1. Introduction
Aliphatic polyesters of high molecular weight were synthesized using highly effective transesterification catalysts and the vacuum technique as well as their chain extension reagents such as diisocyanates. However, a broader use of synthetic aliphatic polyesters is limited by their poor thermal properties such as low melting temperatures [
1]. In addition, the price of crude oil has increased in recent years, and the conservation of fossil resources is very important for establishing a sustainable society and reducing carbon dioxide emissions [
2,
3]. Biobased aliphatic polyesters produced from renewable biomass resources, such as poly(butylene succinate), are attractive with respect to replacing petroleum derived synthetic polyesters. Such green polymers should be produced by environmentally benign processes, e.g., enzymatic and solvent-free processes, and by avoiding the use and generation of hazardous materials such as metal catalyst.
Among the renewable raw materials for polymer production, some of the plant oils are the most promising candidates due to their characteristic molecular structure, non-volatility and biodegradability [
4,
5]. Edible plant oils are essentially used as a food source; however, the supply-demand balance for this market would break down if these oils were used for large-scale polymer production. Thus, the use of inedible resources has attracted much attention from the standpoint of global sustainability and the replacement of fossil resources in the polymer industries [
6]. Inedible castor oil has attracted much attention as a building block for the preparation of functional materials due to its characteristic molecular structure and its abundant occurrence and relatively high purity comparable to a seed oil. Castor oil is obtained from the bean of the castor plant,
Ricinus communi of the family Euphorbiaceae. Approximately 85–90% of the triglyceride-derived fatty acid in castor oil is 12-hydroxy-
cis-9-octadecenoic acid (ricinoleic acid). We recently reported that ricinoleic acid can be enzymatically polymerized to yield high molecular weight polyricinoleate with a weight average molecular weight greater than 90,000. The produced polyricinoleate is a viscous liquid at room temperature with a glass transition temperature of −74.8 °C and is biodegraded by activated sludge [
7]. Enzymatic polymerization has attracted attention in recent years as an environmentally benign process. Enzymes are renewable catalysts that exhibit high catalytic activities under mild conditions [
8]. Thus, enzymatic polymerization is suitable for the synthesis of polymers with labile and reactive functional groups such as epoxy groups.
In order to increase the thermal properties of unsaturated polyesters, sulfur is widely used for the crosslinking of polymers. It has been suggested, however, that ill-smelling sulfur components are produced during degradation in the environment. Furthermore, the sulfide linkage is highly resistant to cleavage by microbes, which leads to resistance to biodegradation. It is known that acid anhydrides readily react with epoxy groups to produce hydrolyzable ester linkages, and many commercial epoxy adhesives and resins are produced using diacid anhydrides as curing agents, such as maleic anhydride and succinic anhydride [
9,
10]. Enzymatic epoxidation with lipase and hydrogen peroxide is an attractive oxidation process because of their mild and safe reaction, high conversion rate and easy removal of the catalyst. Furthermore, only water is produced in the reaction [
11]. Miao
et al. reported the enzymatic epoxidation of oleic acid followed by ring-opening polymerization with heating [
12]. Enzymatic epoxidation is regarded as an environmentally benign process, and thus is effective for the synthesis of biobased derivatives.
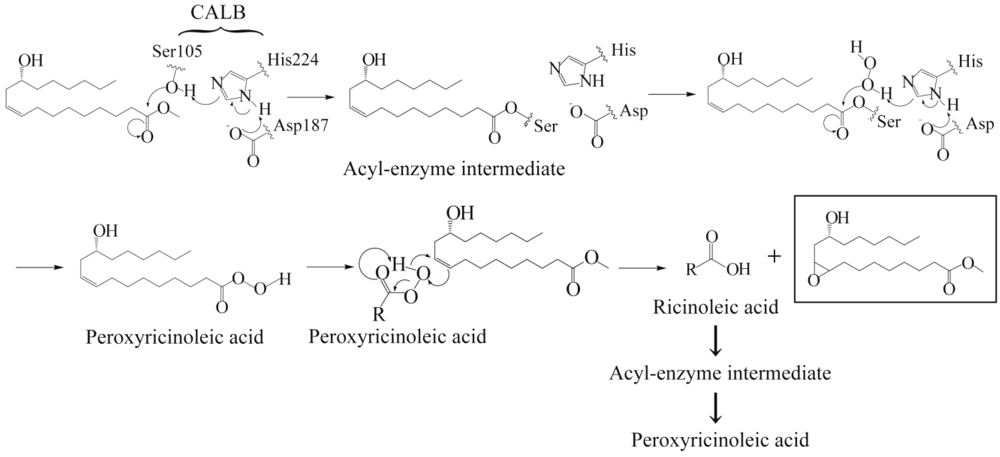
In this report, a series of high molecular weight polyepoxyricinoleate (PER) polymers were prepared by enzymatic epoxidation with subsequent polycondensation of methyl ricinoleate using a lipase. The PER was then crosslinked using diacid anhydrides with the aim of creating novel biobased thermosetting resins (
Scheme I). The thermal and mechanical properties, biodegradability of the PER and crosslinked PER polymers were measured.
Scheme I.
Enzymatic synthesis and crosslinking of high molecular weight polyepoxyricinoleate.
