Synthesis of Hyperbranched Glycoconjugates by the Combined Action of Potato Phosphorylase and Glycogen Branching Enzyme from Deinococcus geothermalis


Abstract
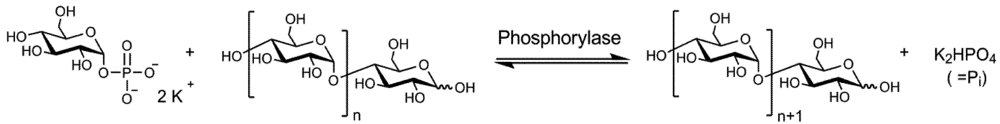
:1. Introduction
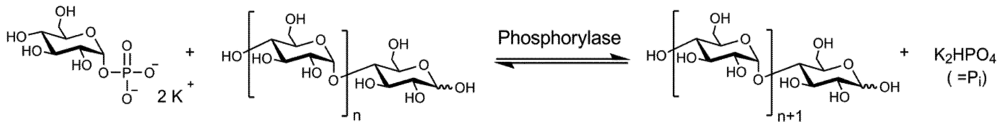
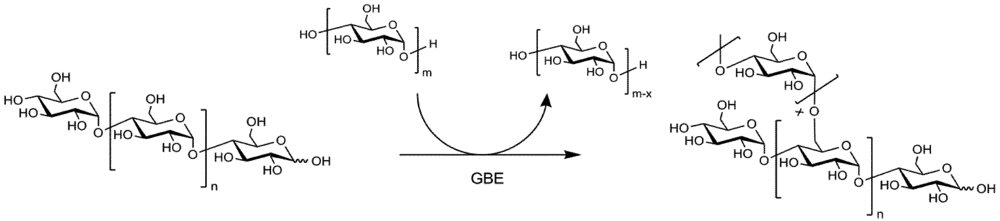

2. Experimental Section
2.1. Materials
2.2. Methods
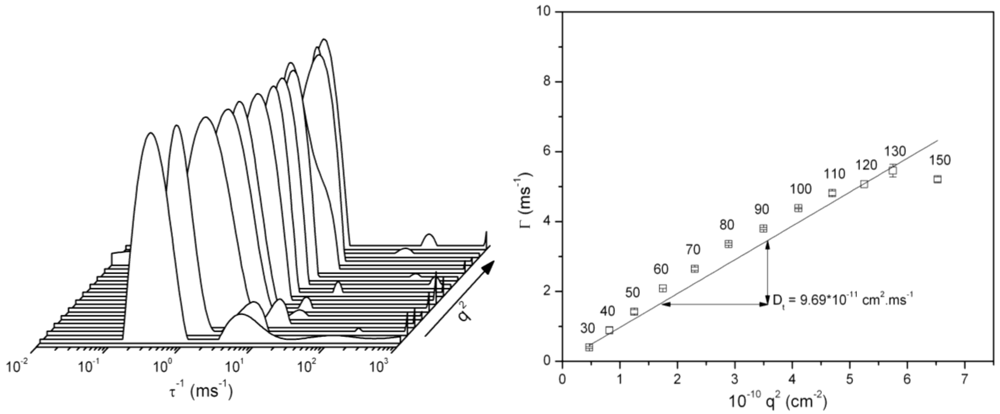
2.2.1. Dynamic Light Scattering (DLS)
2.2.2. Elemental Analysis
2.2.3. 1H-NMR Spectroscopy
2.2.4. ATR Infra-Red Spectroscopy
2.3. Isolation and Purification of the Glycosyltransferases
2.4. Synthesis of Maltoheptaose
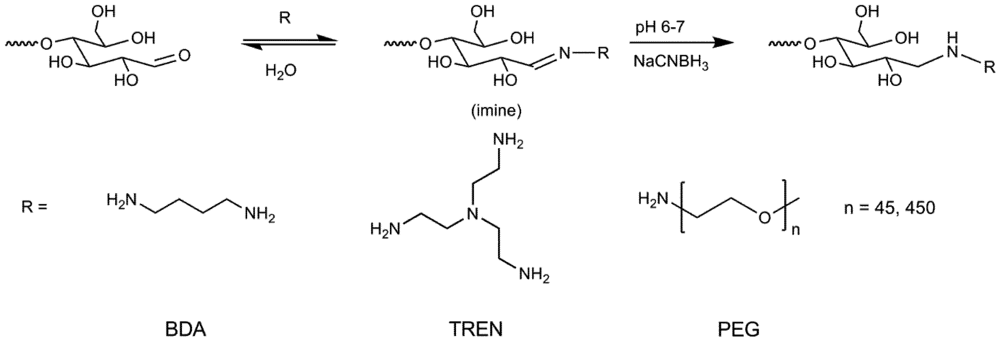
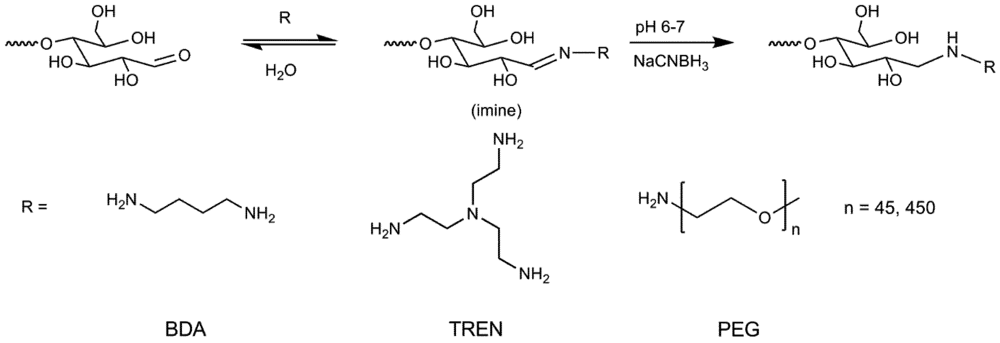
2.5. Synthesis of Primer Adducts
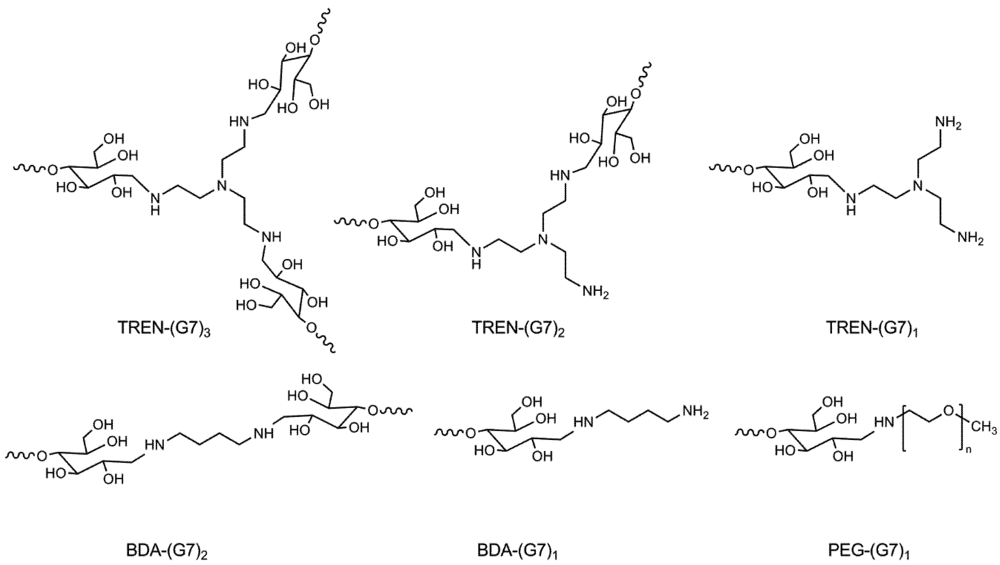
2.5.1. Di-Functional Primer Adduct, BDA-(G7)2
2.5.2. Synthesis of Tri-Functional Primer Adduct, TREN-(G7)3
2.5.3. Synthesis of PEG Phthalimide Intermediate (Mitsunobu)
2.5.4. Synthesis of PEG-NH2 (Hydrazynolysis)
2.5.5. Synthesis of PEG Macro Primer Adduct, PEG-(G7)1
2.6. Typical Enzyme Catalyzed Polymerization
3. Results and Discussion
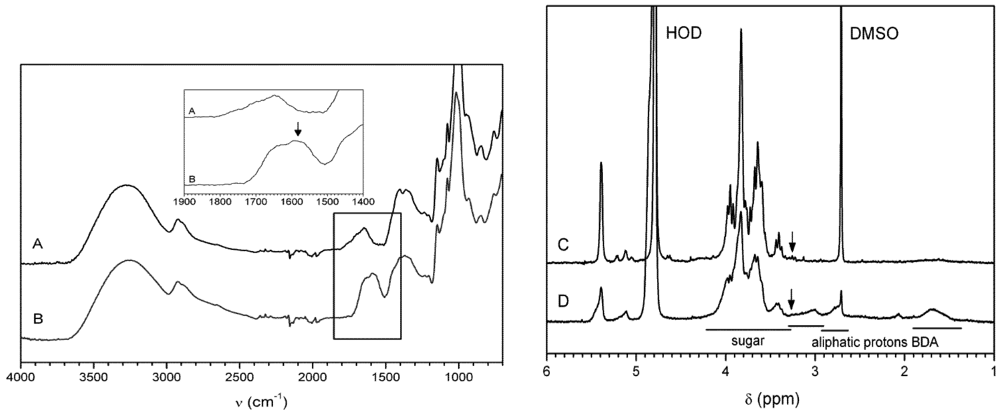
3.1. Synthesis and Purification of Primer Adducts
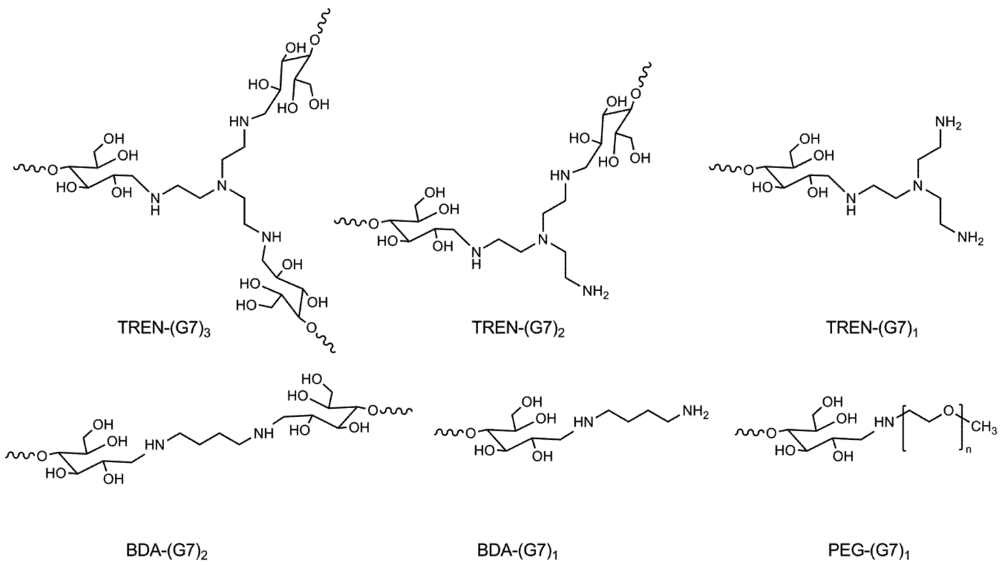

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Eluted with | Theoretical (%) | Observed (%) | ||||
|---|---|---|---|---|---|---|---|
| C | H | N | C | H | N | ||
| BDA-(G7)2 | Water | 44.74 | 6.66 | 1.19 | 39.67 | 5.98 | 1.23 |
| BDA-(G7) | 10% ammonia | 45.10 | 6.91 | 2.29 | 40.79 | 6.35 | 3.67 |
| TREN-(G7)3 | Water | 44.57 | 6.63 | 1.58 | 38.96 | 6.06 | 1.69 |
| TREN-(G7)2, | } 10% ammonia | 44.66 | 6.75 | 2.31 | 38.82 | 6.36 | 3.05 |
| TREN-(G7) | 44.93 | 7.07 | 4.37 | ||||
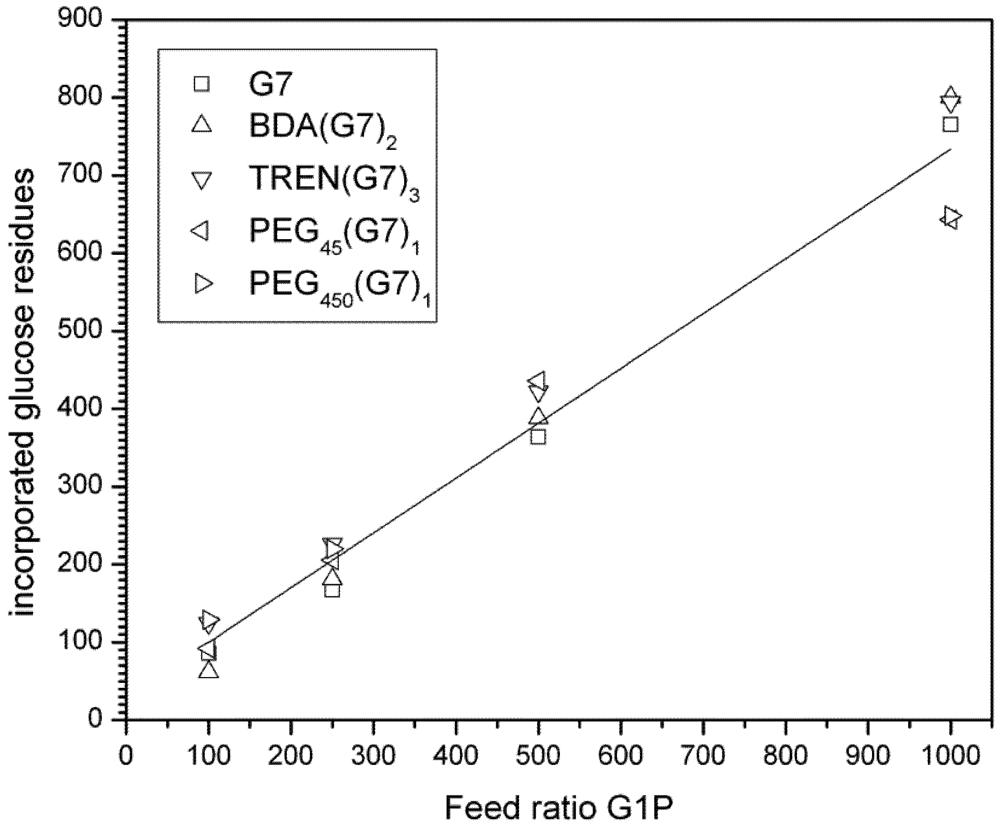
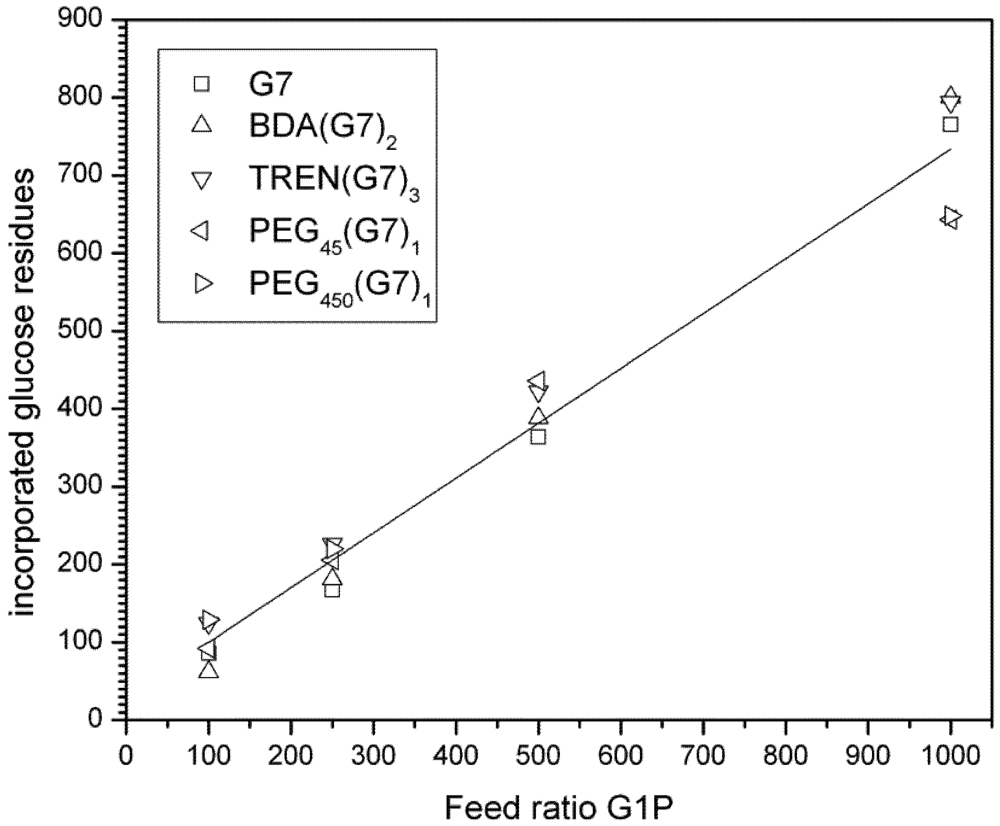
