Controlled Photoradical Polymerization Mediated by 2,2,6,6-Tetramethylpiperidine-1-Oxyl

Department of Environmental and Life Sciences, Toyohashi University of Technology, 1-1 Hibarigaoka, Tempaku-cho, Toyohashi, Aichi 441-8580, Japan
Polymers 2012, 4(2), 1125-1156; https://doi.org/10.3390/polym4021125
Submission received: 1 March 2012
/
Revised: 13 April 2012
/
Accepted: 19 April 2012
/
Published: 2 May 2012
(This article belongs to the Special Issue Living Polymerization Techniques)
Abstract
:In recent years, controlled photoradical polymerization has been established using 2,2,6,6-tetramethylpiperidine-1-oxyl as a mediator. This review article will describe the molecular weight control, polymerization mechanism, influence of initiator structure, effect of substituents supported on photo-acid generator, stability of the propagating chain end, photo-latency of the polymerization, molecular design, and an application to heterogeneous polymerization in an alcoholic medium.
1. Introduction
Controlled/living radical polymerizations have made great progress in the past two decades based on their advantages over ionic polymerizations as a simple procedure without severe conditions and widely applicable monomers [1]. Examples of controlled radical polymerizations include: iniferter polymerization [2]; reversible addition fragmentation chain transfer polymerization (RAFT) [3]; atom transfer radical polymerization (ATRP) using transition metal complexes, such as Cu [4], Ni [5], Co [6], Fe [7], Ru [8], Rh [9], Pd [10], and Re [11]; iodide-transfer polymerization [12,13,14]; and nitroxide-mediated polymerization (NMP) [15,16]. The primary significance of these controlled/living radical polymerizations lies in the fact that the polymerizations can produce precisely designed architectures.
The NMP using 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) has a limited number of monomers [17,18,19,20,21]; however, the scope of the applicable monomers has been extended by improving the structure of the nitroxide [22,23,24] using alkoxyamine adducts [25,26] and utilizing additives [27,28]. The NMP still has advantages in using nonmetallic catalysts, thus creating a great variety of architectures through designing the nitroxide catalysts, and producing uncolored or less-colored polymers. While the nitroxides with the 2,2,5-trimethyl-4-phenyl-3-azahexane-3-oxyl skeleton provide highly controlled molecular weight distributions [25], TEMPO also has been often used as the mediator for the NMP, because TEMPO is easily available with a low cost and can be converted into a variety of derivatives that have functional groups [29,30] and are supported on polymers [31,32,33,34,35]. Recent findings that the TEMPO-mediated NMP is induced by photo irradiation involve the significance of the TEMPO mediator [36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51].
Photopolymerization has significant advantages over thermal polymerization in an energy-saving process that utilizes solar energy, local and medical applications, and photo-specific reactions. Hence, considerable attention has been paid to controlled photoradical polymerization to create macromolecules with well-defined structures. For the purpose of establishing controlled photoradical polymerization to well-control the molecular weight, new photoinitiators have been prepared; the dithiocarbamate derivatives [52,53], trithiocarbonate [54,55], dithiodiethanol [56], xanthate [57], and a benzophenone derivative [58]. The potential of photopolymerization has also been explored for thermal RAFT and ATRP polymerizations using photosensitive initiators [59,60,61,62] and a catalyst containing dithiocarbamate [63]. More recently, it was found that TEMPO-mediated controlled/living photoradical polymerization provided comparatively narrow molecular weight distribution (MWD) for methyl methacrylate (MMA) [36,37,41,43]. This paper describes TEMPO-mediated controlled/living photoradical polymerization, focusing MMA polymerization and the molecular design through this polymerization.
2. Results and Discussion
2.1. Molecular Weight Control of Polymers
Thermal NMP at high temperatures often causes hydrogen abstraction by the nitroxide mediator from the propagating radical [64,65]. Photopolymerization carried out at room temperature can eliminate this side reaction.
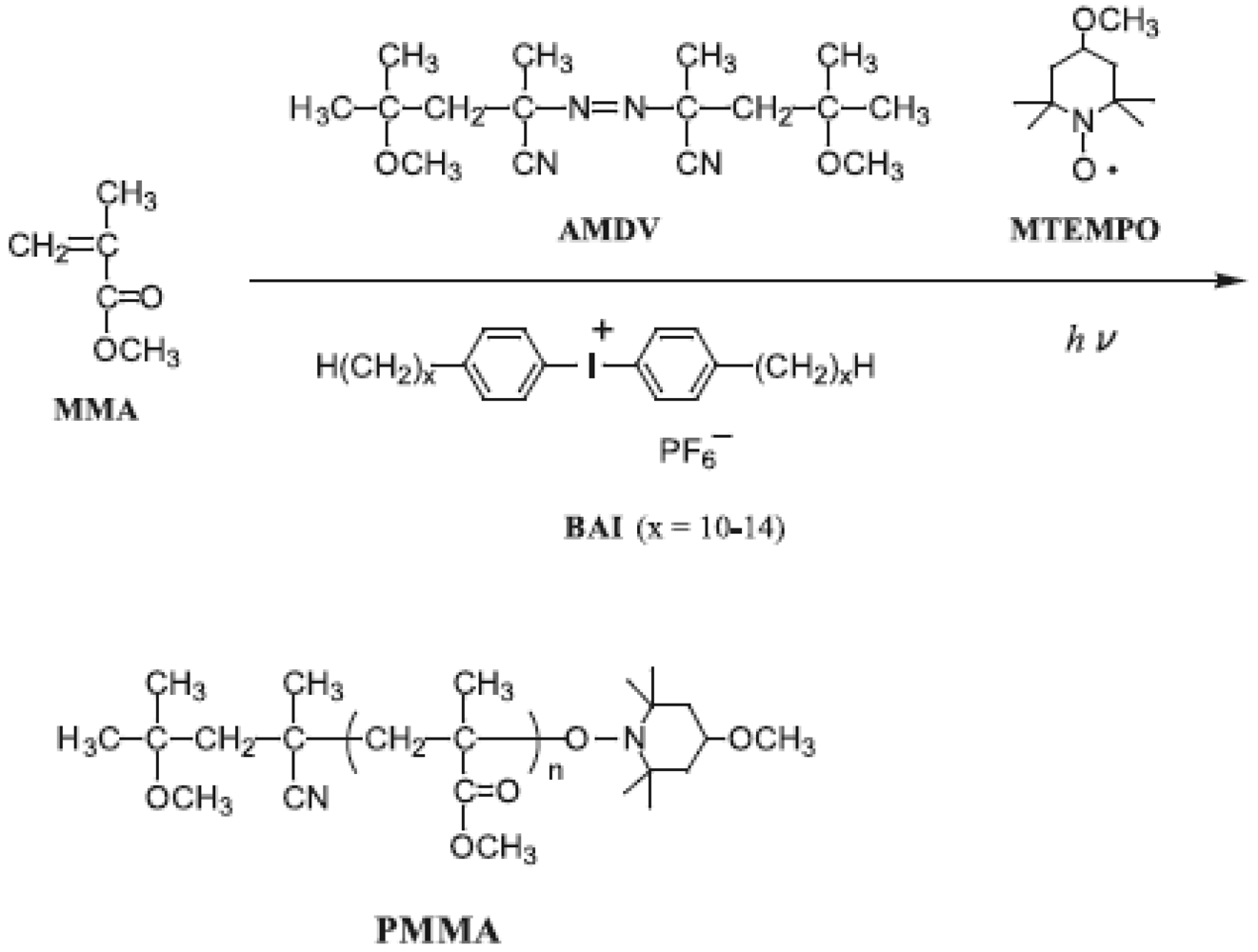
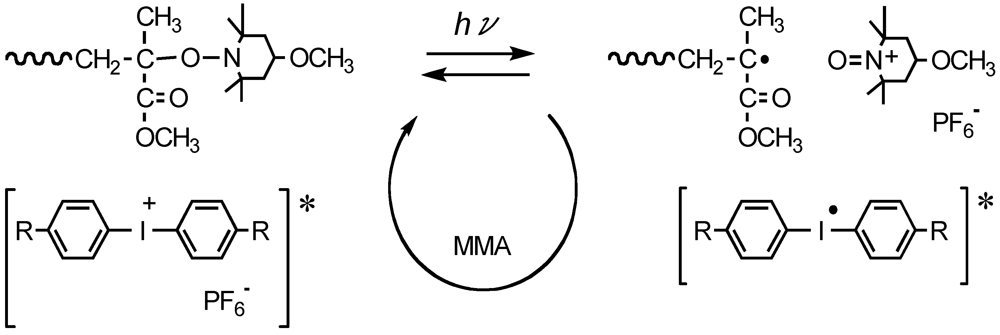
Photoradical polymerization of MMA was performed by azobis(4-methoxy-2,4-dimethylvaleronitrile) (AMDV) as the initiator and 4-methoxy-TEMPO (MTEMPO) as the mediator in the presence of bis(alkylphenyl)iodonium hexafluorophosphate (BAI) as the photo-acid generator [37]. The bulk polymerization was carried out at room temperature by irradiation with a high-pressure mercury lamp. The orange-colored monomer solution turned colorless after polymerization. These results are shown in Table 1. Polymerization in the absence of MTEMPO produced poly(MMA) (PMMA) with a broad MWD. BAI had a slight effect of decreasing the MWD, although the MWD was still broad. It was found that the MWD dramatically decreased in the presence of MTEMPO.

| MTEMPO/AMDV | BAI/MTEMPO | Time (h) | Conv. (%) | Mn a | Mw/Mn a |
|---|---|---|---|---|---|
| – b | – | 1 | 85 | 33,000 | 6.94 |
| – | – c | 1 | 87 | 32,300 | 5.86 |
| 1.1 | – | 31 | 40 | 11,600 | 1.47 |
| 1.1 | 0.5 | 3 | 68 | 16,200 | 1.66 |
| 1.1 | 0.75 | 2.5 | 88 | 19,800 | 2.88 |
| 1.1 | 1.5 | 2 | 95 | 20,600 | 3.16 |
| 1.4 | 0.5 | 9 | 68 | 13,600 | 1.41 |
| 2.0 | 0.5 | 12 | 68 | 10,600 | 1.55 |
a Estimated by GPC based on PMMA standards; b [AMDV]0 = 0.0454 M; c [BAI]0 = 0.0249 M.
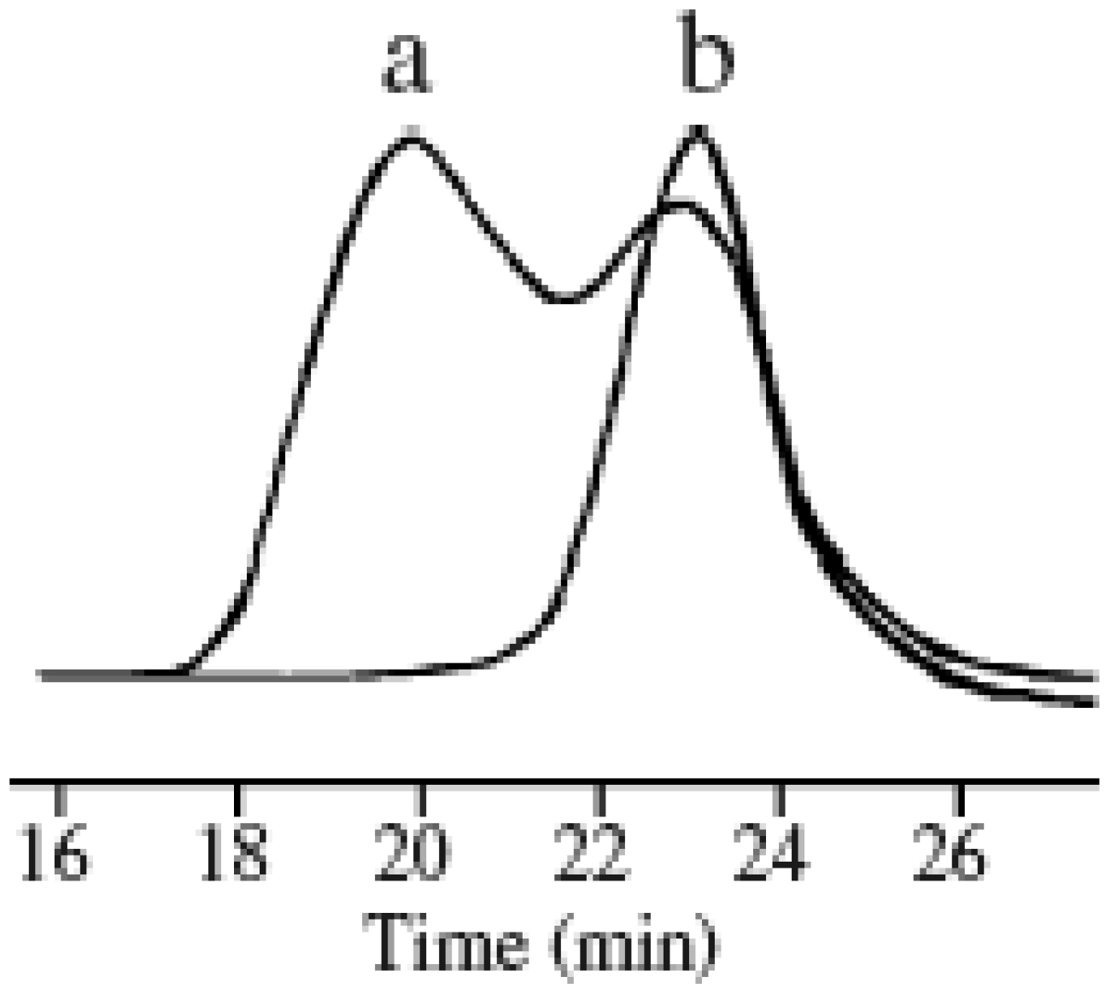
Figure 1 shows the GPC profiles of the PMMA obtained in the presence and absence of MTEMPO. The PMMA in the presence of MTEMPO provided a unimodal GPC with a comparatively narrow MWD, whereas the PMMA prepared in its absence showed a bimodal GPC. BAI also affected the MWD in the presence of MTEMPO. As a result of increasing of the molar ratio of BAI to MTEMPO (BAI/MTEMPO), the polymerization was accelerated, so that BAI served as the accelerator of the polymerization. However, the increase in BAI caused the broadening of the MWD of the resulting polymer. On the other hand, an increase in the molar ratio of MTEMPO to AMDV (MTEMPO/AMDV) decelerated the polymerization.
Figure 1.
GPC profiles of the poly(MMA) (PMMA) obtained by photoradical polymerization in the (a) absence; and (b) presence of 4-methoxy-TEMPO (MTEMPO).
Figure 1.
GPC profiles of the poly(MMA) (PMMA) obtained by photoradical polymerization in the (a) absence; and (b) presence of 4-methoxy-TEMPO (MTEMPO).

