the donor from the acceptor [135,136,137]. The efficiency of these photophysical processes is a combination of the reactivity of the reactants towards each other and the internal segmental dynamics of the branches constituting the constructs. However, the time scale over which the photophysical processes take place is usually much faster than that over which segmental diffusive motion occurs due to the small dendrimer size. Furthermore segmental motion of the branches can be completely halted by working with rigid backbones such as those afforded by phenylacetylene or pyrene building blocks [50,117,118,119].
The study of the internal segmental dynamics of dendrimers can only be achieved with dendrimers constituted of flexible segments such as those made of a backbone of poly(amido amine) (PAMAM dendrimers), polyamide, poly(benzyl ether), poly(benzyl ester), poly(propylene imine) (PPI dendrimers), or poly(2,2-bis(hydroxymethyl)-propionic acid) to name but a few. Over the years, the synthesis of several pyrene-labeled dendrimers has been reported in the literature [36,37,44,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134], but in many instances, the pyrene-labeled constructs were synthesised as pyrene-labeled dendrons to be attached onto a focal point bearing an acceptor molecule capable of communicating with the pyrenyl pendants [36,44,120,121,122,123]. Consequently, these investigations focused on the photophysical process taking place between the pyrene donor and the acceptor rather than on excimer formation between pyrene pendants covalently attached to the chain ends of dendrimers. In effect, pyrene excimer formation has been investigated in earnest in only seven reports [44,120,126,127,128,130,134] and the results of these are now described.
To date, poly(benzyl ether) dendrimers constitute the dendrimer family that has been the most decorated with pyrenyl groups. However, out of the eight reports presenting data on pyrene-labeled poly(benzyl ether) dendrimers [36,37,44,120,121,122,123,124,125], only two [44,120] present some information about the process of pyrene excimer formation, in particular via the ratio (IE/IM)SS. Stewart and Fox [120] chose a 1-pyrenylbenzylether substituent to label a first generation dendrimer (see chemical structure in Table 1) whereas Cicchi et al. [44] chose a 1-pyrenebutyltriazol moiety to label generations 1–3 of their poly(benzyl ether) dendrimers (Table 1). The finding that excimer formation occurs for these pyrene-labeled poly(benzyl ether) dendrimers in solution actually contradicts the results of three other studies that used either 1-pyrenemethylene [123] or diarylamino-1-pyrene [36,122] to label generation 1–3 dendrimers and reported no pyrene excimer formation. Gu et al. [37] also observed excimer formation when four 1st generation poly(benzyl ether) dendrimers labeled with pyrene where covalently attached onto a cyclam core that was introduced as a copper ligand. No fluorescence data was reported in the study by Morales-Espinoza et al. [125].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 Stewart, G.M., et al. [120] | 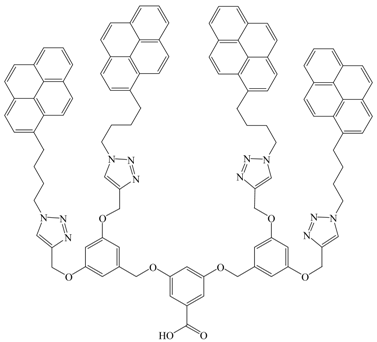 Cicchi, S., et al. [44] |
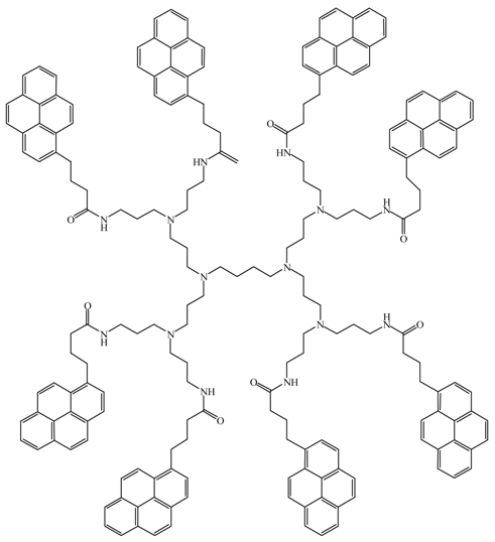 Baker, L.A., et al. [127] |  Wang, B.-B., et al. [128] |
 Yip, J., et al. [126] |  Wilken, R., et al. [134] |
 Brauge, L., et al. [130] | |
Considering the two reports where excimer formation was observed for pyrene-labeled poly(benzyl ether) dendrons, the (IE/IM)SS ratios corresponding to the fluorescence spectra reported by Stewart and Fox [120] and Cicchi et al. [44] were determined herein by taking the ratio of the maximum fluorescence intensity of the excimer over that of the pyrene monomer at the 0–0 transition in the fluorescence spectra given in the respective publications. To account for differences in solvent viscosity, these ratios were multiplied by the viscosity of the solvent. The product η × (IE/IM) was plotted as a function of the number of pyrenyl pendants found in the dendrimer. The dendrimer prepared by Stewart and Fox [120] yielded a product η × (IE/IM)SS that was 10 times larger than the first generation dendrimer prepared by Cicchi et al. [44]. This might be a consequence of the use of a 1-pyrenemethyl versus a 1-pyrenebutyl derivative to label the dendrimers of Stewart and Fox and Cicchi et al., respectively. The overall longer linker used to attach the 1-pyrenebutyl group to the dendrons decreases the local concentration of pyrene inside the dendrimer volume resulting in weaker excimer formation. Also the 0–0 transition is allowed for the 1-pyrenebutyl linker so that IM taken as the fluorescence intensity of the first peak is larger contributing to making (IE/IM)SS smaller in the Cicchi et al. study. The presence of a methoxi-group in the 1-position of the pyrenyl substituent of the Stewart and Fox study partially restores the symmetry of pyrene, thus making the 0–0 transition symmetry forbidden, decreasing IM, and increasing the (IE/IM)SS ratio [98,100,114]. Finally, in the Cicchi et al. study, η × (IE/IM)SS increases continuously with increasing pyrene content.
An increase in η × (IE/IM)SS with increasing pyrene content was also found by Duhamel et al. [126] for a series of four poly(2,2-bis(hydroxymethyl)-propionic acid) dendrimers of generations 1–4 which were labeled with 1-pyrenebutyl pendants (Table 1). As can be seen in Figure 2, the increase in η × (IE/IM)SS obtained by Duhamel et al. parallels closely the increase observed by Cicchi et al. [44] The larger η × (IE/IM)SS values obtained in the Duhamel study reflect the more flexible backbone of the poly(2,2-bis(hydroxymethyl)-propionic acid) dendrimers as well as their smaller dimension.
Figure 2.
Plot of η × (IE/IM)SS as a function of the number of pyrenes per dendrimer. (◊) Reference [120]; (×) Reference [134] assuming that each chain end of the dendrimer is labeled with a pyrenyl pendant; (▲) Reference [127]; (∆) Reference [130], dendrimer in acetonitrile; (♦) Reference [128]; (■) Reference [44]; (□) Reference [126].
Figure 2.
Plot of η × (IE/IM)SS as a function of the number of pyrenes per dendrimer. (◊) Reference [120]; (×) Reference [134] assuming that each chain end of the dendrimer is labeled with a pyrenyl pendant; (▲) Reference [127]; (∆) Reference [130], dendrimer in acetonitrile; (♦) Reference [128]; (■) Reference [44]; (□) Reference [126].

As it turns out, the studies by Duhamel et al. and Cicchi et al. constitute the only two examples where η × (IE/IM)SS was found to increase monotonously and substantially with increasing pyrene content for dendrimers where all ends were labelled with pyrene. The PPI dendrimers labeled with 1-pyrenebutyric acid by Baker and Crook [127] (Table 1) showed a much smaller increase of η × (IE/IM)SS with increasing pyrene content. Similarly, the PAMAM dendrimers labeled with 1-pyrenylsuccinimide groups by Wei et al. [128] (Table 1) showed a more or less flat η × (IE/IM)SS profile with pyrene content with an unexplained spike for the third generation PAMAM dendrimer. The analysis of the fluorescence data obtained with the PPI and PAMAM dendrimers might be affected by the presence of tertiary amines in the dendrimer interior that are known quenchers of pyrene. Also, the pyrene derivative used by Wei et al. [128] bears a nitrogen atom in the 1-position instead of an alkyl group as was used in the Stewart and Fox [120], Cicchi et al. [44], and Duhamel et al. [126] studies. The presence of a heteroatom or other complex function in the 1-position of pyrene instead of an alkyl chain has been found to profoundly affect the fluorescence of pyrene and should be avoided [131]. For instance, a PAMAM dendrimer labeled with a pyrene derivative bearing an imine double bond in the 1-position yielded excimer that had a maximum emission located as far as 650 nm in the fluorescence spectrum instead of 480 nm which is typically observed [133]. With a pyrene label behaving in a manner that is so different from what is known about pyrene excimer formation, the process of excimer formation with this label would have to be fully characterized before any conclusion could be drawn from the fluorescence data obtained with these pyrene-labeled PAMAM dendrimers about their internal segmental dynamics.
Phosphorus containing dendrimers have been labeled internally with 12 1-pyrenebutyric acid groups by Majoral et al. [130]. The ratio (IE/IM)SS of the dendrimer was compared to that of a model compound bearing two pyrene groups in acetonitrile. The product η × (IE/IM)SS was found to decrease slightly with increasing pyrene content, a trend opposite to that observed by Cicchi et al. [44] and Duhamel et al. [126].