3.2. Enzyme Catalyzed Synthesis of Hyperbranched Glycoconjugates

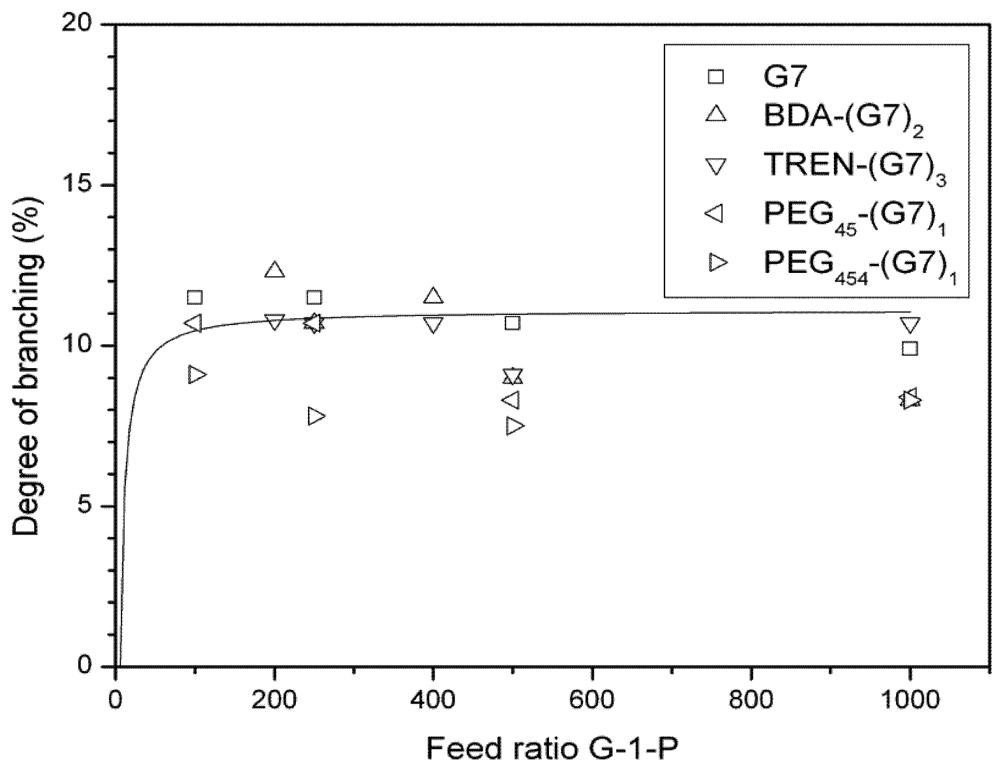
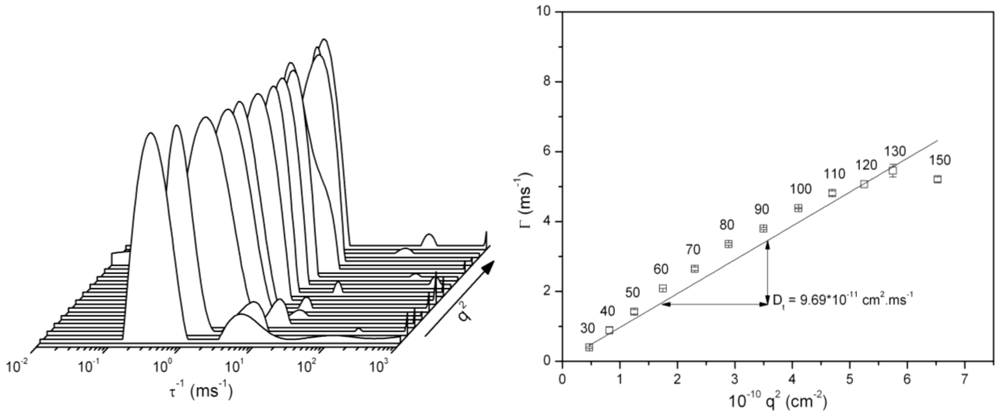
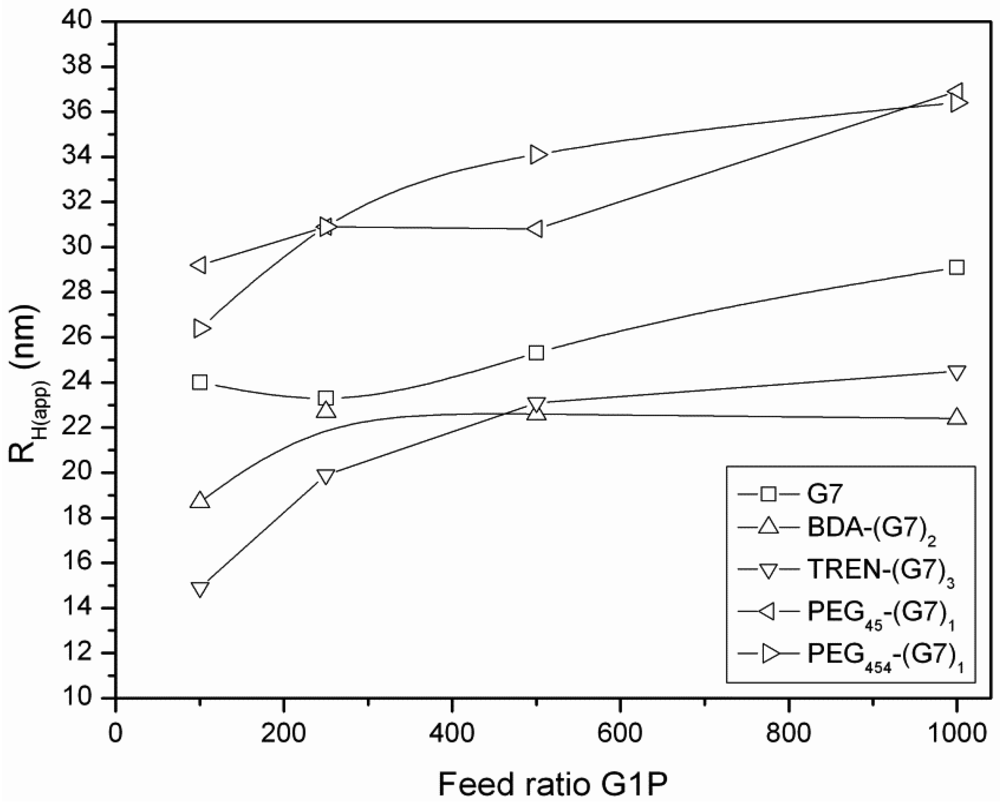
3.3. Dynamic Light Scattering (DLS) Analysis of Hyperbranched Glycoconjugates


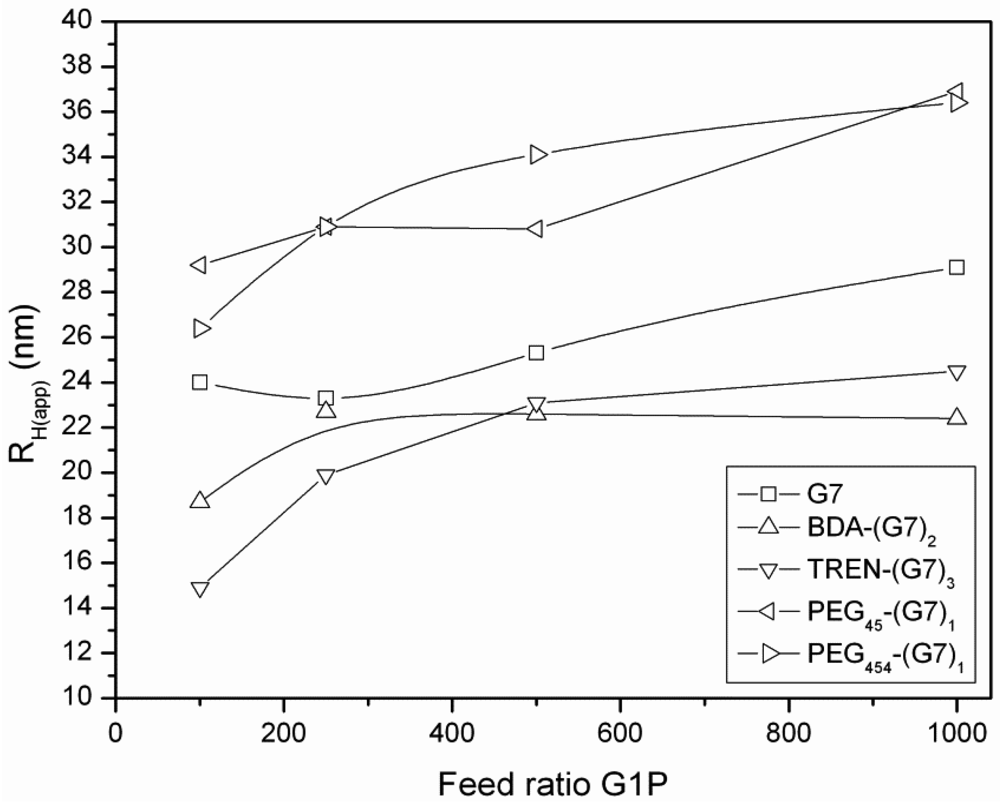
4. Conclusions
Acknowledgments
References
- Houga, C.; Giermanska, J.; Lecommandoux, S.; Borsali, R.; Taton, D.; Gnanou, Y.; Le Meins, J.F. Micelles and polymersomes obtained by self-assembly of dextran and polystyrene based block copolymers. Biomacromolecules 2009, 10, 32–40. [Google Scholar]
- Akiyoshi, K.; Kohara, M.; Ito, K.; Kitamura, S.; Sunamoto, J. Enzymatic synthesis and characterization of amphiphilic block copolymers of poly(ethylene oxide) and amylose. Macromol. Rapid Commun. 1999, 20, 112–115. [Google Scholar]
- Liu, J.Y.; Zhang, L.M. Preparation of a polysaccharide-polyester diblock copolymer and its micellar characteristics. Carbohydr. Polym. 2007, 69, 196–201. [Google Scholar] [CrossRef]
- Hernandez, O.S.; Soliman, G.M.; Winnik, F.M. Synthesis, reactivity, and pH-responsive assembly of new double hydrophilic block copolymers of carboxymethldextran and poly(ethylene glycol). Polymer 2007, 48, 921–930. [Google Scholar]
- Spain, S.G.; Gibson, M.I.; Cameron, N.R. Recent advances in the synthesis of well-defined glycopolymers. J. Polym. Sci. Part A 2007, 45, 2059–2072. [Google Scholar]
- Shoda, S.; Izumi, R.; Fujita, M. Green process in glycotechnology. Bull. Chem. Soc. Jpn. 2003, 76, 1–13. [Google Scholar]
- Kadokawa, J. Precision polysaccharide synthesis catalyzed by enzymes. Chem. Rev. 2011, 111, 4308–4345. [Google Scholar] [CrossRef]
- Loos, K. Biocatalysis in Polymer Chemistry; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Palmans, A.R.A.; Heise, A. Enzymatic Polymerisation; Springer: Berlin, Germany, 2010. [Google Scholar]
