Immobilization of Poly(1,1-dimethysilacyclobutane) by Means of Anionic Ring-Opening Polymerization on Organic Nanoparticles and Reinvestigation of Crystallization

 ,
,
Abstract
:1. Introduction
2. Experimental Section
2.1. Instrumentation
2.2. Materials
2.3. Anionic Polymerization of 1,1-Dimethylsilacyclobutane (DMSB)
2.4. Synthesis of Cross-Linked PS Particles
2.5. Surface-Initiated Anionic Ring Opening Polymerization of DMSB
3. Results and Discussion
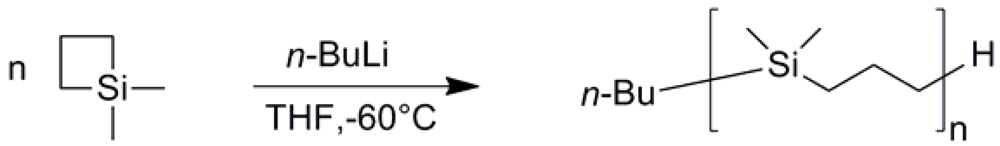
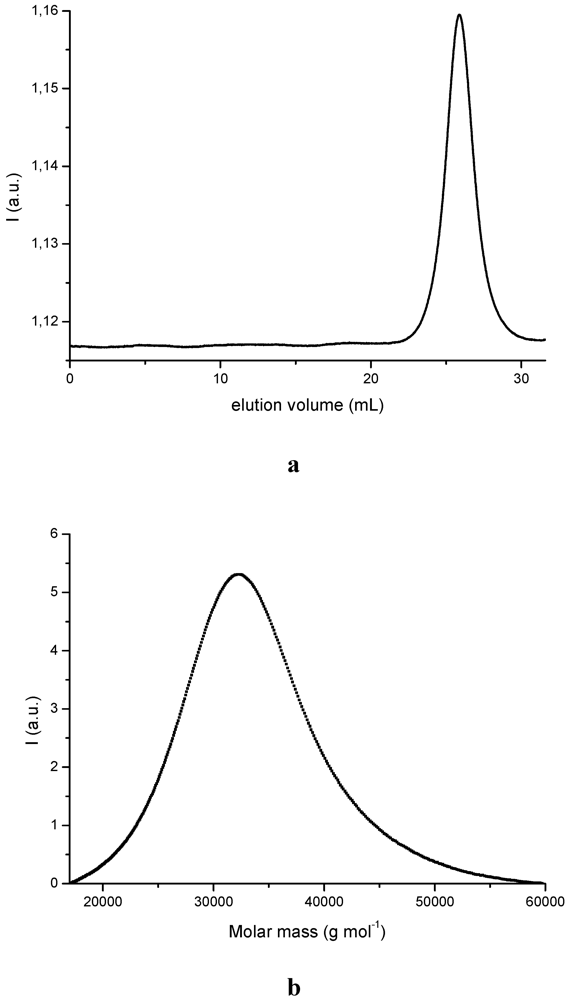
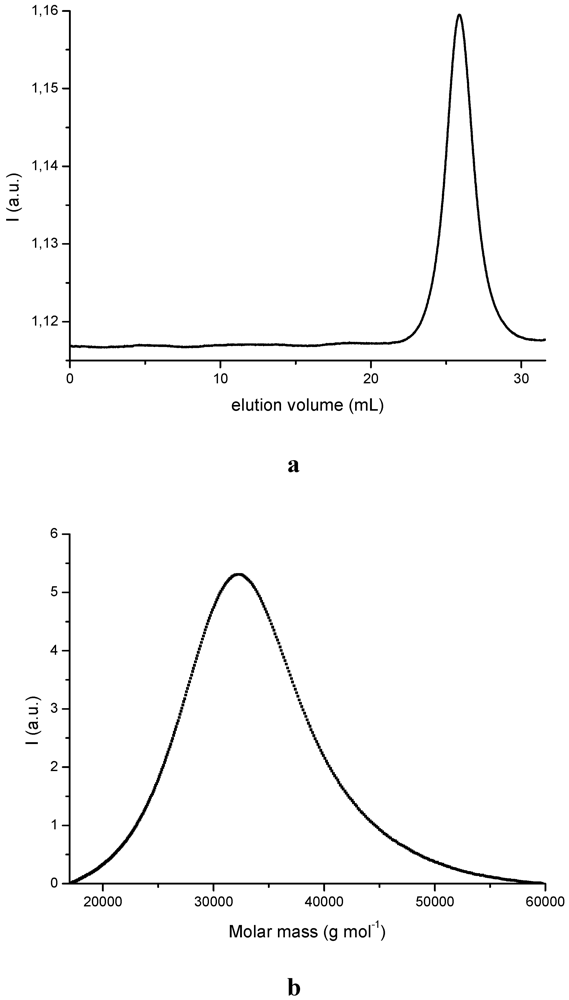
3.1. PDMSB Synthesis
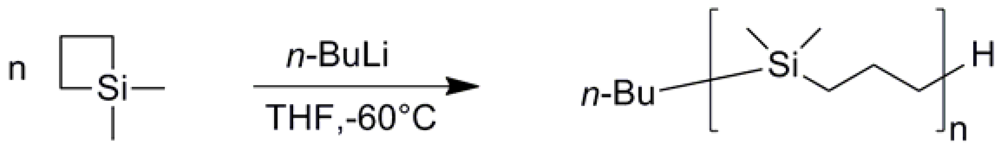

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mna (g mol−1) | Mwa (g mol−1) | PDIa (g mol−1) | Mnb (g mol−1) | Mwb (g mol−1) | PDIb |
|---|---|---|---|---|---|
| 2 250 | 2 590 | 1.15 | 4 110 | 4 650 | 1.13 |
| 5 200 | 5 620 | 1.08 | 7 220 | 8 190 | 1.13 |
| 8 210 | 8 920 | 1.09 | 9 920 | 10 480 | 1.06 |
| 28 680 | 32 900 | 1.15 | 31 450 | 32 610 | 1.04 |
| 52 250 | 59 760 | 1.14 | 57 590 | 59 700 | 1.04 |
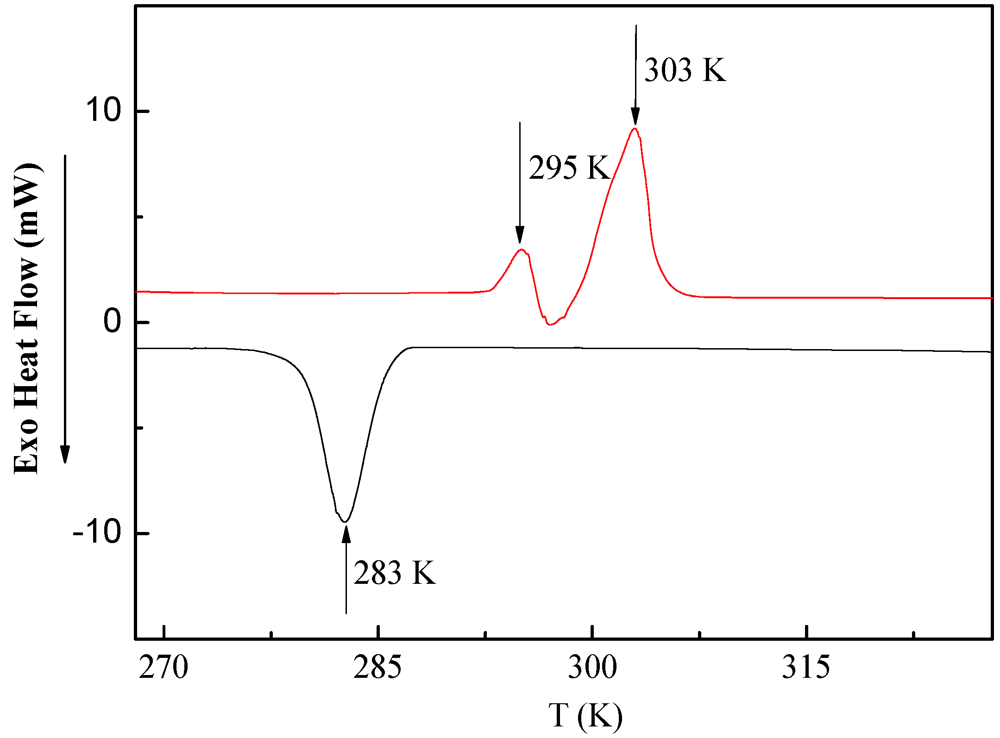
3.2. DSC Measurements of PDMSB Homopolymers
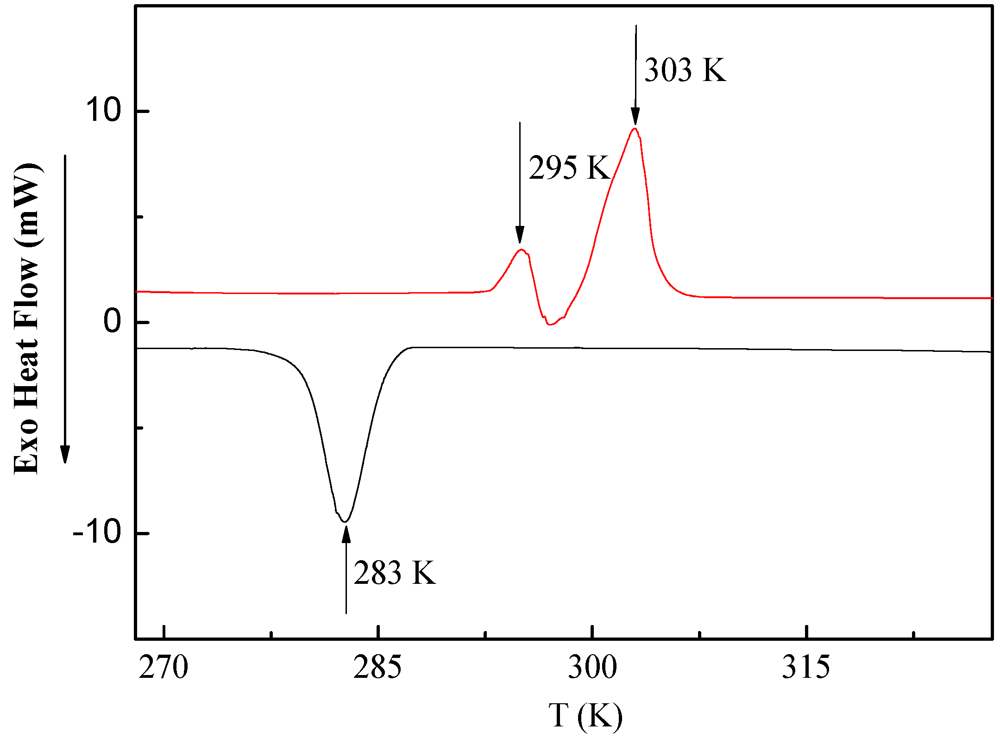
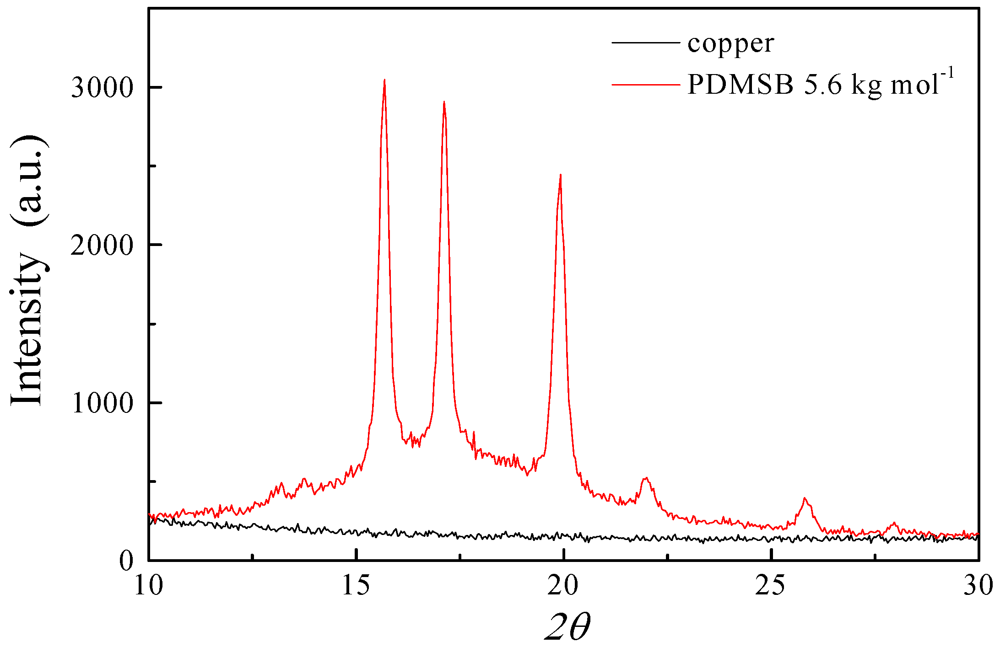
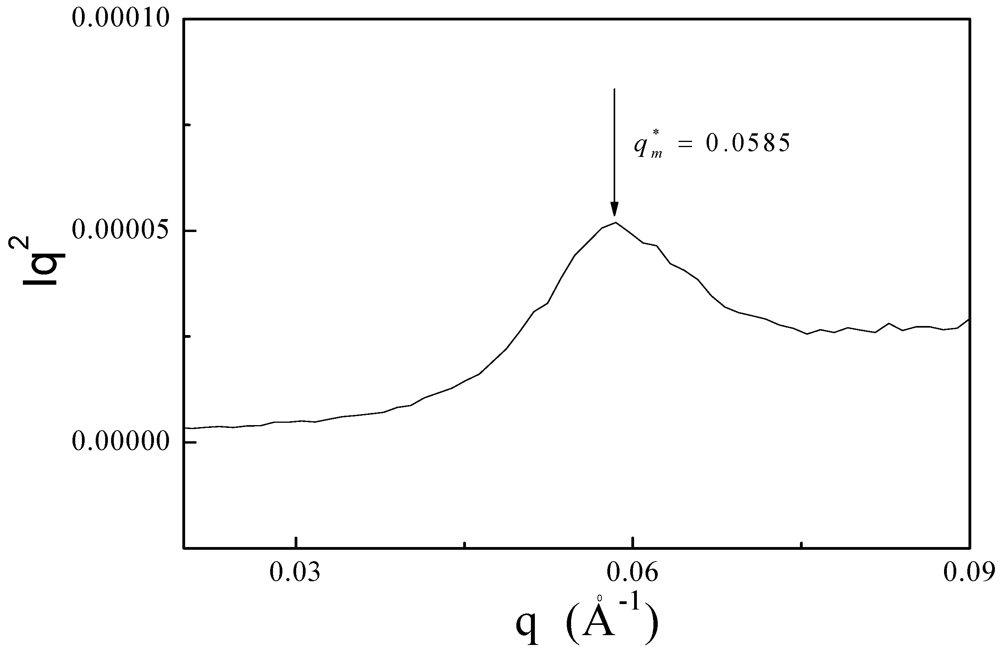
3.3. XRD and SAXS Measurements of PDMSB Homopolymers
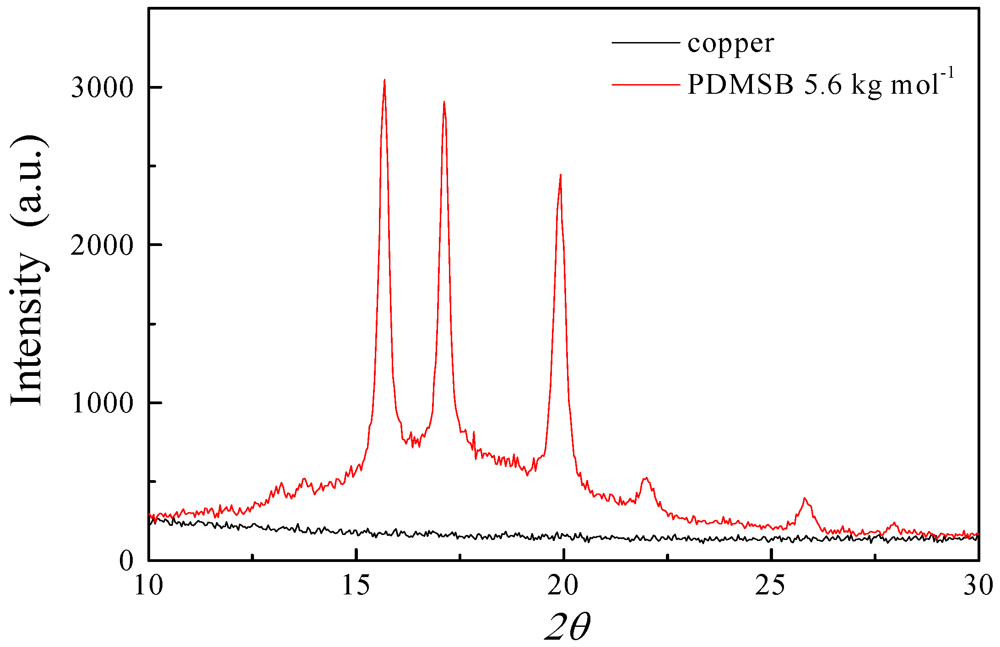
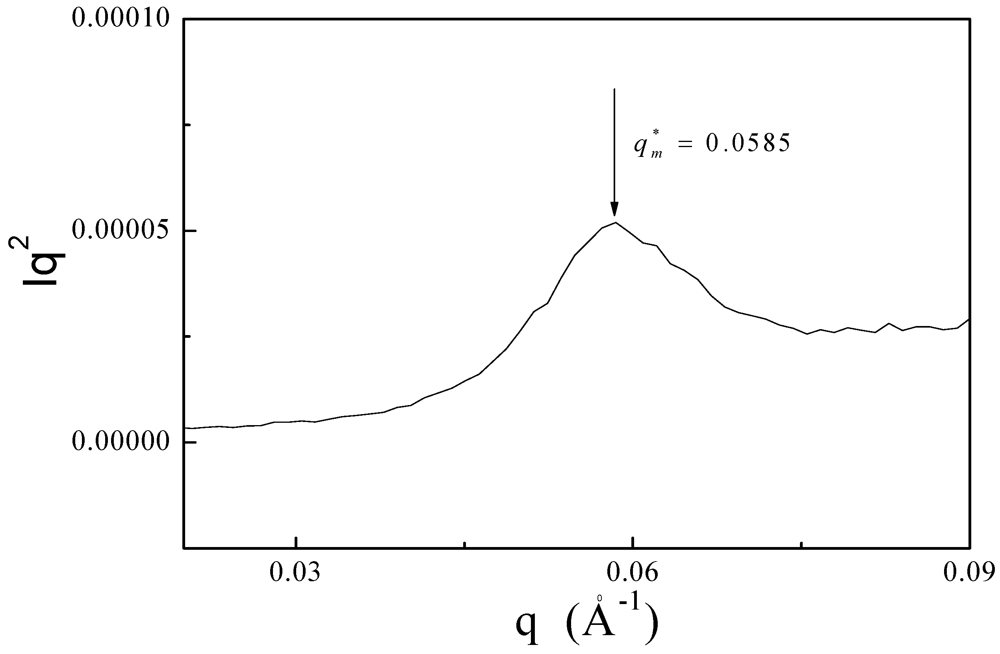
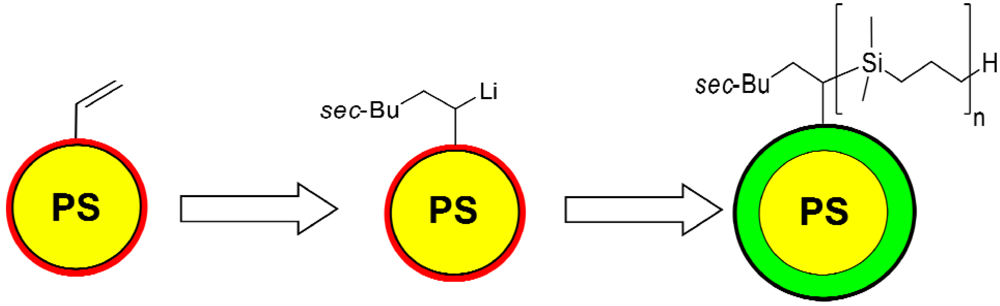
3.4. Surface-Initiated Anionic Polymerization of DMSB on Cross-Linked PS Nanoparticles
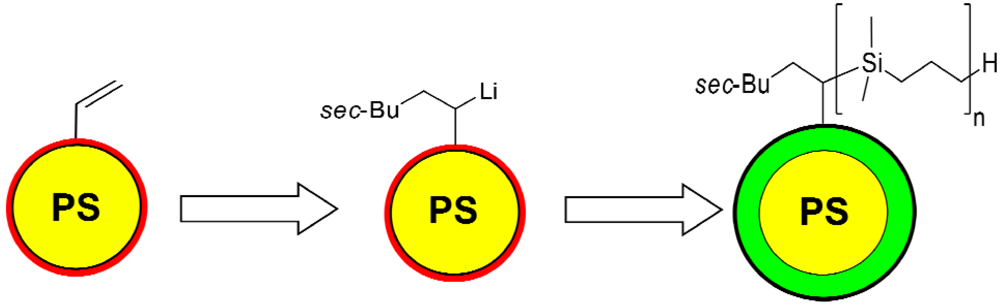
3.5. Characterization of PDMSB-Grafted PS Nanoparticles
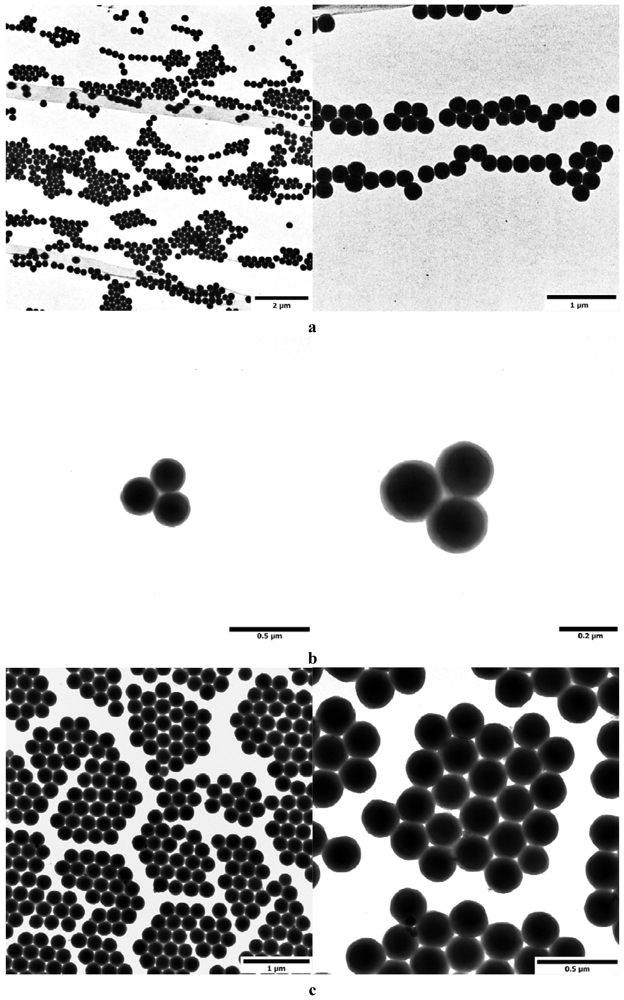
3.5.1. TEM Investigations

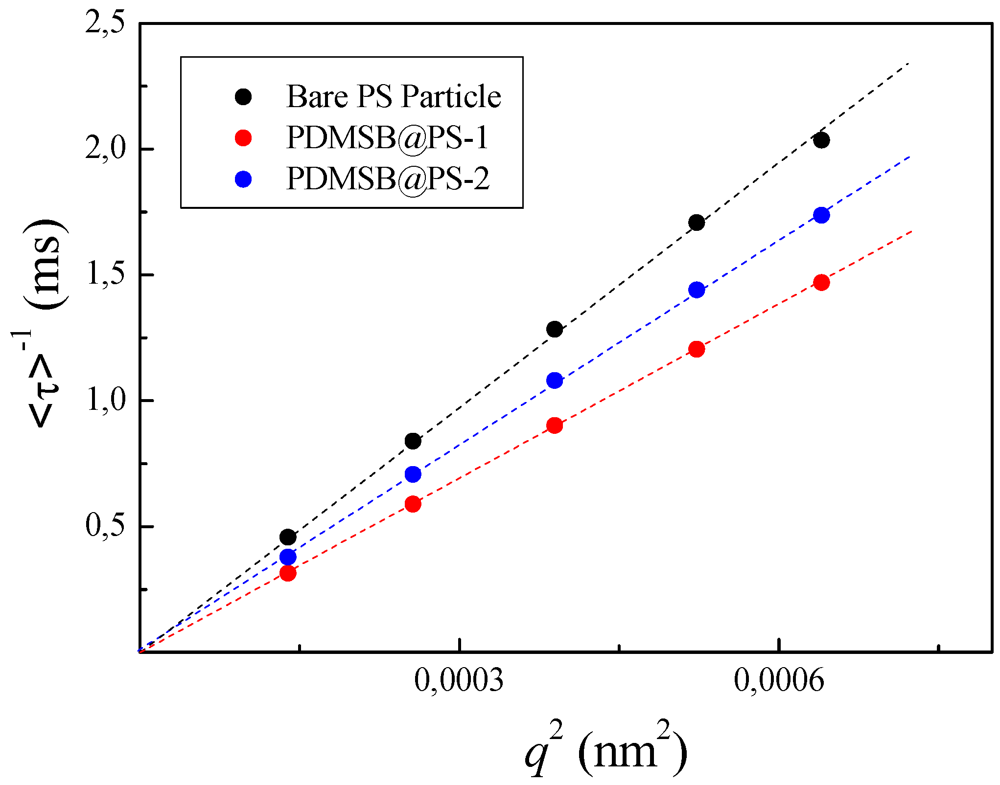
3.5.2. DLS Measurements of PDMSB-Grafted PS Particles
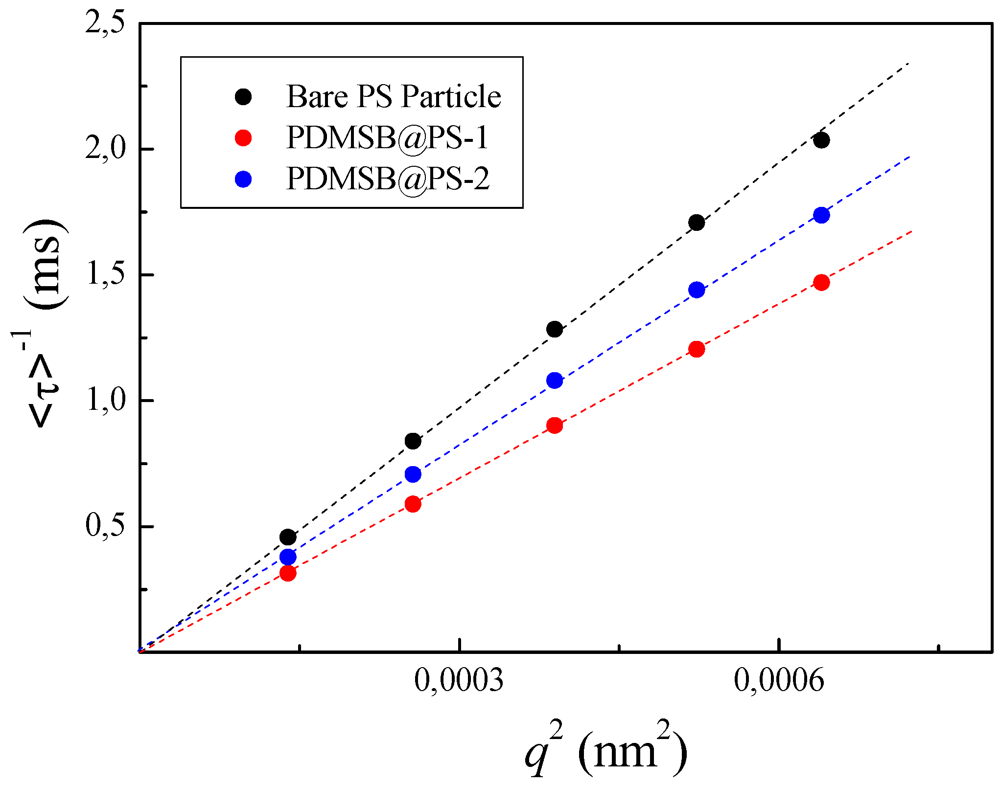
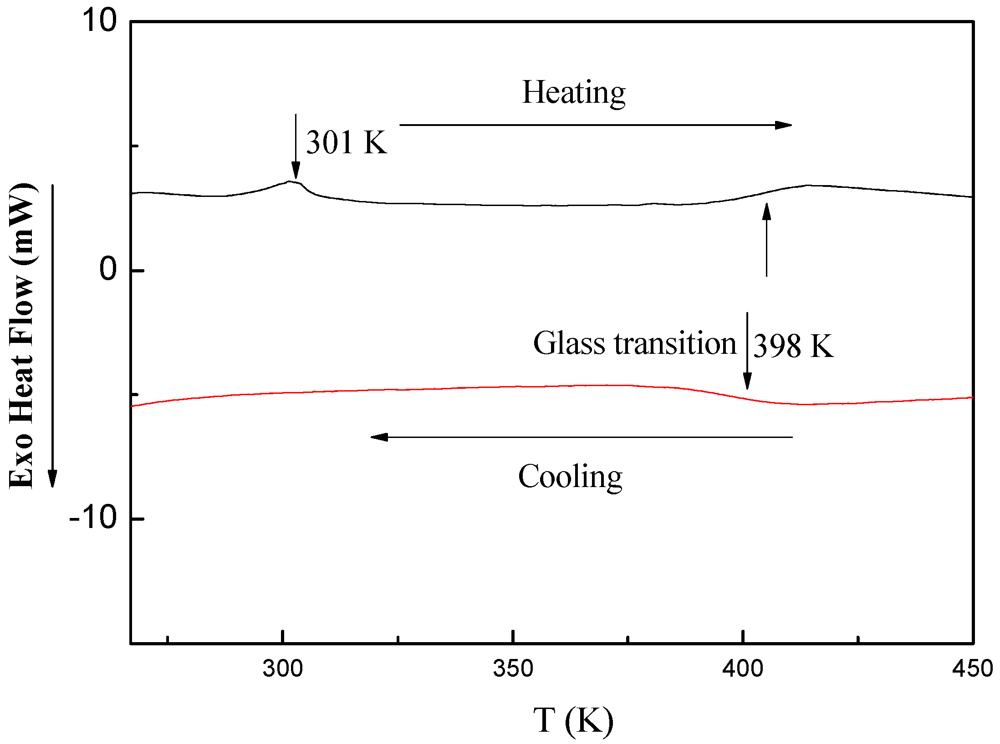
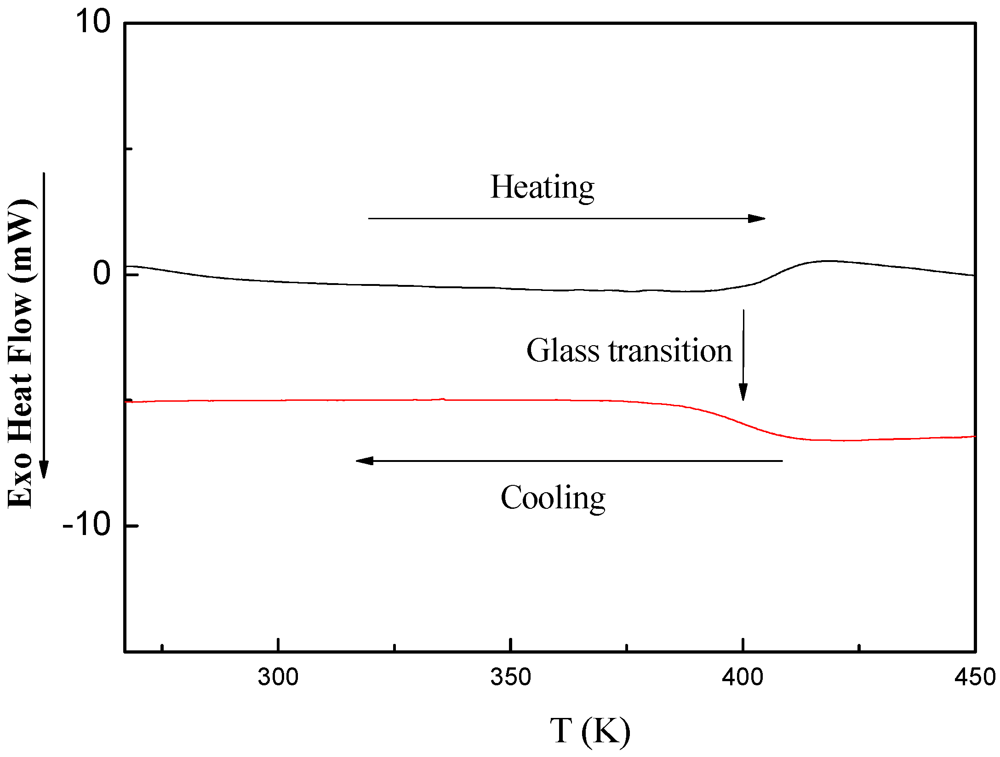
3.5.3. DSC Investigations of PDMSB-Grafted PS Particles
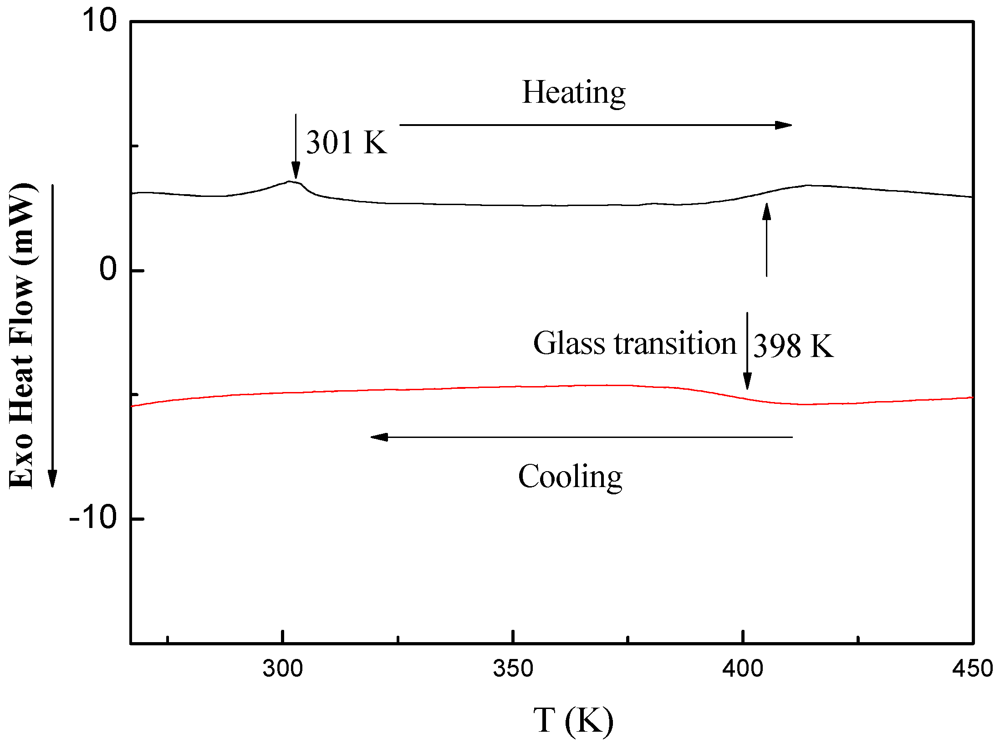
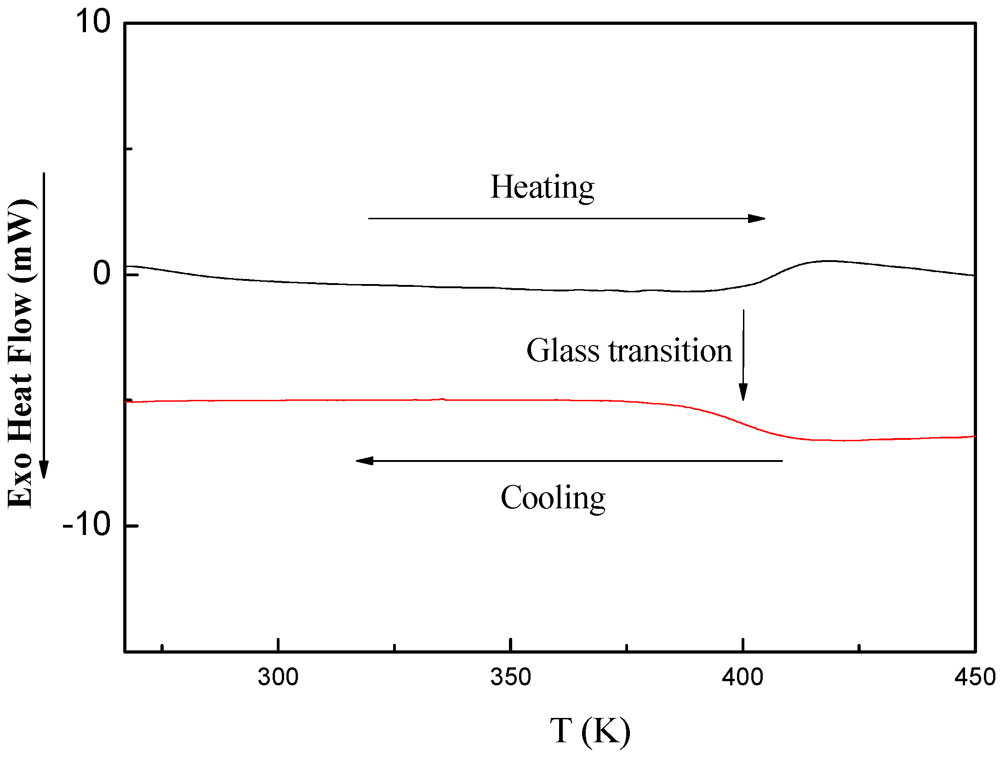
4. Conclusions
Acknowledgments