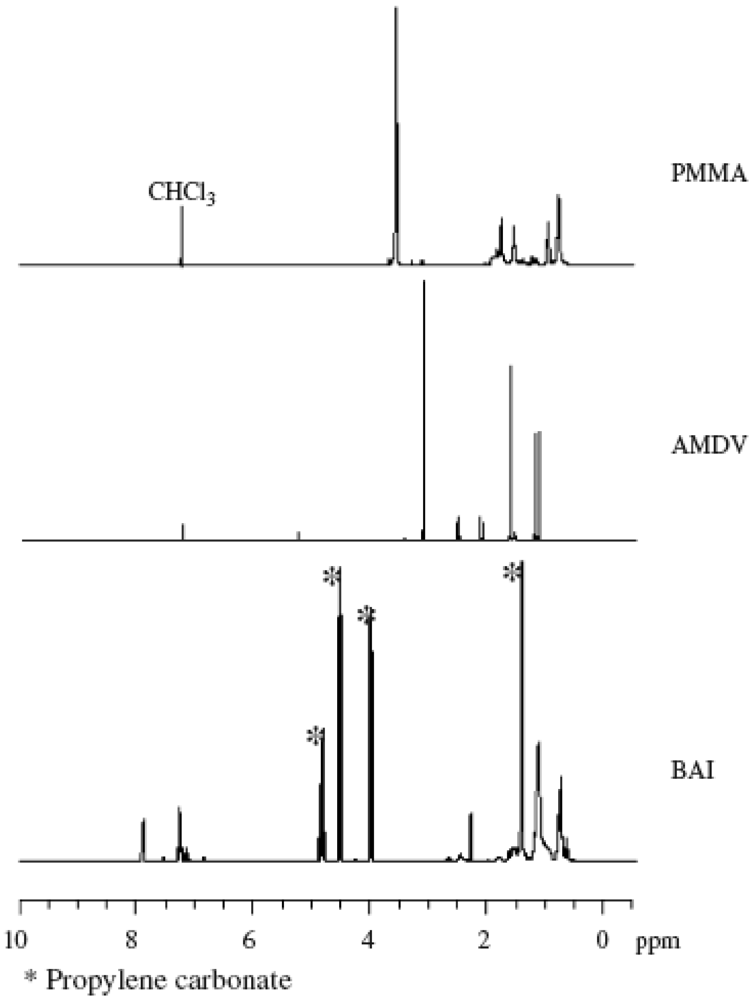
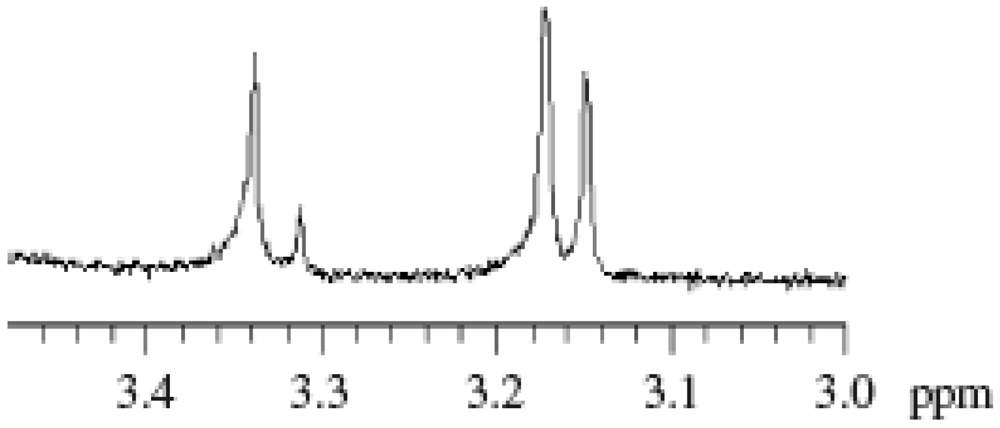
Figure 2 shows the 1H NMR spectra of BAI, AMDV, and the PMMA (Mn = 7,030, Mw/Mn = 1.60, conversion = 23%) prepared by the MTEMPO-mediated photopolymerization at MTEMPO/AMDV = 1.1 and BAI/MTEMPO = 0.5. No observed signals originating from BAI in the spectrum of the PMMA indicates that no fragments of BAI were contained in the polymer structure. It is suggested that the BAI fragments did not combine with the growing polymer chain end or MTEMPO, but just interacted with MTEMPO. The polymerization had no effect on the tacticity of PMMA based on the observation of the signals at 0.84 ppm (syndiotactic), 1.01 ppm (atactic), and 1.25 ppm (isotactic) [66]. It was found that the PMMA involved the 1-cyano-1,3-dimethyl-3-methoxybutyl group (CDM) at the chain head and MTEMPO at the chain end. Signals of the methoxy protons originating from CDM were discerned at 3.13–3.22 ppm, while those from MTEMPO were observed at 3.29–3.39 ppm (Figure 3). The molar ratio of MTEMPO to CDM in the PMMA was estimated to be 0.952 based on their respective methoxy protons, indicating that the PMMA had the CDM and MTEMPO at the 1:1 molar ratio. This good agreement in the molar ratio of MTEMPO/CDM indicates that the growing radical generated by the initiation with the CDM radical was completely captured by MTEMPO (Scheme 1). The molecular weight of the PMMA was estimated by 1H NMR to be Mn = 6500 based on the methoxy protons of CDM and the methyl ester protons of the MMA units. The theoretical molecular weight when the initiator efficiency (IE) determined by UV analysis (IE = 0.378) was taken into account was calculated to be Mn = 6270, being in close agreement with the molecular weight estimated by 1H NMR.
Figure 2.
1H NMR spectra of the PMMA, bis(alkylphenyl)iodonium hexafluorophosphate (BAI), and azobis(4-methoxy-2,4-dimethylvaleronitrile) (AMDV). Solvent: CDCl3.
Figure 2.
1H NMR spectra of the PMMA, bis(alkylphenyl)iodonium hexafluorophosphate (BAI), and azobis(4-methoxy-2,4-dimethylvaleronitrile) (AMDV). Solvent: CDCl3.

Figure 3.
1H NMR spectra of the methoxy protons originating from 1-cyano-1,3-dimethyl-3-methoxybutyl group (CDM) and MTEMPO attached to the PMMA chain ends.
Figure 3.
1H NMR spectra of the methoxy protons originating from 1-cyano-1,3-dimethyl-3-methoxybutyl group (CDM) and MTEMPO attached to the PMMA chain ends.

Scheme 1.
The photoradical polymerization of MMA by AMDV and MTEMPO in the presence of BAI.

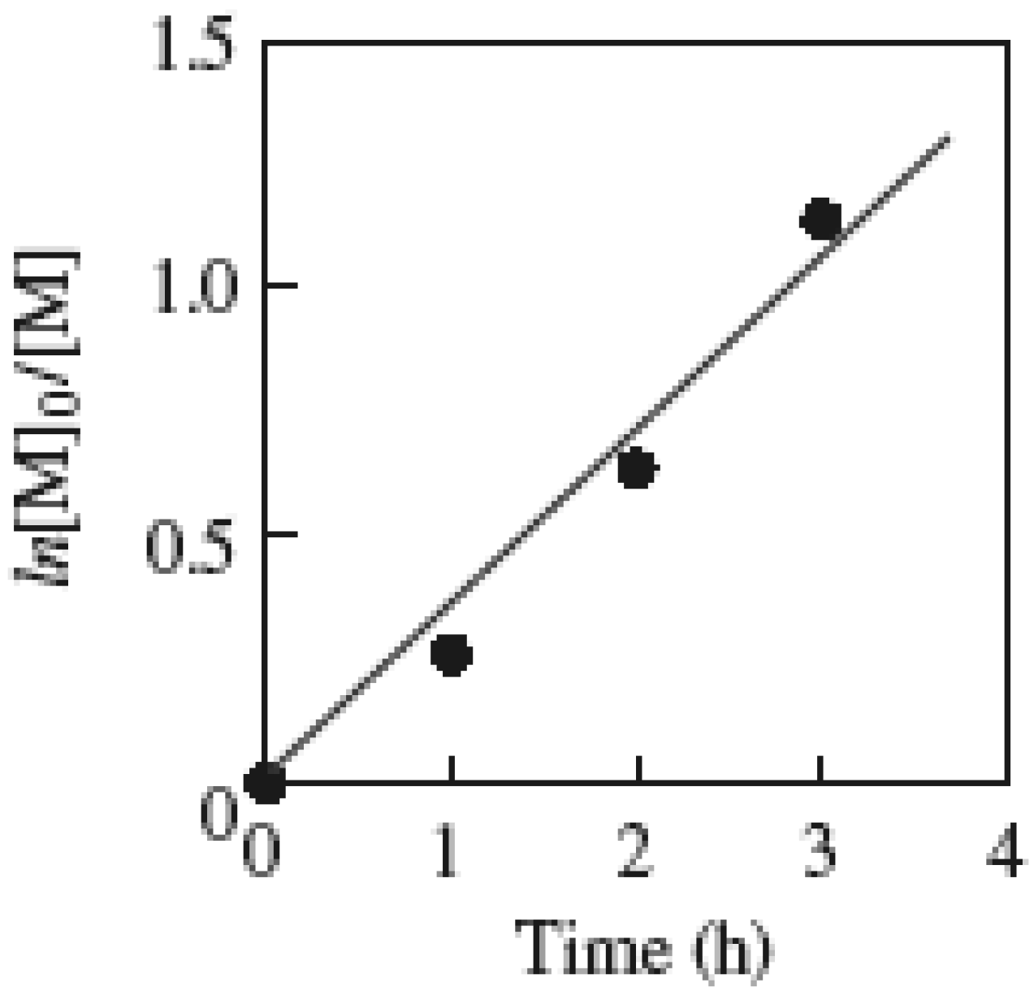
The first order time-conversion plots for the polymerization demonstrated that the number of polymer chains was constant throughout the course of the polymerization. The ln([M]0/[M]) linearly increased over time (Figure 4). As can be seen in Figure 5, the plots of the molecular weight of the resulting PMMA vs. the monomer conversion also linearly increased and the molecular weights were in good agreement with the theoretical molecular weight calculated on the basis on the initiator efficiency (IE = 0.378), indicating the livingness of the polymerization. The MWD of the PMMA was maintained at ca. 1.6 throughout the polymerization. The GPC curve was shifted to the higher side of the molecular weight with an increase in the conversion (Figure 6), also supporting the living mechanism.
Figure 4.
The first order time-conversion plots for the polymerization of MMA. MTEMPO/AMDV = 1.1, BAI/MTEMPO = 0.5. [M] denotes the monomer concentration.
Figure 4.
The first order time-conversion plots for the polymerization of MMA. MTEMPO/AMDV = 1.1, BAI/MTEMPO = 0.5. [M] denotes the monomer concentration.

Figure 5.
The plots of the molecular weight of the PMMA vs. the conversion. MTEMPO/AMDV = 1.1, BAI/MTEMPO = 0.5.
Figure 5.
The plots of the molecular weight of the PMMA vs. the conversion. MTEMPO/AMDV = 1.1, BAI/MTEMPO = 0.5.

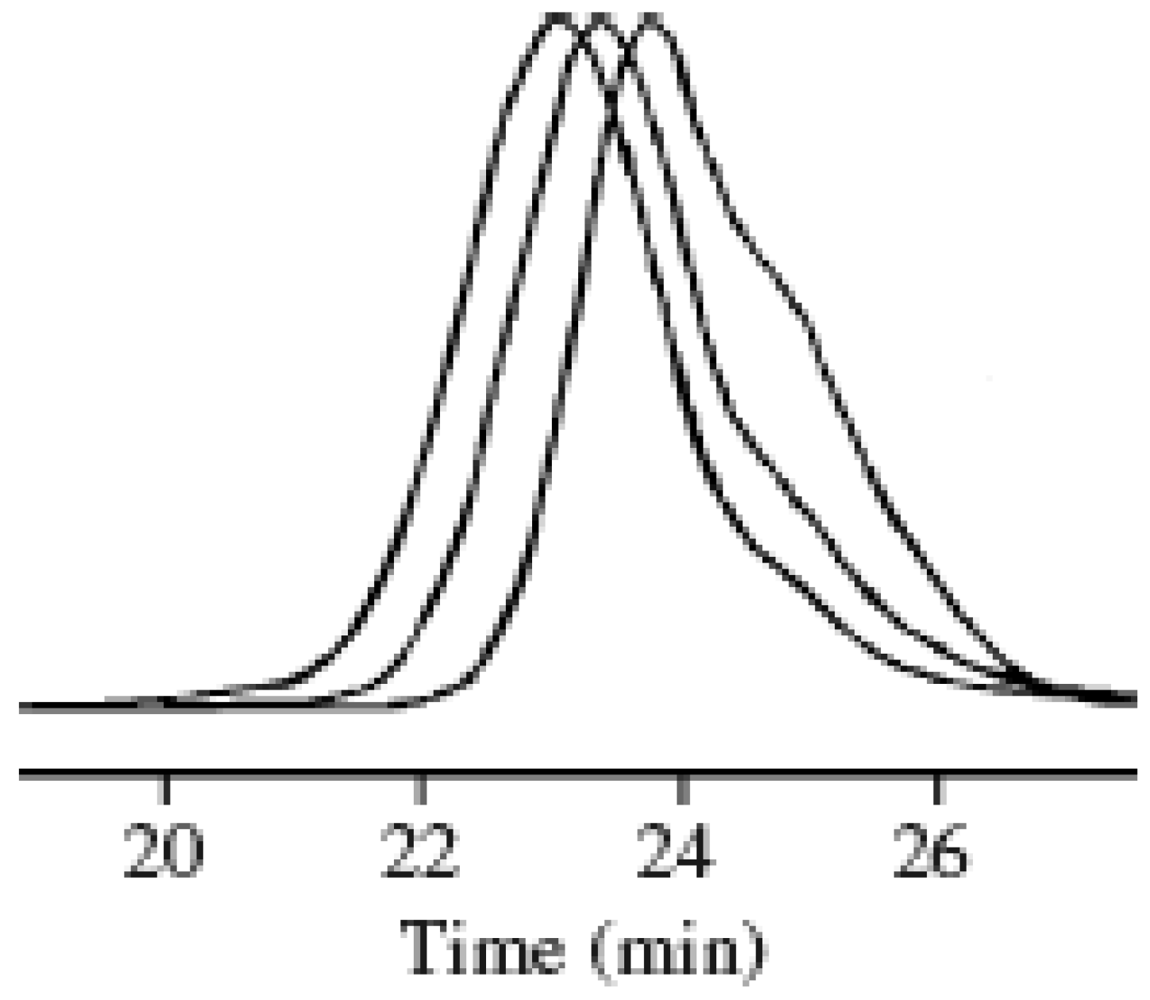
Figure 6.
The variation in the GPC curves vs. the conversion: 23% (1 h), 47% (2 h), and 68% (3 h) from the right.
Figure 6.
The variation in the GPC curves vs. the conversion: 23% (1 h), 47% (2 h), and 68% (3 h) from the right.

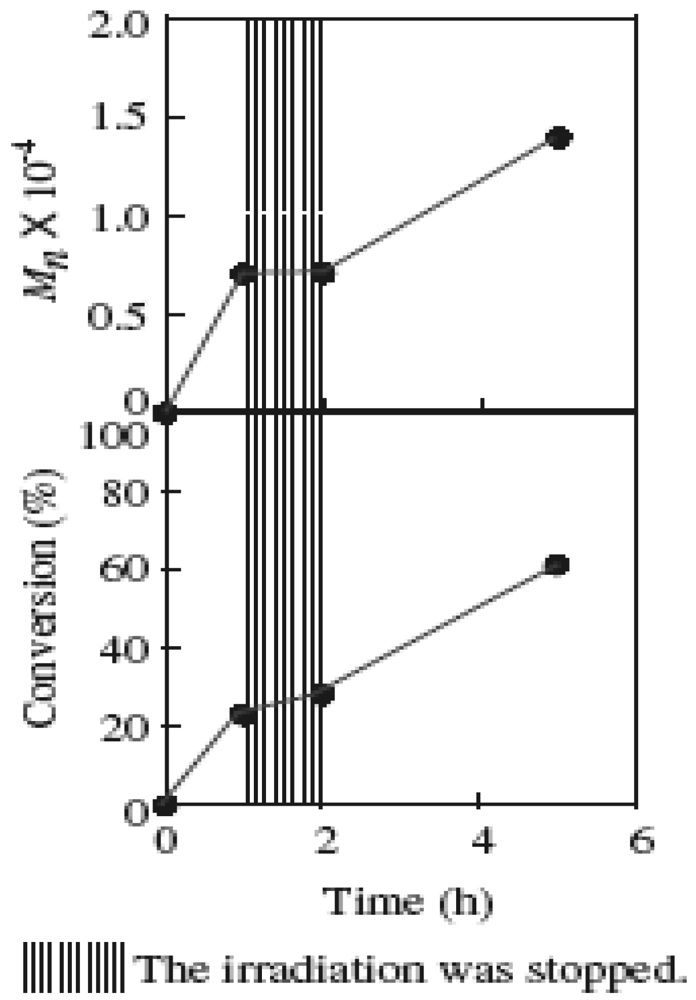
The polymerization was revealed to have a photoswitching ability based on the investigation of the polymerization in the dark. Figure 7 shows the variation in the monomer conversion and molecular weight of PMMA when the irradiation was interrupted for a time during the polymerization. There were negligible changes in the conversion and molecular weight during the dark reaction. However, the conversion and molecular weight increased again by continued irradiation. The progress of the polymerization can be controlled by switching the irradiation on or off.
Figure 7.
The variation in the molecular weight of PMMA and conversion when the light was turned off in the middle of the polymerization. MTEMPO/AMDV = 1.1, BAI/MTEMPO = 0.5.
Figure 7.
The variation in the molecular weight of PMMA and conversion when the light was turned off in the middle of the polymerization. MTEMPO/AMDV = 1.1, BAI/MTEMPO = 0.5.