- Cheng, H.N.; Gross, R.A. Green Polymer Chemistry: Biocatalysis and Biomaterials; American Chemical Society: New York, NY, USA, 2010. [Google Scholar]
- Kobayashi, S.; Makino, A. Enzymatic polymer synthesis: An opportunity for green polymer chemistry. Chem. Rev. 2009, 109, 5288–5353. [Google Scholar] [CrossRef]
- Van der Vlist, J.; Loos, K. Enzymatic polymerizations of polysaccharides. In Biocatalysis in Polymer Chemistry, 1st; Loos, K., Ed.; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Van der Vlist, J.; Loos, K. Transferases in polymer chemistry. Adv. Polym. Sci. 2010, 237, 21–54. [Google Scholar]
- Van der Vlist, J.; Reixach, M.P.; van der Maarel, M.; Dijkhuizen, L.; Schouten, A.J.; Loos, K. Synthesis of branched polyglucans by the tandem action of potato phosphorylase and deinococcus geothermalis glycogen branching enzyme. Macromol. Rapid Commun. 2008, 29, 1293–1297. [Google Scholar]
- Ciric, J.; Loos, K. Synthesis of branched polysaccharides with tunable degree of branching. 2012. submitted. [Google Scholar]
- Kajiura, H.; Kakutani, R.; Akiyama, T.; Takata, H.; Kuriki, T. A novel enzymatic process for glycogen production. Biocatal. Biotransform. 2008, 26, 133–140. [Google Scholar] [CrossRef]
- Fujii, K.; Takata, H.; Yanase, M.; Terada, Y.; Ohdan, K.; Takaha, T.; Okada, S.; Kuriki, T. Bioengineering and application of novel glucose polymers. Biocatal. Biotransformation 2003, 21, 167–172. [Google Scholar]
- Seibel, J.; Joerdening, H.; Buchholz, K. Glycosylation with activated sugars using glycosyltransferases and transglycosidases. Biocatal. Biotransformation 2006, 24, 311–342. [Google Scholar] [CrossRef]
- Kitaoka, M.; Hayashi, K. Carbohydrate-processing phosphorolytic enzymes. Trends Glycosci. Glycotechnol. 2002, 14, 35–50. [Google Scholar] [CrossRef]