The pyrene-labeled poly(benzyl ester) dendrimers prepared by Wilken and Adams [134] had an ingenious design. The generation 1–3 dendrimers contained the same number of three 1-pyrenebutyric acid groups for each generation. Since the poly(benzyl ester) dendrimers bore the same number of pyrenes equal to 3, η × (IE/IM)SS for the pyrene-labeled dendrimers prepared by Wilken and Adams were plotted in Figure 2 as a function of the number of pyrene groups that would have been found in the fully labeled dendrimers, namely 3, 6, and 12 for the dendrimers of generation 1, 2, and 3. As the dendrimer volume increased with increasing generation, η × (IE/IM)SS increased with increasing generation number. Since the number of pyrene labels was constant and equal to 3, the increase in η × (IE/IM)SS with increasing dendrimer volume could only be due to the increased flexibility of the dendrimer arms. But the trend in η × (IE/IM)SS found by Wilken and Adams [134] leads to further questions. Indeed, if increasing generation number increases the internal dynamics of dendrimers for small generation numbers, then why would η × (IE/IM)SS hardly change with generation number for three [127,128,130] out of the seven studies presented in Figure 2?
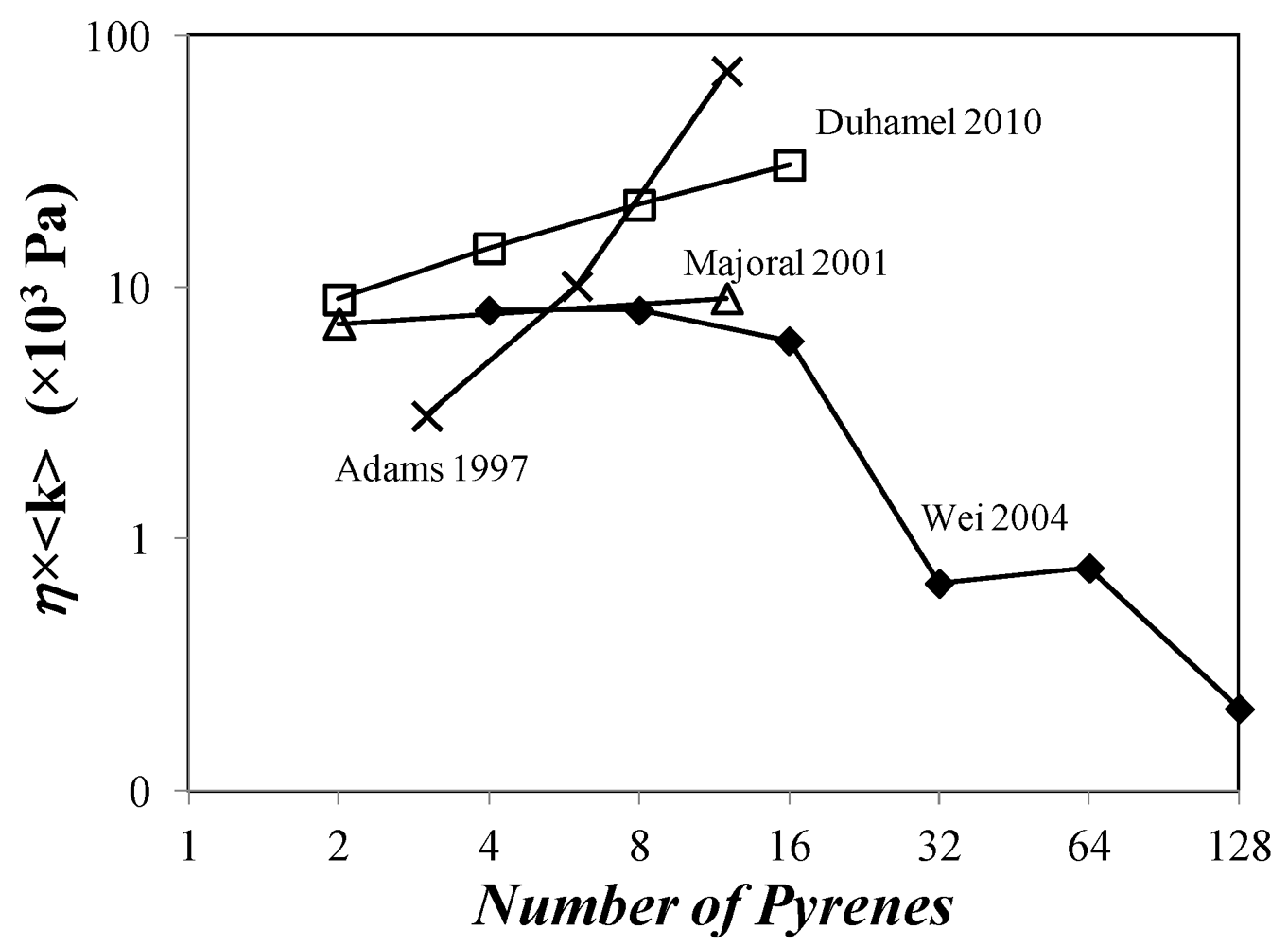
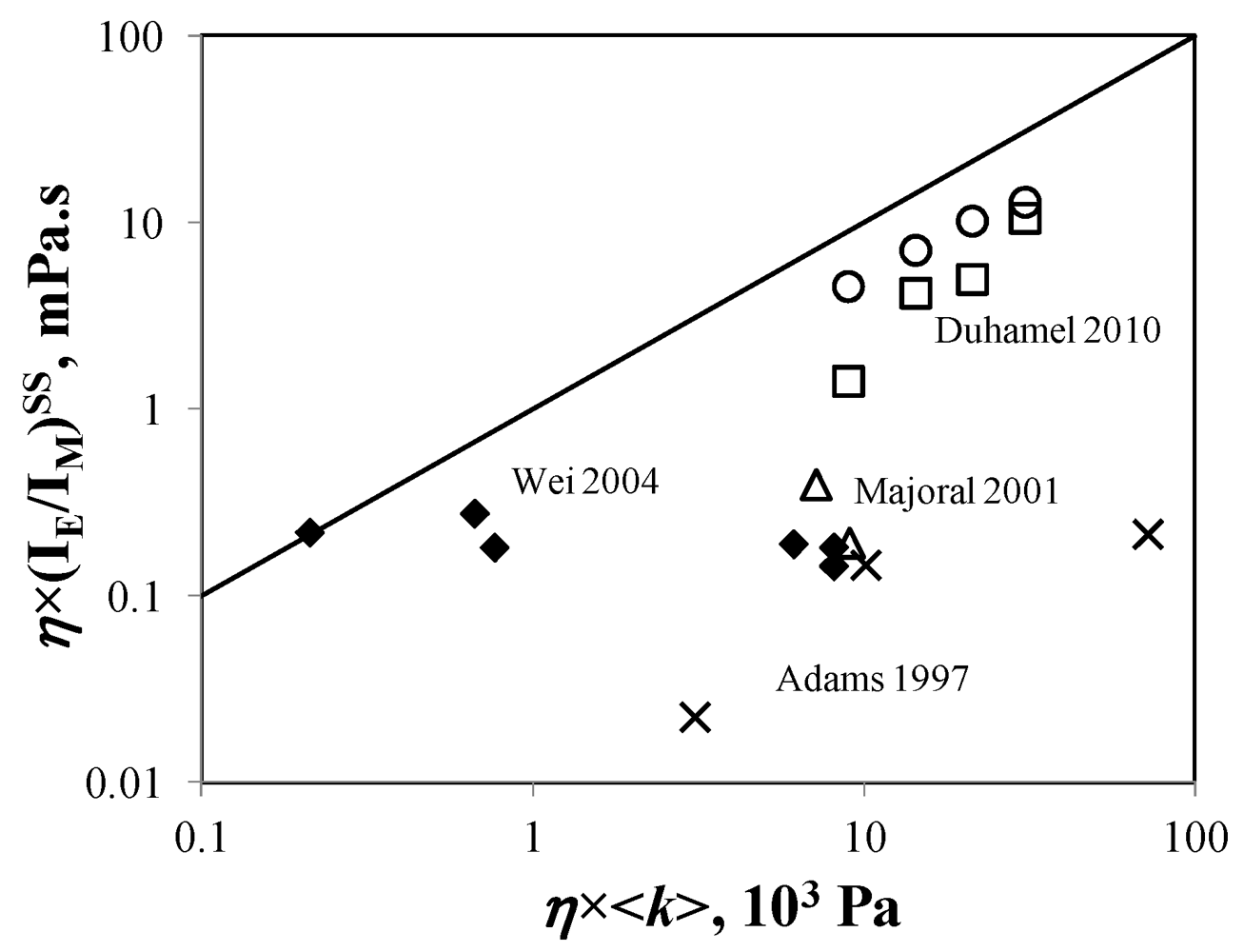
The contradicting nature of the trends obtained for η × (IE/IM)SS in Figure 2 by seven different studies are further confirmed by considering the average rate constant of pyrene excimer formation <k> which was determined in four of the seven studies by using Equation (17). The trends obtained for η × <k> are shown in Figure 3. Duhamel et al. [126] and Wilken and Adams [134] found that the product η × <k> increased with increasing pyrene content and generation number, respectively. Majoral et al. [131] found a slight increase for η × <k> as a function of pyrene content, whereas Wei et al. [128] obtained a decreasing η × <k> product. Obviously the trends shown in Figure 3 are inconsistent. This lack of consensus between the different studies is further exposed in Figure 4 by noting that η × (IE/IM)SS and η × <k> vary with generation number in a manner that is independent of each other. In a log-log plot of η × (IE/IM)SS versus η × <k>, Log[η × (IE/IM)SS] should increase linearly with increasing Log[η × <k>] with a slope equal to 1.0. Even with the data obtained by Duhamel et al. [125] where Log[η × (IE/IM)SS] increases linearly with increasing Log[η × <k>], the slope takes a value of 2.0 which is different from unity. All trends shown in Figure 4. contradict a tenet of pyrene excimer formation that states that (IE/IM)SS and <k> are two equivalent quantities that should scale in a similar manner and which has been found experimentally in a number of examples [73,74,98].
Figure 3.