2.2. Effect of Initiators
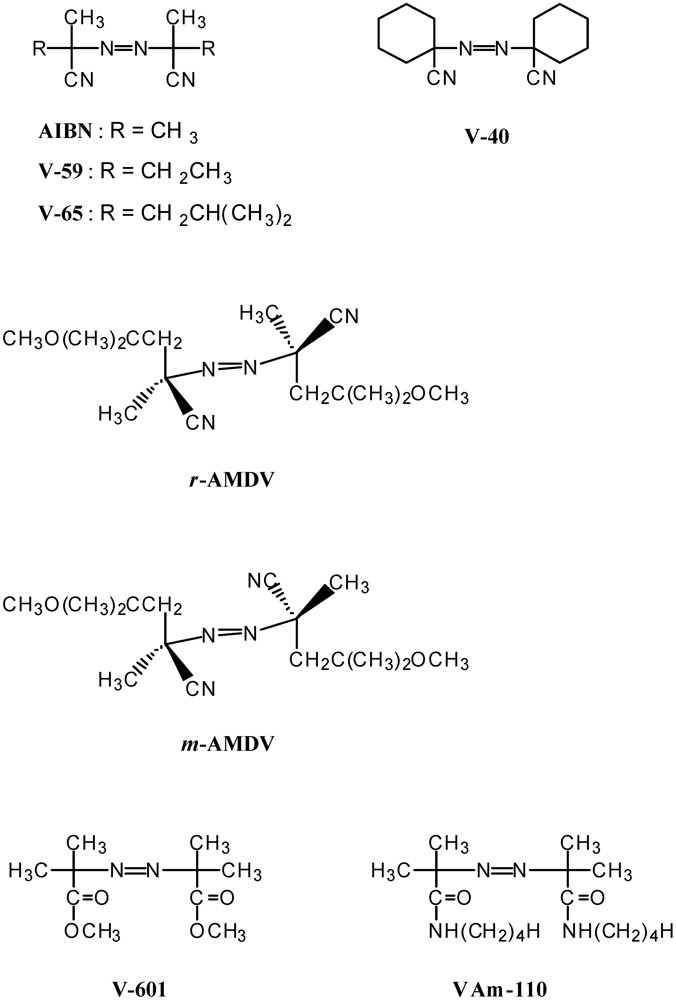
The MTEMPO-mediated controlled/living photoradical polymerization was performed using the azoinitiator in the presence of the BAI photo-acid generator. In order to clarify which characteristic of the azoinitiator dominates the MWD of the PMMA, the MTEMPO-mediated photopolymerization of MMA was performed using eight different kinds of azoinitiators in the presence of BAI [42]. The azoinitiators are shown in Scheme 2 and the results are summarized in Table 2.
| Initiators | MTEMPO/Initiator | Conv. (%) | Mn b | Mw/Mn b | [P] (mM) | IE |
|---|---|---|---|---|---|---|
| AIBN | 1 | 95 | 39,700 | 3.37 | 22.4 | 0.131 |
| 2 | 36 | 15,300 | 1.68 | 22.0 | 0.129 | |
| V-59 | 1 | 97 | 31,600 | 3.37 | 28.8 | 0.173 |
| 2 | 46 | 15,700 | 1.66 | 27.4 | 0.165 | |
| V-65 | 1 | 96 | 18,000 | 2.33 | 50.0 | 0.296 |
| 2 | 62 | 9,410 | 1.53 | 61.7 | 0.365 | |
| V-40 | 1 | 98 | 38,200 | 3.24 | 24.0 | 0.140 |
| 2 | 67 | 26,300 | 1.65 | 23.9 | 0.139 | |
| r-AMDV | 1 | 60 | 7,640 | 1.52 | 73.5 | 0.436 |
| 2 | 37 | 3,530 | 1.28 | 98.2 | 0.582 | |
| m-AMDV | 1 | 68 | 10,100 | 1.50 | 63.3 | 0.375 |
| 2 | 40 | 4,580 | 1.34 | 81.8 | 0.485 | |
| V-601 | 1 | 84 | 26,300 | 1.99 | 29.9 | 0.181 |
| 2 | 35 | 10,700 | 1.68 | 30.6 | 0.185 | |
| VAm-110 | 1 | 25 | 16,600 | 1.53 | 14.1 | 0.0847 |
a [Initiator]0 = 84.3 mM, BAI/MTEMPO = 0.52. Irradiated for 3 h; b Estimated by GPC based on the PMMA standard.
Scheme 2.
Azoinitiators used for the MTEMPO-mediated photopolymerization.

The concentration of the growing polymer chain ([P]) was estimated on the basis of the conversion and the molecular weight of the resulting PMMA. Based on the [P], the IE was also determined. Racemic-AMDV (r-AMDV), meso-AMDV (m-AMDV), V-601, and VAm-110 provided much narrower MWDs with moderate conversions, although VAm-110 produced only a 25% conversion. In particular, r-AMDV and m-AMDV produced the PMMA with the narrowest MWD. r-AMDV had a higher [P] and IE than m-AMDV, being in good agreement with the fact that r-AMDV has a higher reactivity than m-AMDV [67]. As a result of doubling MTEMPO to the initiator, all the initiators produced PMMA with a MWD below 1.7, and the resulting PMMAs showed sharp GPC curves. There were negligible differences in the [P] and IE values between the MTEMPO/initiator ratios of unity and 2.
The characteristics of the UV absorption spectra of the initiators, coupled with their 10-h-half-life temperatures are listed in Table 3. Initiators with higher ε values tended to more strictly control the molecular weight and provide a higher IE. The reason that r-AMDV, m-AMDV, V-601, and VAm-110 provided much narrower MWDs than the other initiators may partly be due to the fact that these initiators exhibit the n → σ* transition in addition to the high ε. It was found that the half-lives of the initiators had little effect on the molecular weight control. The [P] and IE were also independent of the half-life temperature.
| Initiators | λmax (nm) | Absorbance | ε | T1/2 (10 h) |
|---|---|---|---|---|
| AIBN | 345 | 0.176 | 12.3 | 65 |
| V-59 | 348 | 0.226 | 15.8 | 67 |
| V-65 | 348 | 0.291 | 20.4 | 51 |
| V-40 | 350 | 0.236 | 16.5 | 88 |
| r-AMDV | 348
253 | 0.404
0.050 | 28.3
3.50 | 30 |
| m-AMDV | 341
253 | 0.245
0.078 | 17.2
5.47 | 30 |
| V-601 | 363
253 | 0.273
0.164 | 19.1
11.5 | 66 |
| VAm-110 | 376
258 | 0.455
2.392 | 31.9
167.7 | 110b |
2.3. Mechanisms
In order to clarify the mechanism of the MTEMPO-mediated photopolymerization, the polymerization was performed using 1-(cyano-1-methylethoxy)-4-methoxy-2,2,6,6-tetramethylpiperidine (CMTMP), the alkoxyamine adduct as the initiator [39]. CMTMP was prepared by the reaction of MTEMPO and AIBN in methanol. CMTMP had a slight absorption at 257 nm as λmax (ε = 3.43) and almost no overlap with the illumination of the mercury lamp, implying a low photo activity compared to AIBN. The photopolymerization of MMA was performed using CMTMP as the initiator, at room temperature. The results are shown in Table 4.
Table 4.
Photo-radical polymerization of MMA by 1-(cyano-1-methylethoxy)-4-methoxy-2,2,6,6-tetramethylpiperidine (CMTMP) in the presence of BAI.
| [CMTMP]0 (mM) | [BAI]0 (mM) | BAI/CMTMP | Time (h) | Conv. (%) | Mn a | Mw/Mn a | [P] (mM) | I.E. |
|---|---|---|---|---|---|---|---|---|
| 47.2 | 0 | 0 | 24 | 2 | − | − | − | − |
| 47.2 | 5.24 | 0.11 | 5 | 32 | 27,300 | 1.68 | 11.0 | 0.23 |
| 47.2 | 11.8 | 0.25 | 5 | 51 | 29,300 | 1.66 | 13.7 | 0.29 |
| 47.2 | 24.9 | 0.53 | 5 | 50 | 22,600 | 1.69 | 17.4 | 0.37 |
| 47.2 | 47.2 | 1.0 | 5 | 55 | 16,800 | 1.78 | 30.6 | 0.65 |
| 47.2 | 47.2 | 1.0 | 10 | 94 | 25,700 | 2.30 | 34.2 | 0.73 |
| 31.5 | 31.5 | 1.0 | 11 | 82 | 33,400 | 1.88 | 23.0 | 0.73 |
| 94.4 | 94.4 | 1.0 | 3.5 | 92 | 17,300 | 2.67 | 49.8 | 0.53 |
a Estimated by GPC based on the PMMA.
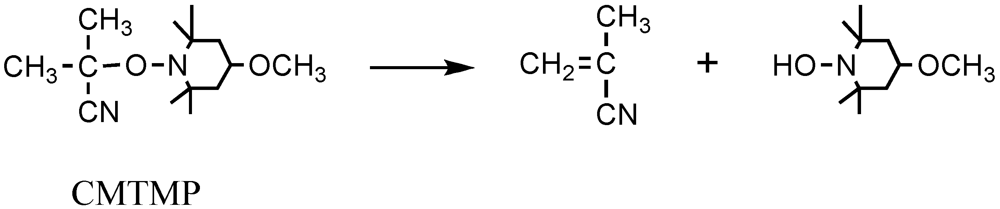
The polymerization only slightly occurred using CMTMP itself; however, it smoothly proceeded in the presence of BAI. The monomer conversion increased with the increasing molar ratio of BAI to CMTMP (BAI/CMTMP). On the other hand, the molecular weight of the resulting polymer decreased with the increasing BAI/CMTMP ratio, suggesting that the BAI/CMTMP ratio affected the initiator efficiency. The IE of CMTMP was determined on the basis of the concentration of the [P] calculated by the monomer conversion and the molecular weight of the resulting polymer. The IE increased with the increase in the BAI/CMTMP ratio, although it was not quantitative even at 1.0 of BAI/CMTMP. This nonquantitative IE was probably caused by the cage effect and the deactivation of CMTMP by the disproportionation in the hydroxylamine and methyl methacrylonitrile (Scheme 3). In fact, the formation of a significant amount of hydroxylamine lowered the yield of CMTMP in its preparation by the reaction of MTEMPO and AIBN. The molecular weight distributions of the resulting polymers were somewhat broadened as compared to those of the polymers obtained by the photopolymerization by AIBN and 4-methoxy-TEMPO in the presence of BAI [36,67]. This broad molecular weight distribution was probably due to the slow initiation by CMTMP having a low photo activity.
Scheme 3.
The disporportionation of CMTMP.

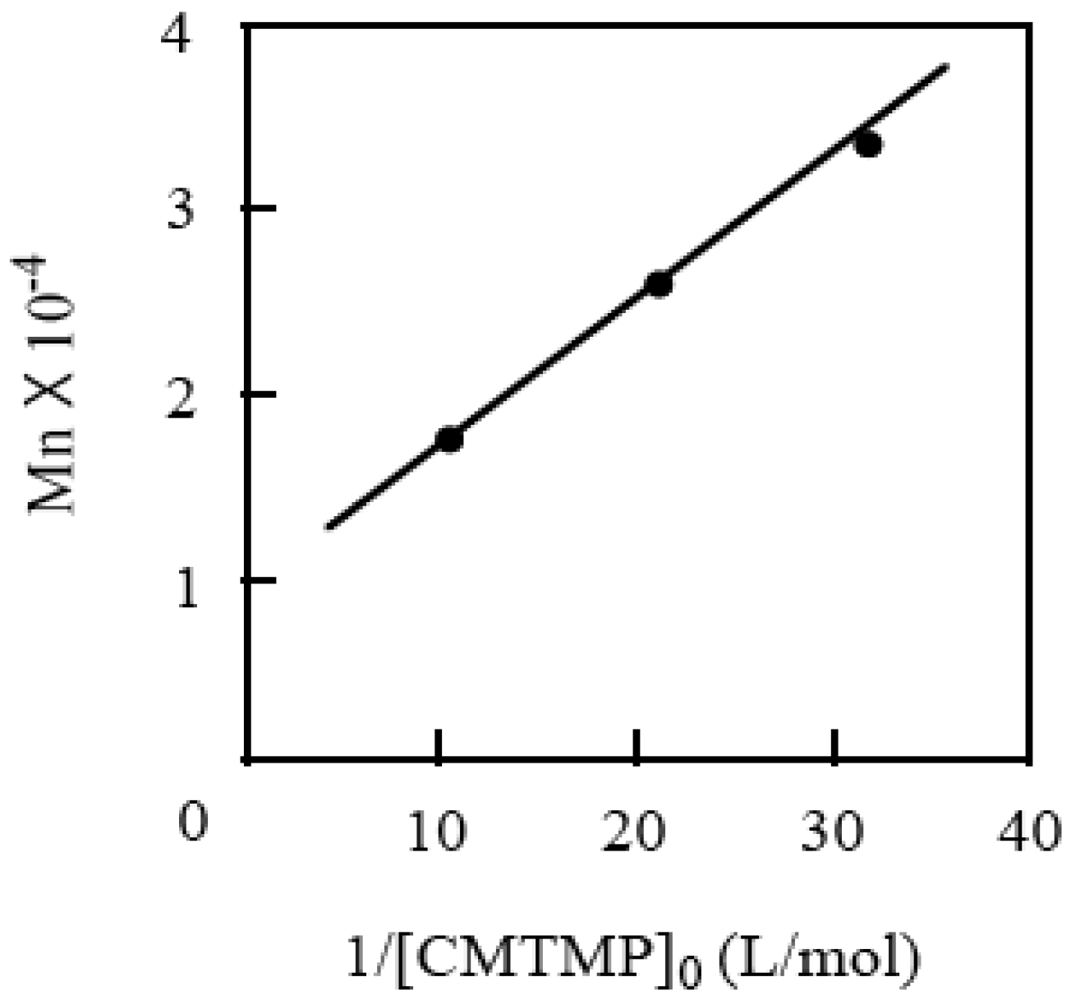
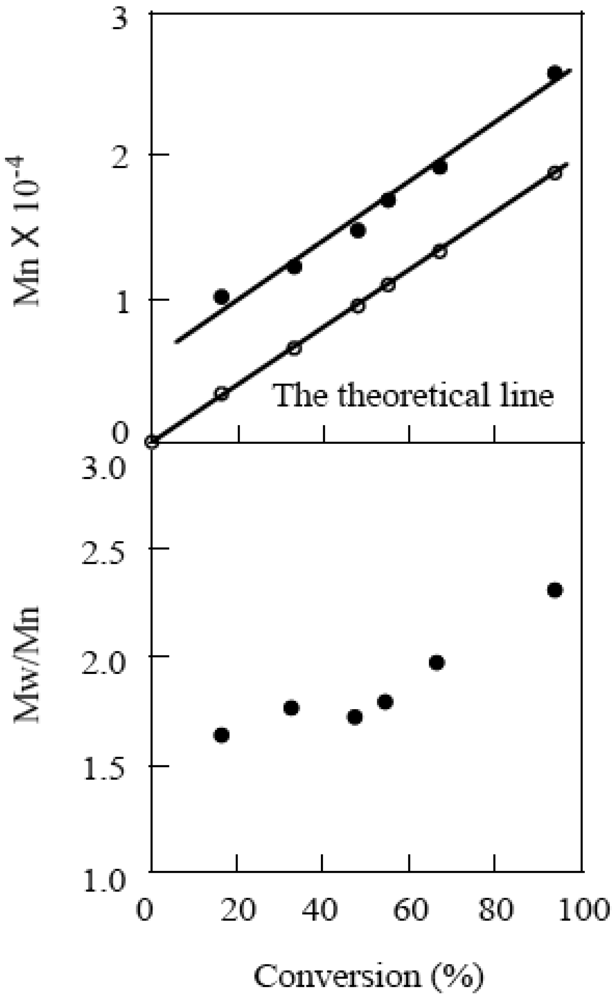
The polymerization was carried out for the different initial concentrations of CMTMP ([CMTMP]0) at a constant BAI/CMTMP ratio, resulting in the molecular weight of the polymer decreasing with an increase in [CMTMP]0. The plots of the molecular weight vs. the reciprocal of [CMTMP]0 provided a linear correlation, implying the living nature of the polymerization (Figure 8). Figure 9 shows the GPC profiles of the polymers produced for each conversion. The curves shifted to the higher molecular weight side with an increase in the conversion. The plots of the molecular weight of the polymer vs. the conversion are shown in Figure 10. The molecular weight linearly increased with an increase in the conversion; however, the line did not pass through the origin and was almost parallel to the theoretical line. This phenomenon can be accounted for by the fact that a small amount of a polymer with a lower molecular weight was produced during the very early stage before the system reached the steady state of the living polymerization.
Figure 8.
The plots of the molecular weight of the resulting polymer vs. the reciprocal of [CMTMP]0. BAI/CMTMP = 1.0.
Figure 8.
The plots of the molecular weight of the resulting polymer vs. the reciprocal of [CMTMP]0. BAI/CMTMP = 1.0.