Scheme I.
Enzymatic synthesis and crosslinking of high molecular weight polyepoxyricinoleate.
2. Experimental Section
2.1. Materials
Methyl ricinoleate [methyl (R)-12-hydroxy-cis-9-octadecenoate] and molecular sieves 4A (MS4A) were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). The MS4A were dried at 150 °C for 2 h. Maleic anhydride (MA) was purchased from TCI Co., Inc. (Tokyo, Japan). Hydrogen peroxide (H2O2, 30 wt% aqueous solution) and succinic anhydride were purchased from Wako Pure Chemical Industries Ltd. (Osaka, Japan). Immobilized lipase from Candida antarctica (CALB: Novozym 435, a lipase (B lipase) produced by submerged fermentation of a genetically modified Aspergillus oryzae microorganism and adsorbed on a macroporous acrylic resin and having 10,000 PLU/g (propyl laurate units: lipase activity based on ester synthesis)) was kindly supplied by Novozymes Japan, Ltd. (Chiba, Japan). Immobilized lipase from Burkholderia cepacia (lipase PS-IM Amano I: a lipase from B. cepacia immobilized on diatomaceous earth) was purchased from Wako Pure Chemical Industries Ltd. (Osaka, Japan). The enzyme was dried under vacuum (3 mmHg) over P2O5 at 25 °C for 2 h before use.
2.2. Measurements
The weight average (Mw) and number average (Mn) molecular weights, as well as the polydispersity (Mw/Mn) of the polymers, were determined by size exclusion chromatography (SEC) (Shodex K-G + K-805 columns, Showa Denko Co., Ltd., Tokyo, Japan) using a refractive index detector. Chloroform was used as the eluent at 1.0 mL·min−1. The SEC system was calibrated with polystyrene standards having a narrow molecular weight distribution. The 1H and 13C-NMR spectra were recorded with an ECA-500 Fourier transform spectrometer (JEOL, Ltd., Tokyo, Japan) operating at 500 MHz and 125 MHz, respectively.
