Photochemical Production of Interpenetrating Polymer Networks; Simultaneous Initiation of Radical and Cationic Polymerization Reactions

Abstract
: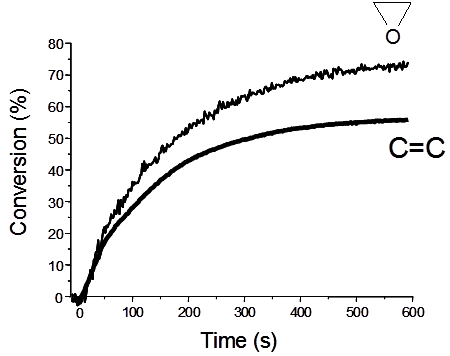
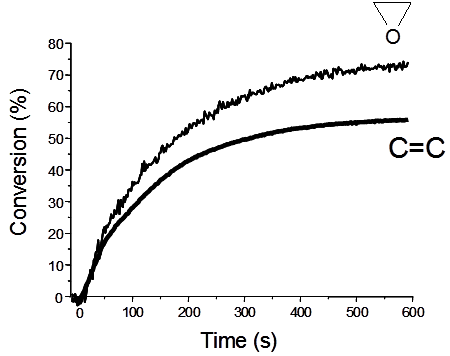{kind=link}
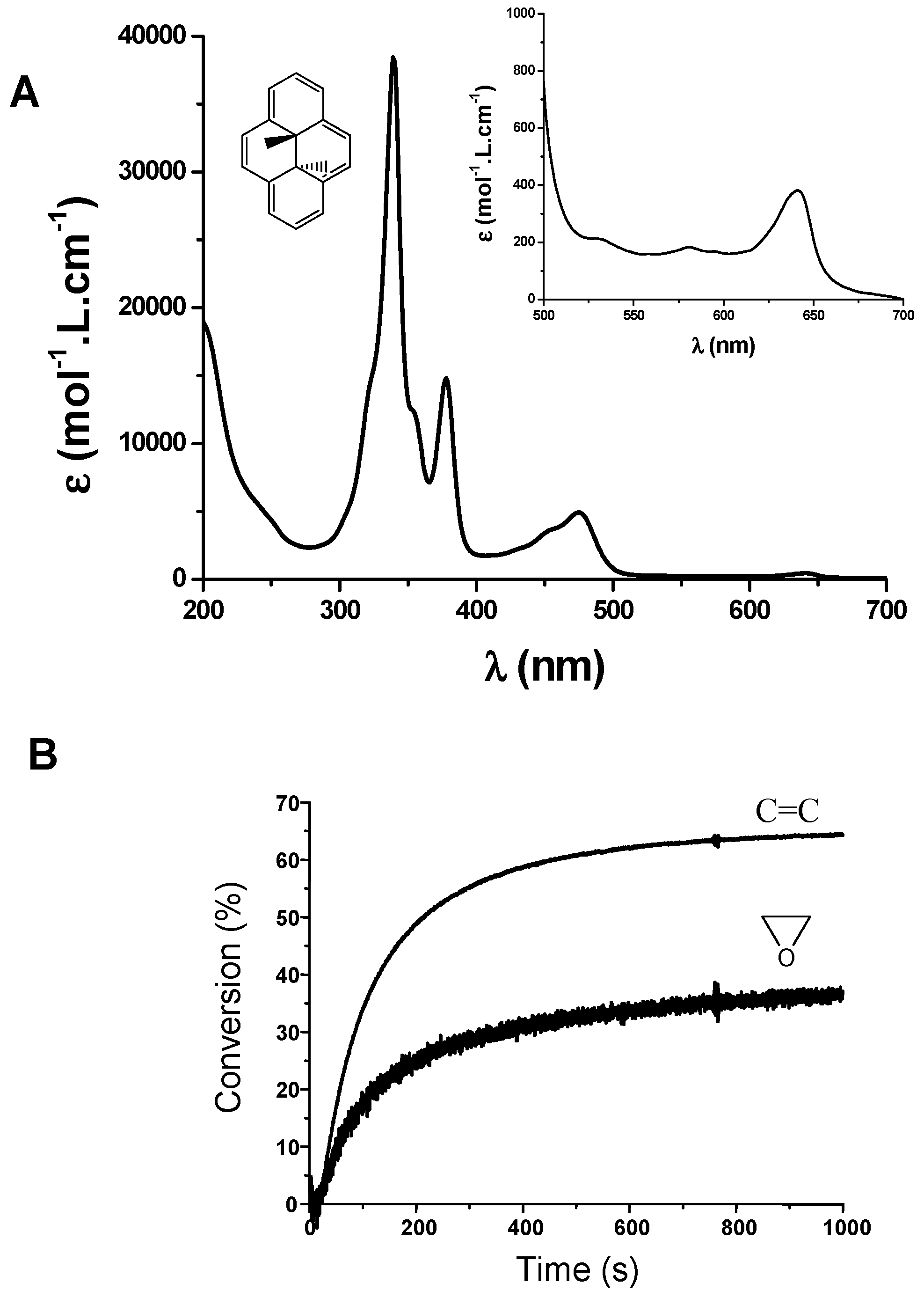{kind=link}
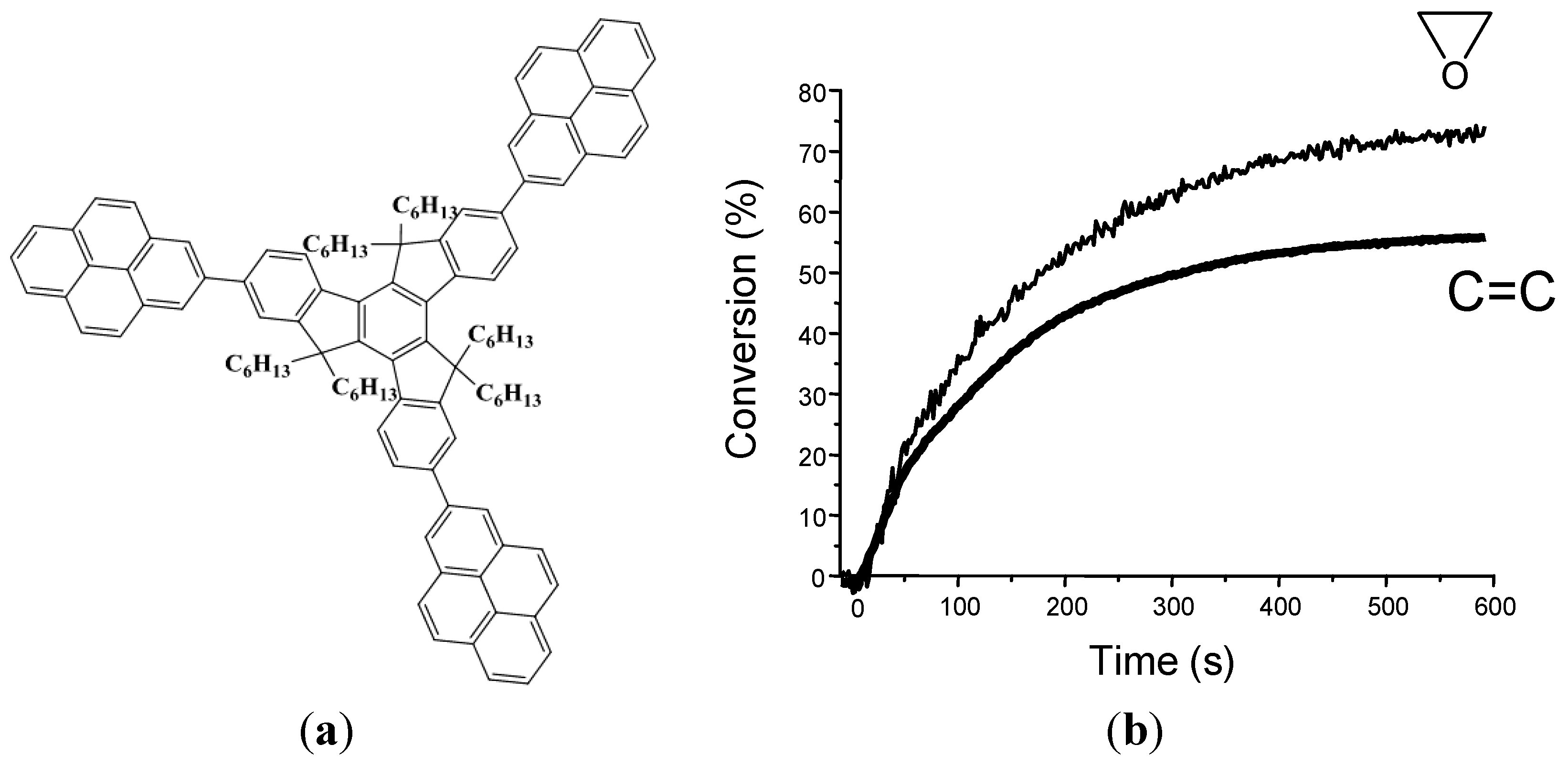{kind=link}
{kind=link}
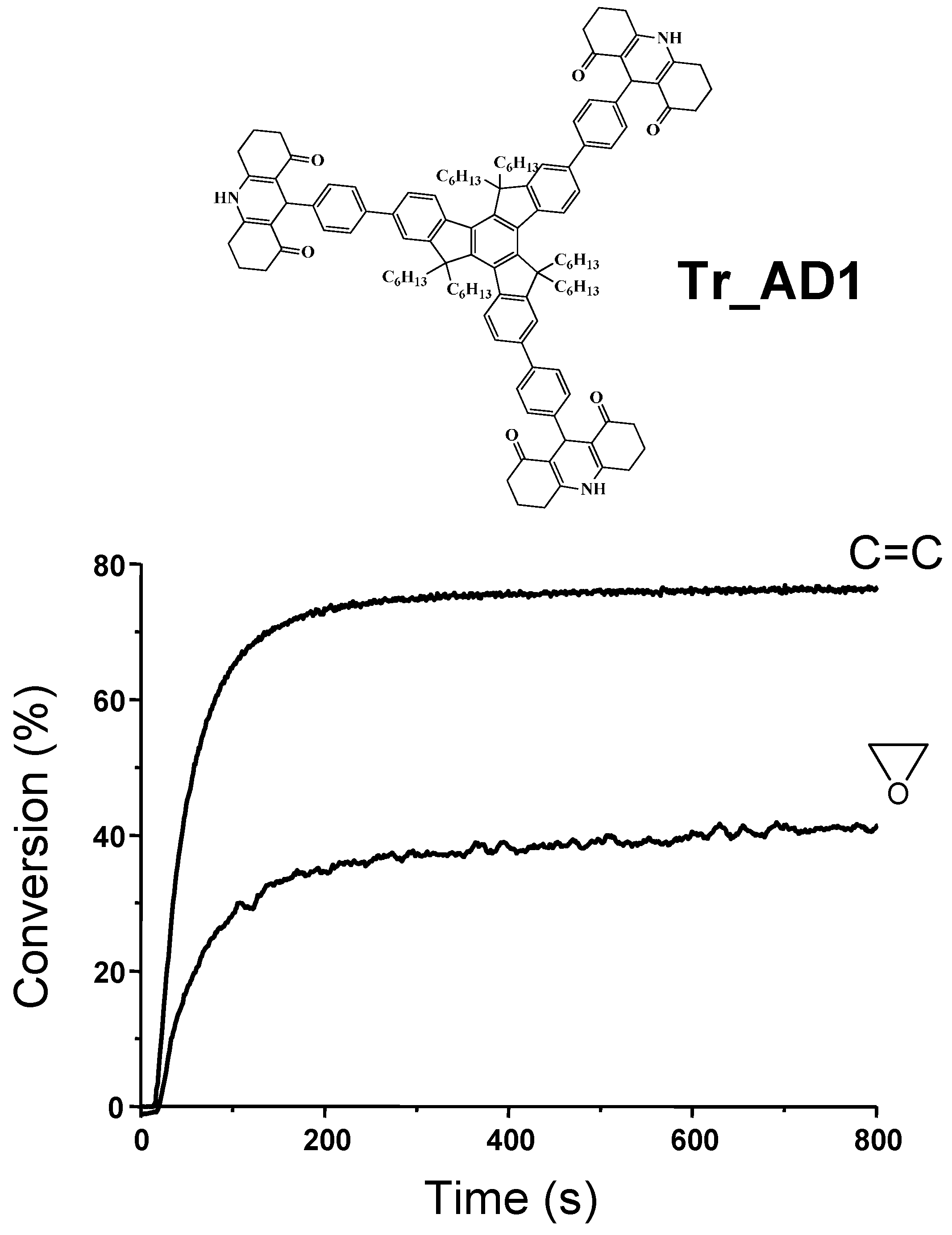{kind=link}
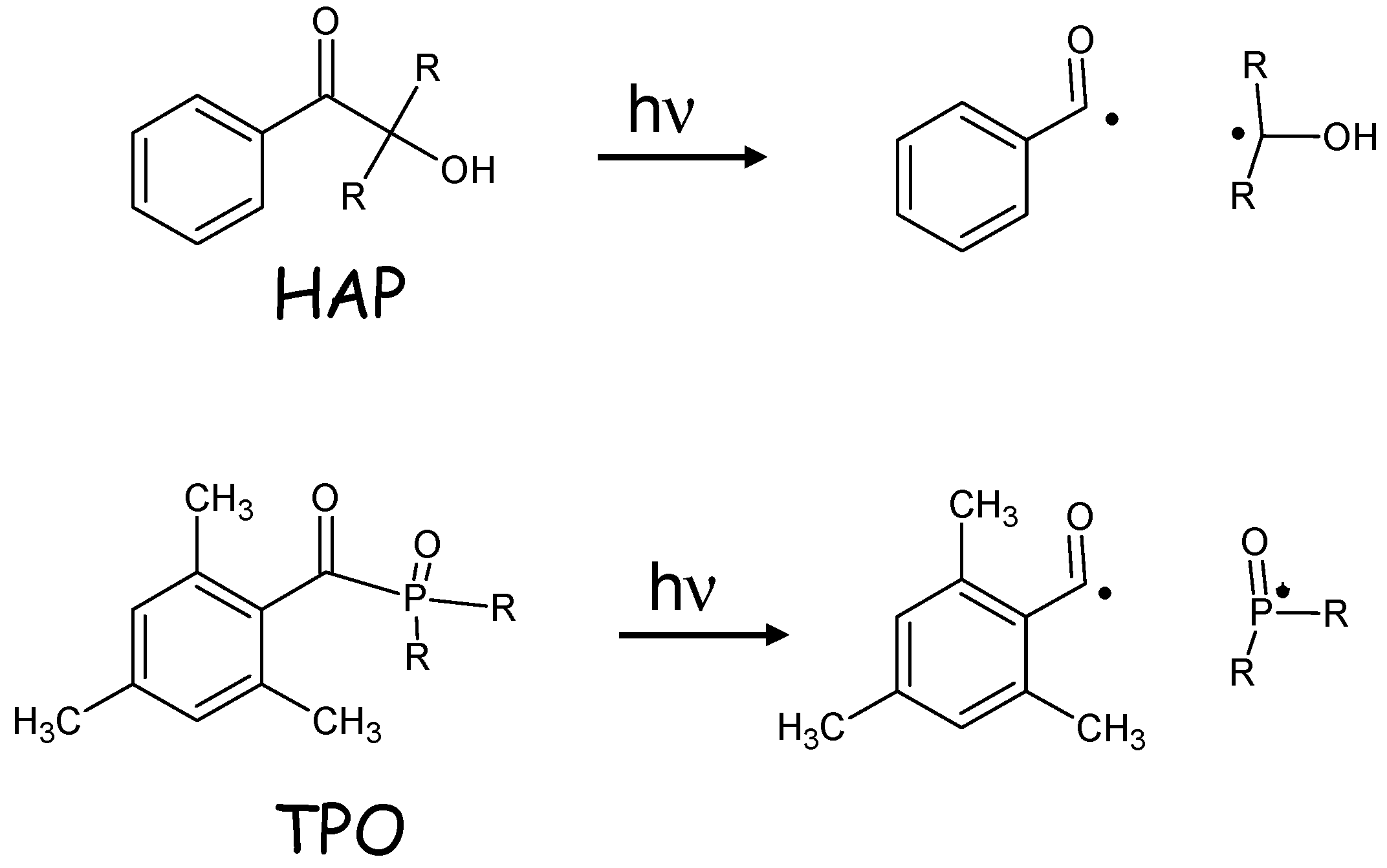{kind=link}
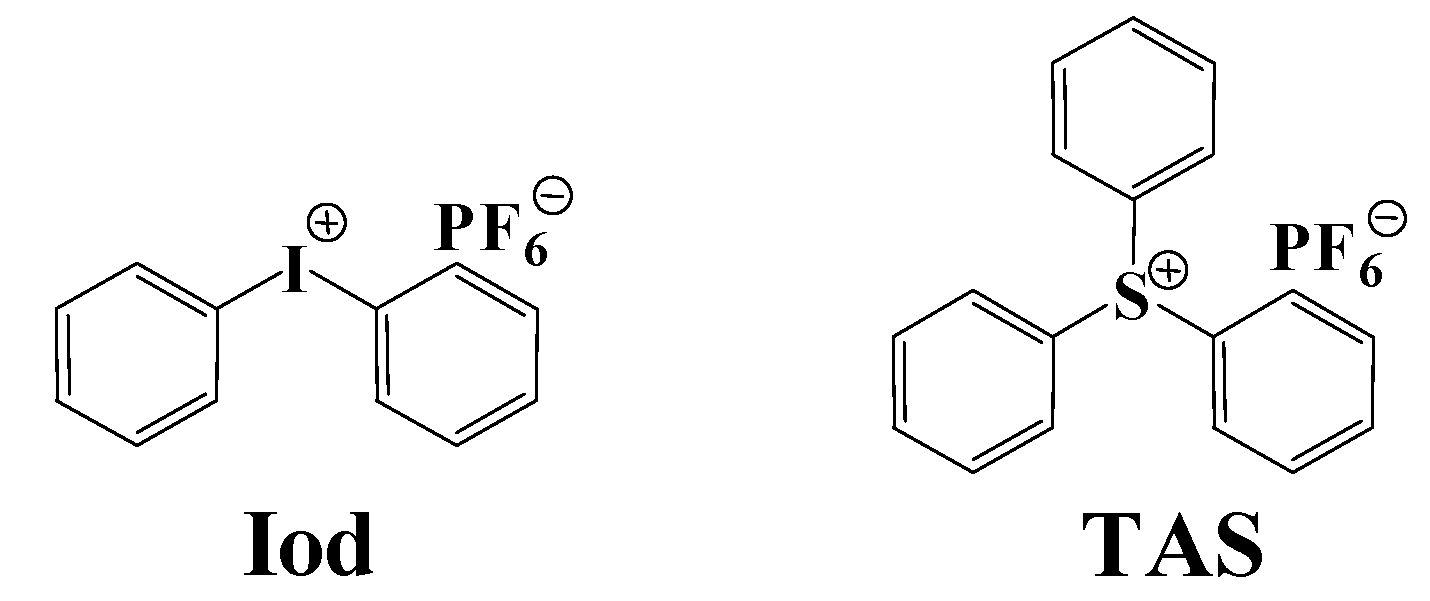{kind=link}
{kind=link}
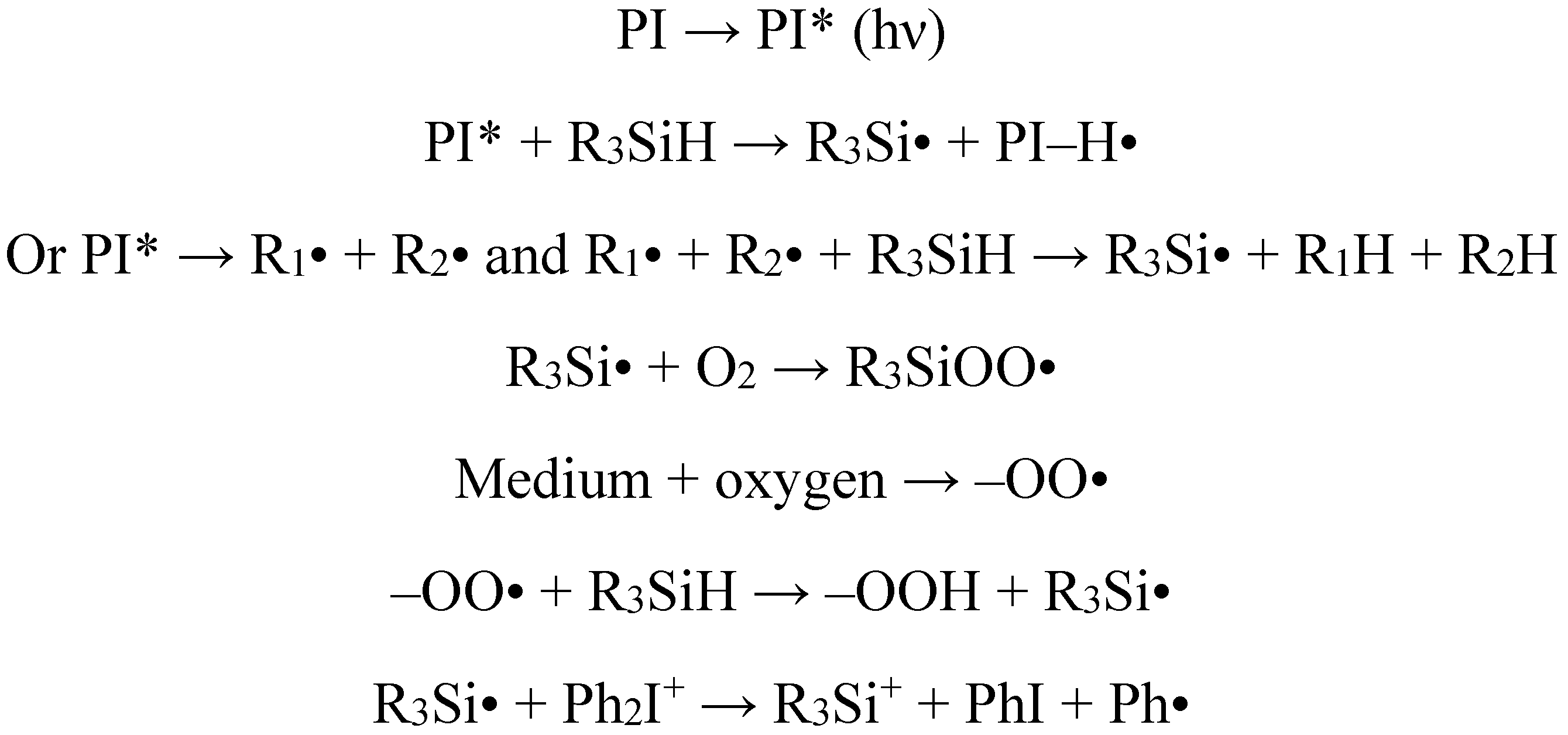{kind=link}
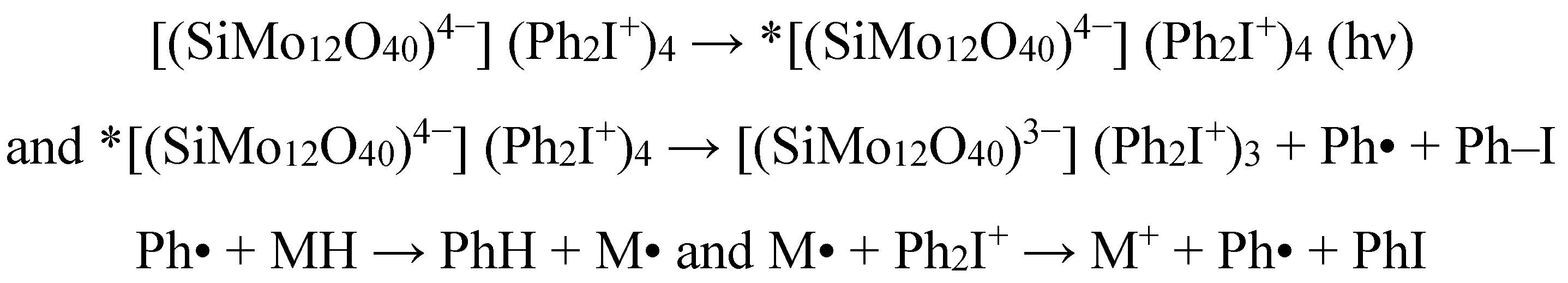{kind=link}
1. Introduction
2. Monomers for the Manufacture of IPNs through Photopolymerization
3. Examples of Final Properties for Photochemically Produced IPNs
4. Short Backgrounds on PIs and PISs


5. Photochemical Production of IPNs: Mechanisms
5.1. Intense UV Light Exposure
5.1.1. One-Photoinitiator Containing System

5.1.2. Two-Photoinitiator Containing Systems and One-Step Exposure
5.1.3. Sequential Production of IPNs Using Two Different Lights
5.2. UV-Thermal Dual Curing
5.3. Visible Light Curing of IPNs
5.4. Recent Design of Dual Photoinitiating Systems Operating under Visible Lights



5.5. Photocuring of IPNs under Soft Conditions



6. Conclusions
Glossary of Terms