References
- Birot, M.; Pillot, J.-P.; Dunogues, J. Comprehensive chemistry of polycarbosilanes, polysilazanes, and polycarbosilazanes as precursors of ceramics. Chem. Rev. 1995, 95, 1443–1477. [Google Scholar] [CrossRef]
- Sanchez, J.C.; Trogler, W.C. Hydrosilylation of diynes as a route to functional polymers delocalized through silicon. Macromol. Chem. Phys. 2008, 209, 1527–1540. [Google Scholar] [CrossRef]
- Vakifahmetoglu, C. Fabrication and properties of ceramic 1D nanostructures from preceramic polymers: a review. Adv. Appl. Ceram. 2011, 110, 188–204. [Google Scholar] [CrossRef]
- Fukushima, M.; Colombo, P. Silicon carbide-based foams from direct blowing of polycarbosilane. J. Eur. Ceram. Soc. 2012, 32, 503–510. [Google Scholar] [CrossRef]
- Schawaller, D.; Clauß, B.; Buchmeiser, M.R. Ceramic filament fibers—a review. Macromol. Mater. Eng. 2012, 297, 502–522. [Google Scholar] [CrossRef]
- Yamashita, H.; Suzuki, Y.; Rao, T.V.; Uchimaru, Y. Synthesis and electrochemical, optical, and thermal properties of polycarbosilanes with silylene–vinylene–phenylene–vinylene backbones and triphenylamine or carbazole unit-containing side chains. J. Organomet. Chem. 2012, 710, 59–67. [Google Scholar] [CrossRef]
- Gupta, R.K.; Mishra, R.; Tiwari, R.K.; Ranjan, A.; Saxena, A.K. Studies on the rheological behavior of polycarbosilane part i: Effect of time, temperature and atmosphere. silicon 2010, 3, 27–35. [Google Scholar]
- Vakifahmetoglu, C.; Balliana, M.; Colombo, P. Ceramic foams and micro-beads from emulsions of a preceramic polymer. J. Eur. Ceram. Soc. 2011, 31, 1481–1490. [Google Scholar] [CrossRef]
- Friebe, L.; Liu, K.; Obermeier, B.; Petrov, S.; Dube, P.; Manners, I. Pyrolysis of Polycarbosilanes with pendant nickel clusters: Synthesis and characterization of magnetic ceramics containing nickel and nickel silicide nanoparticles. Chem. Mater. 2007, 19, 2630–2640. [Google Scholar] [CrossRef]
- Chen, J.; He, G.; Liao, Z.; Zeng, B.; Ye, J.; Chen, L.; Xia, H.; Zhang, L. Control of structure formation of polycarbosilane synthesized from polydimethylsilane by Kumada rearrangement. J. Appl. Polym. Sci. 2008, 108, 3114–3121. [Google Scholar] [CrossRef]