The glass transition temperature (Tg), melting temperature (Tm) and crystallization temperature (Tc) were determined by differential scanning calorimetry (DSC-60, Shimadzu, Kyoto, Japan). The measurements were made with a 10 mg sample on a DSC plate. The polymer samples were heated to 200 °C at the rate of 10 °C·min−1, cooled to −100 °C at the rate of 20 °C·min−1 and then scanned with heating at the rate of 10 °C·min−1 from −100 to 200 °C.
The mechanical properties (Young’s modulus, tensile strength and elongation at break) of the film samples were determined with an Autograph instrument (Shimadzu, Tokyo, Japan). The viscoelastic properties (storage elastic modulus) of the crosslinked polymers were measured by dynamic mechanical analysis (DMA) using an ARES viscoelastic measurement system (TA Instruments, Co., Ltd, New Castle, DE, USA). The temperature ramp tests were conducted in the range of −30–100 °C at the rate of 3 °C·min−1at 1 Hz at a constant value of shear strain (3%). The Tg of the crosslinked polymers was also determined by DMA.
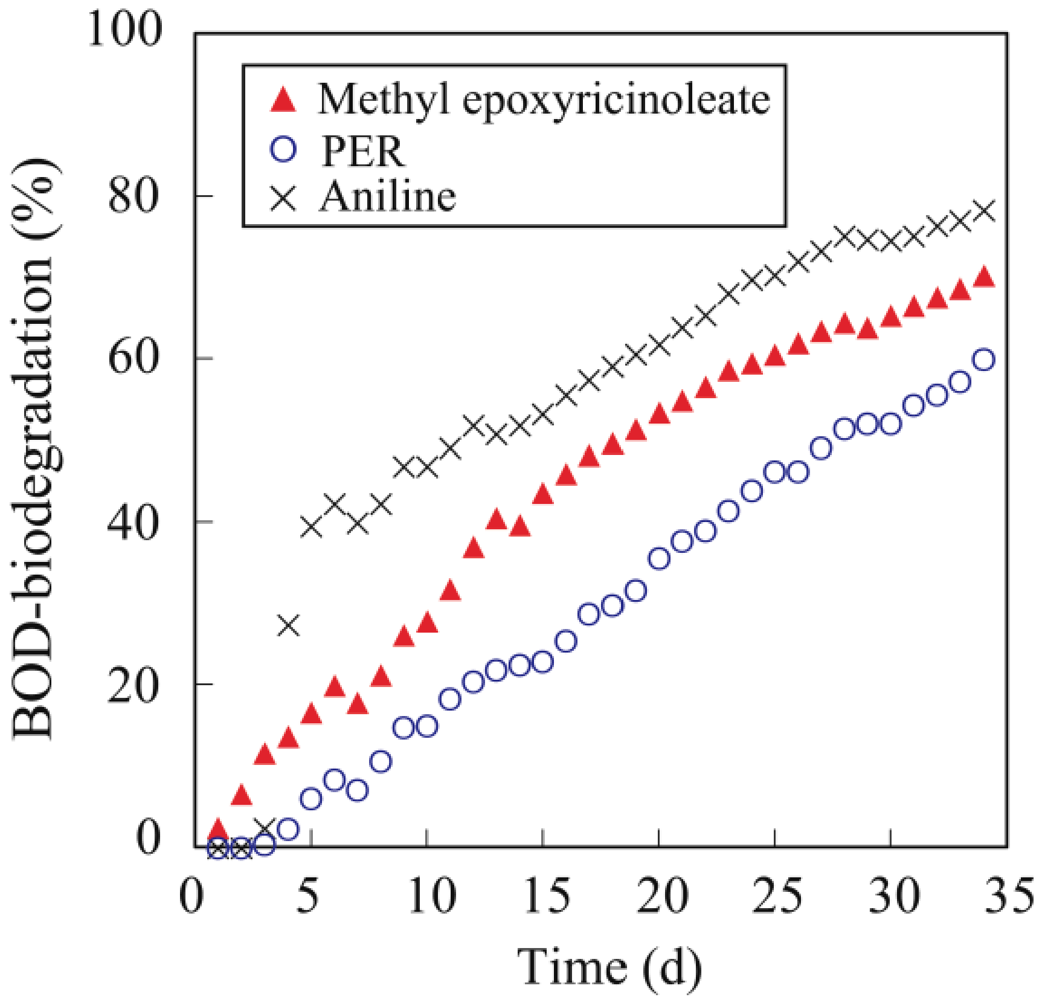
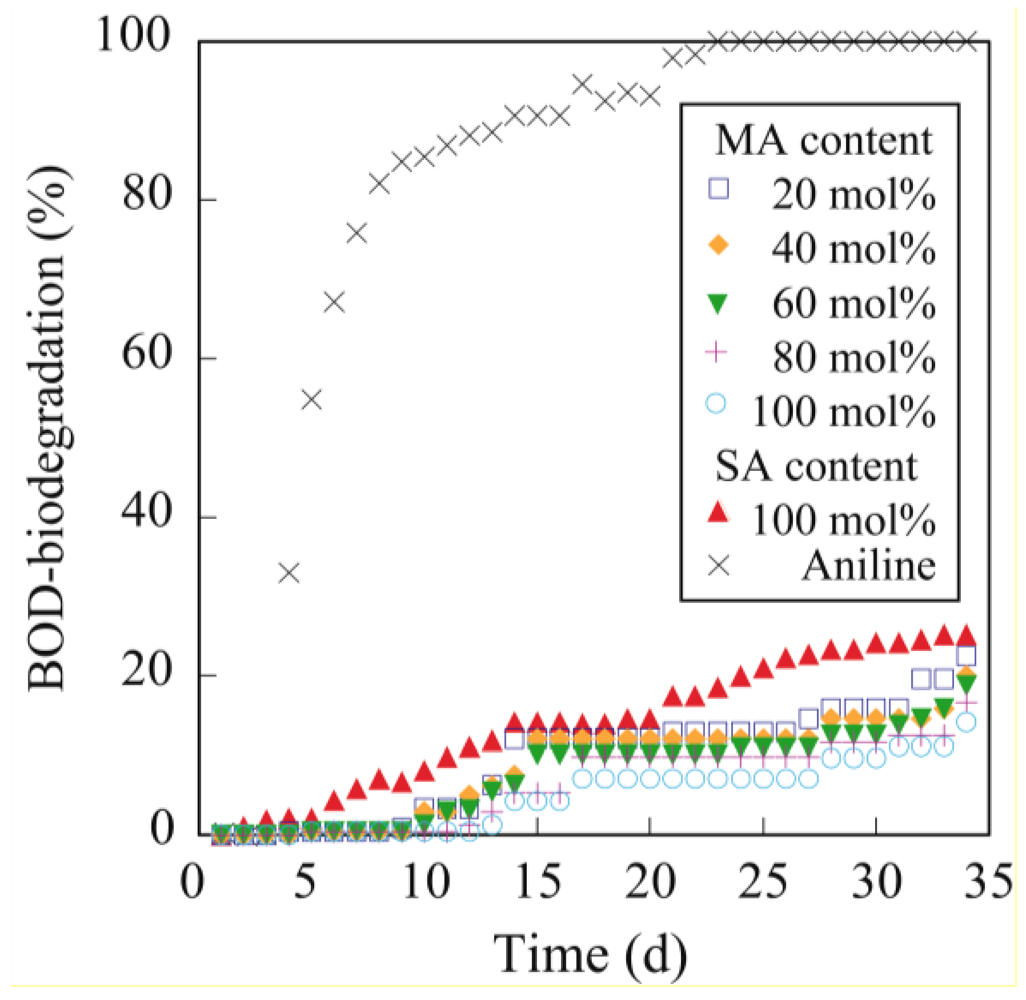
The biodegradability of methyl epoxyricinoleate, PER and PER-MA was evaluated using biochemical oxygen demand (BOD) measurements. The BOD was determined with a BOD tester (VELP Scientifica s.r.l., Usmate, MI, Italy) using the oxygen consumption method according to the modified MITI test [
13]. The activated sludge was obtained from a municipal sewage plant in Yokohama City, Japan. The BOD biodegradation (BOD/Theoretical Oxygen Demand × 100) was measured for 34 d.