| LED | Light Emitting Diode |
| IPNs | Interpenetrating polymer networks |
| SIPN | Semi-interpenetrating polymer networks |
| HDDA | Hexanediol diacrylate |
| EPOX | 3,4-epoxycyclohexylmethyl-3'4' epoxycyclohexyl carboxylate |
| TMPTA | Trimethylolpropane triacrylate |
| DVE-3 | Tri(ethylene glycol) divinyl ether |
| CHVE | 4-cyclo-hexane dimethanol divinyl ether |
| EP | diglycidyl ether of bisphenol A based epoxy resin |
| PEGDA | Poly(ethylene glycol) diacrylate |
| PIS | Photoinitiating systems |
| DPI | Diphenyliodonium salts |
| TAS | Triarylsulfonium salts |
| CP | Cationic Polymerization |
| FRP | Free Radical Polymerization |
| PIS | Photoinitiating system |
| PI | Photoinitiator |
Conflicts of Interest
References
- Sperling, L.H. Interpenetrating Polymer Networks and Related Materials; Plenum Press: New York, NY, USA, 1981. [Google Scholar]
- Sperling, L.H. Interpenetrating polymer networks. In Encyclopedia of Polymer Science and Technology; Wiley: New York, NY, USA, 2004. [Google Scholar]
- Selvaraj, M. Interpenetrating polymer networks, high performance. In Wiley Encyclopedia of Composites; Wiley: New York, NY, USA, 2012. [Google Scholar]
- Stepto, R.F.T. Polymer Networks: Principles of Their Formation, Structure and Properties; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1997; Chapter 6. [Google Scholar]