Plot of η× <k> as a function of the number of pyrenes per dendrimer. (×) Reference [134] assuming that each chain end of the dendrimer is labeled with a pyrenyl pendant; (∆) Reference [130], dendrimer in acetonitrile; (♦) Reference [128] assuming τM = 72.6 ns; (□) Reference [126].

Figure 4.
Log-log plot of η× (IE/IM)SS as a function of η× <k>. (×) Reference [134]; (∆) Reference [130], dendrimer in acetonitrile; (♦) Reference [128]; (□) Reference [126]; (○) ![Polymers 04 00211 i033]() versus <k> for Reference [126].
versus <k> for Reference [126].
 versus <k> for Reference [126].
versus <k> for Reference [126].
Work carried out by this laboratory suggests that the main reason for the inconsistencies exposed in Figure 4 for the Duhamel study resides in the presence of unaccounted for free pyrene labels in the solution [126,176]. Because excimer formation is so efficient inside pyrene-labeled dendrimers, the fluorescence intensity of the pyrene monomer (IM) is extremely small so that the presence of minute amounts of unattached pyrene labels offsets the (IE/IM)SS ratio dramatically. For instance, the presence of 3 mol % of 1-pyrenebutyric acid in the fourth generation of pyrene-labeled poly(2,2-bis(hydroxymethyl)-propionic acid) dendrimer resulted in a 75% drop in the (IE/IM)SS ratio [126].
Indeed, analysis of the monomer and excimer fluorescence decays with Equations (13) and (15) yielded an ffree value of 0.03 [126], suggesting that 3 mol% of unattached pyrene label was present in solution. Plotting (IE/IM)SPC calculated with Equation (16) as a function of generation number led to the same conclusion reached by the analysis of the steady-state fluorescence spectra, namely that the (IE/IM)SPC value for the 4th generation dendrimer was 75% smaller than expected [126]. However, setting ffree = 0 in Equation (16) yielded ![Polymers 04 00211 i023]() which brought the (IE/IM)SPC ratio back to its expected value. Purification of the 4th generation dendrimer by gel permeation chromatography demonstrated the presence of unattached 1-pyrenebutyric acid and the fluorescence spectrum acquired with the purified dendrimer free of unattached pyrene label yielded the expected (IE/IM)SS and (IE/IM)SPC ratios in agreement with the
which brought the (IE/IM)SPC ratio back to its expected value. Purification of the 4th generation dendrimer by gel permeation chromatography demonstrated the presence of unattached 1-pyrenebutyric acid and the fluorescence spectrum acquired with the purified dendrimer free of unattached pyrene label yielded the expected (IE/IM)SS and (IE/IM)SPC ratios in agreement with the ![Polymers 04 00211 i023]() value.
value.
which brought the (IE/IM)SPC ratio back to its expected value. Purification of the 4th generation dendrimer by gel permeation chromatography demonstrated the presence of unattached 1-pyrenebutyric acid and the fluorescence spectrum acquired with the purified dendrimer free of unattached pyrene label yielded the expected (IE/IM)SS and (IE/IM)SPC ratios in agreement with the value. The effect that a non-zero ffree value has on the IE/IM ratio was quantified experimentally by adding known amounts of a fluorescent impurity and comparing the ratios (IE/IM)SS and (IE/IM)SPC obtained with Equation (16) by applying the MF analysis to the pyrene monomer and excimer decays [176]. Both the (IE/IM)SS and (IE/IM)SPC ratios were found to scale in an identical manner decreasing upon addition of a fluorescent impurity, whereas the quantity ![Polymers 04 00211 i023]() remained constant and equal to its original value. This study demonstrated the ability of the MF analysis to probe quantitatively the presence of minute amounts of unattached pyrene label and perhaps more importantly, to account for it by setting ffree to equal zero in Equation (16) [176]. Indeed plotting
remained constant and equal to its original value. This study demonstrated the ability of the MF analysis to probe quantitatively the presence of minute amounts of unattached pyrene label and perhaps more importantly, to account for it by setting ffree to equal zero in Equation (16) [176]. Indeed plotting ![Polymers 04 00211 i031]() as a function of η × <k> in Figure 4 for the pyrene-labeled dendrimers characterized by Duhamel et al. [126] yielded a perfect straight line of slope unity (see hollow circles in Figure 4).
as a function of η × <k> in Figure 4 for the pyrene-labeled dendrimers characterized by Duhamel et al. [126] yielded a perfect straight line of slope unity (see hollow circles in Figure 4).
remained constant and equal to its original value. This study demonstrated the ability of the MF analysis to probe quantitatively the presence of minute amounts of unattached pyrene label and perhaps more importantly, to account for it by setting ffree to equal zero in Equation (16) [176]. Indeed plotting  as a function of η × <k> in Figure 4 for the pyrene-labeled dendrimers characterized by Duhamel et al. [126] yielded a perfect straight line of slope unity (see hollow circles in Figure 4).
as a function of η × <k> in Figure 4 for the pyrene-labeled dendrimers characterized by Duhamel et al. [126] yielded a perfect straight line of slope unity (see hollow circles in Figure 4).Whether all trends obtained with pyrene-labeled dendrimers and pertaining to pyrene excimer formation are affected by the presence of unattached pyrene labels remains to be seen, but its detrimental effect has been shown to be so important [126,176] that any study aiming at studying the process of pyrene excimer formation in pyrene-labeled dendrimers should pay careful attention to the issue. To date, no study beside that of Duhamel et al. [126] has considered this problem. Furthermore, the MF analysis presented earlier has been shown to be a powerful analytical tool that can detect and account for the presence of residual amount of free pyrene labels and provides quantitative information about the internal segmental dynamics of dendrimers, information that has been elusive up to this date.
The data obtained by fitting the fluorescence decays for the pyrene-labeled poly(2,2-bis(hydroxymethyl)-propionic acid) dendrimers with the MF analysis resulted in surprisingly simple trends [126]: Both ![Polymers 04 00211 i023]() and <k> were found to increase linearly with increasing generation number. Considering that the mobility of the chain ends has been found to be slightly affected by increasing generation number, these trends led to the conclusion that the pyrene groups did not probe the entire volume of the dendrimer while being excited. Also <k> took very large values ranging between 10 and 70 × 107 s−1, much larger than those found for pyrene end-labeled polymers, reflecting the expected flexibility of the backbone of these dendrimers and their large local pyrene concentration.
and <k> were found to increase linearly with increasing generation number. Considering that the mobility of the chain ends has been found to be slightly affected by increasing generation number, these trends led to the conclusion that the pyrene groups did not probe the entire volume of the dendrimer while being excited. Also <k> took very large values ranging between 10 and 70 × 107 s−1, much larger than those found for pyrene end-labeled polymers, reflecting the expected flexibility of the backbone of these dendrimers and their large local pyrene concentration.
and <k> were found to increase linearly with increasing generation number. Considering that the mobility of the chain ends has been found to be slightly affected by increasing generation number, these trends led to the conclusion that the pyrene groups did not probe the entire volume of the dendrimer while being excited. Also <k> took very large values ranging between 10 and 70 × 107 s−1, much larger than those found for pyrene end-labeled polymers, reflecting the expected flexibility of the backbone of these dendrimers and their large local pyrene concentration.5. Recommendations for Future Studies on the Internal Segmental Dynamics of Pyrene-Labeled Dendrimers and Conclusions
Considering how complicated it already is to study the process of excimer formation in pyrene-labeled dendrimers (cf. contradicting trends in Figure 2–Figure 4) [36,37,44,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134], studies should focus on dendrimers whose backbone is not expected to quench the excited pyrene or its excimer. Chemical motives that should be avoided at all cost include the tertiary amines such as those found in PAMAM [128,129,133] and PPI [127] dendrimers which are known quenchers of pyrene. Also, the photophysical properties of pyrene are so sensitive to its substituent in the 1-position that pyrene-labeled dendrimers should be prepared with pyrene derivatives that possess an alkyl spacer which seems to act as an insulator [131]. In terms of availability, 1-pyrenemethanol, 1-pyrenemethylamine, 1-pyrenebutanol, and 1-pyrenebutyric acid are all suitable pyrene derivatives which are commercially available at a reasonable price. Finally, utmost care should be paid to minimize the presence of unattached pyrene labels, and if possible account for it as is being done with the MF analysis.
Another advantage of using the MF analysis comes from the fact that quantitative analysis of the fluorescence decays acquired with pyrene-labeled dendrimers is only possible if a mathematical expression exists for f(t) in Scheme 2. To date, only two mathematical expressions of f(t) are known to deal with pyrene-labeled macromolecules. If two (and only two) pyrene moieties are covalently attached at two well-defined positions of the macromolecule and if these two positions are separated by a fixed chain length, then f(t) equals a constant such as kEEC when the pyrenyl groups are attached at the ends of a monodisperse polymer [73,74,174,175]. When the pyrenyl groups are randomly distributed along a polymer, an expression of f(t) has been derived by applying the Fluorescence Blob Model (FBM) [75]. Any other configuration of the pyrene pendants inside the macromolecule requires a specific expression of f(t) and such an expression has not been derived yet for dendrimers. In this respect, the MF analysis is quite versatile as it applies to any possible distribution of the pyrene pendants covalently attached onto a macromolecule, such as for dendrimers labeled with pyrene at the chain ends.