2.3. General Enzymatic Epoxidation Procedure
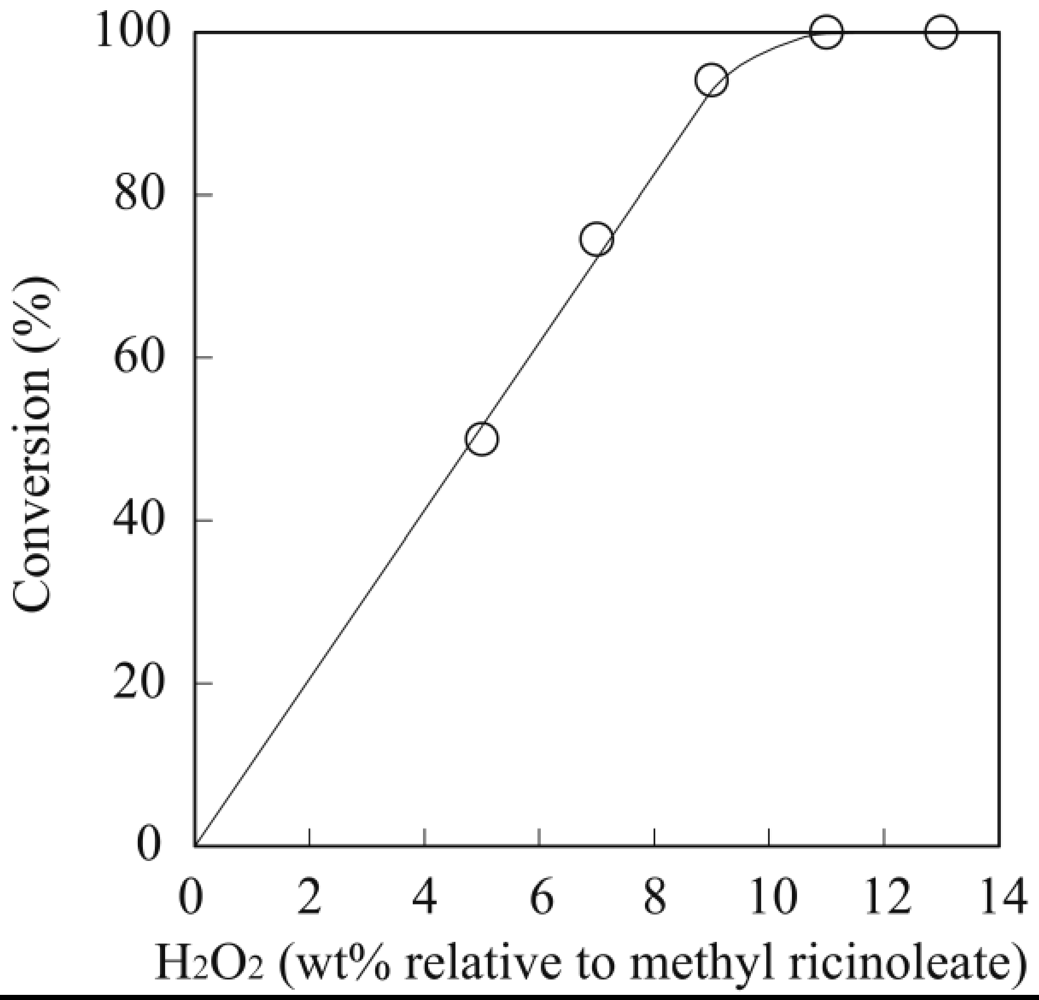
The general procedure for the enzymatic epoxidation of methyl ricinoleate was carried out in a screw-capped vial with MS4A placed at the top of the vial (vapor phase). Methyl ricinoleate (200 mg) and toluene (2 mL) were mixed in a vial at room temperature and then lipase (28 mg) was added followed by H2O2 (70 μL, 30 wt% aqueous solution), and the mixture was stirred at room temperature for 24 h. After the reaction, lipase was removed by filtration, and the filtrate was washed four times with water to remove any unreacted H2O2. The solvent was then evaporated under reduced pressure, and the residue was purified by silica gel column chromatography (n-hexane/ethyl acetate = 4/1) to obtain methyl epoxyricinoleate in 77% yield as a colorless syrup. The molecular structure was analyzed by FT-IR, 1H and 13C-NMR spectroscopy.
FT-IR: 3449 (OH), 2928 (CH2), 1740, 1171 (ester C=O), 831 (epoxy C-O-C) cm−1.
1H-NMR (CDCH3): δ = 0.88 (t, J = 6.6 Hz, 3H, CH3), 1.21–1.83 (m, 20H, CH2), 2.31 (t, J = 7.5 Hz, 2H, 2-H2), 2.92 (m, 1H, 9-H), 3.14 (m, 1H, 10-H), 3.66 (s, 3H, COOCH3), 3.87 (m, 1H, CHOH).
13C-NMR (CDCH3): δ = 14.08 (C-18 CH3), 22.61 (C-17 CH2), 24.88 (C-3 CH2), 25.51/25.61 (C-14 CH2), 26.43 (C-7 CH2), 27.90/28.02 (C-8 CH2), 29.01 (C-4 CH2), 29.15 (C-15 CH2), 29.26 (C-5 CH2), 29.31 (C-6 CH2), 31.81 (C-16), 34.06 (C-2 CH2), 34.72/35.17 (C-11 CH2), 37.46/37.77(C-13 CH2), 51.47 (COOCH3), 54.43/55.47 (C-10 HC-O), 56.31/57.10 (C-9 HC-O), 70.20/70.96 (C-12 CH),174.3 (C-1 C-O).