- Sperling, L.H. Interpenetrating polymer networks. In Advances in Chemistry; Klepner, D., Utracki, L.A., Eds.; American Chemical Society (ACS): Washington, DC, USA, 1994. [Google Scholar]
- Allen, N.S. Photochemistry and Photophysics of Polymer Materials; Wiley: New York, NY, USA, 2010. [Google Scholar]
- Belfied, K.D.; Crivello, J.V. Photoinitiated. Polymerization; American Chemical Society (ACS): Washington, DC, USA, 2003. [Google Scholar]
- Davidson, S. Exploring the science. In Technology and Application of UV and EB Curing; Sita Technology Ltd.: London, UK, 1999. [Google Scholar]
- Neckers, D.C.; Jager, W. UV and EB at the Millenium; Sita Technology: London, UK, 1999. [Google Scholar]
- Fouassier, J.P. Photoinitiation, Photopolymerization, and Photocuring: Fundamentals and Applications; Hanser: Münich, Germany, 1995. [Google Scholar]
- Scranton, A.B.; Bowman, C.N.; Peiffer, R.W. Photopolymerization: Fundamentals and applications. In American Chemical Society Symposium Series; American Chemical Society (ACS): Washington, DC, USA, 1997. [Google Scholar]
- Fouassier, J.P.; Rabek, J.F. Lasers in Polymer Science and Technology: Applications; CRC Press: Boca Raton, FL, USA, 1990. [Google Scholar]
- Pappas, S.P. UV-Curing: Science and Technology; Plenum Press: New York, NY, USA, 1992. [Google Scholar]