- Fang, Y.; Huang, M.; Yu, Z.; Xia, H.; Chen, L.; Zhang, Y.; Zhang, L. Synthesis, Characterization, and Pyrolytic Conversion of a Novel Liquid Polycarbosilane. J. Am. Ceram. Soc. 2008, 91, 3298–3302. [Google Scholar] [CrossRef]
- Li, H.; Zhang, L.; Cheng, L.; Wang, Y.; Yu, Z.; Huang, M.; Tu, H.; Xia, H. Polymer–ceramic conversion of a highly branched liquid polycarbosilane for SiC-based ceramics. J. Mater. Sci. 2008, 43, 2806–2811. [Google Scholar] [CrossRef]
- Yu, Z.; Li, R.; Zhan, J.; Zhou, C.; Yang, L.; He, G.; Xia, H. Synthesis and characterization of a propargyl-substituted polycarbosilane with high ceramic yield. J. Appl. Polym. Sci. 2011, 121, 3400–3406. [Google Scholar]
- Will, U.; Veljanovski, D.; Harter, P.; Rieger, B. Hyperbranched Polycarbosilanes of Homogeneous Architecture: Regioselective Hydrosilylation of AB2Monomers and Consecutive Functionalization. Macromolecules 2010, 43, 934–938. [Google Scholar] [CrossRef]
- Liao, C.X.; Weber, W.P. Synthesis and Properties of Novel Functionally Substituted Carbosilane Polymers. Macromolecules 1993, 26, 563–566. [Google Scholar] [CrossRef]
- Ganicz, T.; Staficzyk, W.; Biatecka-Florjaficzyk, E.; Sledzifiska, I. Synthesis and characterization of liquid crystal polycarbosilanes: poly(1-methyl-1-silaethylene), poly(1-methyl-1-silabutane)and poly(1-silabutane) with pendant mesogenic groups. Polymer 1996, 37, 4167–4174. [Google Scholar] [CrossRef]
- Shankar, R.; Saxena, A.; Brar, A.S. Synthesis and reactivity of novel oligosilanes bearing functional carbosilane side chains. J. Organomet. Chem. 2001, 628, 262–270. [Google Scholar] [CrossRef]
- Wurm, F.; Schule, H.; Frey, H. Amphiphilic Linear-Hyperbranched Block Copolymers with Linear Poly(ethylene oxide) and Hyperbranched Poly(carbosilane) Block. Macromolecules 2008, 41, 9602–9611. [Google Scholar] [CrossRef]
- Wurm, F.; Hilf, S.; Frey, H. Electroactive linear-hyperbranched block copolymers based on linear poly(ferrocenylsilane)s and hyperbranched poly(carbosilane)s. Chem. Eur. J. 2009, 15, 9068–9077. [Google Scholar] [CrossRef]