Considering the parlous state of the understanding of pyrene excimer formation in pyrene-labeled dendrimers such as illustrated in Figure 2–Figure 4, the scientific community should focus its efforts toward building a clear consensus on how ![Polymers 04 00211 i023]() and <k> vary with increasing generation number beginning with dendrimers whose chain ends are fully labeled with pyrenyl pendants. The simpler synthetic protocols associated with the full labeling of the dendrimer terminals should make the preparation of such dendrimers more achievable. In each study, the fluorescence data must be shown to yield internally consistent trends, particularly with respect to
and <k> vary with increasing generation number beginning with dendrimers whose chain ends are fully labeled with pyrenyl pendants. The simpler synthetic protocols associated with the full labeling of the dendrimer terminals should make the preparation of such dendrimers more achievable. In each study, the fluorescence data must be shown to yield internally consistent trends, particularly with respect to ![Polymers 04 00211 i023]() and <k> as was done in the study carried out by Duhamel et al. Only after such precautions have been taken will a clear consensus emerge on how pyrene excimer formation takes place in pyrene end-labeled dendrimers. In turn, the information retrieved about the long range motion of the chain ends of a dendrimer will prove valuable to predict the time scale over which the chain ends are expected to exchange location between the dendrimer interior and its surface.
and <k> as was done in the study carried out by Duhamel et al. Only after such precautions have been taken will a clear consensus emerge on how pyrene excimer formation takes place in pyrene end-labeled dendrimers. In turn, the information retrieved about the long range motion of the chain ends of a dendrimer will prove valuable to predict the time scale over which the chain ends are expected to exchange location between the dendrimer interior and its surface.
and <k> vary with increasing generation number beginning with dendrimers whose chain ends are fully labeled with pyrenyl pendants. The simpler synthetic protocols associated with the full labeling of the dendrimer terminals should make the preparation of such dendrimers more achievable. In each study, the fluorescence data must be shown to yield internally consistent trends, particularly with respect to and <k> as was done in the study carried out by Duhamel et al. Only after such precautions have been taken will a clear consensus emerge on how pyrene excimer formation takes place in pyrene end-labeled dendrimers. In turn, the information retrieved about the long range motion of the chain ends of a dendrimer will prove valuable to predict the time scale over which the chain ends are expected to exchange location between the dendrimer interior and its surface.References
- Newkome, G.R.; Yao, Z.; Baker, G.R.; Gupta, V.K. Micelles. Part 1. Cascade molecules: A new approach to micelles. A [27]-arborol. J. Org. Chem. 1985, 50, 2003–2004. [Google Scholar] [CrossRef]
- Tomalia, D.A.; Baker, H.; Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. A new class of polymers: Starburst-dendritic macromolecules. Polym. J. 1985, 17, 117–132. [Google Scholar] [CrossRef]
- Balzani, V.; Ceroni, P.; Maestri, M.; Saudan, C.; Vicinelli, V. Luminescent dendrimers. Recent advances. Top. Curr. Chem. 2003, 228, 159–191. [Google Scholar] [CrossRef]
- D’Ambruoso, G.D.; McGrath, D.V. Energy harvesting in synthetic dendrimer materials. Adv. Polym. Sci. 2008, 214, 87–147. [Google Scholar]
- Nanjwade, B.K.; Bechra, H.M.; Derkar, G.K.; Manvi, F.V.; Nanjwade, V.K. Dendrimers: Emerging polymers for drug-delivery systems. Eur. J. Pharma. Sci. 2009, 38, 185–196. [Google Scholar] [CrossRef]
- Gillies, E.R.; Fréchet, J.M.J. Dendrimer and dendritic polymers in drug delivery. Drug Discov. Today 2005, 10, 35–43. [Google Scholar] [CrossRef]
- Helms, B.; Fréchet, J.M.J. The dendrimer effect in homogeneous catalysis. Adv. Synth. Catal. 2006, 348, 1125–1148. [Google Scholar] [CrossRef]
- Fréchet, J.M.J. Dendrimers and other dendritic macromolecules: From building blocks to functional assemblies in nanoscience and nanotechnology. J. Polym. Sci. A 2003, 41, 3713–3725. [Google Scholar] [CrossRef]
- Astruc, D.; Boisselier, E.; Ornelas, C. Dendrimers designed for functions: From physical, photophysical, and supramolecular properties to applications in sensing, catalysis, molecular electronics, photonics, and nanomedicine. Chem. Rev. 2010, 110, 1857–1959. [Google Scholar] [CrossRef]
- Hwang, S.-H.; Schreiner, C.D.; Moorefield, C.N.; Newkome, G.R. Recent progress and applications for metallodendrimers. New J. Chem. 2007, 31, 1192–1217. [Google Scholar] [CrossRef]
- Buhleier, E.; Wehner, W.; Vögtle, F. “Cascade”- and “nonskid-chain-like” syntheses of molecular cavity topologies. Synthesis 1978, 2, 155–158. [Google Scholar]
- Mansfield, M.L.; Klushin, L.I. Monte carlo studies of dendrimer molecules. Macromolecules 1993, 26, 4262–4268. [Google Scholar] [CrossRef]
- Murat, M.; Grest, G.S. Molecular dynamic study of dendrimer molecules in solvents of varying quality. Macromolecules 1996, 29, 1278–1285. [Google Scholar] [CrossRef]
- Scherrenberg, R.; Coussens, B.; Vliet, P.V.; Edouard, G.; Brackman, J.; Brabander, E.D.; Mortensen, K. The molecular characteristics of poly(propyleneimine) dendrimers as studied with small-angle neutron scattering, viscosimetry, and molecular dynamics. Macromolecules 1998, 31, 456–461. [Google Scholar]
- Cavallo, L.; Fraternali, F. A molecular dynamics study of the first five generations of poly(propylene imine) dendrimers modified with N-tBoc-L-phenylalinine. Chem. Eur. J. 1998, 4, 927–934. [Google Scholar] [CrossRef]
- Mansfield, M.L. Monte carlo studies of dendrimers. additional results for the diamond lattice model. Macromolecules 2000, 33, 8043–8049. [Google Scholar] [CrossRef]
- Mansfield, M.L.; Jeong, M. Simulation of lattice dendrimers by a monte carlo technique with detailed balance. Macromolecules 2002, 35, 9794–9798. [Google Scholar] [CrossRef]
- Giupponi, G.; Buzza, D.M.A. A monte carlo simulation scheme for nonideal dendrimers satisfying details balance. Macromolecules 2002, 35, 9799–9812. [Google Scholar] [CrossRef]
- Pavlov, G.M.; Korneeva, E.V.; Meijer, E.W. Molecular characteristics of poly(propylene imine) dendrimers as studied with translational diffusion and viscometry. Colloid Polym. Sci. 2002, 280, 416–423. [Google Scholar] [CrossRef]
- Rathgeber, S.; Monkenbush, M.; Kreitschmann, M.; Urban, V.; Brulet, A. Dynamics of star-burst dendrimers in solution in relation to their structural properties. J. Chem. Phys. 2002, 117, 4047–4062. [Google Scholar] [CrossRef]
- Rathgeber, S.; Pakula, T.; Urban, V. Structure of star-burst dendrimers: A comparison between small angle x-ray scattering and computer simulation results. J. Chem. Phys. 2004, 121, 3840–3853. [Google Scholar]
- De Gennes, P.G.; Hervet, H. Statistics of <<Starburst>> polymers. J. Phys. Lett. 1983, 44, 351–360. [Google Scholar] [CrossRef]
- Lescanec, R.L.; Muthukumar, M. Configurational characteristics and scaling behavior of starburst molecules: A computational study. Macromolecules 1990, 23, 2280–2288. [Google Scholar] [CrossRef]
- Evmenenko, G.; Bauer, B.J.; Kleppinger, R.; Forier, B.; Dehaen, W.; Amis, E.J.; Mischenko, N.; Reynaers, H. The influence of molecular architecture and solvent type of the size and structure of poly(benzyl ether) dendrimers by SANS. Macromol. Chem. Phys. 2001, 202, 891–899. [Google Scholar]
- Rosenfeldt, S.; Dingenouts, N.; Ballauff, M.; Werner, N.; Vögtle, F.; Lindner, P. Distribution of end groups within a dendritic structure: A sans study including contrast variation. Macromolecules 2002, 35, 8098–8105. [Google Scholar]
- Mansfield, M.L. Dendron segregation in model dendrimers. Polymer 1994, 35, 1827–1830. [Google Scholar] [CrossRef]
- Boris, D.; Rubinstein, M. A self-consistent mean field model of a starburst dendrimer: Dense core vs. dense shell. Macromolecules 1996, 29, 7251–7260. [Google Scholar] [CrossRef]