- Fouassier, J.P.; Rabek, J.F. Radiation Curing in Polymer Science and Technology; Chapman & Hall: London, UK, 1993. [Google Scholar]
- Fouassier, J.P.; Allonas, X. Basics and Applications of Photopolymerization Reactions; Research Signpost: Trivandrum, India, 2010. [Google Scholar]
- Fouassier, J.P. Photochemistry and UV Curing: New Trends; Research Signpost: Trivandrum, India, 2006. [Google Scholar]
- Mishra, M.K.; Yagci, Y. Handbook of Vinyl Polymers; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- Sangermano, M.; Carbonaro, W.; Bongiovanni, R.; Thomas, R.; Kausch, C.M. Interpenetrating polymer networks of hydrocarbon. Macromol. Mater. Eng. 2010, 295, 469–475. [Google Scholar] [CrossRef]
- Decker, C. Photoinitiated cross-linking polymerization of monomer blends. In Photoinitiated Polymerization; Belfied, K.D., Crivello, J.V., Eds.; American Chemical Society (ACS): Washington, DC, USA, 2003; Chapter 8; pp. 92–104. [Google Scholar]
- Decker, C.; Nguyen Thi Viet, T.; Decker, D.; Weber-Koehl, E. UV-radiation curing of acrylate/epoxide systems. Polymer 2001, 42, 5531–5541. [Google Scholar] [CrossRef]
- Decker, C.; Le Xuan, H.; Nguyen Thi Viet, T. Photocrosslinking of functionalized rubber III. Polymerization of multifunctional monomers in epoxidized liquid natural rubber. J. Polym. Sci. A Polym. Chem. 1996, 34, 1771–1788. [Google Scholar] [CrossRef]