- Van der Vlist, J.; Schonen, I.; Loos, K. Utilization of glycosyltransferases for the synthesis of a densely packed hyperbranched polysaccharide brush coating as artificial glycocalyx. Biomacromolecules 2011, 12, 3728–3732. [Google Scholar] [CrossRef]
- Narumi, A.; Kawasaki, K.; Kaga, H.; Satoh, T.; Sugimoto, N.; Kakuchi, T. Glycoconjugated polymer 6. Synthesis of poly[styrene-block-(styrene-graft-amylose) via potato phosphorylase catalyszed polymerization. Polym. Bull. 2003, 49, 405–410. [Google Scholar] [CrossRef]
- Loos, K.; Müller, A.H.E. New routes to the synthesis of amylose-block-polystyrene rod-coil block copolymers. Biomacromolecules 2002, 3, 368–373. [Google Scholar]
- Loos, K.; vonBraunmühl, V.; Stadler, R.; Landfester, K.; Spiess, H.W. Saccharide modified silica particles by enzymatic grafting. Macromol. Rapid Commun. 1997, 18, 927–938. [Google Scholar] [CrossRef]
- Ziegast, G.; Pfannemüller, B. Linear and star-shaped hybrid polymers. 1. A new method for the conversion of hydroxyl end groups of poly(oxyethylene) and other polyols into amino end groups. Makromol. Chem. Rapid Commun. 1984, 5, 363–371. [Google Scholar] [CrossRef]
- Ziegast, G.; Pfannemüller, B. Linear and star-shaped hybrid polymers. 2. Coupling of monosaccharide and oligosaccharide to alpha,omega-diamino substituted poly(oxyethylene) and multifunctional amines by amide linkage. Makromol. Chem. Rapid Commun. 1984, 5, 373–379. [Google Scholar] [CrossRef]
- Ziegast, G.; Pfannemüller, B. Linear and star-shaped hybrid polymers. 3. An improved purification procedure for coupling products of oligosaccharides by amide linkage. Makromol. Chem. Macromol. Chem. Phys. 1984, 185, 1855–1866. [Google Scholar]
- Ziegast, G.; Pfannemüller, B. Linear and star-shaped hybrid polymers. 4. Phosphorolytic syntheses with di-functional, oligo-functional and multifunctional primers. Carbohydr. Res. 1987, 160, 185–204. [Google Scholar] [CrossRef]
