Synthesis of Well-Defined, Water-Soluble Hyperbranched Polyamides by Chain-Growth Condensation Polymerization of AB2 Monomer

Abstract
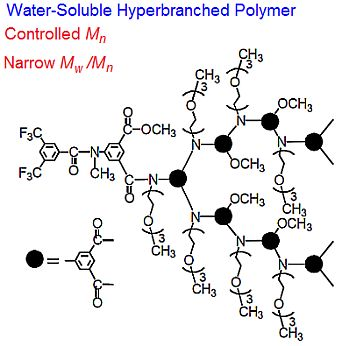
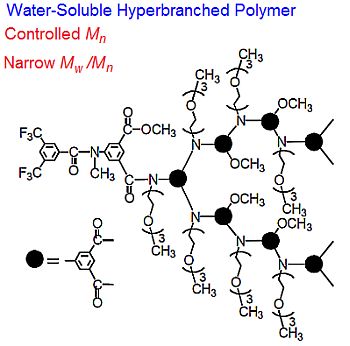
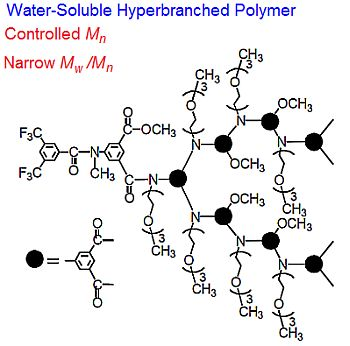
:
1. Introduction

2. Results and Discussion
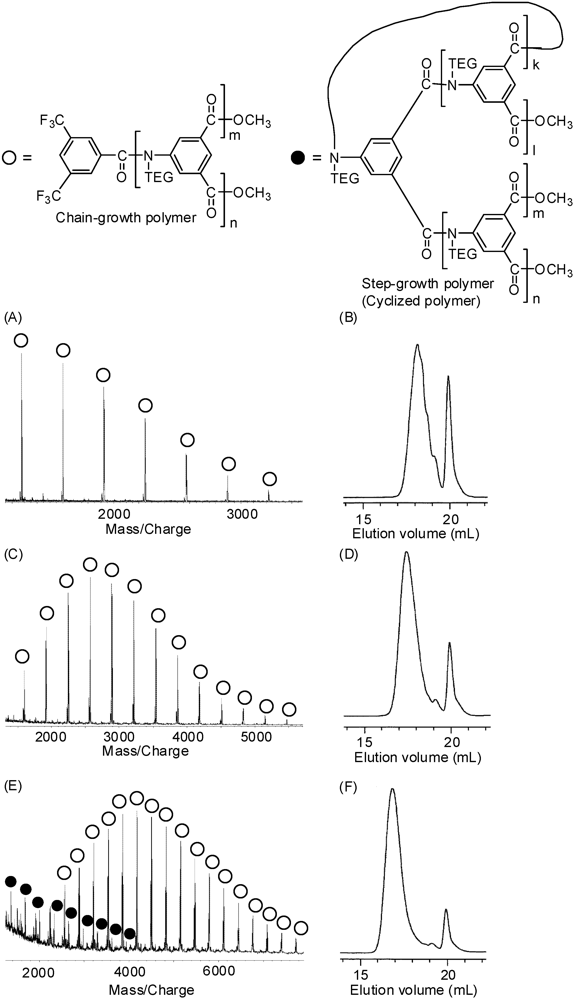
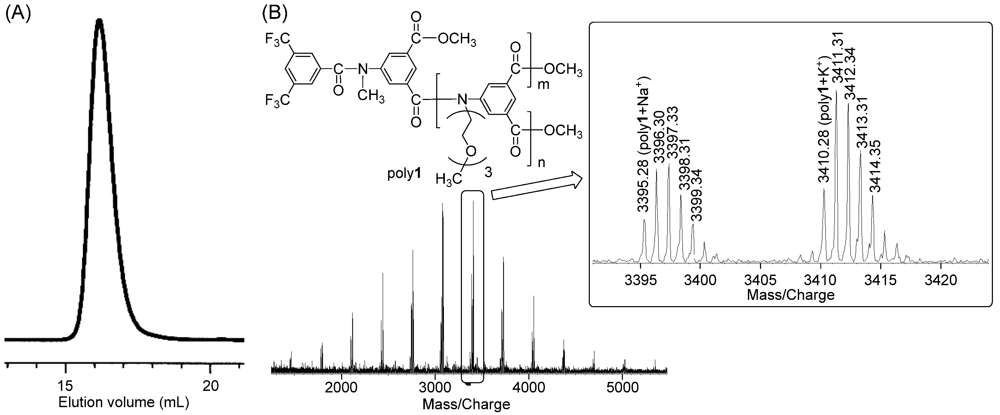
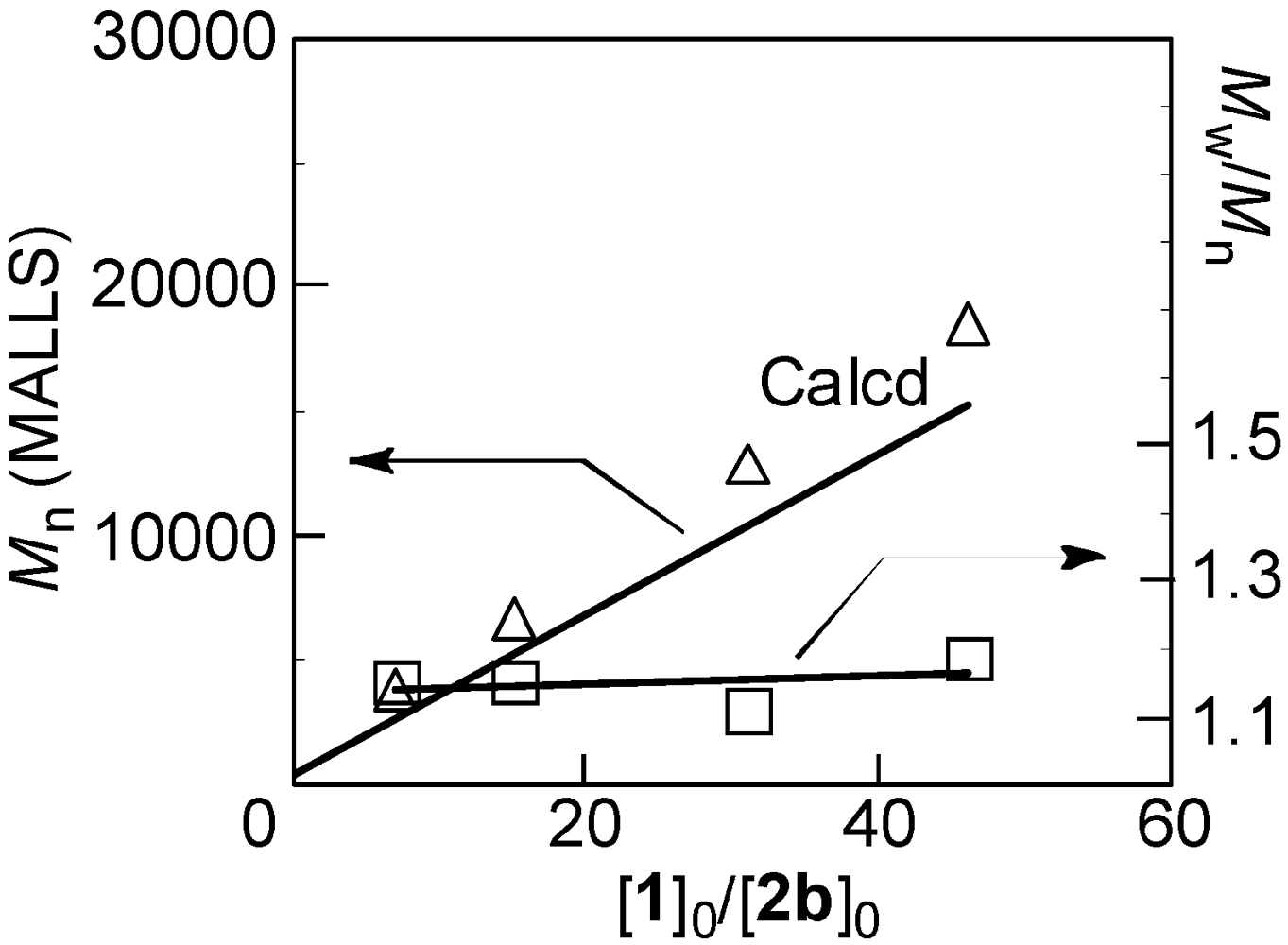
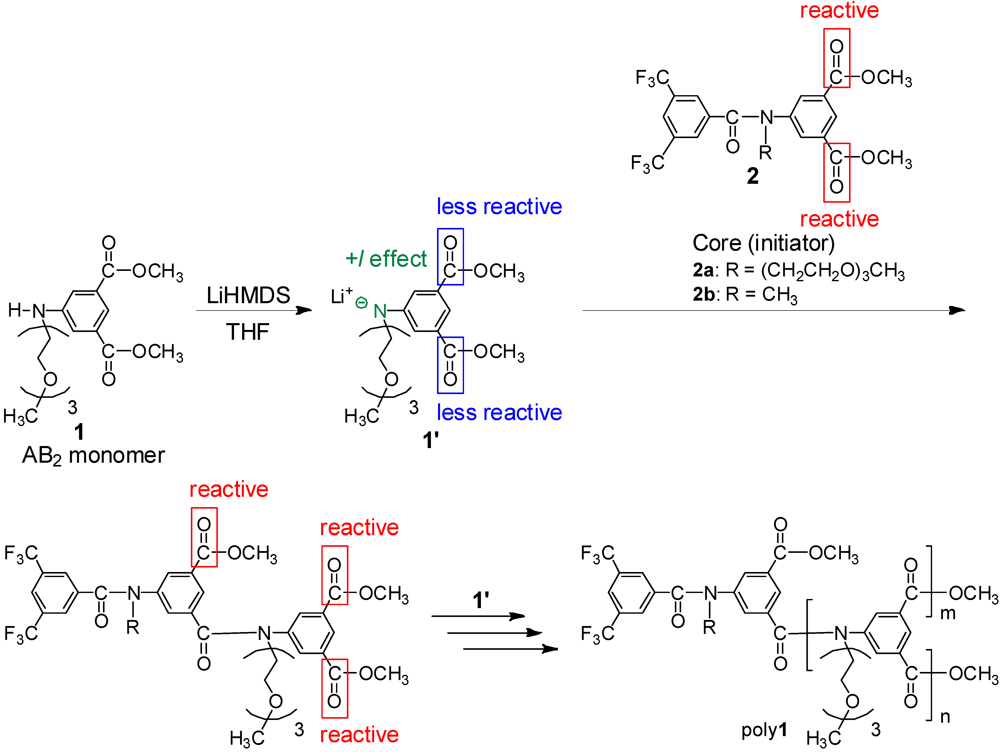
2.1. Polymerization Conditions

| Entry | Additive | [1]0/[2a]0 | Temp (°C) | Reaction time (h) | Mn (b) | Mw/Mn (b) |
|---|---|---|---|---|---|---|
| 1 | TMEDA | 31 | −30 | 15 | 4760 | 1.33 |
| 2 | LiCl | 35 | −30 | 24 | 5370 | 1.32 |
| 3 | LiCl | 31 | −20 | 3 | 4970 | 1.32 |
| 4 | LiCl | 30 | −10 | 1 | 5250 | 1.29 |
| 5 | LiCl | 33 | 0 | 0.17 | 4850 | 1.47 |
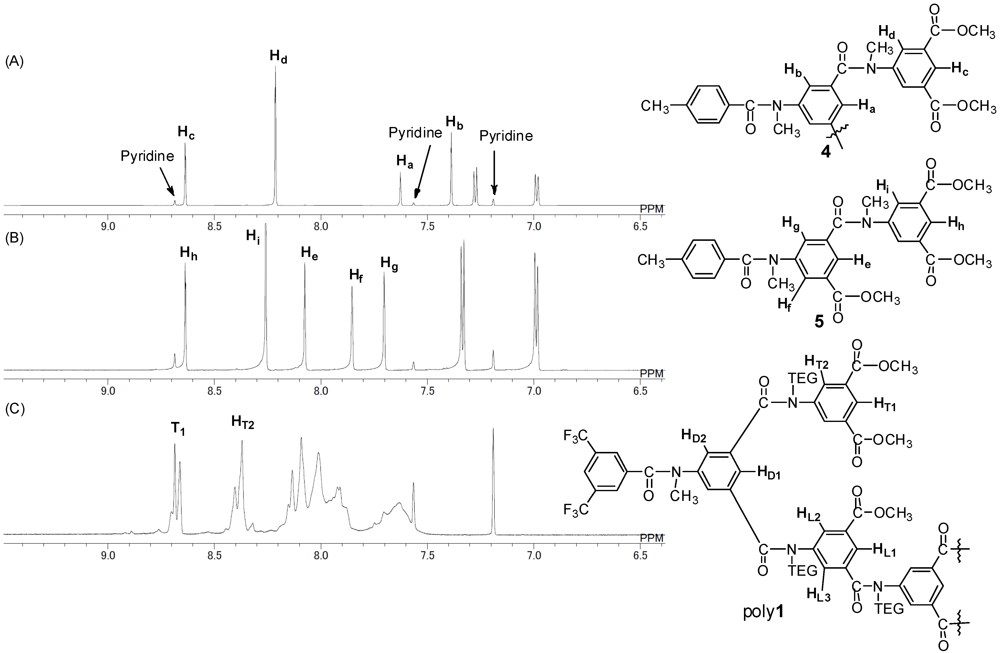
2.2. Effect of N-TEG Chain on Polymerization

2.3. Effect of Initiator



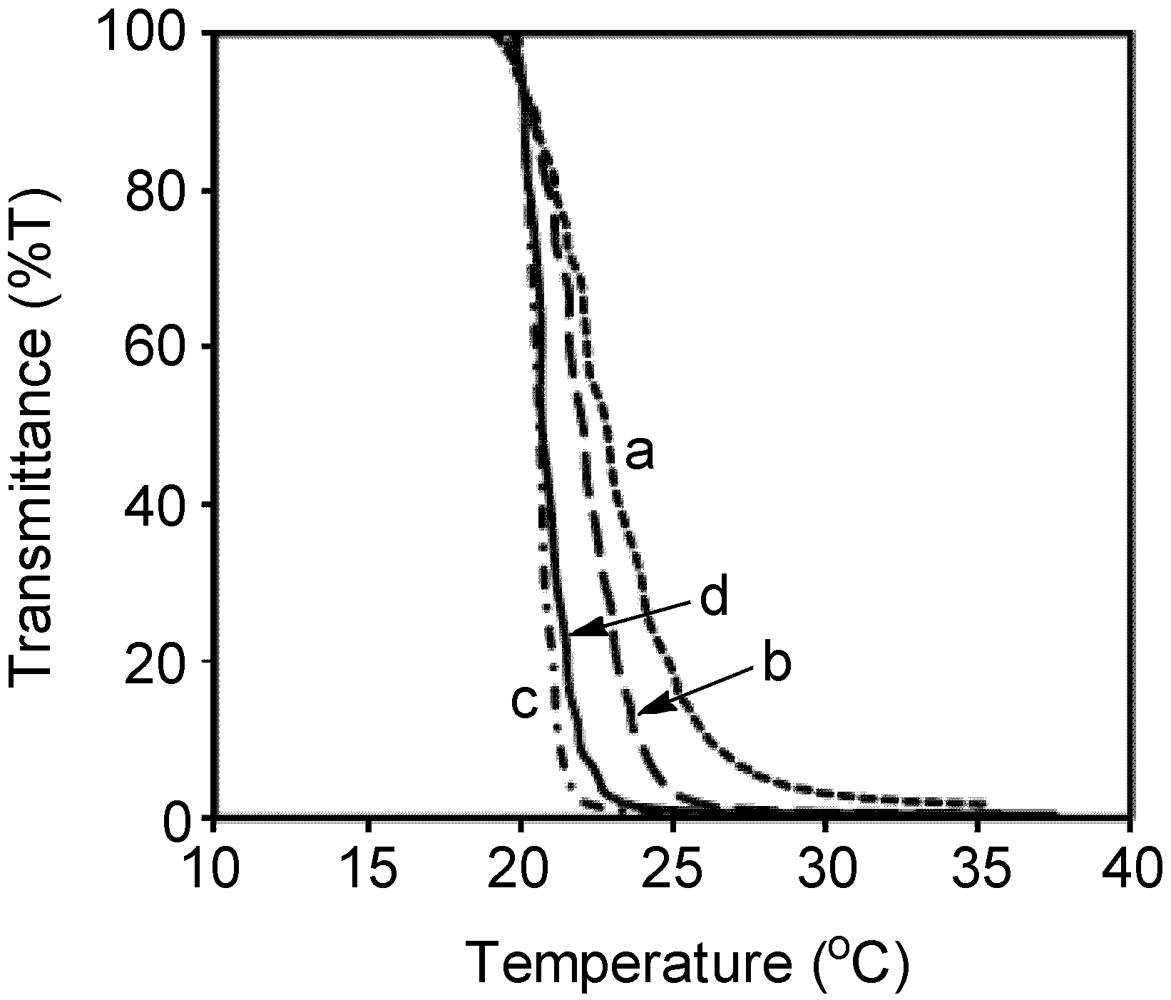
2.4. Solubility and Aqueous Solution Behavior of Poly1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | poly1 | poly( N-TEG-m-benzamide) |