- Lecamp, L.; Pavillon, C.; Lebaudy, P.; Bunel, C. Influence of temperature and nature of photoinitiator on the formation kinetics of an interpenetrating network photocured from an epoxide/methacrylate system. Eur. Polym. J. 2005, 41, 169–175. [Google Scholar] [CrossRef]
- Lin, Y.; Stansbury, J.W. Kinetics studies of hybrid structure formation by controlled photopolymerization. Polymer 2003, 44, 4781–4788. [Google Scholar] [CrossRef]
- Choe, J.-D.; Hong, J.W. UV-initiated free radical and cationic photopolymerizations of acrylate/epoxide and acrylate/vinyl ether hybrid systems with and without photosensitizer. J. Appl. Polym. Sci. 2004, 93, 1473–1483. [Google Scholar] [CrossRef]
- Rajaraman, C.; Mowers, W.A.; Crivello, J.V. Novel hybrid monomers bearing cycloaliphatic epoxy and 1-propenyl ether groups. Macromolecules 1999, 32, 36–41. [Google Scholar] [CrossRef]
- Sangermano, M.; Malucelli, G.; Bongiovanni, R.; Priola, A. Photopolymerization of oxetanes based systems. Eur. Polym. J. 2004, 40, 353–358. [Google Scholar] [CrossRef]
- Sangermano, M.; Malucelli, G.; Priola, A.; Manea, M. Recent advances in elastomeric nanocomposites. Prog. Org. Coat. 2006, 55, 225–230. [Google Scholar] [CrossRef]