2.4. General Enzymatic Polymerization Procedure
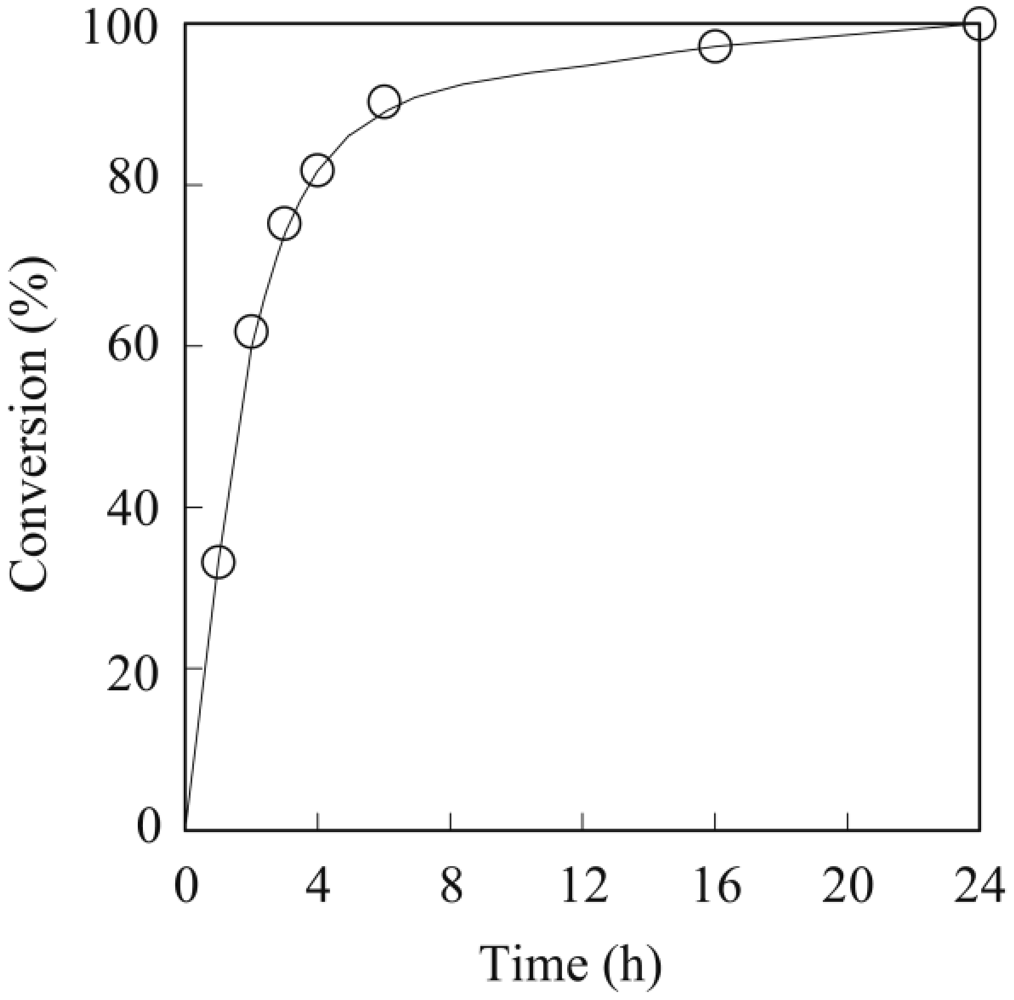
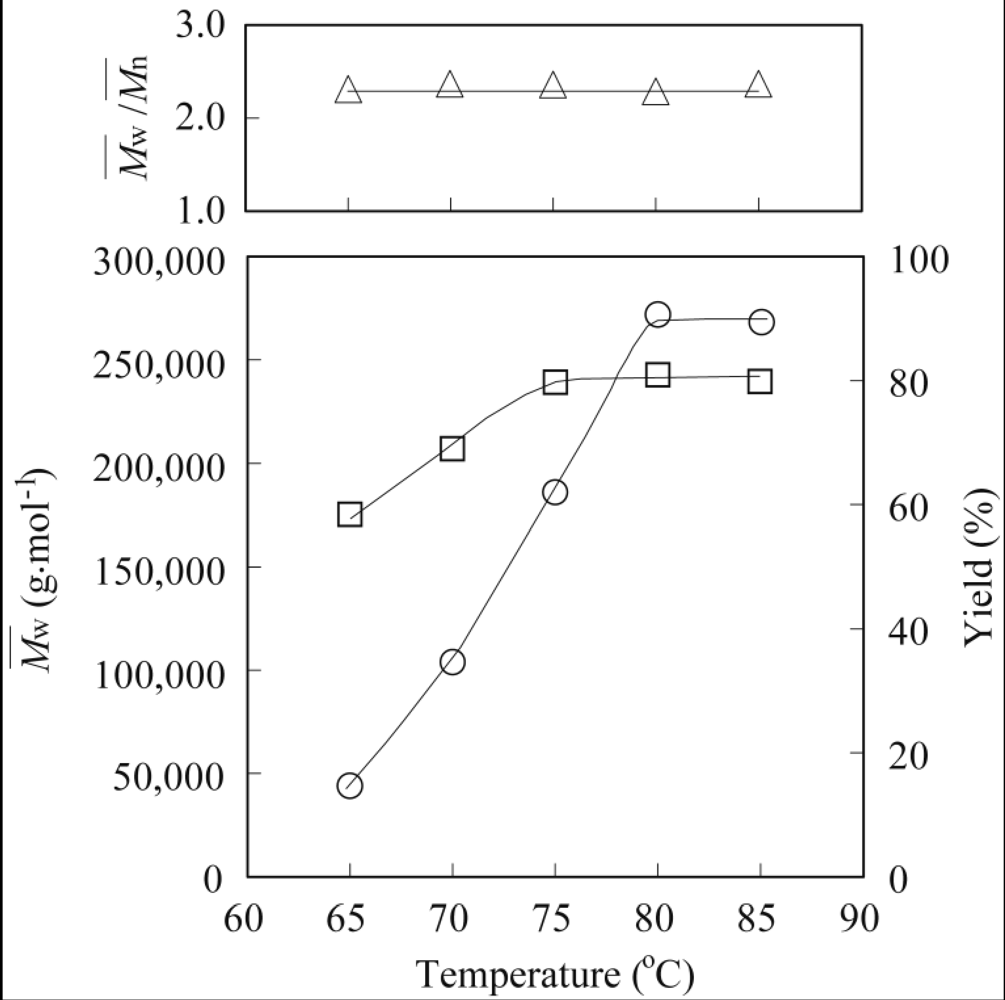
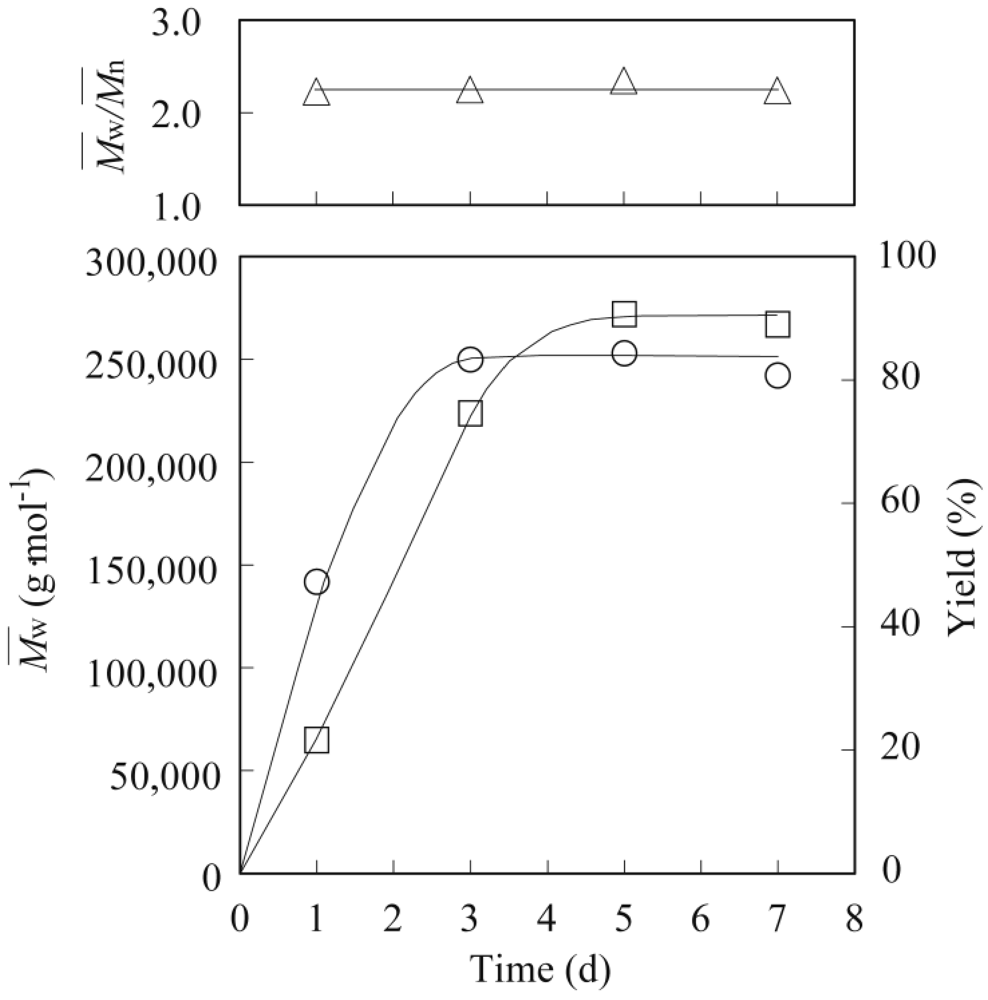
The general procedure for the enzymatic polymerization of methyl epoxyricinoleate was carried out in a screw-capped vial with MS4A placed at the top of the vial (vapor phase) to absorb byproducts, such as water or methanol [
14,
15,
16,
17]. The preparation of PER with a
Mw of 272,000 is described as a typical example. Methyl epoxyricinoleate (50 mg, 0.16 mmol) was polymerized in the presence of lipase PS-IM (50 mg) under a nitrogen atmosphere at 80 °C for 7 d. The polymerization mixture was dissolved in hot chloroform (20 mL), and the insoluble enzyme was removed by filtration. The solvent was next evaporated to obtain the polymer as a colorless viscous syrup, which was then purified by reprecipitation using chloroform (good solvent)–methanol (poor solvent) to remove unreacted monomers. The
Mw and the
Mw/
Mn of the polymer were determined using SEC. The molecular structure was analyzed using FT-IR,
1H and
13C-NMR spectroscopy. The spectral data for PER with a
Mw of 272,000 is shown as an example.