- Bosker, W.T.E.; Agoston, K.; Cohen Stuart, M.A.; Norde, W.; Timmermans, J.W.; Slaghek, T.M. Synthesis and interfacial behavior of polystyrene-polysaccharide diblock copolymers. Macromolecules 2003, 36, 1982–1987. [Google Scholar]
- Houga, C.; Le Meins, J.F.; Borsali, R.; Taton, D.; Gnanou, Y. Synthesis of ATRP-induced dextran-b-polystyrene diblock copolymers and preliminary investigation of their self-assembly in water. Chem. Commun. 2007, 3063–3065. [Google Scholar]
- Hernandez, O.S.; Soliman, G.M.; Winnik, F.M. Synthesis, reactivity, and pH-responsive assembly of new double hydrophilic block copolymers of carboxymethyldextran and poly(ethylene glycol). Polymer 2007, 48, 921–930. [Google Scholar] [CrossRef]
- Schatz, C.; Louguet, S.; le Meins, J.F.; Lecommandoux, S. Polysaccharide-block-polypeptide copolymer vesicles: Towards synthetic viral capsids. Angew. Chem. Int. Ed. 2009, 48, 2572–2575. [Google Scholar]
- Upadhyay, K.K.; le Meins, J.F.; Misra, A.; Voisin, P.; Bouchaud, V.; Ibarboure, E.; Schatz, C.; Lecommandoux, S. Biomimetic doxorubicin loaded polymersomes from hyaluronan-block-poly(gamma-benzyl glutamate) copolymers. Biomacromolecules 2009, 10, 2802–2808. [Google Scholar]
- Yang, Y.L.; Kataoka, K.; Winnik, F.M. Synthesis of diblock copolymers consisting of hyaluronan and poly(2-ethyl-2-oxazoline). Macromolecules 2005, 38, 2043–2046. [Google Scholar]
- Loos, K.; Böker, A.; Zettl, H.; Zhang, A.F.; Krausch, G.; Müller, A.H.E. Micellar aggregates of amylose-block-polystyrene rod-coil block copolymers in water and THF. Macromolecules 2005, 38, 873–879. [Google Scholar]
- Soliman, G.M.; Winnik, F.M. Enhancement of hydrophilic drug loading and release characteristics through micellization with new carboxymethyldextran-PEG block copolymers of tunable charge density. Int. J. Pharm. 2008, 356, 248–258. [Google Scholar] [CrossRef]