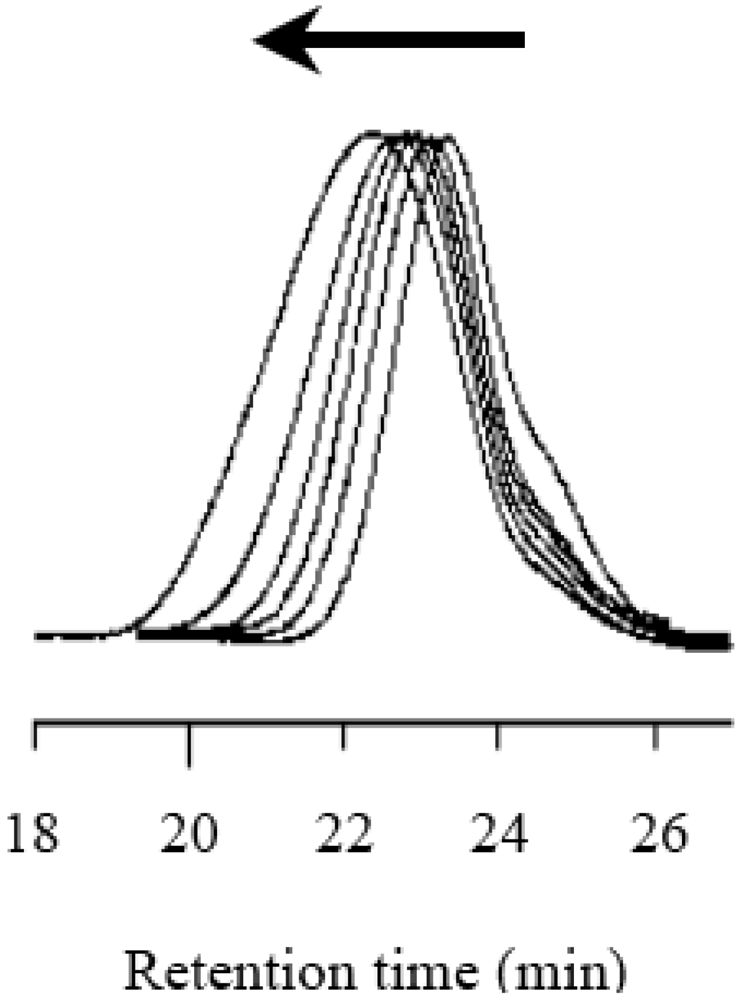
Figure 9.
The GPC profiles of the resulting polymer obtained at each conversion: 17% (1 h), 33% (2 h), 48% (3 h), 55% (5 h), 67% (7 h), and 94% (10 h) from the right.
Figure 9.
The GPC profiles of the resulting polymer obtained at each conversion: 17% (1 h), 33% (2 h), 48% (3 h), 55% (5 h), 67% (7 h), and 94% (10 h) from the right.

Figure 10.
The plots of the molecular weight vs. the conversion. [CMTMP]0 = 47.2 mM, BAI/CMTMP = 1.0.
Figure 10.
The plots of the molecular weight vs. the conversion. [CMTMP]0 = 47.2 mM, BAI/CMTMP = 1.0.

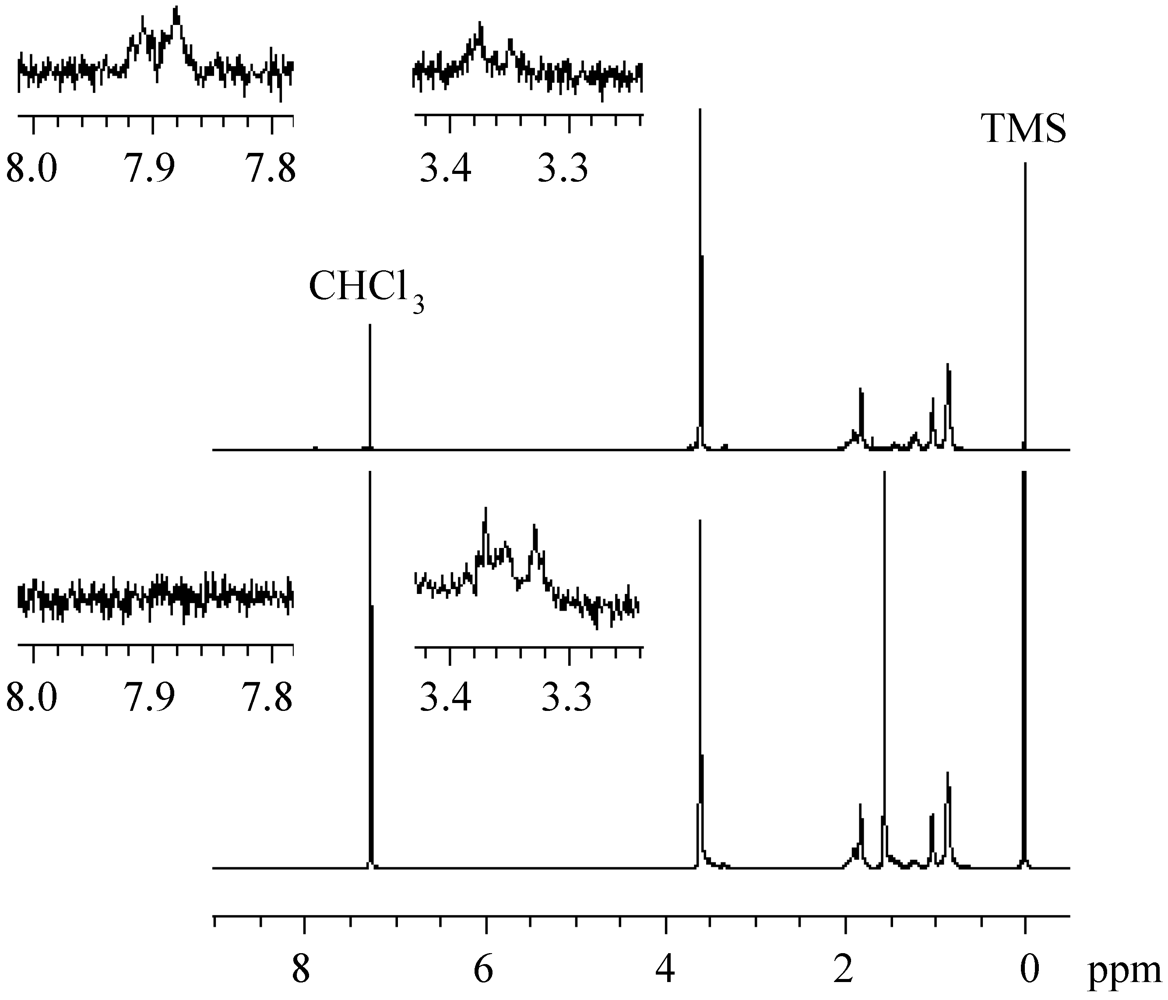
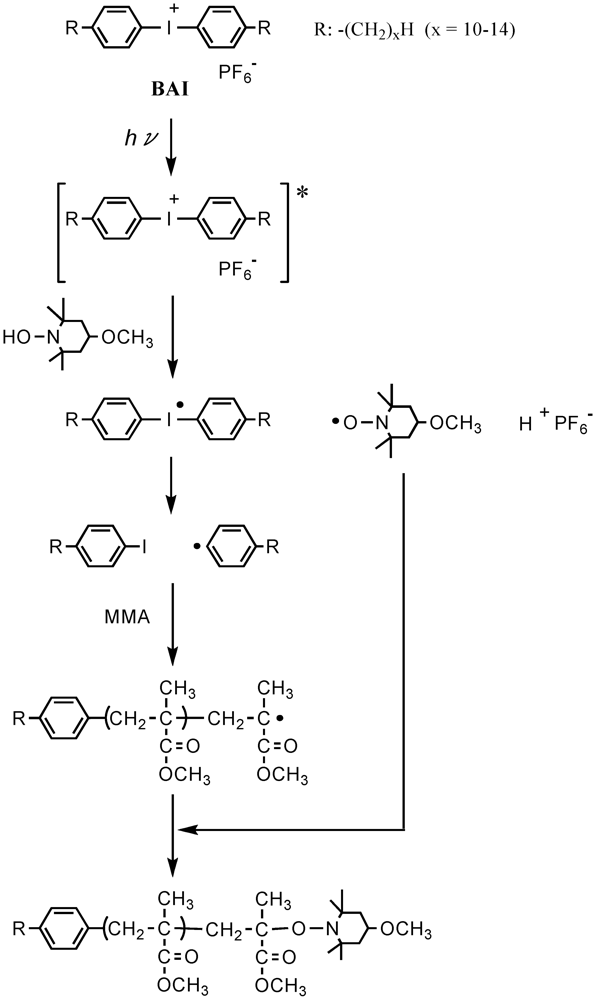
The 1H NMR analysis revealed that the formation of the polymer, with a low molecular weight during the very early stage before the steady state, was caused by the phenyl radicals generated from CMTMP. Figure 11 shows the 1H NMR spectra of the polymers obtained by the MTEMPO-mediated polymerization initiated by CMTMP and AIBN/MTEMPO in the presence of BAI. Whereas no BAI fragments were inserted into the polymer structure prepared from AIBN/MTEMPO, the polymer obtained from CMTMP contained the BAI fragment in its structure, because the aromatic proton signals originating from BAI were discerned at 7.21–7.40 and 7.85–7.92 ppm. The formation of the phenyl radical is supported by the decomposition mechanism of the photo-acid generator [68,69,70]. The initiation by the phenyl radical was also confirmed by the experiment in which the uncontrolled photopolymerization of MMA performed in the presence of BAI without CMTMP produced a polymer with Mn = 62,800 and Mw/Mn = 3.77 at 84% conversion within only 2 h. During the MTEMPO-mediated polymerization, the propagating chains formed by the phenyl radical probably participated in the controlled polymerization by MTEMPO by obtaining the counter radical from the hydroxylamine (Scheme 4) because the GPC curves were shifted to the higher molecular weight side without deactivation with the increasing conversion. In the MTEMPO/AIBN system, the propagation proceeded by repeating the dissociation and recombination of the C-ON bond, as well as the thermal polymerization, since no BAI fragments were contained in the polymer structure. However, electron transfer is expected to occur between MTEMPO and BAI in the excited state during the dissociation and recombination when it is taken into consideration that continued polymerization is difficult in the absence of BAI and that the excited diphenyliodonium salt easily receives an electron [68,69,70]. Consequently, the mechanism of the polymerization was thus proposed (Scheme 5).
Figure 11.
The 1H NMR spectra of the polymers obtained by the MTEMPO-mediated polymerizations initiated by CMTMP (upper, Mn = 10,195 and Mw/Mn = 1.63) and AIBN (lower, Mn = 9,140 and Mw/Mn = 1.57) in the presence of BAI. Solvent: CDCl3.
Figure 11.
The 1H NMR spectra of the polymers obtained by the MTEMPO-mediated polymerizations initiated by CMTMP (upper, Mn = 10,195 and Mw/Mn = 1.63) and AIBN (lower, Mn = 9,140 and Mw/Mn = 1.57) in the presence of BAI. Solvent: CDCl3.

Scheme 4.
The initiation by the phenyl radical.

Scheme 5.
The propagation mechanism.

2.4. Effects of Photo-Acid Generators
2.4.1. Triarylsulfonium Salts
Triarylsulfonium salts are known as efficient photoinitiators for cationic polymerization as are diaryliodonium salts. While triarylsulfonium salts show a lower photosensitivity than the diaryliodonium salts [68,71], the sulfonium salts have advantages over the diaryliodonium salts in the extraordinary thermal stability [72] and easier preparation [73]. In order to precisely control the molecular weight of polymers, the MTEMPO-mediated photopolymerization was performed using the triarylsulfonium salt instead of the diaryliodonium salt.
The polymerization of MMA was performed at room temperature using (4-tert-butylphenyl)diphenylsulfonium triflate (tBuS) as the photo-acid generator by the r-AMDV initiator and the MTEMPO mediator [41]. The results are shown in Table 5. While the polymerization slowly occurred in the absence of tBuS, the polymerization smoothly proceeded in its presence. The tBuS/MTEMPO molar ratio increased [P], resulting in a decrease in the molecular weight of the resulting polymer. There was a tendency that the polymerization was accelerated as the tBuS/MTEMPO ratio increased. The polymerization rate was also dependent on MTEMPO because the large excess of MTEMPO to r-AMDV retarded the polymerization even in the presence of tBuS. The MWD was significantly broadened at a MTEMPO/r-AMDV less than 1, suggesting less control of the polymerization. Furthermore, no polymerization occurred in the absence of r-AMDV, although tBuS solely initiated the polymerization to produce a polymer with a very broad MWD. It is implied that MTEMPO controlled the polymerization.
| [MTEMPO]0 (mM) | [tBuS] 0 (mM) | MTEMPO/AMDV | tBuS/MTEMPO | Time (h) | Conv. (%) | Mn b | Mw/Mnb | [P] (mM) |
|---|---|---|---|---|---|---|---|---|
| 48.3 | − | 1.06 | − | 31 | 40 | 11,600 | 1.47 | 32.3 |
| 48.3 | 12.8 | 1.06 | 0.265 | 9 | 63 | 13,200 | 1.46 | 44.8 |
| 48.3 | 23.5 | 1.06 | 0.487 | 9 | 63 | 11,100 | 1.46 | 53.0 |
| 48.3 | 34.1 | 1.06 | 0.706 | 9 | 64 | 10,200 | 1.44 | 59.0 |
| 48.3 | 47.0 | 1.06 | 0.973 | 6 | 77 | 15,700 | 1.58 | 46.0 |
| 48.3 | 70.4 | 1.06 | 1.46 | 6 | 71 | 11,100 | 1.45 | 60.1 |
| 80.5 | 66.2 | 1.77 | 0.822 | 23 | 74 | 9,350 | 1.51 | 74.1 |
| 91.3 | 91.8 | 2.01 | 1.01 | 23 | 82 | 7,790 | 1.70 | 98.5 |
| 91.3 | 47.0 | 2.01 | 0.515 | 14 | 26 | 4,670 | 1.36 | 52.1 |
| 21.5 | 10.7 | 0.473 | 0.498 | 5 | 71 | 18,300 | 1.76 | 36.4 |
| 48.3 | 25.6 | − c | 0.530 | 6 | 0 | − | − | − |
| − | 94.4 | − c | − | 3 | 73 | 417,000 | 19.6 | 1.64 |
a [AMDV]0 = 45.4 mM; b Estimated by GPC based on the PMMA standard; c Without AMDV.
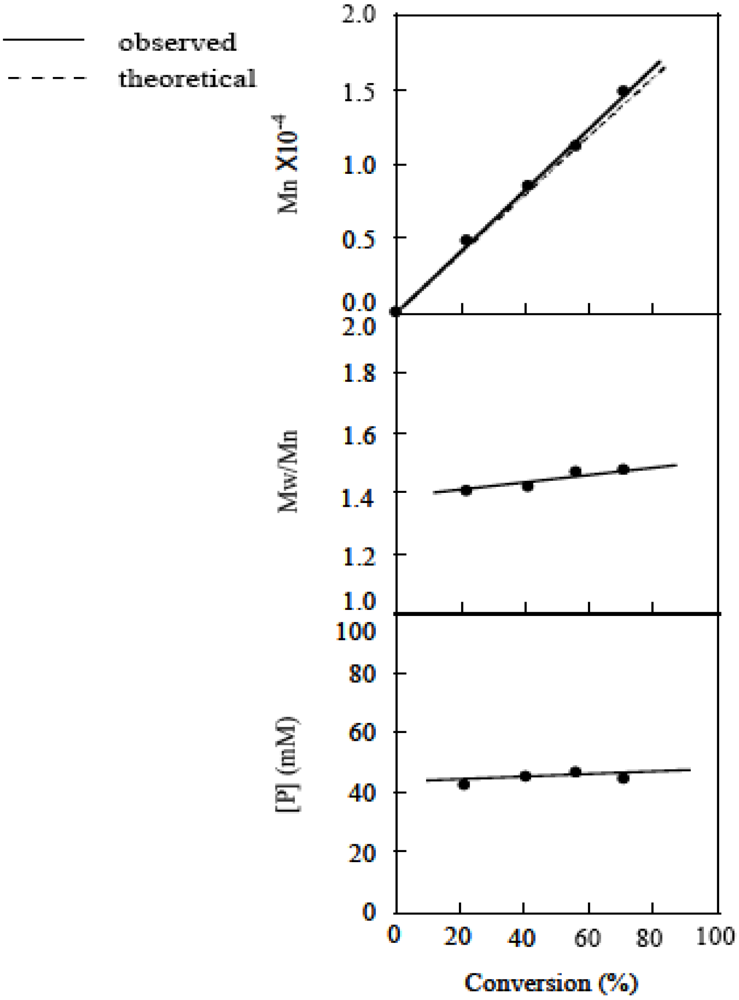
The livingness of the polymerization was evaluated at the MTEMPO/r-AMDV of 1.06 and tBuS/MTEMPO of 0.487. The first order time-conversion plots for the polymerization are shown in Figure 12. The ln([M]0/[M]t) almost linearly increased with time, suggesting that the number of polymer chains was constant throughout the course of the polymerization. Figure 13 shows the plots of the molecular weight, MWD, and [P] vs. the conversion. The molecular weight linearly increased with the increasing conversion and was in a good agreement with the theoretical values. The MWD remained constant at around 1.45. The [P] also remained at ca. 45 mM, indicating the constant number of polymer chains. The GPC profiles of the resulting polymer for each conversion are shown in Figure 14. The curves were shifted to the higher molecular weight side with the increasing conversion. Based on the GPC analysis, coupled with the linear correlations of the first order time-conversion and conversion-molecular weight plots, it can be deduced that the polymerization proceeded by a living mechanism and that tBuS more effectively controlled the molecular weight than BAI.
Figure 12.
The first order time-conversion plots for the polymerization of MMA. MTEMPO/r-AMDV = 1.06, tBuS/MTEMPO = 0.487.
Figure 12.
The first order time-conversion plots for the polymerization of MMA. MTEMPO/r-AMDV = 1.06, tBuS/MTEMPO = 0.487.