- Lyulin, A.V.; Davies, G.R.; Adolf, D.B. Brownian dynamics simulation of dendrimers under shear flow. Macromolecules 2000, 33, 3294–3304. [Google Scholar] [CrossRef]
- Lyulin, A.V.; Davies, G.R.; Adolf, D.B. Location of terminal groups of dendrimers: Brownian dynamics simulation. Macromolecules 2000, 33, 6899–6900. [Google Scholar] [CrossRef]
- Karatasos, K.; Adolf, D.B.; Davies, G.R. Statistics and dynamics of model dendrimers as studied by molecular dynamics simulations. J. Chem. Phys. 2001, 115, 5310–5318. [Google Scholar]
- Zook, T.C.; Pickett, G.T. Hollow-core dendrimers revisited. Phys. Rev. Lett. 2003. [Google Scholar]
- Harreis, H.M.; Likos, C.N.; Ballauff, M. Can dendrimers be viewed as compact colloids? A simulation study of the fluctuations in a dendrimer of fourth generation. J. Chem. Phys. 2003, 118, 1979–1988. [Google Scholar] [CrossRef]
- Timoshenko, E.G.; Kuznetsov, Y.A.; Connolly, R. Conformations of dendrimers in dilute solutions. J. Chem. Phys. 2002, 117, 9050–9062. [Google Scholar] [CrossRef]
- Han, M.; Chen, P.; Yang, X. Molecular dynamics simulation of PAMAM dendrimer in aqueous solution. Polymer 2005, 46, 3481–3488. [Google Scholar] [CrossRef]
- Lee, I.; Athey, B.D.; Wetzel, A.W.; Meixner, W.; Baker, J.R. Structural molecular dynamics studies on polyamidoamine dendrimers for a therapeutic application: Effect of pH and generation. Macromolecules 2002, 35, 4510–4520. [Google Scholar]
- Ahn, T.-S.; Nantalaksakul, A.; Dasari, R.R.; Al-Kaysi, R.O.; Müller, A.M.; Thayumanavan, S.; Bardeen, C.J. Energy and charge transfer dynamics in fully decorated benzyl ether dendrimers and their disubstituted analogues. J. Phys. Chem. B 2006, 110, 24331–24339. [Google Scholar]
- Gu, T.; Whitesell, J.K.; Fox, M.A. Intramolecular charge transfer in 1:1 Cu(II)/pyrenylcyclam dendrimer complexes. J. Phys. Chem. B 2006, 110, 25149–25152. [Google Scholar]
- Li, Y.-Y.; Han, L.; Chen, J.; Zheng, S.; Zen, Y.; Li, Y.; Li, S.; Yang, G. Study on the extent of folding back conformation in poly(aryl ether) dendrimers by intramolecular electron transfer and exciplex formation. Macromolecules 2007, 40, 9384–9390. [Google Scholar]
- Denti, G.; Campagna, S.; Serroni, S.; Ciano, M.; Balzani, V. Decanuclear homo- and heterometallic polypyridine complexes: Syntheses, absorption spectra, luminescence, electrochemical oxidation, and intercomponent energy transfer. J. Am. Chem. Soc. 1992, 114, 2944–2950. [Google Scholar] [CrossRef]
- Gilat, S.L.; Adronov, A.; Fréchet, J.M.J. Light harvesting and energy transfer in novel convergently constructed dendrimers. Angew. Chem. Int. Ed. 1999, 38, 1422–1427. [Google Scholar] [CrossRef]
- Adronov, A.; Gilat, S.L.; Fréchet, J.M.J.; Ohta, K.; Neuwahl, F.V.R.; Fleming, G.R. Light harvesting and energy transfer in laser-dye-labeled poly(aryl ether) dendrimers. J. Am. Chem. Soc. 2000, 122, 1175–1185. [Google Scholar]
- Adronov, A.; Fréchet, J.M.J. Light-harvesting dendrimers. Chem. Commun. 2000. [Google Scholar]
- Chen, J.; Li, S.; Zhang, L.; Liu, B.; Han, Y.; Yang, G.; Li, Y. Light-harvesting and photoisomerization in benzophenone and norbornadiene-labeled poly(aryl ether) dendrimers via intramolecular triplet energy transfer. J. Am. Chem. Soc. 2005, 127, 2165–2171. [Google Scholar]
- Cicchi, S.; Fabbrizzi, P.; Ghini, G.; Brandi, A.; Foggi, P.; Marcelli, A.; Righini, R.; Botta, C. Pyrene-excimers-based antenna systems. Chem. Eur. J. 2009, 15, 754–764. [Google Scholar]
- Mourey, T.H.; Turner, S.R.; Rubinstein, M.; Fréchet, J.M.J.; Hawker, C.J.; Wooley, K.L. Unique behavior of dendritic macromolecules: Intrinsic viscosity of polyether dendrimers. Macromolecules 1992, 25, 2401–2406. [Google Scholar] [CrossRef]
- Gorman, C.B.; Hager, M.W.; Parkhurst, B.L.; Smith, J.C. Use of a paramagnetic core to affect longitudinal nuclear relaxation in dendrimers—A tool for probing dendrimer conformation. Macromolecules 1998, 31, 815–822. [Google Scholar]
- Wooley, K.L.; Klug, C.A.; Tasaki, K.; Schaefer, J. Shapes of dendrimers from rotational-echo double-resonance NMR. J. Am. Chem. Soc. 1997, 119, 53–58. [Google Scholar] [CrossRef]
- Malyarenko, D.I.; Vold, R.L.; Hoaston, G.L. Solid state deuteron NMR studies of polyamidoamine dendrimer salts. 2. Relaxation and molecular motion. Macromolecules 2000, 33, 7508–7520. [Google Scholar] [CrossRef]
- Chai, M.; Niu, Y.; Youngs, W.J.; Rinaldi, P.L. Structure and conformation of DAB dendrimers in solution via multidimensional NMR techniques. J. Am. Chem. Soc. 2001, 123, 4670–4678. [Google Scholar]
- Wind, M.; Saalwächter, K.; Wiesler, U.M.; Müllen, K.; Spiess, H.W. Solid-state NMR investigations of molecular dynamics in polyphenylene dendrimers: Evidence of dense-shell packing. Macromolecules 2002, 35, 10071–10086. [Google Scholar]
- Malveau, C.; Baille, W.E.; Zhu, X.X.; Ford, W.T. Molecular dynamics of hydrophilic poly(propylene imine) dendrimers in aqueous solutions by 1H BNR relaxation. J. Polym. Sci. B 2003, 41, 2969–2975. [Google Scholar] [CrossRef]
- Prosa, T.J.; Bauer, B.J.; Amis, E.J.; Tomalia, D.A.; Scherrenberg, R. A SAXS study of the internal structure of dendritic polymer systems. J. Polym. Sci. B 1997, 35, 2913–2924. [Google Scholar] [CrossRef]
- Topp, A.; Bauer, B.J.; Klimash, J.W.; Spindler, R.; Tomalia, D.A.; Amis, E.J. Probing the location of the terminal groups of dendrimers in dilute solution. Macromolecules 1999, 32, 7226–7231. [Google Scholar]
- Pötschke, D.; Ballauf, M.; Lindner, P.; Fischer, M.; Vögtle, F. Analysis of the structure of dendrimers in solution by small-angle neutron scattering including contrast variation. Macromolecules 1999, 32, 4079–4087. [Google Scholar] [CrossRef]
- Ballauff, M.; Likos, C.N. Dendrimers in solution: Insight from theory and simulation. Angew. Chem. Int. Ed. 2004, 43, 2998–3020. [Google Scholar] [CrossRef]
- Welch, P.; Muthukumar, M. Tuning the density profile of dendritic polyelectrolytes. Macromolecules 1998, 31, 5892–5897. [Google Scholar] [CrossRef]
- Lyulin, S.V.; Evers, L.J.; van der Schoot, P.; Darinskii, A.A.; Lyulin, A.V.; Michels, M.A.J. Effect of solvent quality and electrostatic interactions on size and structure of dendrimers. Brownian dynamics simulation and mean-field theory. Macromolecules 2004, 37, 3049–3063. [Google Scholar] [CrossRef]
- Bosman, A.W.; Bruining, M.J.; Kooijman, H.; Speck, A.; Janssen, R.A.J.; Meijer, E.W. Concerning the localization of end groups in dendrimers. J. Am. Chem. Soc. 1998, 120, 8547–8548. [Google Scholar]
- Meltzer, A.D.; Tirrell, D.A.; Jones, A.A.; Inglefield, P.T.; Hedstrand, D.M.; Tomalia, D.A. Chain dynamics in poly(amido amine) dendrimers. A study of 13C NMR relaxation parameters. Macromolecules 1992, 25, 4541–4548. [Google Scholar]
- Meltzer, A.D.; Tirrell, D.A.; Jones, A.A.; Inglefield, P.T. Chain dynamics in poly(amido amine) dendrimers. A study of 2H NMR relaxation parameters. Macromolecules 1992, 25, 4549–4552. [Google Scholar] [CrossRef]
- Rathgeber, S.; Monkenbusch, M.; Hedrick, J.L.; Trolsås, M.; Gast, A.P. Starlike dendrimers in solutions: Structural properties and internal dynamics. J. Chem. Phys. 2006. [Google Scholar]
- Stark, B.; Stühn, B.; Frey, H.; Lach, C.; Lorenz, K.; Frick, B. Segmental dynamics in dendrimers with perfluorinated end groups: A study using quasielastic neutron scattering. Macromolecules 1998, 31, 5415–5423. [Google Scholar] [CrossRef]
- Li, X.; Zamponi, M.; Hong, K.; Porcar, L.; Shew, C.-Y.; Jenkins, T.; Liu, E.; Smith, G.S.; Herwig, K.W.; Liu, Y.; Chen, W.-R. pH responsiveness of polyelectrolyte dendrimers: A dynamical perspective. Soft Matter 2011, 7, 618–622. [Google Scholar]
- Emran, S.K.; Newkome, G.R.; Weis, C.D.; Harmon, J.P. Molecular relaxations in ester-terminated, amine-based dendrimers. J. Polym. Sci. B 1999, 37, 2025–2038. [Google Scholar] [CrossRef]
- Mijović, J.; Ristić, S.; Kenny, J. Dynamics of six generations of PAMAM dendrimers as studied by dielectric relaxation spectroscopy. Macromolecules 2007, 40, 5212–5221. [Google Scholar] [CrossRef]
- Uppuluri, S.; Morrison, F.A.; Dvornic, P.R. Rheology of dendrimers. 2. Bulk polyamidoamine dendrimers under steady shear, creep, and dynamic oscillatory shear. Macromolecules 2000, 33, 2551–2560. [Google Scholar] [CrossRef]