- Sangermano, M.; Carbonaro, W.; Malucelli, G.; Priola, A. Polymer networks: Preparation and characterization. Macromol. Mater. Eng. 2008, 293, 515–520. [Google Scholar] [CrossRef]
- Decker, C. Kinetic study and new applications of UV radiation curing. Macromol. Rapid Commun. 2002, 23, 1063–1093. [Google Scholar] [CrossRef]
- Crivello, J.V.; Narayan, R.; Sternstein, S.S. Fabrication and mechanical characterization of glass fiber reinforced UV-cured composites from epoxidized vegetable oils. J. Appl. Polym. Sci. 1997, 64, 2073–2087. [Google Scholar] [CrossRef]
- Thames, S.F.; Yu, H. Cationic UV-cured coatings of epoxide-containing. Surf. Coat. Technol. 1999, 115, 208–214. [Google Scholar] [CrossRef]
- Decker, C.; Bianchi, C.; Decker, D.; Morel, F. Radiation curing: Coatings and printing inks. Prog. Org. Coat. 2001, 42, 253–266. [Google Scholar] [CrossRef]
- Kim, Y.M.; Kostanski, L.K.; Mac Gregor, J.F. Carboxylate and tri(ethylene glycol) methyl vinyl ether. Polymer 2003, 44, 5103–5109. [Google Scholar] [CrossRef]
- Rajaraman, S.K.; Mowers, W.A.; Crivello, J.V. Interaction of epoxy and vinyl ethers during photoinitiated cationic polymerization. J. Polym. Sci. A Polym. Chem. 1999, 37, 4007–4018. [Google Scholar] [CrossRef]