Figure 13.
The plots of the molecular weight, molecular weight distribution, and [P] vs. the conversion. MTEMPO/r-AMDV = 1.06, tBuS/MTEMPO = 0.487.
Figure 13.
The plots of the molecular weight, molecular weight distribution, and [P] vs. the conversion. MTEMPO/r-AMDV = 1.06, tBuS/MTEMPO = 0.487.

Figure 14.
GPC profiles of the PMMA obtained at each conversion: 22% (2.25 h), 41% (4.00 h), 56% (4.92 h), and 71% (9.50 h) from the right.
Figure 14.
GPC profiles of the PMMA obtained at each conversion: 22% (2.25 h), 41% (4.00 h), 56% (4.92 h), and 71% (9.50 h) from the right.

In order to explore the influence of the substituents attached to the sulfonium salt on the polymerization, the polymerization was performed using 13 different sulfonium salts [43]. These results are shown in Table 6. The solubility of the sulfonium salt in MMA had no influence on the MWD, because the heterogeneous solution including the insoluble sulfonium salt became homogeneous within 1 h of the irradiation. There was a negligible difference in [P] and IE, independent of the substituents. The substituents produced a significant difference in the MWD. The sulfonium salts with the alkyl, methoxy, phenoxy, methylthio, and tert-butoxycarbonylmethoxy groups had no effect on the molecular weight control because the MWDs of the polymers obtained by these sulfonium salts were close to that by the triphenylsulfonium triflate (Run 1). Halogens with the exception of the iodide also had a negligible effect on it. These polymers had similar GPC profiles. On the other hand, the iodide, phenylthio, and naphthyl groups on the sulfonium salts caused broad MWDs with the bimodal GPCs including a peak on the higher molecular weight side. These functional groups should have competitively participated in the electron transfer between the sulfonium salt and MTEMPO during the propagation, resulting in the formation of polymers with uncontrolled high molecular weights.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run no. | R1 | R2 | Solubility b | Conv. (%) | Mn c | Mw/Mn c | [P] (mM) | IE |
|---|---|---|---|---|---|---|---|---|
| 1 |  | I | 63 | 12,200 | 1.42 | 48.2 | 0.531 | |
| 2 | S | 52 | 10,200 | 1.39 | 47.8 | 0.527 | ||
| 3 | S | 58 | 11,000 | 1.43 | 49.5 | 0.545 | ||
| 4 | I | 62 | 11,000 | 1.50 | 52.9 | 0.582 | ||
| 5 | S | 46 | 9,810 | 1.45 | 43.9 | 0.483 | ||
| 6 | S | 80 | 427,000 d 14,000 | 3.64 1.70 | 40.1 | 0.442 | ||
| 7 | S | 66 | 12,200 | 1.45 | 51.0 | 0.562 | ||
| 8 | S | 58 | 12,600 | 1.44 | 43.0 | 0.474 | ||
| 9 | S | 64 | 11,400 | 1.45 | 52.3 | 0.577 | ||
| 10 | I | 53 | 11,000 | 1.44 | 45.0 | 0.496 | ||
| 11 | S | 70 | 12,100 | 1.49 | 54.0 | 0.594 | ||
| 12 | I | 62 | 10,100 | 3.14 | 57.5 | 0.634 | ||
| 13 | I | 73 | 13,200 | 2.40 | 51.9 | 0.571 | ||
a MTEMPO/r-AMDV = 1.1, Sulfonium salt/MTEMPO = 0.5. Irradiated for 6 h; b Solubility of the sulfonium salts. S: Soluble, I: Insoluble; c Estimated by GPC based on the PMMA standard; d Bimodal GPC. The area ratio: Mn(427,000/14,000) = 0.26/0.74.
2.4.2. An Iron-Arene Complex
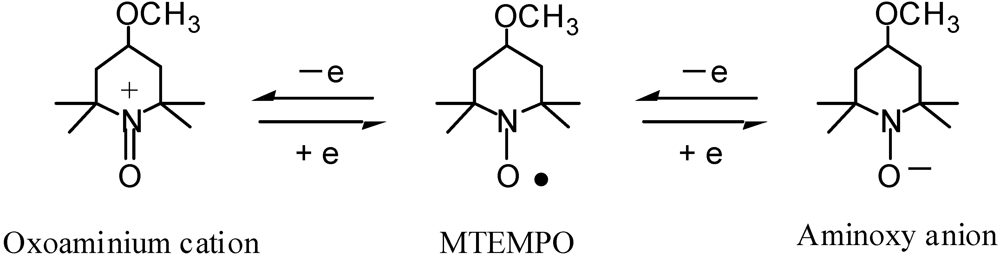
Iron-arene complexes act as photoinitiators for cationic polymerizations as well as diaryliodonium salts and triarylsulfonium salts. For the MTEMPO-mediated polymerization, the diaryliodonium salts and triarylsulfonium salts did not directly engage in controlling the growing polymer radicals, but enhanced the polymerization rate by interacting with MTEMPO accompanied by the growing polymer radical. It was considered that the interaction was attributed to the electron transfer between these onium salts and MTEMPO in their excited states. This electron transfer mechanism was based on the fact that MTEMPO forms the redox systems in which MTEMPO is converted into the oxoaminium cation by the one-electron oxidation, while it is converted into the aminoxy anion by the one-electron reduction (Scheme 6) [74].
Scheme 6.
The redox systems of MTEMPO.

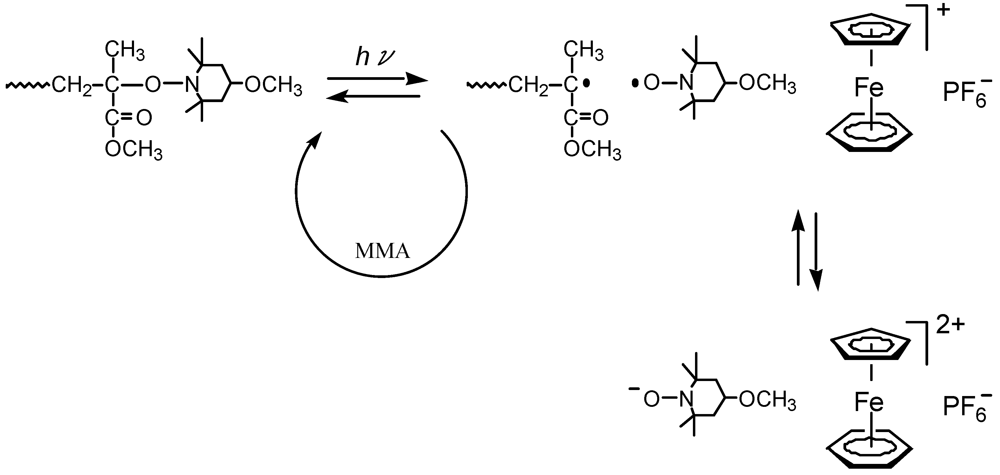
The iron-arene complexes are more stable toward the electron transfer due to the redox formation of the iron than the iodonium salts and sulfonium salts [75]. The MTEMPO-mediated photopolymerization of MMA was performed using (η6-benzene)(η5-cyclopentadienyl)FeII hexafluorophosphate (BzCpFeII) as the accelerator (Table 7) [46]. The conversion reached 73% at 3 h, however, it did not increase over this time. It is likely that the conversion reached its plateau at 3 h. It was found that an increase in the amount of BzCpFeII retarded the polymerization rate. BzCpFeII showed the opposite tendency against BAI and tBuS that enhanced the polymerization rate [36,37,41]. It is suggested that BzCpFeII is different from the diaryliodonium salts and triarylsulfonium salts regarding the molecular weight control mechanism. For the BzCpFeII redox system, the reduction redox system between MTEMPO and the aminoxy anion is involved in the electron transfer interaction, while the oxidation redox system between MTEMPO and the oxoaminium cation is used for the onium salt redox systems (Scheme 7). The polymerization using BzCpFeII also proceeded by a living mechanism, since the first-order time-conversion plots and conversion-molecular weight plots showed linear increases. The MWDs retained Mw/Mn = 1.4–1.5 during the polymerization.
| r-AMDV (μmol) | MTEMPO (μmol) | BzCpFeII (μmol) | Time (h) | Conv. (%) | Mn a | Mw/Mn a |
|---|---|---|---|---|---|---|
| 45.4 | 48.3 | 48.3 | 3 | 73 | 17,500 | 1.54 |
| 45.4 | 48.3 | 48.3 | 4 | 66 | 16,700 | 1.58 |
| 45.4 | 48.3 | 96.5 | 7 | 46 | 11,500 | 1.47 |
| − | − | 48.3 | 3 | 6 | 90,500 | 3.09 |
| − | 48.3 | 48.3 | 3 | 20 | 48,300 | 2.45 |
| 45.4 | − | 48.3 | 1 | 80 | 15,100 | 7.21 |
a Estimated by GPC based on PMMA standards.
Scheme 7.
The electron transfer between MTEMPO and BzCpFeII.

2.5. Stability of the Growing Polymer Chain End
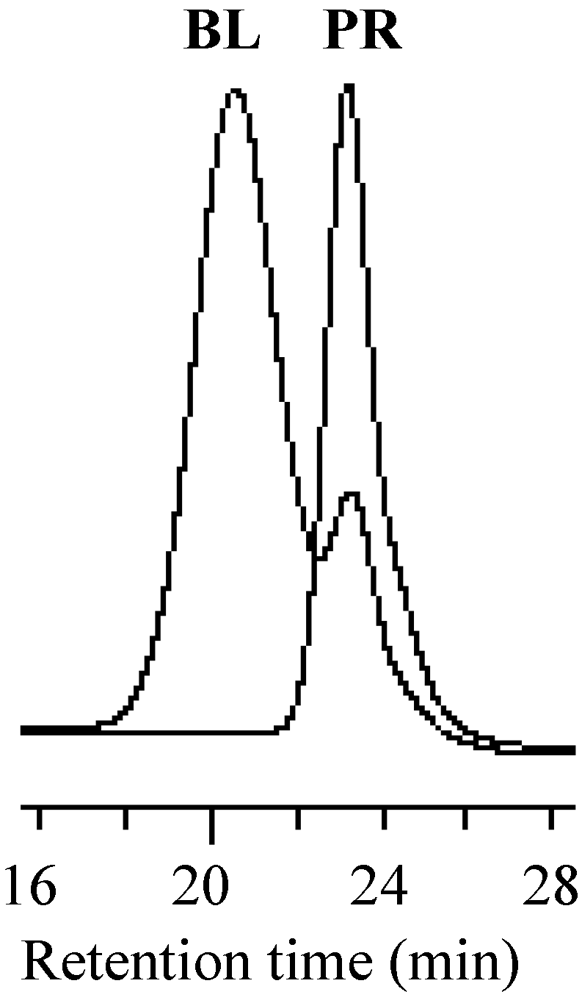
The thermal NMP is often deactivated through the hydrogen elimination from the growing polymer radical by the nitroxide at the end stage of the polymerization [64,76,77]. The stability of the growing polymer chain ends for the MTEMPO-mediated photopolymerization of MMA was investigated by the block copolymerization with isopropyl methacrylate (iPMA) [44]. The block copolymerization of iPMA was performed using the PMMA prepolymer prepared through the photopolymerization of MMA by the r-AMDV initiator, the MTEMPO mediator, and the tBuS photo-acid generator. The block copolymerization continued until the reaction mixture was completely solidified. For the MMA polymerization by this system, the monomer conversion linearly increased up to 5 h, followed by a slight increase over 5 h to 10 h [41]. The GPC profiles of the prepolymer prepared by the MMA polymerization for 6.5 h and the block copolymer are shown in Figure 15. The resulting block copolymer contained the prepolymer due to the deactivation of the propagating chain end. It was found that ca. 75% of the prepolymer was deactivated on the basis of the area ratio of the respective molecular weights in the GPC curve of the block copolymer. The MMA polymerization was shortened to 5 h in order to prevent the deactivation of the growing chain end. As can be seen in Figure 16, the GPC curve of the block copolymer was shifted to the higher molecular weight side and contained no prepolymer. The molecular weight and its distribution of the block copolymer were estimated to be Mn = 43,200 and Mw/Mn = 2.14, while those of the prepolymer were Mn = 10,100 and Mw/Mn = 1.63. It was found that the deactivation of the propagating chain ends was caused by a decrease in the monomer concentration and occurred during the end stage of the polymerization. The absolute molecular weight of the PMMA-b-PiPMA diblock copolymer obtained by the MMA polymerization for 5 h was determined by 1H NMR to be Mn = 36,000 based on the signal intensity of the methyl ester protons at 3.63 ppm and the methine protons at 4.87 ppm.
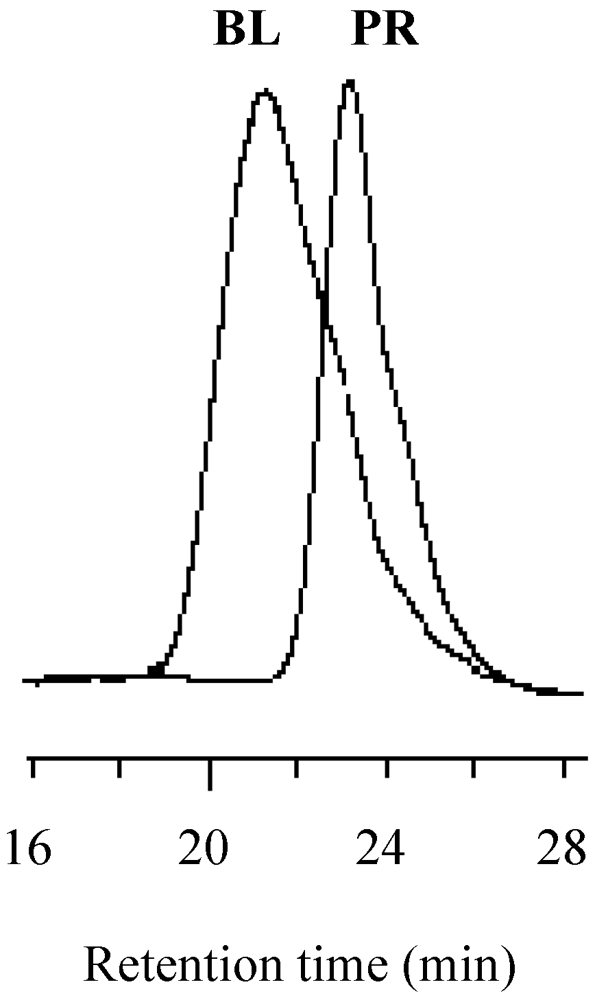
Figure 15.
The GPC profiles of the resulting block copolymer (BL) and prepolymer (PR) prepared by the MMA polymerization for 6.5 h.
Figure 15.
The GPC profiles of the resulting block copolymer (BL) and prepolymer (PR) prepared by the MMA polymerization for 6.5 h.

Figure 16.
The GPC profiles of the resulting block copolymer (BL) and prepolymer (PR) prepared by the MMA polymerization for 5 h.
Figure 16.
The GPC profiles of the resulting block copolymer (BL) and prepolymer (PR) prepared by the MMA polymerization for 5 h.