|---|---|---|
| Ether | – | – |
| Toluene | + | + |
| CH2Cl2 | + | + |
| Ethyl acetate | + | + |
| Acetone | + | + |
| CH3CN | + | + |
| DMF | + | + |
| DMSO | + | + |
| Ethanol | + | + |
| Methanol | + | + |
| H2O | + | + |

3. Experimental Section
3.1. Measurements
3.2. Materials
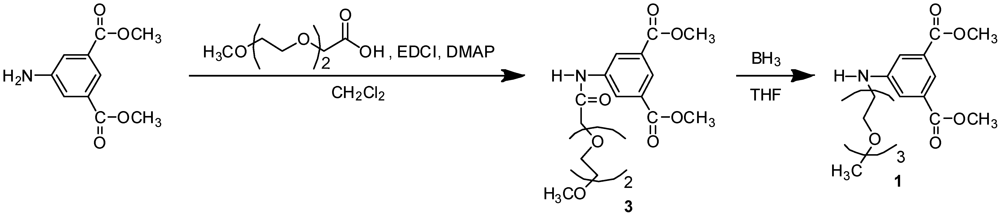
3.3. Synthesis of Monomer 1

3.3.1. Compound 3
3.3.2. Monomer 1
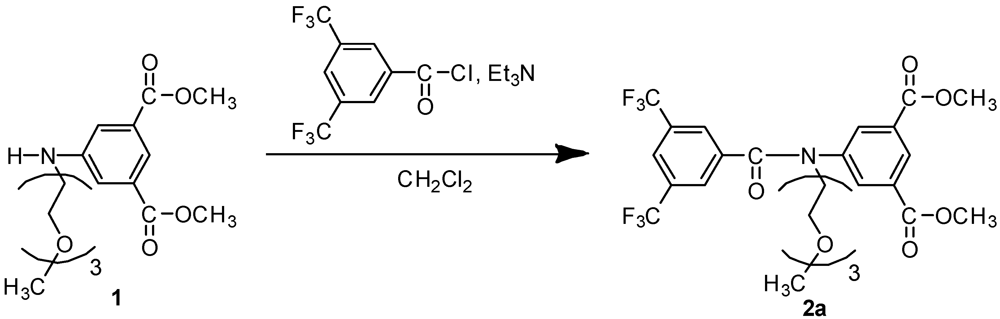
3.4. Synthesis of Initiator 2a

3.5. Polymerization of Monomer 1 with Initiator 2b in the Presence of LiCl
4. Conclusions
Acknowledgments
References
- Zeng, F.; Zimmerman, S.C. Dendrimers in supramolecular chemistry: From molecular recognition to self-assembly. Chem. Rev. 1997, 97, 1681–1712. [Google Scholar]
- Bosman, A.W.; Janssen, H.M.; Meijer, E.W. About dendrimers: Structure, physical properties, and applications. Chem. Rev. 1999, 99, 1665–1688. [Google Scholar]
- Grayson, S.M.; Fréchet, J.M.J. Convergent dendrons and dendrimers: From synthesis to applications. Chem. Rev. 2001, 101, 3819–3868. [Google Scholar]