- Kudo, H.; Fujiwara, Y.; Miyasaka, M.; Nishikubo, T. Synthesis of polycarbosilanes by A2 + Bn (n = 2, 3, and 4) type hydrosilylation reaction and evaluation of their refractive index properties. J. Polym. Sci. A Polym. Chem. 2010, 48, 5746–5751. [Google Scholar] [CrossRef]
- Schacher, F.H.; Rupar, P.A.; Manners, I. Functional Block Copolymers: Nanostructured Materials with Emerging Applications. Angew. Chem. Int. Ed. 2012, 51, 7898–7921. [Google Scholar] [CrossRef]
- Rangou, S.; Shishatskiy, S.; Filiz, V.; Abetz, V. Poly(vinyl trimethylsilane) and block copolymers of vinyl trimethylsilane with isoprene: Anionic polymerization, morphology and gas transport properties. Eur. Polym. J. 2011, 47, 723–729. [Google Scholar] [CrossRef]
- Zhou, S.Q.; Park, Y.T.; Manuel, G.; Weber, W.P. Anionic ring opening polymerization of 1-silacyclopent-3-ene. Polym. Bull. 1990, 23, 491–496. [Google Scholar] [CrossRef]
- Liao, C.X.; Weber, W.P. Synthesis and characterization of poly(1-methyl-l-silabutane), poly(1-phenyl-l-silabutane) and poly(1-silabutane). Polym. Bull. 1992, 28, 281–286. [Google Scholar] [CrossRef]
- Theurig, M.; Weber, W.P. Stereoselective anionic ring opening polymerization of 1,1-dimethyl-l-silacyclobutene. Polym. Bull. 1992, 28, 17–21. [Google Scholar] [CrossRef]
- Rulkens, R.; Lough, A.J.; Manners, I. Anionic Ring-Opening Oligomerization and Polymerization of Silicon-Bridged [1]Ferrocenophanes: Characterization of Short-Chain Models for Poly(ferrocenylsilane) High Polymers. J. Am. Chem. Soc. 1994, 116, 797–798. [Google Scholar] [CrossRef]
- Suzuki, M.; Kotani, J.; Gyobu, S.; Kaneko, T.; Saegusa, T. Synthesis of Sequence-Ordered Polysilane by Anionic Ring-Opening Polymerization of Phenylnonamethylcyclopentasilane. Macromolecules 1994, 27, 2360–2363. [Google Scholar] [CrossRef]
- Kulbaba, K.; Manners, I. Polyferrocenylsilanes: Metal-Containing Polymers for Materials Science, Self-Assembly and Nanostructure Applications. Macromol. Rap. Comm. 2001, 22, 711–724. [Google Scholar] [CrossRef]