2.6. Solution Polymerization
The deactivation of the growing polymer chain ends occurred during the MTEMPO-mediated photopolymerization, as well as the thermal NMP [44]. The growing polymer chain ends should be more stabilized in a solution than in the bulk, because their deactivation occurs at the end stage of the polymerization where most monomers are consumed [64,76,77], although the solution polymerization retards the propagation rate. In order to obtain more stable growing polymer chain ends, the solution polymerization was explored [45]. The polymerization was performed in acetonitrile at room temperature using the r-AMDV initiator and the MTEMPO mediator, and the tBuS photo-acid generator. The results are shown in Table 8. The solution polymerization allowed the conversion to reach 97%, whereas it was difficult for the bulk polymerization to increase the conversion over 85% [41].
| MTEMPO/r-AMDV | tBuS/MTEMPO | Conv. (%) | Mn a | Mw/Mn a |
|---|---|---|---|---|
| 1.06 | 0.53 | 80 | 7,930 | 1.72 |
| 2.13 | 0.53 | 67 | 10,100 | 1.67 |
| 2.13 | 1.02 | 97 | 11,700 | 1.78 |
Irradiation time: 24 h. [MMA]0 = 9.35 M, [MTEMPO]0 = 0.0483 M. a Estimated by GPC based on PMMA standards.
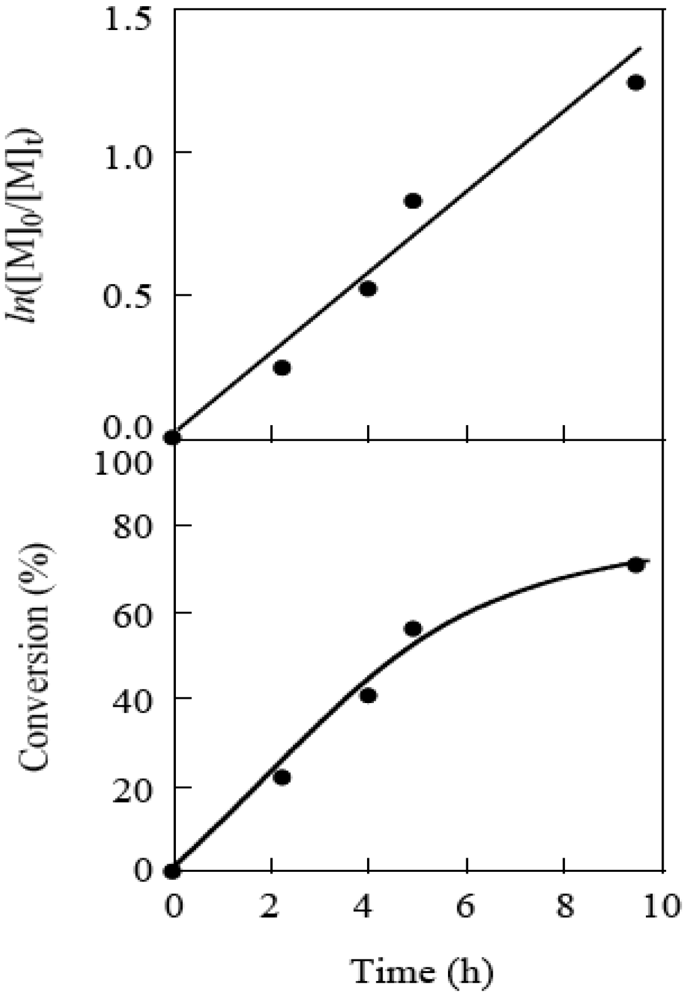
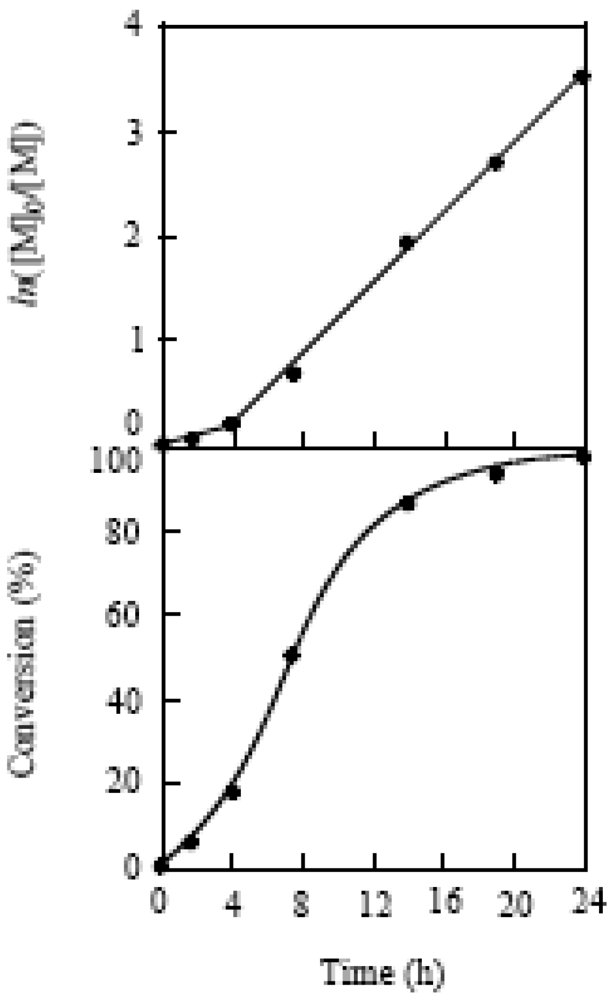
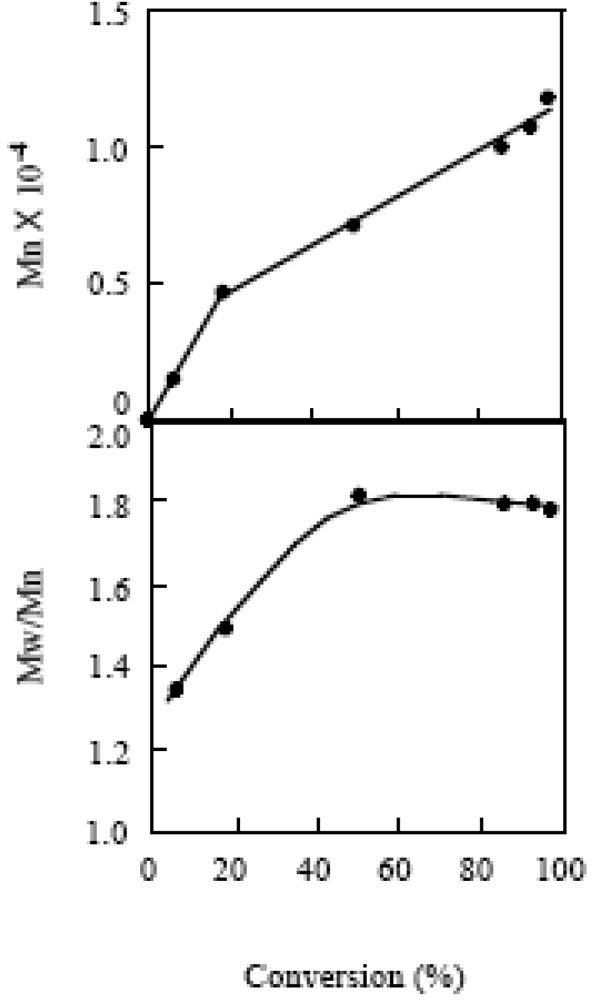
The livingness of the polymerization was investigated at the MTEMPO/r-AMDV of 2.13 and tBuS/MTEMPO of 1.02. The time-conversion and its first-order plots for the polymerization are shown in Figure 17. The ln[M]0/[M] plots showed different lines before and after 4 h, indicating that the radical concentrations were different for these two lines. The polymerization should be under the non-steady-state below 4 h and reached the steady-state over this time. The non-steady-state was not observed in the bulk polymerization [41]. It is suggested that MTEMPO could not effectively trap the propagating radical due to its low concentration in the solution polymerization, causing the occurrence of a normal termination between the propagating radicals under the non-steady-state. However, the proportion of the polymers produced by the normal termination is low based on its conversion (<18%). Figure 18 shows the plots of the molecular weight vs. the conversion for the polymerization. It was observed that oligomers with several thousand molecular weights were formed under the non-steady-state below the 18% conversion. However, the molecular weight linearly increased with the conversion under the steady-state, indicating that the polymerization proceeded by the living mechanism. The MWD was around 1.8, somewhat higher than that for the bulk polymerization. This broader MWD should be due to the presence of the oligomers produced under the non-steady-state.
Figure 17.
The time-conversion and its first order plots for the polymerization of MMA.

Figure 18.
The plots of the molecular weight and MWD vs. the conversion for the polymerization of MMA in acetonitrile. [MMA]0 = 9.35 M, MTEMPO/r-AMDV = 2.13, tBuS/MTEMPO = 1.02.
Figure 18.
The plots of the molecular weight and MWD vs. the conversion for the polymerization of MMA in acetonitrile. [MMA]0 = 9.35 M, MTEMPO/r-AMDV = 2.13, tBuS/MTEMPO = 1.02.

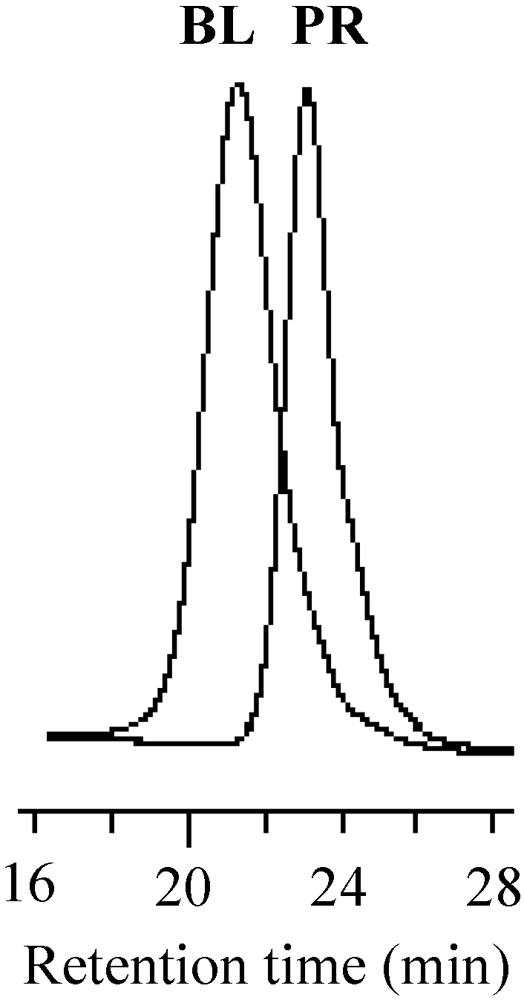
The stability of the growing polymer chain ends for the solution polymerization was also investigated through the block copolymerization. The PMMA prepolymer was prepared by a 14 h polymerization. The block copolymerization was performed for 19 h using iPMA as the second monomer. Figure 19 shows the GPC profiles of the prepolymer and the block copolymer. The curve of the block copolymer was shifted to the higher molecular weight side without deactivation, indicating that the prepolymer efficiently initiated the polymerization of iPMA. The growing polymer chain ends were stabilized even at a high conversion over 85% for the solution polymerization.
Figure 19.
The GPC profiles of the resulting block copolymer (BL) and prepolymer (PR).

2.7. Block Copolymerization Using a TEMPO Macromediator
The MTEMPO-mediated photo controlled/living radical polymerization has the potential to create a variety of architectures through designing TEMPO derivatives. The synthesis of a diblock copolymer using a macromediator of TEMPO is described in this section.
Figure 20.
1H NMR spectra of the block copolymer and the TEMPO-terminated poly(tetrahydrofuran) (PTHF-TEMPO). Solvent: CDCl3.
Figure 20.
1H NMR spectra of the block copolymer and the TEMPO-terminated poly(tetrahydrofuran) (PTHF-TEMPO). Solvent: CDCl3.

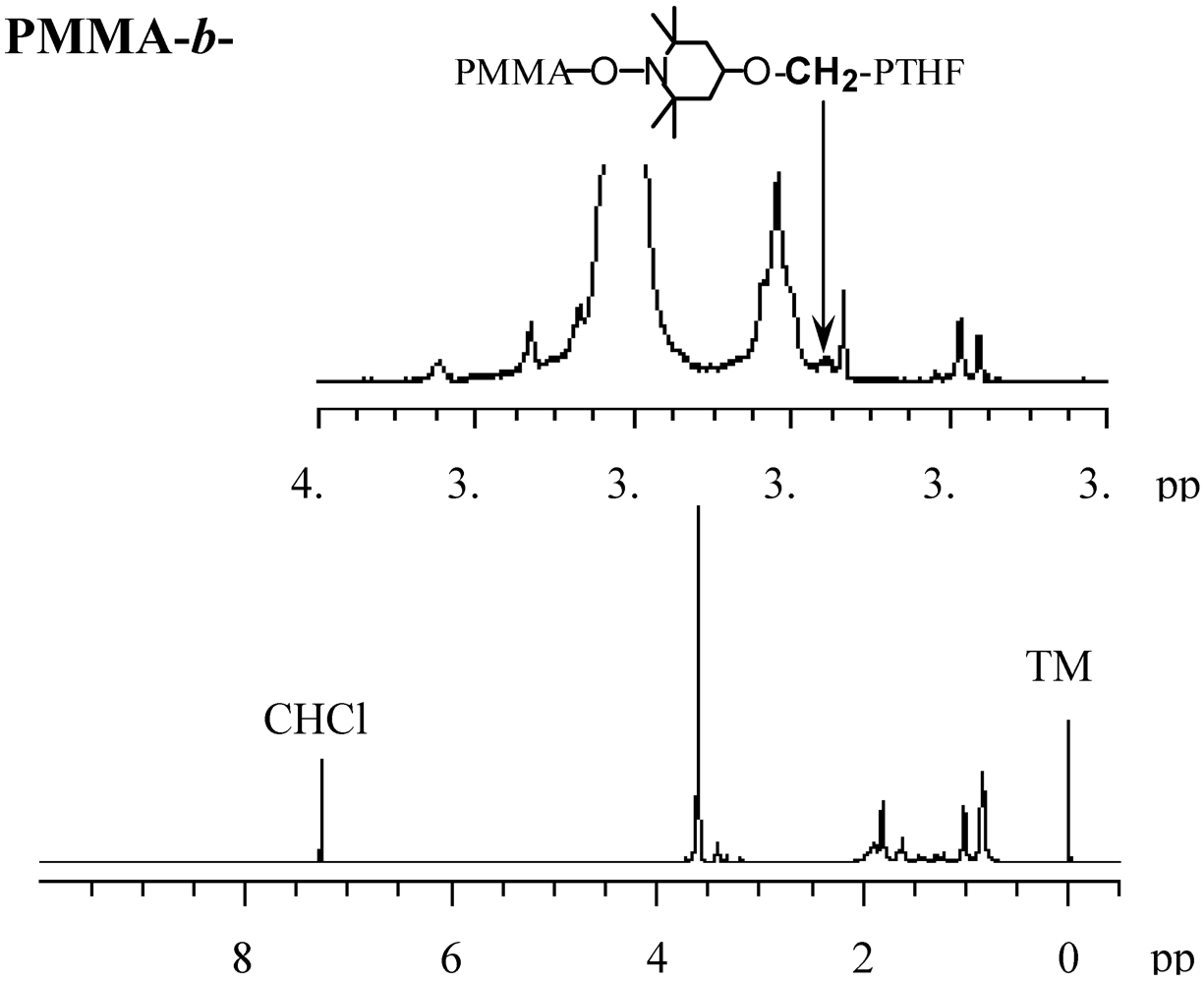
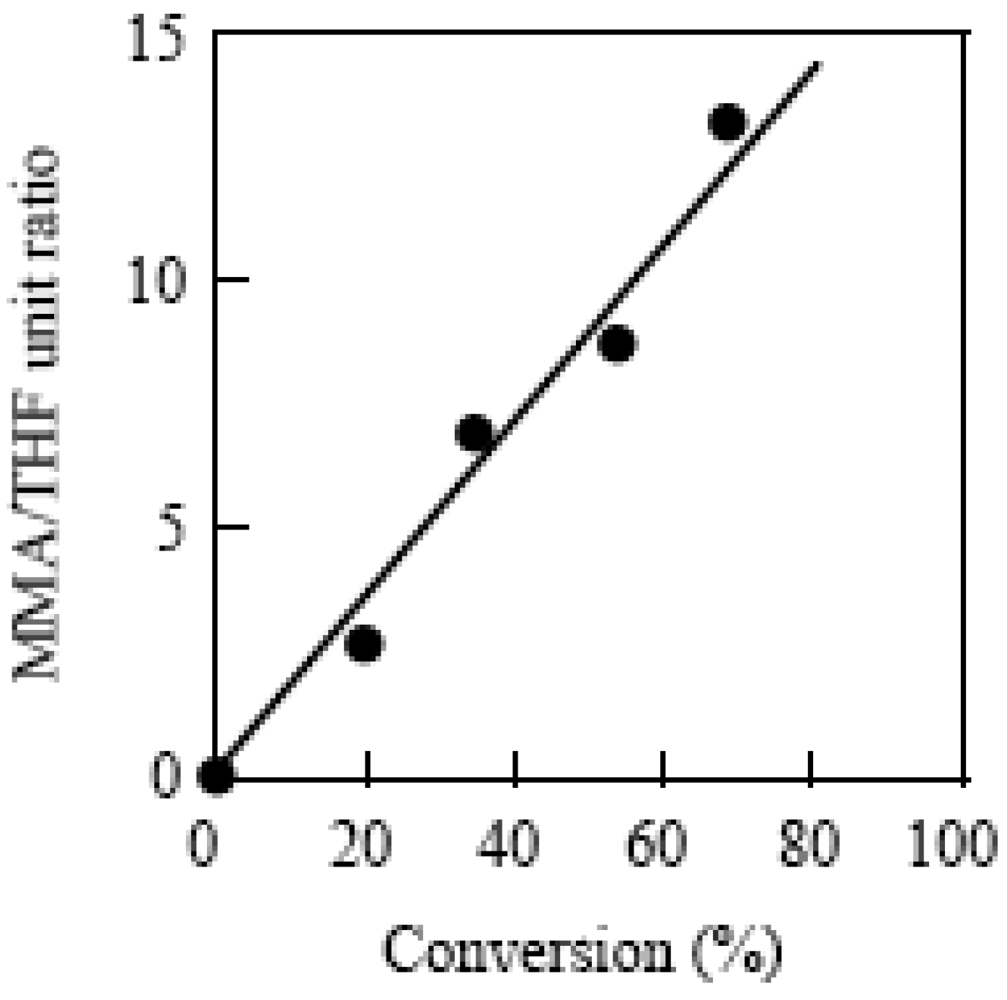
TEMPO-terminated poly(tetrahydrofuran) (PTHF-TEMPO) with Mn = 1,720 and Mw/Mn = 1.91 was used as the macromediator for the polymerization [78]. The photopolymerization of MMA was performed by AMDV, BAI, and PTHF-TEMPO [38]. Figure 20 shows the 1H NMR spectra of the resulting block copolymer with Mn = 11,300 and Mw/Mn = 1.83. The observation of a signal of the methylene attached to the TEMPO at 3.36 ppm confirmed that the PMMA and PTHF blocks were connected through the TEMPO. The plots of the molar ratio of the MMA unit to the THF unit versus the conversion using their signal intensity are shown in Figure 21. The MMA/THF ratio linearly increased with the increase in the conversion, suggesting the living nature of the polymerization.
Figure 21.
The plots of molar ratio of the MMA unit to the THF unit for the copolymers vs. the conversion.
Figure 21.
The plots of molar ratio of the MMA unit to the THF unit for the copolymers vs. the conversion.