- Akiyoshi, K.; Maruichi, N.; Kohara, M.; Kitamura, S. Amphiphilic block copolymer with a molecular recognition site: Induction of a novel binding characteristic of amylose by self-assembly of poly(ethylene oxide)-block-amylose in chloroform. Biomacromolecules 2002, 3, 280–283. [Google Scholar] [CrossRef]
- Houga, C.; Giermanska, J.; Lecommandoux, S.; Borsali, R.; Taton, D.; Gnanou, Y.; le Meins, J. Micelles and polymersomes obtained by self-assembly of dextran and polystyrene based block copolymers. Biomacromolecules 2009, 10, 32–40. [Google Scholar] [CrossRef]
- Palomo, M.; Pijning, T.; Booiman, T.; Dobruchowska, J.M.; van der Vlist, J.; Kralj, S.; Planas, A.; Loos, K.; Kamerling, J.P.; Dijkstra, B.W.; et al. Thermus thermophilus glycoside hydrolase family 57 branching enzyme crystal structure, mechanism of action and products formed. J. Biol. Chem. 2011, 286, 3520–3530. [Google Scholar]
- Geddes, R.; Harvey, J.D.; Wills, P.R. Molecular-size and shape of liver-glycogen. Biochem. J. 1977, 163, 201–209. [Google Scholar]
- Geddes, R. Glycogen: A metabolic viewpoint. Biosci. Rep. 1986, 6, 415–428. [Google Scholar] [CrossRef]
- Smythe, C.; Cohen, P. The discovery of glycogenin and the priming mechanism for glycogen biogenesis. Eur. J. Biochem. 1991, 200, 625–631. [Google Scholar] [CrossRef]
- Miura, Y. Synthesis and biological application of glycopolymers. J. Polym. Sci. Part A 2007, 45, 5031–5036. [Google Scholar] [CrossRef]
- Palomo, R.M.; Kralj, S.; van der Maarel, M.J.E.C.; Dijkhuizen, L. The unique branching patterns of deinococcus glycogen branching enzymes are determined by their N-termnal domains. Appl. Environ. Microbiol. 2009, 75, 1355–1362. [Google Scholar]