- Bellas, V.; Rehahn, M. Polyferrocenylsilan-basierte Polymersysteme. Angew. Chem. 2007, 119, 5174–5197. [Google Scholar] [CrossRef]
- Bellas, V.; Rehahn, M. Polyferrocenylsilane-Based Polymer Systems. Angew. Chem. Int. Ed. 2007, 46, 5082–5104. [Google Scholar] [CrossRef]
- Knischka, R.; Frey, H.; Rapp, U.; Mayer-Posner, F.J. Living anionic ring-opening polymerization of 1,1-dipropylsilacyclobutane. Macromol. Rap. Comm. 1998, 19, 455–459. [Google Scholar] [CrossRef]
- Matsumoto, K.; Yamaoka, H. Living Anionic Ring-Opening Polymerization of 1,1-Dimethylsilacyclobutane. Macromolecules 1995, 28, 7029–7031. [Google Scholar] [CrossRef]
- Matsumoto, K.; Shimazu, H.; Deguchi, M.; Yamaoka, H. Anionic Ring-Opening Polymerization of Silacyclobutane Derivatives. J. Polym. Sci. A Poly. Chem. 1997, 35, 3207–3216. [Google Scholar] [CrossRef]
- Kawahara, S.; Nagai, A.; Kazama, T.; Takano, A.; Isono, Y. Preparation of Poly(1,1-dimethyl silabutane) by Anionic Polymerization and Its Crystallization. Macromolecules 2004, 37, 315–321. [Google Scholar] [CrossRef]
- Nghiem, Q.D.; Kim, D.-P. Direct Preparation of High Surface Area Mesoporous SiC-Based Ceramic by Pyrolysis of a Self-Assembled Polycarbosilane-block-Polystyrene Diblock Copolymer. Chem. Mater. 2008, 20, 3735–3739. [Google Scholar] [CrossRef]
- Gallei, M.; Tockner, S.; Klein, R.; Rehahn, M. Silacyclobutane-Based Diblock Copolymers with Vinylferrocene, Ferrocenylmethyl Methacrylate, and [1]Dimethylsilaferrocenophane. Macromol. Rap. Comm. 2010, 31, 889–896. [Google Scholar] [CrossRef]
- Ungar, G.; Putra, E.G. R.; de Silva, D.S. M.; Shcherbina, M.A.; Waddon, A.J. The effect of self-poisoning on crystal morphology and growth rates. Adv. Polym. Sci. 2005, 180, 45–87. [Google Scholar]
- Keller, N.; Pham-Huu, C.; Crouzet, C.; Ledoux, M.J.; Savin-Poncet, S.; Nougayrede, J.-B.; Bousquet, J. Direct oxidation of H2S into S. New catalysts and processes based on SiC support. Catal. Today 1999, 53, 535–542. [Google Scholar]