The living mechanism of the polymerization was also confirmed on the basis of plots of linear correlation of the molecular weight of the copolymer vs. the initial concentration of AMDV ([AMDV]0). The MWD of the copolymers was the same as, or less than, that of PTHF-TEMPO. Linear increases in the first order time-conversion plots and the conversion-molecular weight plots endorsed the controlled/living mechanism.
2.8. Photo Dispersion Polymerization
The heterogeneous polymerization using the controlled/living radical polymerization has the potential to simultaneously control the molecular weight and particle size of a polymer. A number of the thermal controlled/living radical heterogeneous polymerization systems have been studied, and there is an outstanding review article on them [79]. The MTEMPO-mediated photo dispersion polymerization of MMA was performed at room temperature using r-AMDV, tBuS, and polyvinylpyrrolidone (PVP) as the surfactant in a mixed solvent of MeOH/water = 3/1 (v/v) [49]. Figure 22 shows SEM images of the resulting polymer particles. The photo dispersion polymerization in the absence of MTEMPO provided nonspecific particles. High molecular weight polymers should be produced by very fast uncontrolled polymerization, precipitated before being stabilized by PVP. On the other hand, the MTEMPO-mediated photo dispersion polymerization produced spherical particles of PMMA. The particle size distribution decreased as the PVP concentration increased. It was found that the size distributions were below 1.1 for the PVP concentrations of 60 and 75 wt% (Table 9). The spherical particles showed a comparatively narrow molecular weight distribution, indicating the simultaneous control of the molecular weight and particle size.
Figure 22.
SEM images of the PMMA particles obtained by uncontrolled polymerization (a) without MTEMPO and tBuS; (b) without MTEMPO; and (c) without r-AMDV and MTEMPO; and the MTEMPO-mediated polymerization at different PVP concentrations: (d) 30 wt%; (e) 45 wt%; (f) 60 wt%; and (g) 75 wt%.
Figure 22.
SEM images of the PMMA particles obtained by uncontrolled polymerization (a) without MTEMPO and tBuS; (b) without MTEMPO; and (c) without r-AMDV and MTEMPO; and the MTEMPO-mediated polymerization at different PVP concentrations: (d) 30 wt%; (e) 45 wt%; (f) 60 wt%; and (g) 75 wt%.

| r-AMDV (μmol) | MTEMPO (μmol) | tBuS (μmol) | PVP (wt%) | Conv. (%) | Mn a | Mw/Mn a | Morphology b | Dn b (μm) | Dw/Dn b |
|---|---|---|---|---|---|---|---|---|---|
| 45.4 | – | – | 45 | 70 | 78,900 | 3.26 | nonspecific | – | – |
| 45.4 | – | 23.5 | 45 | 34 | 57,600 | 3.64 | nonspecific | – | – |
| – | – | 23.5 | 45 | 69 | 175,000 | 2.50 | nonspecific | – | – |
| 45.4 | 48.3 | 23.5 | 30 | 20 | 18,200 | 1.62 | spherical | 3.87 | 2.80 |
| 45.4 | 48.3 | 23.5 | 45 | 23 | 17,700 | 1.61 | spherical | 5.66 | 1.27 |
| 45.4 | 48.3 | 23.5 | 60 | 37 | 20,500 | 1.65 | spherical | 3.99 | 1.06 |
| 45.4 | 48.3 | 23.5 | 75 | 28 | 18,000 | 1.64 | spherical | 4.14 | 1.04 |
Polymerization for 6 h in the mixed solvent (MeOH/water = 3/1). a Estimated by GPC based on PMMA standards; b Determined by the SEM observation.
Figure 23.
The time-conversion plots, the first order time-conversion plots, and the variation in turbidity of the polymerization system with time.
Figure 23.
The time-conversion plots, the first order time-conversion plots, and the variation in turbidity of the polymerization system with time.