- Tehfe, M.A.; Dumur, F.; Vilas, N.; Graff, B.; Mayer, C.R.; Fouassier, J.P.; Gigmes, D.; Lalevée, J. A multicolor photoinitiator for cationic polymerization and interpenetrated polymer network synthesis: 2,7-di-tert-butyldimethyldihydropyrene. Macromol. Rapid Commun. 2013, 34, 1104–1109. [Google Scholar] [CrossRef]
- Nakanishi, H.; Satoh, M.; Norisuye, T.; Tran-Cong-Miyata, Q. Phase separation of interpenetrating polymer networks synthesized by using an autocatalytic reaction. Macromolecules 2006, 39, 9456–9466. [Google Scholar] [CrossRef]
- Vancaeyzeele, C.; Fichet, O.; Laskar, J.; Boileau, S.; Teyssié, D. Polyisobutene/polystyrene interpenetrating polymer networks: Effects of network formation order and composition on the IPN architecture. Polymer 2006, 47, 2046–2060. [Google Scholar] [CrossRef]
- Lu, C.-H.; Su, Y.-C.; Wang, C.-F.; Huang, C.-F.; Sheen, Y.-F.; Chang, F.-C. Thermal properties and surface energy characteristics of interpenetrating polyacrylate and polybenzoxazine networks. Polymer 2008, 49, 4852–4860. [Google Scholar] [CrossRef]
- Boudraa, K.; Bouchaour, T.; Maschke, U. Swelling of acrylic interpenetrating polymer networks in liquid crystals. Macromol. Symposia 2008, 273, 33–37. [Google Scholar] [CrossRef]
- Boudraa, K.; Bouchaour, T.; Maschke, U. Equilibrium phase diagrams of interpenetrating polymer networks and liquid crystals. Macromol. Symposia 2011, 303, 95–99. [Google Scholar] [CrossRef]