- Connolly, E.J.; O'Halloran, G.M.; Pham, H.T.M.; Sarro, P.M.; French, P.J. Comparison of porous silicon, porous polysilicon and porous silicon carbide as materials for humidity sensing applications. Sens. Actuators A 2002, 99, 25–30. [Google Scholar] [CrossRef]
- TamilSelvan, S.; Aldeyab, S.S.; Zaidi, J.S. M.; Arivuoli, D.; Ariga, K.; Mori, T.; Vinu, A. Preparation and characterization of highly ordered mesoporous SiC nanoparticles with rod shaped morphology and tunable pore diameters. Journal of Materials Chemistry 2011, 21, 8792–8799. [Google Scholar]
- Kovacs, A.J.; Gonthier, A. Crystallization and fusion of self-seeded polymers. Colloid Polym. Sci. 1972, 250, 530–551. [Google Scholar]
- Kovacs, A.J.; Gonthier, A.; Straupe, C. Isothermal growth, thickening, and melting of poly(ethylene oxide) singel crystals in the bulk. J. Polym. Sci. Polym. Symp. 1975, 50, 283–325. [Google Scholar] [CrossRef]
- Kovacs, A.J.; Straupe, C.; Gonthier, A. Isothermal growth, thickening, and melting of poly(ethylene oxide) singel crystals in the bulk. J. Polym. Sci. Polym. Symp. 1977, 59, 31–54. [Google Scholar] [CrossRef]
- Kovacs, A.J.; Straupe, C. Isothermal growth, thickening and melting of poly(ethylene oxide) single crystals in the bulk. Part 4: Dependence of pathological crystal habits on temperature and thermal history. Faraday Discuss. Chem. Soc. 1979, 68, 225–239. [Google Scholar]
- Kovacs, A.J.; Straupe, C. Isothermal growth, thickening and melting of poly(ethylene oxide) single crystals in the bulk. J. Cryst. Growth 1980, 48, 210–226. [Google Scholar] [CrossRef]
- Cheng, S.Z. D.; Zhang, A.; Chen, J. Existence of a Transient Nonintegral Folding Lamellar Crystal in a Low Molecular Mass Poly (Ethylene Oxide) Fraction Crystallized from the Melt. J. Polym. Sci. C Polym. Lett. 1990, 28, 233–239. [Google Scholar] [CrossRef]
- Cheng, S.Z. D.; Zhang, A.; Barley, J.S.; Chen, J.; Habenschuss, A.; Zschack, P.R. Isothermal Thickening and Thinning Processes in Low Molecular Weight Poly(ethy1ene oxide) Fractions. 1. From Nonintegral-Folding to Integral-Folding Chain Crystal Transitions. Macromolecules 1991, 24, 3973–3944. [Google Scholar]