- Aumanen, J.; Kesti, T.; Sundström, V.; Teobaldi, G.; Zerbetto, F.; Werner, N.; Richardt, G.; van Heyst, J.; Vögtle, F.; Korppi-Tommola, J. Internal dynamics and energy transfer in dansylated POPAM dendrimers and their eosin complexes. J. Phys. Chem. B 2010, 114, 1548–1558. [Google Scholar]
- Gorman, C.B.; Smith, J.C. Effect of repeat unit flexibility on dendrimer conformation as studied by atomistic molecular dynamics simulations. Polymer 2000, 41, 675–683. [Google Scholar] [CrossRef]
- Mazo, M.A.; Shamaev, M.Y.; Balabaev, N.K.; Darinskii, A.A.; Neelov, I.M. Conformational mobility of carbosilane dendrimer: Molecular dynamics simulation. Phys. Chem. Chem. Phys. 2004, 6, 1285–1289. [Google Scholar]
- Lyulin, S.V.; Darinskii, A.A.; Lyulin, A.V.; Michels, M.A.J. Computer simulation of the dynamics of neutral and charged dendrimers. Macromolecules 2004, 37, 4676–4685. [Google Scholar]
- Markelov, D.A.; Lyulin, S.V.; Gotlib, Y.Y.; Lyulin, A.V.; Matveev, V.V.; Lahderanta, E.; Darinski, A.A. Orientational mobility and relaxation spectra of dendrimers: Theory and computer simulation. J. Chem. Phys. 2009. [Google Scholar]
- Tuschl, T.; Gohlke, C.; Jovin, T.M.; Westhof, E.; Eckstein, F. A three-dimensional model for the hammerhead ribozyme based on fluorescence measurements. Science 1994, 266, 785–789. [Google Scholar]
- Cuniberti, C.; Perico, A. Intramolecular diffusion-controlled reactions and polymer dynamics. Prog. Polym. Sci. 1984, 10, 271–316. [Google Scholar] [CrossRef]
- Winnik, M.A. End-to-end cyclization of polymer chains. Acc. Chem. Res. 1985, 18, 73–79. [Google Scholar] [CrossRef]
- Duhamel, J. Polymer chain dynamics in solution probed with a fluorescence blob model. Acc. Chem. Res. 2006, 39, 953–960. [Google Scholar] [CrossRef]
- Winnik, F.M. Photophysics of preassociated pyrenes in aqueous polymer solutions and in other organized media. Chem. Rev. 1993, 93, 587–614. [Google Scholar]
- Tjipangandjara, K.F.; Somasundaran, P. Effects of charges in adsorbed polyacrylic acid conformation on alumina flocculation. Colloids Surf. 1991, 55, 245–255. [Google Scholar] [CrossRef]
- Cuniberti, C.; Perico, A. Intramolecular excimers and microbrownian motion of flexible polymer molecules in solution. Eur. Polym. J. 1977, 13, 369–374. [Google Scholar] [CrossRef]
- Winnik, M.A.; Redpath, T.; Richards, D.H. The dynamics of end-to-end cyclization in polystyrene probed by pyrene excimer formation. Macromolecules 1980, 13, 328–335. [Google Scholar] [CrossRef]
- Cheung, S.-T.; Winnik, M.A.; Redpath, A.E.C. cyclization dynamics of polymers, 5. The effect of solvent on end-to-end cyclization of poly(ethylene oxide) probed by intramolecular pyrene excimer formation. Macromol. Chem. Phys. 1982, 183, 1815–1824. [Google Scholar] [CrossRef]
- Svirskaya, P.; Danhelka, J.; Redpath, A.E.C.; Winnik, M.A. Cyclization dynamics of polymers: 7. Application of the pyrene excimer technique to the internal dynamics of poly(dimethylsiloxane) chains. Polymer 1983, 24, 319–322. [Google Scholar] [CrossRef]
- Redpath, A.E.C.; Winnik, M.A. Temperature dependence of excimer formation between pyrenes at the ends of a polymer in a good solvent. Cyclization dynamics of polymers. 9. J. Am. Chem. Soc. 1982, 104, 5604–5607. [Google Scholar]
- Winnik, M.A.; Redpath, A.E.C.; Paton, K.; Danhelka, J. Cyclization dynamics of polymers: 10 Synthesis, fractionation, and fluorescent spectroscopy of pyrene end-capped polystyrenes. Polymer 1984, 25, 91–99. [Google Scholar] [CrossRef]
- Boileau, S.; Méchin, F.; Martinho, J.M.G.; Winnik, M.A. End-to-end cyclization of a pyrene end-capped poly(bisphenol A-diethylene glycol carbonate). Macromolecules 1989, 22, 215–220. [Google Scholar]
- Ghiggino, K.P.; Snare, M.J.; Thistlethwaite, P.J. Cyclization dynamics in poly(ethylene oxide). chain length and temperature dependence. Eur. Polym. J. 1985, 21, 265–272. [Google Scholar] [CrossRef]
- Slomkowski, S.; Winnik, M.A. fluorescence quenching in pyrene/benzil end-labeled poly(tetramethylene oxide). Cyclization dynamics in polymers 19. Macromolecules 1986, 19, 500–501. [Google Scholar] [CrossRef]
- Xu, H.; Martinho, J.M.G.; Winnik, M.A.; Beinert, G. Transient effects in diffusion-controlled polymer cyclization. Makromol. Chem. 1989, 190, 1333–1343. [Google Scholar] [CrossRef]
- Farinha, J.P.S.; Martinho, J.M.G.; Xu, H.; Winnik, M.A.; Quirk, R.P. Influence of intrachain hydrogen bonding on the cyclization of a polystyrene chain. J. Polym. Sci. Part B 1994, 32, 1635–1642. [Google Scholar] [CrossRef]
- Martinho, J.M.G.; Reis e Sousa, A.T.; Winnik, M.A. Effect of temperature and solvent on the cyclization of a polystyrene chain. Macromolecules 1993, 26, 4484–4488. [Google Scholar] [CrossRef]
- Duhamel, J.; Khayakin, Y.; Hu, Y.Z.; Winnik, M.A.; Boileau, S.; Méchin, F. End-to-end cyclization of a pyrene end-capped poly(bisphenol AF-diethylene glycol carbonate) in solution. Eur. Polym. J. 1994, 30, 129–134. [Google Scholar] [CrossRef]
- Lee, S.; Winnik, M.A. Cyclization rates for two points in the interior of a polymer chain. Macromolecules 1997, 30, 2633–2641. [Google Scholar] [CrossRef]
- Lee, S.; Duhamel, J. Monitoring the hydrophobic interactions of internally pyrene-labeled poly(ethylene oxide)s in water by fluorescence spectroscopy. Macromolecules 1998, 31, 9193–9200. [Google Scholar] [CrossRef]
- Reis e Sousa, A.T.; Castanheira, E.M.S.; Fedorov, A.; Martinho, J.M.G. Polystyrene cyclization using pyrene excimer formation. Effect of geminate pairs in good solvents. J. Phys. Chem. A 1998, 102, 6406–6411. [Google Scholar]
- Picarra, S.; Gomes, P.T.; Martinho, J.M.G. Fluorescence study of the coil-globule transition of a poly(ε-caprolactone) chain labeled with pyrenes at both ends. Macromolecules 2000, 33, 3947–3950. [Google Scholar] [CrossRef]
- Farinha, J.P.S.; Picarra, S.; Miesel, K.; Martinho, J.M.G. Fluorescence study of the coil-globule transition of a PEO chain in toluene. J. Phys. Chem. B 2001, 105, 10536–10545. [Google Scholar]
- Kim, S.D.; Torkelson, J.M. Nanoscale confinement and temperature effects on associative polymers in thin films: Fluorescence study of a telechelic, pyrene-labeled poly(dimethylsiloxane). Macromolecules 2002, 35, 5943–5952. [Google Scholar]
- Gardinier, W.E.; Bright, F.V. Temperature-dependent tail-tail dynamics of pyrene-labeled poly(dimethylsiloxane) oligomers dissolved in ethyl acetate. J. Phys. Chem. B 2005, 109, 14824–14829. [Google Scholar] [CrossRef]
- Ingratta, M.; Hollinger, J.; Duhamel, J. A case for using randomly labeled polymers to study long-range polymer chain dynamics by fluorescence. J. Am. Chem. Soc. 2008, 130, 9420–9428. [Google Scholar]
- Costa, T.; Seixas de Melo, J.; Burrows, H.D. Fluorescence behavior of a pyrene-end-capped poly(ethylene oxide) in organic solvents and in dioxane-water mixtures. J. Phys. Chem. B 2009, 113, 618–626. [Google Scholar]
- Chen, S.; Duhamel, J.; Winnik, M.A. Probing end-to-end cyclization beyond willemski and fixmann. J. Phys. Chem. B 2011, 115, 3289–3302. [Google Scholar]
- Winnik, M.A.; Egan, L.S.; Tencer, M.; Croucher, M.D. Luminescence studies on sterically stabilized polymer colloid particles: Pyrene excimer formation. Polymer 1987, 28, 1553–1560. [Google Scholar] [CrossRef]
- Wang, F.W.; Lowry, R.E. Excimer fluorescence technique for study of polymer-segment mobility: Applications to pyrene-labelled poly(methyl methacrylate) and poly(methyl acrylate) in solution. Polymer 1985, 26, 1046–1052. [Google Scholar] [CrossRef]
- Mathew, A.; Siu, H.; Duhamel, J. A blob model to study chain folding by fluorescence. Macromolecules 1999, 32, 7100–7108. [Google Scholar] [CrossRef]
- Kanagalingam, S.; Ngan, C.F.; Duhamel, J. Effect of solvent quality on the level of association and encounter kinetics of hydrophobic pendants covalently attached onto a water-soluble polymer. Macromolecules 2002, 35, 8560–8570. [Google Scholar] [CrossRef]
- Kanagalingam, S.; Spartalis, J.; Cao, T.-C.; Duhamel, J. Scaling relations related to the kinetics of excimer formation between pyrene groups attached onto poly(N, N-dimethylacrylamide)s. Macromolecules 2002, 35, 8571–8577. [Google Scholar] [CrossRef]