- Tomalia, D.A.; Fréchet, J.M.J. Discovery of dendrimers and dendritic polymers: A brief historical perspective. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 2719–2728. [Google Scholar]
- Fréchet, J.M.J. Dendrimers and other dendritic macromolecules: From building blocks to functional assemblies in nanoscience and nanotechnology. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 3713–3725. [Google Scholar]
- Kim, Y.H. Hyperbranched polymers 10 years after. J. Polym. Sci. Part A Polym. Chem. 1998, 36, 1685–1698. [Google Scholar]
- Gao, C.; Yan, D. Hyperbranched polymers: From synthesis to applications. Prog. Polym. Sci. 2004, 29, 183–275. [Google Scholar]
- Voit, B. Hyperbranched polymers-all problems solved after 15 years of research? J. Polym. Sci. Part A Polym. Chem. 2005, 43, 2679–2699. [Google Scholar]
- Voit, B.I.; Lederer, A. Hyperbranched and highly branched polymer architectures—Synthetic strategies and major characterization aspects. Chem. Rev. 2009, 109, 5924–5973. [Google Scholar]
- Sunder, A.; Hanselmann, R.; Frey, H.; Mulhaupt, R. Controlled synthesis of hyperbranched polyglycerols by ring-opening multibranching polymerization. Macromolecules 1999, 32, 4240–4246. [Google Scholar]