- Lowry, O.H.; Lopez, J.A. The Determination of inorganic phosphate in the presence of labile phosphate esters. J. Biol. Chem. 1946, 162, 421–428. [Google Scholar]
- Fiske, C.H.; Subbarow, Y. The colorimetric determination of phosphorus. J. Biol. Chem. 1925, 66, 375–400. [Google Scholar]
- Lane, C.F. Sodium cyanoborohydride—Highly selective reducing agent for organic functional groups. Synth. Stuttg. 1975, 135–146. [Google Scholar] [CrossRef]
- Borch, R.F.; Bernstei, M.D.; Durst, H.D. Cyanohydridoborate anion as a selective reducing agent. J. Am. Chem. Soc. 1971, 93, 2897–2904. [Google Scholar]
- Yalpani, M.; Brooks, D.E. Selective chemical modifications of dextran. J. Polym. Sci. Part A 1985, 23, 1395–1405. [Google Scholar]
- Ryoo, S.J.; Kim, J.; Kim, J.S.; Lee, Y.S. Efficient methods of converting hydroxyl groups into amino groups in poly(ethylene glycol)-grafted polystyrene resin. J. Comb. Chem. 2002, 4, 187–190. [Google Scholar]
- Mitsunobu, O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural-products. Synth. Stuttg. 1981, 1–28. [Google Scholar] [CrossRef]
- Sisu, E.; Bosker, W.T.E.; Norde, W.; Slaghek, T.M.; Timmermans, J.W.; Peter-Katalinic, J.; Cohen-Stuart, M.A.; Zamfir, A.D. Electrospray ionization quadrupole time-of-flight tandem mass spectrometric analysis of hexamethylenediamine-modified maltodextrin and dextran. Rapid Commun. Mass Spectrom. 2006, 20, 209–218. [Google Scholar]
- Perdivara, I.; Sisu, E.; Sisu, I.; Dinca, N.; Tomer, K.B.; Przybylski, M.; Zamfir, A.D. Enhanced electrospray ionization fourier transform ion cyclotron resonance mass spectrometry of long-chain polysaccharides. Rapid Commun. Mass Spectrom. 2008, 22, 773–782. [Google Scholar]
- Mcintyre, D.D.; Ho, C.; Vogel, H.J. One-dimensional nuclear-magnetic-resonance studies of starch and starch products. Starch 1990, 42, 260–267. [Google Scholar] [CrossRef]