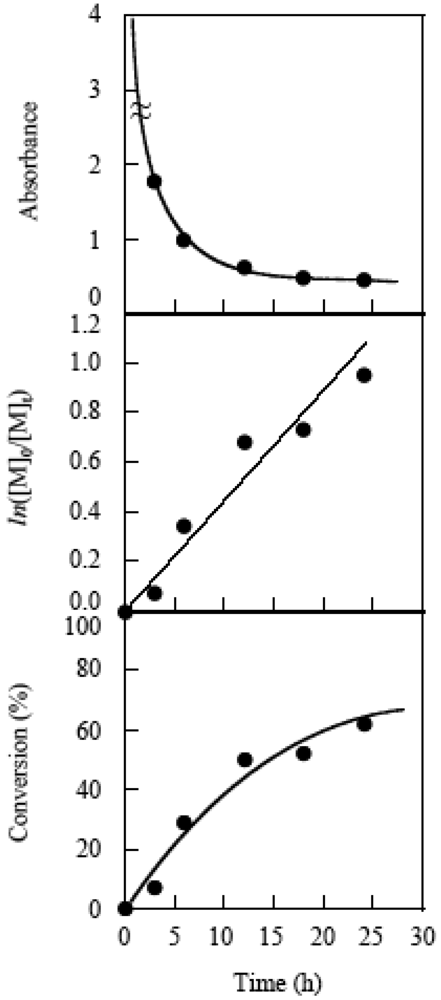
Figure 23 shows the time-conversion plots, the first order time-conversion plots, and the variation in turbidity of the polymerization system. The turbidity was determined by UV based on the absorbance at 230 nm. The turbidity of the system rapidly increased as the polymerization was initiated and became almost constant over 12 h. The constant turbidity suggests the limitation of the light passing though the system. The continued increase in the conversion in spite of the constant turbidity implies that over 12 h, the polymerization occurred outside of the particles. This inference was supported by a decrease in the molecular weight of the particles and the formation of a large amount of small particles outside of the secondary particles by the further extension of the polymerization time. The small particles had ca. a 1-μm diameter. It is considered that polymerization outside of the secondary particles did not occur by the transfer of the active species in the secondary particles, but by the remaining initiator in the solution or the autopolymerization. However, polymerization outside of the secondary particles should be slow because of the low monomer concentration at the later stage of polymerization. Therefore, the simultaneous control of the molecular weight and particle size was possible as long as light penetrated into the secondary particles.
3. Conclusions
TEMPO-mediated photopolymerization is a novel method to control molecular weight by photo irradiation. This polymerization also expanded the range of applicable monomers for thermal NMP, because photopolymerization has already been applied to other vinyl monomers, such as vinyl acetate [40] and ethyl acrylate [48]. This TEMPO-mediated controlled/living photoradical polymerization should expand the scope of molecular design and its industrial applications.
4. Experimental Section
Instrumentation The photopolymerization was carried out using a Wacom HX-500 illuminator with a 500 W high-pressure mercury lamp and an Ushio optical modulex BA-H502, an illuminator OPM2–502H with a high-illumination lens UI-OP2SL, and a 500 W super high-pressure UV lamp (USH-500SC2, Ushio Co. Ltd.). Gel permeation chromatography (GPC) was performed using a Tosoh GPC-8020 instrument equipped with a DP-8020 dual pump, a CO-8020 column oven, and a RI-8020 refractometer. Three polystyrene gel columns, Tosoh TSKGEL G2000HXL, G4000HXL, and G6000HXL were used with THF as the eluent at 40 °C. UV spectra were obtained using a Shimadzu UV-160A UV-Vis recording spectrophotometer. Gas chromatography (GC) was performed with Shimadzu GC-8A. Differential scanning calorimetry (DSC) was performed by a Shimadzu DSC-60 instrument equipped with a TA-60WS system controller and a FC-60 nitrogen flow controller. Scanning electron microscopy (SEM) measurements were performed using a Hitachi SU8000 scanning electron microscope. Centrifugation of polymer particles was carried out using a Tomy LC-200 centrifugal separator.
Materials All the azoinitiators were purified by recrystallization in methanol. Racemic-(2RS,2’RS)-azobis(4-methoxy-2,4-dimethylvaleronitrile) (r-AMDV) and meso-(2RS,2’SR)-azobis(4-methoxy-2,4-dimethylvaleronitrile) (m-AMDV) were obtained by separation from their mixture by recrystallization from ether [67]. 4-Methoxy-TEMPO was prepared as reported previously [80]. All the photo-acid generators were used as received. MMA and iPMA were washed with 5 wt% sodium hydroxide solution and water and then distilled over calcium hydride. PTHF-TEMPO was prepared as reported previously [78]. PVP (K30) with the average molecular weight of Mw ° 40,000 was purchased from Wako Pure Chemicals Co. Ltd and was used without further purification.
Photopolymerization General Procedure MMA (936 mg, 9.35 mmol), r-AMDV (14 mg, 0.0454 mmol), MTEMPO (9 mg, 0.0483 mmol), and tBuS (11 mg, 0.0341 mmol) were placed in an ampoule. After degassing the contents, the ampoule was sealed under vacuum. The bulk polymerization was carried out at room temperature for 9.5 h with irradiation by reflective light using a mirror with a 500W high-pressure mercury lamp at 7.0 A. The resulting mass was dissolved in dichloromethane (10 mL). The solution was concentrated by an evaporator to remove the dichloromethane and the monomer unreacted and was freeze-dried with benzene (30 mL) at 40 °C to obtain the product as white powder (664 mg). The conversion was estimated with the weight. The product was dissolved in dichloromethane (5 mL) and poured into hexane (500 mL). The precipitate was collected by filtration and dried in vacuo for several hours to be subjected to GPC analysis.
Photopolymerization of MMA by PTHF-TEMPO: General Procedure A mixture of MMA (936 mg, 9.35 mmol), PTHF-TEMPO (24 mg, 0.0240 mmol as the TEMPO content), AMDV (7 mg, 0.0227 mmol), and BAI in 50 wt% propylene carbonate solution (19 mg, 0.0125 mmol) was placed in an ampoule. After degassing the contents, the ampoule was sealed under vacuum. The polymerization was carried out at room temperature for 6 h with irradiation by reflective light using a mirror with a 500 W high-pressure mercury lamp at 7.0 A. The resulting mixture was dissolved in dichloromethane (10 mL). Ethylbenzene was added as a standard to the dichloromethane solution, and the solution was subjected to GC. The solution was concentrated to 3 mL with an evaporator and was poured into hexane (500 mL) to remove the monomer and PTHF-TEMPO unreacted. The precipitate was collected by filtration and dried in vacuo for several hours. The resulting polymer was freeze-dried with benzene to obtain the PMMA-b-PTHF (472 mg, 50% in yield).
Dispersion polymerization: General procedure MMA (936 mg, 9.35 mmol), r-AMDV (14 mg, 0.0454 mmol), MTEMPO (9 mg, 0.0483 mmol), tBuS (11 mg, 0.0235 mmol), PVP (421.6 mg, 45 wt% to MMA), MeOH (7.5 mL), and water (2.5 mL) were placed in an ampoule. After degassing the contents, the ampoule was sealed under vacuum. The polymerization was carried out at room temperature at 400 rpm for 12 h with irradiation by reflective light using a mirror with the high-pressure UV lamp. The resulting particles of PMMA were cleaned with MeOH by a repeated sedimentation-redispersion process. The conversion was estimated gravimetrically. The polymer particles were dried in vacuo for several hours and were obtained as white powder (465 mg).
References
- Odian, G. Radical chain polymerization. In Principles of Polymerization, 4th ed; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004. [Google Scholar]
- Otsu, T. Organic polysulfides as polymerization initiators. J. Polym. Sci. 1956, 21, 559–561. [Google Scholar]
- Moad, G.; Rizzardo, E.; Thang, S.H. Toward living radical polymerization. Accounts Chem. Res. 2008, 41, 1133–1142. [Google Scholar]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/living radical polymerization: Features, developments, and perspectives. Prog. Polym. Sci. 2007, 32, 93–146. [Google Scholar] [CrossRef]
- Uegaki, H.; Kotani, Y.; Kamigaito, M.; Sawamoto, M. Nickel-mediated living radical polymerization of methyl methacrylate. Macromolecules 1997, 30, 2249–2253. [Google Scholar]
- Wayland, B.B.; Poszmik, G.; Mukerjee, S.L. Living radical polymerization of acrylates by organocobalt porphyrin complexes. J. Am. Chem. Soc. 1994, 116, 7943–7944. [Google Scholar]
- Ando, T.; Kamigaito, M.; Sawamoto, M.; Higashimura, T. Iron(II) chloride complex for living radical polymerization of methyl methacrylate. Macromolecules 1995, 30, 4507–4510. [Google Scholar]
- Kato, M.; Kamigaito, M.; Sawamoto, M.; Higashimura, T. Polymerization of methyl methacrylate with the carbon tetrachloride/dichlorotris-(triphenylphosphine)ruthenium(II)/methylaluminum bis(2,6-di-tert-butylphenoxide) initiating system: Possibility of living radical polymerization. Macromolecules 1995, 28, 1721–1723. [Google Scholar] [CrossRef]
- Percec, V.; Barboiu, B.; Neumann, A.; Ronda, J.C.; Zhao, M. Metal-catalyzed “living” radical polymerization of styrene initiated with arenesulfonyl chlorides. From heterogeneous to homogeneous catalysis. Macromolecules 1996, 29, 3665–3668. [Google Scholar] [CrossRef]
- Lecmote, P.; Drapier, I.; Dubois, P.; Teyssie, P.; Jerome, R. Controlled radical polymerization of methyl methacrylate in the presence of palladium acetate, triphenylphosphine, and carbon tetrachloride. Macromolecules 1997, 30, 7631–7633. [Google Scholar] [CrossRef]
- Kotani, Y.; Kamigaito, M.; Sawamoto, M. Re(V)-mediated living radical polymerization of styrene: ReO2I(PPh3)2/R−I initiating systems. Macromolecules 1999, 32, 2420–2424. [Google Scholar] [CrossRef]
- Oka, M.; Tatemoto, M. Vinylidene fluoride–Hexafluoropropylene copolymer having terminal iodines. In Contemporary Topics in Polymer Science; Plenum Press: New York, NY, USA, 1984. [Google Scholar]
- Lacroix-Desmazes, P.; Severac, R.; Boutevin, B. Reverse iodine transfer polymerization of methyl acrylate and n-butyl acrylate. Macromolecules 2005, 38, 6299–6309. [Google Scholar] [CrossRef]
- Pouget, E.; Tonnar, J.; Eloy, C.; Lacroix-Desmazes, P.; Boutevin, B. Synthesis of poly(styrene)-b-poly(dimethylsiloxane)-b-poly(styrene) triblock copolymers by iodine transfer polymerization in miniemulsion. Macromolecules 2006, 39, 6009–6016. [Google Scholar] [CrossRef]
- Hawker, C.J.; Bosman, A.W.; Harth, E. New polymer synthesis by nitroxide mediated living radical polymerizations. Chem. Rev. 2001, 101, 3661–3688. [Google Scholar]
- Sciannamea, V.; Jérôme, R.; Detrembleur, C. In situ nitroxide-mediated radical polymerization (NMP) processes: Their understanding and optimization. Chem. Rev. 2008, 108, 1104–1126. [Google Scholar] [CrossRef]
- Yousi, Z.; Jian, L.; Rongchuan, Z.; Jianliang, Y.; Lizong, D.; Lansun, Z. Synthesis of block copolymer from dissimilar vinyl monomer by stable free radical polymerization. Macromolecules 2000, 33, 4745–4749. [Google Scholar]
- Devonport, W.; Michalak, L.; Malmstrom, E.; Mate, M.; Kurdi, B.; Hawker, C.J.; Barclay, G.G.; Sinta, R. “Living” free-radical pin the absence of initiators: Controlled autopolymerization. Macromolecules 1997, 30, 1929–1934. [Google Scholar]
- Mardare, D.; Matyjaszewski, K. “Living” radical polymerization of vinyl acetate. Macromolecules 1994, 27, 645–649. [Google Scholar] [CrossRef]
- Granel, C.; Jerime, R.; Teyssie, P.; Jasieczek, C.B.; Shooter, A.J.; Haddleton, D.M.; Hastings, J.J.; Gigmes, D.; Grimaldi, S.; Tordo, P.; Greszta, D.; Matyjaszewski, K. Investigation of methyl methacrylate and vinyl acetate polymerization promoted by Al(iBu)3/2,2’-bipyridine and Al(iBu)3/2,2’-bipyridine/TEMPO complexes. Macromolecules 1998, 31, 7133–7141. [Google Scholar]
- Matyjaszewski, K.; Gaynor, S.; Greszta, D.; Mardare, D.; Shigemoto, T. “Living” and controlled radical polymerization. J. Phys. Org. Chem. 1995, 8, 306–315. [Google Scholar] [CrossRef]
- Hawker, C.J.; Barclay, G.G.; Orellana, A.; Dao, J.; Devonport, W. Initiating systems for nitroxide-mediated “living” free radical polymerizations: Synthesis and evaluation. Macromolrcules 1996, 29, 5245–5254. [Google Scholar]
- Dao, J.; Benoit, D.; Hawker, C.J. A versatile and efficient synthesis of alkoxyamine LFR initiators via manganese based asymmetric epoxidation catalysts. J. Polym. Sci. A Polym. Chem. 1998, 36, 2161–2167. [Google Scholar] [CrossRef]
- Banoit, D.; Chaplinski, V.; Braslau, R.; Hawker, C.J. Development of a universal alkoxyamine for “living” free radical polymerizations. J. Am. Chem. Soc. 1999, 121, 3904–3920. [Google Scholar]
- Veregin, R.P.N.; Georges, M.K.; Hamer, G.K.; Kazmaier, P.M. mechanism of living free radical polymerizations with narrow polydispersity: Electron spin resonance and kinetic studies. Macromolecules 1995, 28, 4391–4398. [Google Scholar] [CrossRef]
- Greszta, D.; Matyjaszewski, K. Comments on the paper “living radical polymerization: Kinetic results”. Macromolecules 1996, 29, 5239–5240. [Google Scholar]
- Braslau, R.; Burrill, L.C., II; Siano, M.; Naik, N.; Howden, R.K.; Mahal, L.K. Low-temperature preparations of unimolecular nitroxide initiators for “living” free radical polymerizations. Macromolecules 1997, 30, 6445–6450. [Google Scholar]
- Li, I.Q.; Howell, B.A.; Koster, R.A.; Priddy, D.B. Mono- and dinitroxide styrene polymerization initiators. Macromolecules 1996, 29, 8554–8555. [Google Scholar]
- Mercier, C.L.; Acerbis, S.; Bertin, D.; Chauvin, F.; Gigmes, D.; Guerret, O.; Lansalot, M.; Marque, S.; Moigne, F.L.; Fischer, H.; Tordo, P. Design and use of β-phosphorus nitroxides and alkoxyamines in controlled/”living” free radical polymerizations. Macromol. Symposia 2002, 182, 225–247. [Google Scholar]
- Yoshida, E.; Ishizone, T.; Hirao, A.; Nakahama, S.; Takata, T.; Endo, T. Synthesis of polystyrene having an aminoxy terminal by the reactions of living polystyrene with an oxoaminium salt and with the corresponding nitroxyl radical. Macromolecules 1994, 27, 3119–3124. [Google Scholar]
- Yoshida, E.; Nakamura, K.; Takata, T.; Endo, T. Oxoaminium salts as initiators for cationic polymerization of vinyl monomers. J. Polym. Sci. A Polym. Chem. 1993, 31, 1505–1512. [Google Scholar]
- Pyun, J.; Tang, C.; Kowalewski, T.; Fréchet, J.M.J.; Hawker, C.J. Synthesis and direct visualization of block copolymers composed of different macromolecular architectures. Macromolecules 2005, 38, 2674–2685. [Google Scholar]
- Hawker, C.J.; Hedrick, J.L. Accurate control of chain ends by a novel “living” free-radical polymerization process. Macromolecules 1995, 28, 2993–2995. [Google Scholar]
- Hawker, C.J. Architectural Control in “Living” free radical polymerizations: Preparation of star and graft polymers. Angew. Chem. Int. Ed. Eng. 1995, l34, 1456–1459. [Google Scholar]
- Tsoukatos, T.; Pispas, S.; Hadjichristidis, N. Complex macromolecular architectures by combining TEMPO living free radical and anionic polymerization. Macromolecules 2000, 33, 9504–9511. [Google Scholar]
- Yoshida, E. Photo-living radical polymerization of methyl methacrylate by a nitroxide mediator. Colloid Polym. Sci. 2008, 286, 1663–1666. [Google Scholar]
- Yoshida, E. Photo-living radical polymerization of methyl methacrylate by 2,2,6,6-tetramethylpiperidine-1-oxyl in the presence of a photo-acid generator. Colloid Polym. Sci. 2009, 287, 767–772. [Google Scholar]
- Yoshida, E. Synthesis of poly(methyl methacrylate)-block-poly(tetrahydrofuran) by photo-living radical polymerization using a 2,2,6,6-tetramethylpiperidine-1-oxyl macromediator. Colloid Polym. Sci. 2009, 287, 1417–1424. [Google Scholar] [CrossRef]
- Yoshida, E. Photo-living radical polymerization of methyl methacrylate using alkoxyamine as an initiator. Colloid Polym. Sci. 2010, 288, 7–13. [Google Scholar]
- Yoshida, E. Nitroxide-mediated photo-living radical polymerization of vinyl acetate. Colloid Polym. Sci. 2010, 288, 73–78. [Google Scholar]
- Yoshida, E. Nitroxide-mediated photo-living radical polymerization of methyl methacrylate using (4-tert-butylphenyl)diphenylsulfonium triflate as a photo-acid generator. Colloid Polym. Sci. 2010, 288, 239–243. [Google Scholar] [CrossRef]
- Yoshida, E. Effect of azoinitiators on nitroxide-mediated photo-living radical polymerization of methyl methacrylate. Colloid Polym. Sci. 2010, 288, 341–345. [Google Scholar]
- Yoshida, E. Effects of initiators and photo-acid generators on nitroxide-mediated photo-living radical polymerization of methyl methacrylate. Colloid Polym. Sci. 2010, 288, 901–905. [Google Scholar]
- Yoshida, E. Stability of growing polymer chain ends for nitroxide-mediated photo-living radical polymerization. Colloid Polym. Sci. 2010, 288, 1027–1030. [Google Scholar]
- Yoshida, E. Nitroxide-mediated photo-living radical polymerization of methyl methacrylate in solution. Colloid Polym. Sci. 2010, 288, 1639–1643. [Google Scholar]
- Yoshida, E. Nitroxide-mediated photo-living radical polymerization of methyl methacrylate in the presence of (η6-benzene)(η5-cyclopentadienyl)FeII hexafluorophosphate. Colloid Polym. Sci. 2010, 288, 1745–1749. [Google Scholar]
- Yoshida, E. Graft copolymerization of methyl methacrylate on polystyrene backbone through nitroxide-mediated photo-living radical polymerization. Colloid Polym. Sci. 2011, 289, 837–841. [Google Scholar]
- Yoshida, E. Nitroxide-mediated photo-controlled/living radical polymerization of ethyl acrylate. Colloid Polym. Sci. 2011, 289, 1127–1132. [Google Scholar]
- Yoshida, E. Nitroxide-mediated photo-controlled/living radical dispersion polymerization of methyl methacrylate. Colloid Polym. Sci. 2011, 289, 1625–1630. [Google Scholar]
- Guillaneuf, Y.; Bertin, D.; Gigmes, D.; Versace, D.; Lalevee, J.; Fouassier, J. Toward nitroxide-mediated photopolymerization. Macromolecules 2010, 43, 2204–2212. [Google Scholar]
- Goto, A.; Scaiano, J.C.; Maretti, L. Photolysis of an alkoxyamine using intramolecular energy transfer from a quinoline antenna—Towards photo-induced living radical polymerization. Photochem. Photobiol. Sci. 2007, 6, 833–853. [Google Scholar]
- Lalevee, J.; Allonas, X.; Fouassier, J.P. A new efficient photoiniferter for living radical photopolymerization. Macromolecules 2006, 39, 8216–8218. [Google Scholar]
- Nemoto, Y.; Nakayama, Y. Optimal irradiation wavelength in iniferter-based photocontrolled radical polymerization. J Polym. Sci. A Polym. Chem. 2008, 46, 4505–4512. [Google Scholar]
- You, Y.; Hong, C.; Bai, R.; Pan, C.; Wang, J. Photo-initiated living free radical polymerization in the presence of dibenzyl trithiocarbonate. Macromol. Chem. Phys. 2002, 203, 477–483. [Google Scholar]
- Ran, R.; Yu, Y.; Wan, T. Photoinitiated RAFT polymerization in the presence of trithiocarbonate. J. Appl. Polym. Sci. 2007, 105, 398–404. [Google Scholar]
- Baruah, S.R.; Kakati, D.K. Photopolymerization of methyl methacrylate by 2,2'-dithiodiethanol: Effect of reaction conditions. J. Appl. Polym. Sci. 2006, 100, 1601–1606. [Google Scholar]
- Ajayaghosh, A.; Francis, R. A xanthate-derived photoinitiator that recognizes and controls the free radical polymerization pathways of methyl methacrylate and styrene. J. Am. Chem. Soc. 1999, 121, 6599–6606. [Google Scholar]
- Yang, W.; Ranby, B. Radical living graft polymerization on the surface of polymeric materials. Macromolecules 1996, 29, 3308–3310. [Google Scholar]
- Tasdelen, M.A.; Durmaz, Y.Y.; Karagoz, B.; Bicak, N.; Yagci, Y. A new photoiniferter/RAFT agent for ambient temperature rapid and well-controlled radical polymerization. J. Polym. Sci. A Polym. Chem. 2008, 46, 3387–3395. [Google Scholar]
- Ajayaghosh, A.; Francis, R. Narrow polydispersed reactive polymers by a photoinitiated free radical polymerization approach. Controlled polymerization of methyl methacrylate. Macromolecules 1998, 31, 1436–1438. [Google Scholar] [CrossRef]
- Yin, H.; Zheng, H.; Lu, L.; Liu, P.; Cai, Y. Highly efficient and well-controlled ambient temperature RAFT polymerization of glycidyl methacrylate under visible light radiation. J. Polym. Sci. A Polym. Chem. 2007, 45, 5091–5102. [Google Scholar]
- Cohen, N.A.; Tillman, E.S.; Thakur, S.; Smith, J.R.; Eckenhoff, W.T.; Pintauer, T. Effect of the ligand in atom transfer radical polymerization reactions initiated by photodimers of 9-bromoanthracene. Macromol. Chem. Phys. 2009, 210, 263–268. [Google Scholar]
- Li, P.; Qiu, K. Cu(S2CNEt2)Cl-catalyzed reverse atom-transfer radical polymerization of vinyl monomers. Macromol. Rapid Commun. 2002, 23, 1124–1129. [Google Scholar]
- Yoshida, E.; Okada, Y. Living radical polymerization of styrene in the presence of 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl, and radical transformation of the resulting polymers by other radicals. Bull. Chem. Soc. Jpn. 1997, 70, 275–281. [Google Scholar]
- McHale, R.; Aldabbagh, F.; Zetterlund, P.B. The role of excess nitroxide in the SG1 (N-tert-butyl-N-[1-diethlphosphono-(2,2-dimethylpropyl)] nitroxide)-mediated polymerization of methyl methacrylate. J. Polym. Sci. A Polym. Chem. 1999, 45, 2194–2203. [Google Scholar]
- Bovey, F.A.; Tiers, G.V.D. Polymer NSR spectroscopy. II. The high resolution spectra of methyl methacrylate polymers prepared with free radical and anionic initiators. J. Polym. Sci. 1960, 44, 173–182. [Google Scholar] [CrossRef]
- Kita, Y.; Gotanda, K.; Murata, K.; Suemura, M.; Sano, A.; Yamaguchi, T.; Oka, M.; Matsugi, M. Practical radical additions under mild conditions using 2,2'-azobis(2,4-dimethyl-4-methoxyvaleronitrile) [V-70] as an initiator. Org. Process Res. Dev. 1998, 2, 250–254. [Google Scholar]
- Crivello, J.V.; Sangermano, M. Visible and long-wavelength photoinitiated cationic polymerization. J. Polym. Sci. A Polym. Chem. 2001, 39, 343–356. [Google Scholar]
- Crivello, J.V. The discovery and development of onium salt cationic photoinitiators. J. Polym. Sci. A Polym. Chem. 1999, 37, 4241–4254. [Google Scholar]
- Devoe, R.J.; Sahyunn, M.R.V.; Sepone, D.; Sharmac, D.K. Transient intermediates in the photolysis of iodonium cations. Can. J. Chem. 1987, 65, 2342–2349. [Google Scholar]
- Crivello, J.V.; Lam, J.H. W.; Moore, J.E.; Schroeter, S.H. Triarylsulfonium salts: A new class of photoinitiators for cationic polymerization. J. Radiat. Curing 1978, 5, 2–17. [Google Scholar]
- Crivello, J.V.; Lam, J.H.W. Complex triarylsulfonium salt photoinitiators. I. The identification, characterization, and syntheses of a new class of triarylsulfonium salt photoinitiators. J. Polym. Sci. Polym. Chem. Ed. 1980, 18, 2677–2695. [Google Scholar] [CrossRef]
- Crivello, J.V.; Lam, J.H.W. Photoinitiated cationic polymerization with triarylsulfonium salts. J. Polym. Sci. Polym. Chem. Ed. 1979, 17, 977–999. [Google Scholar]
- Liu, Y.C.; Wu, L.M.; Chen, P. A facile generation of radical cations via the action of nitroxides. Tetrahedron Lett. 1985, 26, 4201–4202. [Google Scholar]
- Meier, K.; Zweifel, H. Photoinitiated cationic polymerization of epoxides with iron-arene complexes. J. Radiat. Curing 1986, 13, 26–32. [Google Scholar]
- Li, I.; Howell, B.A.; Matyjaszewski, K.; Shigemoto, T.; Smith, P.B.; Priddy, D.B. Kinetics of decomposition of 2,2,6,6-tetramethyl-1-(1-phenylethoxy)piperidine and its implications on nitroxyl-mediated styrene polymerization. Macromolecules 1995, 28, 6692–6693. [Google Scholar] [CrossRef]
- Yoshida, E.; Sugita, A. Synthesis of poly(styrene-b-tetrahydrofuran-b-styrene) triblock copolymers by transformation from living cationic into living radical polymerization using 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl as a transforming agent. J. Polym. Sci. Polym. Chem. Ed. 1998, 36, 2059–2068. [Google Scholar]
- Yoshida, E.; Sugita, A. Synthesis of poly(tetrahydrofuran) with a nitroxyl radical at the chain end, and its application to living radical polymerization. Macromolecules 1996, 29, 6422–6426. [Google Scholar]
- Zetterlund, P.B.; Kagawa, Y.; Okubo, M. Controlled/living radical polymerization in dispersed systems. Chem. Rev. 2008, 108, 3747–3794. [Google Scholar]
- Miyazawa, T.; Endo, T.; Shiihashi, S.; Ogawara, M. Selective oxidation of alcohols by oxoaminium salts (R2N:O+ X-). J. Org. Chem. 1985, 50, 1332–1334. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Yoshida, E. Controlled Photoradical Polymerization Mediated by 2,2,6,6-Tetramethylpiperidine-1-Oxyl. Polymers 2012, 4, 1125-1156. https://doi.org/10.3390/polym4021125
AMA Style
Yoshida E. Controlled Photoradical Polymerization Mediated by 2,2,6,6-Tetramethylpiperidine-1-Oxyl. Polymers. 2012; 4(2):1125-1156. https://doi.org/10.3390/polym4021125
Chicago/Turabian StyleYoshida, Eri. 2012. "Controlled Photoradical Polymerization Mediated by 2,2,6,6-Tetramethylpiperidine-1-Oxyl" Polymers 4, no. 2: 1125-1156. https://doi.org/10.3390/polym4021125