- Irondi, K.; Zhang, M.; Duhamel, J. Study of the semidilute solutions of poly(N,N-dimethylacrylamide) by fluorescence and its implications to the kinetics of coil-to-globule transitions. J. Phys. Chem. B 2006, 110, 2628–2637. [Google Scholar]
- Ingratta, M.; Duhamel, J. Correlating pyrene excimer formation with polymer chain dynamics in solution. Possibilities and limitations. Macromolecules 2007, 40, 6647–6657. [Google Scholar] [CrossRef]
- Seixas de Melo, J.; Costa, T.; Francisco, A.; Macanita, A.L.; Gago, S.; Goncalves, I.S. Dynamics of short as compared with long poly(acrylic acid) chains hydrophobically modified with pyrene, as followed by fluorescence techniques. Phys. Chem. Chem. Phys. 2007, 9, 1370–1385. [Google Scholar]
- Picarra, S.; Relogio, P.; Afonso, C.A.M.; Martinho, J.M.G.; Farinha, J.P.S. Coil-globule transition of poly(dimethylacrylamide): Fluorescence and light scattering study. Macromolecules 2003, 36, 8119–8129. [Google Scholar] [CrossRef]
- Picarra, S.; Duhamel, J.; Fedorov, A.; Martinho, J.M.G. Coil-globule transition of pyrene-labeled polystyrene in cyclohexane: Determination of polymer chain radii by fluorescence. J. Phys. Chem. B 2004, 108, 12009–12015. [Google Scholar]
- Teertstra, S.J.; Lin, W.Y.; Gauthier, M.; Ingratta, M.; Duhamel, J. Comparison of the long range polymer chain dynamics of polystyrene and cis-polyisoprene using polymers randomly labeled with pyrene. Polymer 2009, 50, 5456–5466. [Google Scholar]
- Ingratta, M.; Duhamel, J. Effect of viscosity on long-range polymer chain dynamics in solution studied with a fluorescence blob model. Macromolecules 2009, 42, 1244–1251. [Google Scholar] [CrossRef]
- Ingratta, M.; Duhamel, J. Effect of time on the rate of long range polymer segmental intramolecular encounters. J. Phys. Chem. B 2009, 113, 2284–2292. [Google Scholar] [CrossRef]
- Ingratta, M.; Mathew, M.; Duhamel, J. How switching the substituent of a pyrene derivative from a methyl to a butyl affects the fluorescence response of polystyrene randomly labeled with pyrene. Can. J. Chem. 2010, 88, 217–227. [Google Scholar]
- Yip, J.; Duhamel, J.; Qiu, X.; Winnik, F.M. Fluorescence studies of a series of monodisperse telechelic α,ω-dipyrenyl poly(N-isopropylacrylamide)s in ethanol and in water. Can. J. Chem. 2011, 89, 163–172. [Google Scholar] [CrossRef]
- Yip, J.; Duhamel, J.; Qiu, X.P.; Winnik, F.M. Long-range polymer chain dynamics of pyrene-labelled poly(N-isopropylacrylamide)s studied by fluorescence. Macromolecules 2011, 44, 5363–5372. [Google Scholar]
- Modrakowski, C.; Camacho Flores, S.; Beinhoff, M.; Schlüter, A.D. Synthesis of pyrene containing building blocks for dendrimer synthesis. Synthesis 2001, 14, 2143–2155. [Google Scholar]
- Figueira-Duarte, T.M.; Simon, S.C.; Wagner, M.; Druzhinin, S.I.; Zachariasse, K.A.; Müllen, K. Polypyrene dendrimers. Angew. Chem. Int. Ed. 2008, 47, 10175–10178. [Google Scholar]
- Figueira-Duarte, T.M.; Müllen, K. Pyrene-based materials for organic electronics. Chem. Rev. 2011, 111, 7260–7314. [Google Scholar] [CrossRef]
- Stewart, G.M.; Fox, M.A. Fluorophore-labeled dendrons as light harvesting antennae. J. Am. Chem. Soc. 1996, 118, 4354–4360. [Google Scholar] [CrossRef]
- Giovanella, U.; Mróz, W.; Foggi, P.; Fabbrizzi, P.; Cicchi, S.; Botta, C. Multi-colour electroluminescence of dendronic antennae containing pyrenes as light harvesters. ChemPhysChem 2010, 11, 683–688. [Google Scholar] [CrossRef]
- Thomas, R.R.J.; Thompson, A.L.; Sivakumar, A.V.; Bardeen, C.J.; Thayumanavan, S. Energy and electron transfer in bifunctional non-conjugated dendrimers. J. Am. Chem. Soc. 2005, 127, 373–383. [Google Scholar]
- Vanjinathan, M.; Lin, H.-C.; Nasar, A.S. Synthesis, characterization and photophysical properties of DCM-based light-harvesting dendrimers. Macromol. Chem. Phys. 2011, 212, 849–859. [Google Scholar] [CrossRef]
- Gu, T.; Whitesell, J.K.; Fox, M.A. Intramolecular charge transfer in 1:1 Cu(II)/pyrenylcyclam dendrimer complexes. J. Phys. Chem. B 2006, 110, 25149–25152. [Google Scholar]
- Morales-Espinoza, E.G.; Lijanova, I.V.; Morales-Saavedra, O.G.; Torres-Zuñiga, V.; Hernandez-Ortega, S.; Martines-Garcia, M. Synthesis of porphyrin-dendrimers with a pyrene in the periphery and their cubic nonlinear optical properties. Molecules 2011, 16, 6950–6968. [Google Scholar]
- Yip, J.; Duhamel, J.; Bahun, G.J.; Adronov, A. A study of the branch ends of a series of pyrene-labeled dendrimers based on pyrene excimer formation. J. Phys. Chem. B 2010, 114, 10254–10265. [Google Scholar]
- Baker, L.A.; Crooks, R.M. Photophysical properties of pyrene-functionalized poly(propylene imine) dendrimers. Macromolecules 2000, 33, 9034–9039. [Google Scholar] [CrossRef]
- Wang, B.-B.; Zhang, X.; Jia, X.-R.; Li, Z.-C.; Ji, Y.; Yang, L.; Wei, Y. Fluorescence and aggregation behaviour of poly(amidoamine) dendrimers peripherally modified with aromatic fluorophores: The effect of dendritic architecture. J. Am. Chem. Soc. 2004, 126, 15180–15194. [Google Scholar]
- Wang, B.-B.; Zhang, X.; Yang, L.; Jia, X.-R.; Ji, Y.; Li, W.-S.; Wei, Y. Poly(amisoamine) dendrimers bearing electron-donating fluorophores: Fluorescence and electrochemical properties. Polym. Bull. 2006, 56, 63–74. [Google Scholar] [CrossRef]
- Brauge, L.; Caminade, A.-M.; Majoral, J.-P.; Slomkowski, S.; Wolszczak, M. Segmental mobility in phosphorus-containing dendrimers. studies by fluorescent spectroscopy. Macromolecules 2001, 34, 5599–5606. [Google Scholar]
- Brauge, L.; Vériot, G.; Franc, G.; Deloncle, R.; Caminade, A.-M.; Majoral, J.-P. Synthesis of phosphorus dendrimers bearing chromophoric end groups: Toward organic blue light-emitting diodes. Tetrahedron 2006, 62, 11891–11899. [Google Scholar]
- Caminade, A.-M.; Hameau, A.; Turrin, C.-O.; Ianchuk, M.; Delavaux-Nicot, B.; Majoral, J.-P. fluorescent phosphorus dendrimers and their role in supramolecular interactions. Phosphorus Sulfur 2011, 186, 860–868. [Google Scholar] [CrossRef]
- Lekha, P.K.; Prasad, E. Tunable emission of static excimer in a pyrene-modified polyamidoamine dendrimer aggregate through positive solvatochromism. Chem. Eur. J. 2011, 17, 8609–8617. [Google Scholar] [CrossRef]
- Wilken, R.; Adams, J. End-group dynamics of fluorescently labelled dendrimers. Macromol. Rapid. Commun. 1997, 18, 659–665. [Google Scholar] [CrossRef]
- Sharma, A.; Schulman, S.G. Introduction to Fluorescence Spectroscopy; Wiley: New York, NY, USA, 1999. [Google Scholar]
- Valeur, B. Molecular Fluorescence. Principles and Applications; Wiley: New York, NY, USA, 2002. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed; Springer: New York, NY, USA, 2006. [Google Scholar]
- Karstens, T.; Kobs, K. Rhodamine B and rhodamine 101 as reference substances for fluorescence quantum yield measurements. J. Phys. Chem. 1980, 84, 1871–1872. [Google Scholar] [CrossRef]
- Birks, J.B. Photophysics of Aromatic Molecules; Wiley: New York, NY, USA, 1970; p. 301. [Google Scholar]
- Nakajima, A. Fluorescence spectra of pyrene in chlorinated aromatic solvents. J. Lumin. 1976, 11, 429–432. [Google Scholar] [CrossRef]
- Kalyanasundaram, K.; Thomas, J.K. Environmental effects on vibronic band intensities in pyrene monomer fluorescence and their application in studies of micellar systems. J. Am. Chem. Soc. 1977, 99, 2039–2044. [Google Scholar] [CrossRef]
- Lianos, P.; Georghiou, S. Solute-solvent interaction and its effect on the vibronic and vibrational structure of pyrene spectra. Photochem. Photobiol. 1979, 30, 355–362. [Google Scholar] [CrossRef]
- Dong, D.C.; Winnik, M.A. The Py scale of solvent polarities. Solvent effects on the vibronic fine structure of pyrene fluorescence and empirical correlations with ET and Y values. Photochem. Photobiol. 1982, 35, 17–21. [Google Scholar] [CrossRef]
- Dong, D.C.; Winnik, M.A. The Py scale of solvent polarities. Can. J. Chem. 1984, 62, 2560–2565. [Google Scholar] [CrossRef]
- Jones, A.; Dickson, T.J.; Wilson, B.E.; Duhamel, J. Fluorescence properties of poly(ethylene terephthalate-co-2,6-naphthalene dicarboxylate) with naphthalene contents ranging from 0.01 to 100 mole%. Macromolecules 1999, 32, 2956–2961. [Google Scholar]