- Bharathi, P.; Moore, J.S. Controlled synthesis of hyperbranched polymers by slow monomer addition to a core. Macromolecules 2000, 33, 3212–3218. [Google Scholar]
- Möck, A.; Burgath, A.; Hanselmann, R.; Frey, H. Synthesis of hyperbranched aromatic homo- and copolyesters via the slow monomer addition method. Macromolecules 2001, 34, 7692–7698. [Google Scholar]
- Kainthan, R.K.; Muliawan, E.B.; Hatzikiriakos, S.G.; Brooks, D.E. Synthesis, characterization, and viscoelastic properties of high molecular weight hyperbranched polyglycerols. Macromolecules 2006, 39, 7708–7717. [Google Scholar]
- Bharathi, P.; Moore, J.S. Solid-supported hyperbranched polymerization: Evidence for self-limited growth. J. Am. Chem. Soc. 1997, 119, 3391–3392. [Google Scholar]
- Bernal, D.P.; Bedrossian, L.; Collins, K.; Fossum, E. Effect of core reactivity on the molecular weight, polydispersity, and degree of branching of hyperbranched poly(arylene ether phosphine oxide)s. Macromolecules 2003, 36, 333–338. [Google Scholar]
- Suzuki, M.; Ii, A.; Saegusa, T. Multibranching polymerization: Palladium-catalyzed ring-opening polymerization of cyclic carbamate to produce hyperbranched dendritic polyamine. Macromolecules 1992, 25, 7071–7072. [Google Scholar]
- Suzuki, M.; Yoshida, S.; Shiraga, K.; Saegusa, T. New ring-opening polymerization via a π-allylpalladium complex. 5. multibranching polymerization of cyclic carbamate to produce hyperbranched dendritic polyamine. Macromolecules 1998, 31, 1716–1719. [Google Scholar] [CrossRef]
- Ohta, Y.; Fujii, S.; Yokoyama, A.; Furuyama, T.; Uchiyama, M.; Yokozawa, T. Synthesis of well-defined hyperbranched polyamides by condensation polymerization of ab2 monomer through changed substituent effects. Angew. Chem. Int. Ed. 2009, 48, 5942–5945. [Google Scholar]
- Ohta, Y.; Kamijyo, Y.; Fujii, S.; Yokoyama, A.; Yokozawa, T. Synthesis and properties of a variety of well-defined hyperbranched n-alkyl and n-h polyamides by chain-growth condensation polymerization of ab2 monomers. Macromolecules 2011, 44, 5112–5122. [Google Scholar] [CrossRef]
- Hirai, T; Huan, L; Ohta, Y; Yokozawa, T; Tanaka, K. Surface segregation of well-defied n-substituted hyperbranched polyamides in linear polymer matrix. Chem. Lett. 2011, 40, 366–367. [Google Scholar] [CrossRef]