- Cheng, S.Z.D.; Zhang, A.; Chen, J.; Heberer, D.P. Nonintegral and Integral Folding Crystal Growth in Low-Molecular Mass Poly (ethylene Oxide) Fractions. I. Isothermal lamellar Thickening and Thinning. J. Polym. Sci. B Polym. Phys. 1991, 29, 287–297. [Google Scholar] [CrossRef]
- Cheng, S.Z.D.; Chen, J.; Zhang, A.; Heberer, D.P. Nonintegral and Integral Folding Crystal Growth in Low-Molecular Mass Poly (ethylene Oxide) Fractions. II. End-Group Effect: α,ω-Methoxy-Poly(ethylene Oxide). J. Polym. Sci. B Polym. Phys. 1991, 29, 299–310. [Google Scholar]
- Cheng, S.Z.D.; Chen, J. Nonintegral and Integral Folding Crystal Growth in Low-Molecular Mass Poly (ethylene Oxide) Fractions,III. Linear Crystal Growth Rates and Crystal Morphology. J. Polym. Sci. B Polym. Phys. 1991, 29, 311–327. [Google Scholar] [CrossRef]
- Cheng, S.Z.D.; Chen, J.; Barley, J.S.; Zhang, A. Isothermal Thickening and Thinning Processes in Low Molecular Weight Poly(ethy1ene oxide) Fractions Crystallized from the Melt.3. Molecular Weight Dependence. Macromolecules 1992, 25, 1453–1460. [Google Scholar] [CrossRef]
- Cheng, S.Z.D.; Wu, S.S.; Chen, J.; Zhuo, Q.; Quirk, R.P.; von Meerwall, E.D.; Hsiao, B.S.; Habenschuss, A.; Zschack, P.R. Isothermal Thickening and Thinning Processes in Low Molecular Weight Poly(ethy1ene oxide) Fractions Crystallized from the Melt.4. End-Group Dependence. Macromolecules 1993, 26, 5105–5117. [Google Scholar]
- Cheng, S.Z.D.; Chen, J.; Wu, S.X.; Zhang, A.; Yandrasits, M.A.; Zhuo, Q.; Quirk, R.P.; Habenschuss, A.; Zschack, P.R. Nonintegral and Integral Folding Crystal Growth in Low Molecular Weight Poly(Ethylene Oxide) Fractions. Cryst. Polym. 1993, 405, 51–62. [Google Scholar]
- Lee, S.-W.; Chen, E.; Zhang, A.; Yoon, Y.; Moon, B.S.; Lee, S.; Harris, F.W.; Cheng, S.Z. D. Isothermal Thickening and Thinning Processes in Low Molecular Weight Poly(ethylene oxide) Fractions Crystallized from the Melt. 5. Effect of Chain Defects. Macromolecules 1996, 29, 8816–8823. [Google Scholar]