- Genest, D.; Wahl, P. Time-Resolved Fluorescence Spectroscopy in Biochemistry and Biology; Cundall, R., Ed.; Springer: Berlin, Germany, 1980; pp. 523–539. [Google Scholar]
- Strickler, S.J.; Berg, R.A. Relationship between absorption intensity and fluorescence lifetime of molecules. J. Chem. Phys. 1962, 37, 814–822. [Google Scholar] [CrossRef]
- Stevens, B.; Ban, M.I. Spectrophotometric determination of enthalpies and entropies of photoassociation for dissolved aromatic hydrocarbons. Trans. Far. Soc. 1964, 60, 1515–1523. [Google Scholar] [CrossRef]
- Zachariasse, K.A.; Duveneck, G.; Busse, R. Intramolecular excimer formation with 1,3-Di(1-pyrenyl)propane. Decay parameters and influence of viscosity. J. Am. Chem. Soc. 1984, 106, 1045–1051. [Google Scholar]
- Siu, H.; Duhamel, J. Comparison of the association level of a hydrophobically modified associative polymer obtained from an analysis based on two different models. J. Phys. Chem. B 2005, 109, 1770–1780. [Google Scholar]
- Zachariasse, K.; Kühnle, W. Intramolecular excimers with α,ω-diarylalkanes. Z. Phys. Chem. Neue Fol. 1976, 101, 267–276. [Google Scholar] [CrossRef]
- Kanaya, T.; Goshiki, K.; Yamamoto, M.; Nishijima, Y. Intramolecular end-to-end excimer formation of bis((1-pyrenylmethoxy)carbonyl)alkanes. A study of end-to-end collisional frequency on a chain molecule. J. Am. Chem. Soc. 1982, 104, 3580–3587. [Google Scholar]
- Eaton, D.F.; Smart, B.E. Are fluorocarbon chains “stiffer” than hydrocarbon chains? dynamics of end-to-end cyclization in a C8F16 segment monitored by fluorescence. J. Am. Chem. Soc. 1990, 112, 2821–2823. [Google Scholar] [CrossRef]
- Reynders, P.; Kühnle, W.; Zachariasse, K.A. Ground-state dimers in excimer-forming bichromophoric molecules. 1. Bis(pyrenylcarboxy)alkanes. J. Am. Chem. Soc. 1990, 112, 3929–3939. [Google Scholar]
- Snare, M.J.; Thistlethwaite, P.J.; Ghiggino, K.P. Kinetic studies of intramolecular excimer formation in dipyrenylalkanes. J. Am. Chem. Soc. 1983, 105, 3328–3332. [Google Scholar] [CrossRef]
- Zachariasse, K.A.; Duveneck, G.; Busse, R. Intramolecular excimer formation with 1,3-Di(1-pyrenyl)propane. Decay parameters and influence of viscosity. J. Am. Chem. Soc. 1984, 106, 1045–1051. [Google Scholar] [CrossRef]
- Reynders, P.; Kühnle, W.; Zachariasse, K.A. Ground-state dimers in excimer-forming bichromophoric molecules. NMR and single-photon-counting data. 2. Racemic and meso dipyrenylpentanes and dipyrenylalkanes. J. Phys. Chem. 1990, 94, 4073–4082. [Google Scholar] [CrossRef]
- Zachariasse, K.A.; Maçanita, A.L.; Kühnle, W. chain length dependence of intramolecular excimer formation with 1, n-bis(1-pyrenylcarboxy)alkanes for n = 1 – 16, 22, and 32. J. Phys. Chem. B 1999, 103, 9356–9365. [Google Scholar]
- Pandey, S.; Kane, M.A.; Baker, G.A.; Bright, F.V.; Fürstner, A.; Seidel, G.; Leitner, W. The photophysics of 6-(1-pyrenyl)hexyl-1,11(1-pyrenyl)undecanoate dissolved in organic liquids and supercritical carbon dioxide: Impact on olefin metathesis. J. Phys. Chem. B 2002, 106, 1820–1832. [Google Scholar]
- Maçanita, A.L.; Zachariasse, K.A. Viscosity dependence of intramolecular excimer formation with 1,5-bis(1-pyrenylcarboxy)pentane in alkane solvents as a function of temperature. J. Phys. Chem. A 2011, 115, 3183–3195. [Google Scholar]
- Eaton, W.A.; Muñoz, V.; Hagen, S.J.; Jas, G.S.; Lapidus, L.J.; Henry, E.R.; Hofrichter, J. Fast kinetics and mechanisms in protein folding. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 327–359. [Google Scholar] [CrossRef]
- Hagen, S.J.; Hofrichter, J.; Szabo, A.; Eaton, W.A. Diffusion-limited contact formation in unfolded cytochrome C: Estimating the maximum rate of protein folding. Proc. Natl. Acad. Sci. 1996, 93, 11615–11617. [Google Scholar]
- McGimpsey, W.G.; Chen, L.; Carraway, R.; Samaniego, W.N. Singlet-singlet and triplet-triplet energy transfer in bichromophoric peptides. J. Phys. Chem. A 1999, 103, 6082–6090. [Google Scholar] [CrossRef]
- Möglich, A.; Krieger, F.; Kiefhaber, T. Molecular basis for the effect of urea and guanidinium chloride on the dynamics of unfolded polypeptide chains. J. Mol. Biol. 2005, 345, 153–162. [Google Scholar] [CrossRef]
- Fierz, B.; Satzger, H.; Root, C.; Gich, P.; Zinth, W.; Kiefhaber, T. Loop formation in unfolded polypeptide chains on the picoseconds to microseconds time scale. Proc. Natl. Acad. Sci. USA 2007, 104, 2163–2168. [Google Scholar]
- Möglich, A.; Joder, K.; Kiefhaber, T. End-to-end distance distributions and intrachain diffusion constants in unfolded polypeptide chains indicate intramolecular hydrogen bond formation. Proc. Natl. Acad. Sci. USA 2006, 103, 12394–12399. [Google Scholar]
- Hudgins, R.R.; Huang, F.; Gramlich, G.; Nau, W.M. A fluorescence-based method for direct measurement of submicrosecond intramolecular contact formation in biopolymers: An exploratory study with polypeptides. J. Am. Chem. Soc. 2002, 124, 556–564. [Google Scholar]
- Huang, F.; Hudgins, R.R.; Nau, W.M. Primary and secondary structure dependence of peptide flexibility assessed by fluorescence-based measurement of end-to-end collision rates. J. Am. Chem. Soc. 2004, 126, 16665–16675. [Google Scholar]
- Roccatano, D.; Sahoo, H.; Zacharias, M.; Nau, W.M. Temperature dependence of looping rates in a short peptide. J. Phys. Chem. B 2007, 111, 2639–2646. [Google Scholar]
- Bieri, O.; Wirz, J.; Hellrung, B.; Schutkowski, M.; Drewello, M.; Kiefhaber, T. The speed limit for protein folding measured by triplet–triplet energy transfer. Proc. Natl. Acad. Sci. USA 1999, 96, 9597–9601. [Google Scholar]
- Krieger, F.; Fierz, B.; Bieri, O.; Drewello, M.; Kiefhaber, T. Dynamics of unfolded polypeptide chains as model for the earliest steps in protein folding. J. Mol. Biol. 2003, 332, 265–274. [Google Scholar] [CrossRef]
- Lapidus, L.J.; Eaton, W.A.; Hofrichter, J. Measuring the rate of intramolecular contact formation in polypeptides. Proc. Natl. Acad. Sci. USA 2000, 97, 7220–7225. [Google Scholar]
- Neuweiler, H.; Löllmann, M.; Doose, S.; Sauer, M. Dynamics of unfolded polypeptide chains in crowded environment studied by fluorescence correlation spectroscopy. J. Mol. Biol. 2007, 365, 856–869. [Google Scholar] [CrossRef]
- Wilemski, G.; Fixman, M. Diffusion-controlled intrachain reactions of polymers. I theory. J. Chem. Phys. 1974, 60, 866–877. [Google Scholar] [CrossRef]
- Wilemski, G.; Fixman, M. Diffusion-controlled intrachain reactions of polymers. II results for a pair of terminal reactive groups. J. Chem. Phys. 1974, 60, 878–890. [Google Scholar] [CrossRef]
- Chen, S.; Duhamel, J.; Bahun, G.; Adronov, A. Effect of fluorescent impurities in the study of pyrene-labeled macromolecules by fluorescence. J. Phys. Chem. B 2011, 115, 9921–9929. [Google Scholar]
- Keyes-Baig, C.; Duhamel, J.; Wettig, S. Characterization of the behavior of a pyrene substituted gemini surfactant in water by fluorescence. Langmuir 2011, 27, 3361–3371. [Google Scholar]
- Andriessen, R.; Boens, N.; Ameloot, M.; De Schryver, F.C. Non a priori analysis of fluorescence decay surfaces of excited-state processes. 2. Intermolecular excimer formation of pyrene. J. Phys. Chem. 1991, 95, 2047–2058. [Google Scholar]
- Andriessen, R.; Ameloot, M.; Boens, N.; de Schryver, F.C. Non a priori analysis of fluorescence decay surfaces of excited-state processes. 3. Intermolecular excimer formation of pyrene quenched by iodomethane. J. Phys. Chem. 1992, 96, 314–326. [Google Scholar]
- Boens, N.; Ameloot, M. Compartmental modeling and identifiability analysis in photophysics: Review. Int. J. Quantum Chem. 2006, 106, 300–315. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Duhamel, J. Internal Dynamics of Dendritic Molecules Probed by Pyrene Excimer Formation. Polymers 2012, 4, 211-239. https://doi.org/10.3390/polym4010211
AMA Style
Duhamel J. Internal Dynamics of Dendritic Molecules Probed by Pyrene Excimer Formation. Polymers. 2012; 4(1):211-239. https://doi.org/10.3390/polym4010211
Chicago/Turabian StyleDuhamel, Jean. 2012. "Internal Dynamics of Dendritic Molecules Probed by Pyrene Excimer Formation" Polymers 4, no. 1: 211-239. https://doi.org/10.3390/polym4010211