FT-IR: 2930 (CH2), 1732, 1175 (ester C=O), 835 (epoxy C-O-C) cm−1.
1H-NMR: δ = 0.88 (t, J = 6.6 Hz, 3H, CH3), 1.21–1.80 (m, 20H, CH2), 2.30 (t, J = 7.5 Hz, 2H, 2-H2), 2.87 (m, 1H, 9-H), 2.96 (m, 1H, 10-H), 5.06 (m, 1H, HC-O).
13C-NMR: δ = 14.08 (C-18 CH3), 22.57 (C-17 CH2), 25.01 (C-3 CH2), 25.24/25.37 (C-14 CH2), 26.61 (C-7 CH2), 27.96/28.02 (C-8 CH2), 29.06 (C-4 CH2), 29.11 (C-15 CH2), 29.27 (C-5 CH2), 29.39 (C-6 CH2), 31.72 (C-16 CH2), 32.72/32.79 (C-11 CH2), 34.17/34.37 (C-13 CH2), 34.60 (C-2 CH2), 53.70/53.85 (C-10 HC-O), 56.15/56.79 (C-9 HC-O), 72.15/72.19 (C-12 CH), 173.5 (C-1 C-O).
2.5. General Crosslinking Procedure for PER
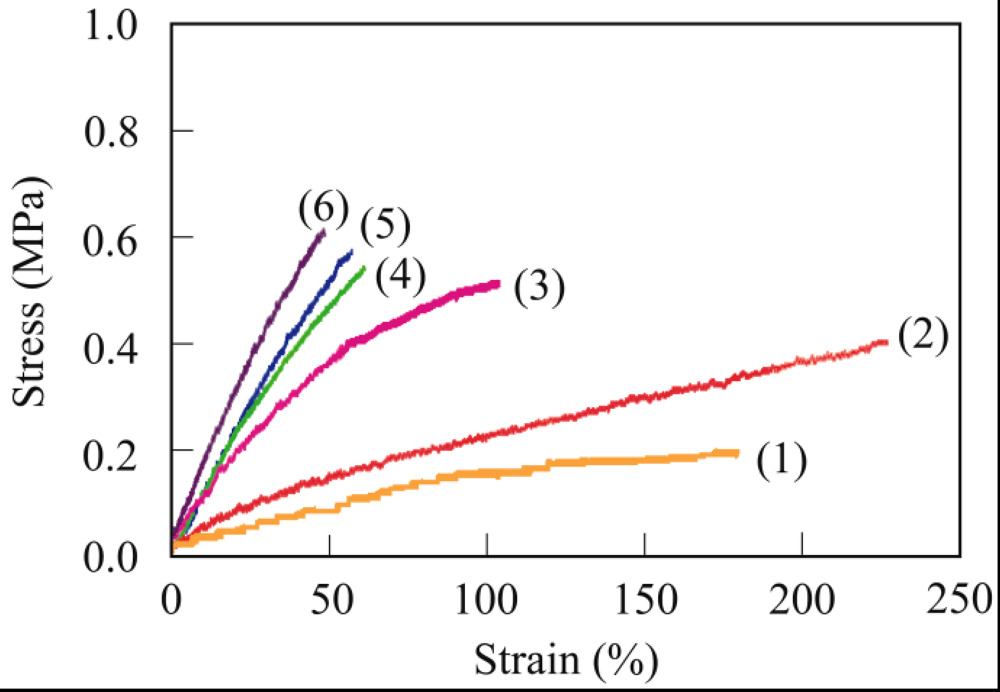
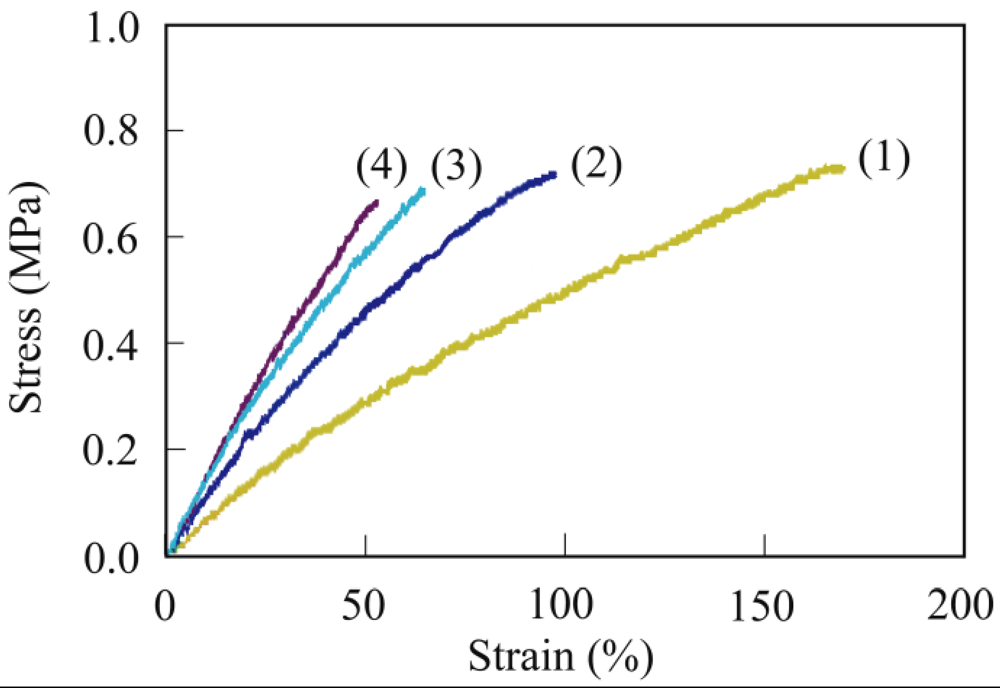
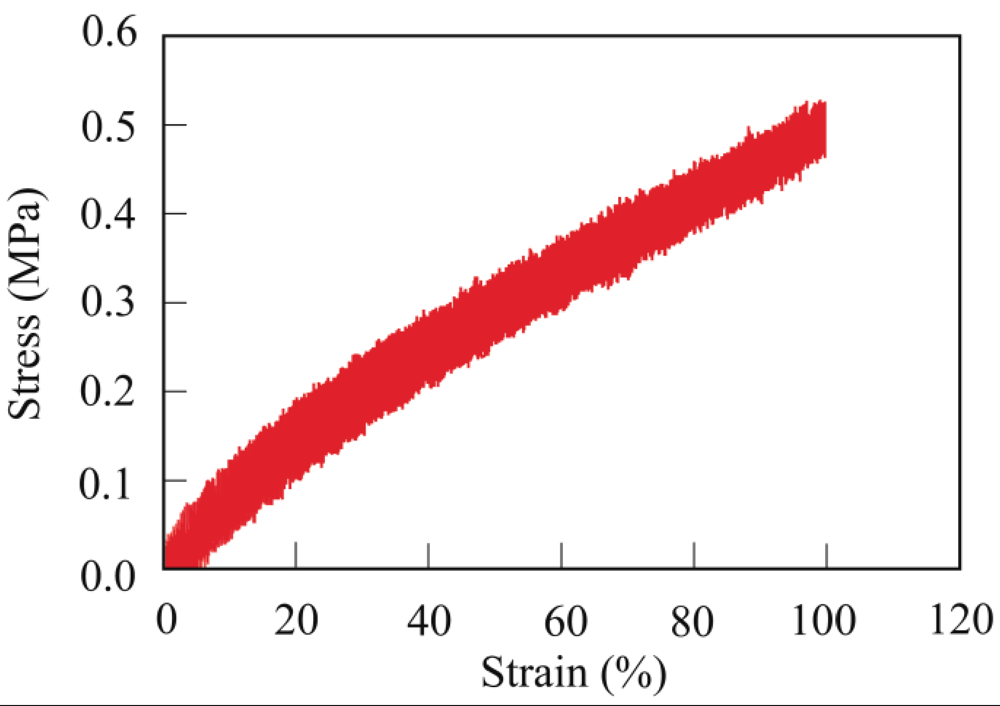
The crosslinking of PER with a Mw of 186,000 is described as a typical example. PER (200 mg) and MA (62.8 mg, 0.64 mmol) as a crosslinking agent were dissolved in hot chloroform (5 mL). The obtained viscous solution was poured onto a Teflon sheet and then dried in air for 12 h to vaporize the chloroform. After vaporizing the chloroform, the viscous liquid reaction mixture was held between the Teflon sheets with a thickness of 0.3 mm, and then cured at 80 °C for 30 min using a hot press machine. The viscous PER was readily cured by MA at 80 °C for 30 min to form a maleic anhydride-crosslinked PER (PER-MA) film with a thickness of 0.3 mm.
The gel fraction of the crosslinked PER was determined from the weight remaining after soaking of the sample in chloroform using the following equation:
Gel fraction (%) = (Wg/W0) × 100
where W0 is the weight of the (dry) crosslinked PER before soaking and Wg is the weight of the crosslinked PER remaining (dry gel component) after soaking in chloroform at room temperature for 48 h. The molecular structure was analyzed using FT-IR spectroscopy.
FT-IR: 2923 (CH2), 1731, 1176 (ester C=O), 1647 (C=C) cm−1
In a similar procedure, succinic anhydride-crosslinked PER (PER-SA) was prepared by the reaction of PER and succinic anhydride.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