- Sugiyama, H.; Nitta, T.; Horii, M.; Motohashi, K.; Sakai, J.; Usui, T.; Hisamichi, K.; Ishiyama, J. The conformation of alpha-(1 -> 4)-linked glucose oligomers from maltose to maltoheptaose and short-chain amylose in solution. Carbohydr. Res. 2000, 325, 177–182. [Google Scholar] [CrossRef]
- Le Meins, J.F.; Houg, C.; Borsali, R.; Taton, D.; Gnanou, Y. Morphological changes induced by addition of polystyrene to dextran-polystyrene block copolymer solutions. Macromol. Symp. 2009, 281, 113–118. [Google Scholar] [CrossRef]
- Boris, D.; Rubinstein, M. A self-consistent mean field model of a starburst dendrimer: Dense core vs. dense shell. Macromolecules 1996, 29, 7251–7260. [Google Scholar]
- Kuga, S. Pore size distribution analysis of gel substances by size exclusion chromatography. J. Chromatogr. A 1981, 206, 449–461. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vlist, J.v.d.; Faber, M.; Loen, L.; Dijkman, T.J.; Asri, L.A.T.W.; Loos, K. Synthesis of Hyperbranched Glycoconjugates by the Combined Action of Potato Phosphorylase and Glycogen Branching Enzyme from Deinococcus geothermalis. Polymers 2012, 4, 674-690. https://doi.org/10.3390/polym4010674
Vlist Jvd, Faber M, Loen L, Dijkman TJ, Asri LATW, Loos K. Synthesis of Hyperbranched Glycoconjugates by the Combined Action of Potato Phosphorylase and Glycogen Branching Enzyme from Deinococcus geothermalis. Polymers. 2012; 4(1):674-690. https://doi.org/10.3390/polym4010674
Chicago/Turabian StyleVlist, Jeroen van der, Martin Faber, Lizette Loen, Teunis J. Dijkman, Lia A. T. W. Asri, and Katja Loos. 2012. "Synthesis of Hyperbranched Glycoconjugates by the Combined Action of Potato Phosphorylase and Glycogen Branching Enzyme from Deinococcus geothermalis" Polymers 4, no. 1: 674-690. https://doi.org/10.3390/polym4010674