- Chen, E.-Q.; Lee, S.-W.; Zhang, A.; Moon, B.-S.; Mann, I.; Harris, F.W.; Cheng, S.Z. D.; Hsiao, B.S.; Yeh, F.; Merrewell, E.von; Grubb, D.T. Isothermal Thickening and Thinning Processes in Low-Molecular-Weight Poly(ethylene oxide) Fractions Crystallized from the Melt. 8. Molecular Shape Dependence. Macromolecules 1999, 32, 4784–4793. [Google Scholar]
- Chen, E.-Q.; Lee, S.-W.; Zhang, A.; Moon, B.-S.; Honigfort, P.S.; Mann, I.; Lin, H.-M.; Harris, F.W.; Cheng, S.Z. D.; Hsiao, B.S.; Yeh, F. Isothermal thickening and thinning processes in low molecular weight poly(ethylene oxide) fractions crystallized from the melt 6. Configurational defects in molecules. Polymer 1999, 40, 4543–4551. [Google Scholar] [CrossRef]
- Mazurowski, M.; Gallei, M.; Li, J.; Didzoleit, H.; Stühn, B.; Rehahn, M. Redox-Responsive Polymer Brushes Grafted from Polystyrene Nanoparticles by Means of Surface Initiated Atom Transfer Radical Polymerization. Macromolecules 2012, 45, 8970–8981. [Google Scholar] [CrossRef]
- Rieger, J. The Glass Transition Temperature of Polystyrene. J. Therm. Anal. 1996, 46, 965–972. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gallei, M.; Li, J.; Elbert, J.; Mazurowski, M.; Schönberger, A.; Schmidt, C.; Stühn, B.; Rehahn, M. Immobilization of Poly(1,1-dimethysilacyclobutane) by Means of Anionic Ring-Opening Polymerization on Organic Nanoparticles and Reinvestigation of Crystallization. Polymers 2013, 5, 284-302. https://doi.org/10.3390/polym5010284
Gallei M, Li J, Elbert J, Mazurowski M, Schönberger A, Schmidt C, Stühn B, Rehahn M. Immobilization of Poly(1,1-dimethysilacyclobutane) by Means of Anionic Ring-Opening Polymerization on Organic Nanoparticles and Reinvestigation of Crystallization. Polymers. 2013; 5(1):284-302. https://doi.org/10.3390/polym5010284
Chicago/Turabian StyleGallei, Markus, Junyu Li, Johannes Elbert, Markus Mazurowski, Astrid Schönberger, Christian Schmidt, Bernd Stühn, and Matthias Rehahn. 2013. "Immobilization of Poly(1,1-dimethysilacyclobutane) by Means of Anionic Ring-Opening Polymerization on Organic Nanoparticles and Reinvestigation of Crystallization" Polymers 5, no. 1: 284-302. https://doi.org/10.3390/polym5010284