- Sugi, R.; Ohishi, T.; Yokoyama, A.; Yokozawa, T. Novel water-soluble poly(m-benzamide)s: Precision synthesis and thermosensitivity in aqueous solution. Macromol. Rapid Commun. 2006, 27, 716–721. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ohta, Y.; Kamijyo, Y.; Yokoyama, A.; Yokozawa, T. Synthesis of Well-Defined, Water-Soluble Hyperbranched Polyamides by Chain-Growth Condensation Polymerization of AB2 Monomer. Polymers 2012, 4, 1170-1182. https://doi.org/10.3390/polym4021170
Ohta Y, Kamijyo Y, Yokoyama A, Yokozawa T. Synthesis of Well-Defined, Water-Soluble Hyperbranched Polyamides by Chain-Growth Condensation Polymerization of AB2 Monomer. Polymers. 2012; 4(2):1170-1182. https://doi.org/10.3390/polym4021170
Chicago/Turabian StyleOhta, Yoshihiro, Yusuke Kamijyo, Akihiro Yokoyama, and Tsutomu Yokozawa. 2012. "Synthesis of Well-Defined, Water-Soluble Hyperbranched Polyamides by Chain-Growth Condensation Polymerization of AB2 Monomer" Polymers 4, no. 2: 1170-1182. https://doi.org/10.3390/polym4021170