- Wang, J.; Sun, F.; Li, X. Preparation and antidehydration of interpenetrating polymer network hydrogels based on 2-hydroxyethyl methacrylate and N-vinyl-2-pyrrolidone. J. Appl. Polym. Sci. 2010, 117, 1851–1858. [Google Scholar]
- Karabanova, L.V.; Boiteux, G.; Seytre, G.; Stevenson, I.; Lloyd, A.W.; Mikhalovsky, S.V.; Helias, M.; Sergeeva, L.M.; Lutsyk, E.D.; Svyatyna, A. Phase separation in the polyurethane/poly(2-hydroxyethyl methacrylate) semi-interpenetrating polymer networks synthesized by different ways. Polym. Eng. Sci. 2008, 48, 588–597. [Google Scholar] [CrossRef]
- Pescosolido, L.; Vermonden, T.; Malda, J.; Censi, R.; Dhert, W.J.A.; Alhaique, F.; Hennink, W.E.; Matricardi, P. In situ forming IPN hydrogels of calcium alginate and dextran-HEMA for biomedical applications. Acta Biomater. 2011, 7, 1627–1633. [Google Scholar] [CrossRef] [PubMed]
- Sangermano, M.; Cook, W.D.; Papagna, S.; Grassini, S. Curing kinetics and morphology of IPNs from a flexible dimethacrylate and a rigid epoxy via sequential photo and thermal polymerization. Eur. Polym. J. 2012, 48, 1796–1804. [Google Scholar] [CrossRef]
- Sangermano, M.; Razza, N.; Crivello, J.V. Cationic UV-curing: Technology and applications. Macromol. Mater. Eng. 2014, 41, 775–793. [Google Scholar] [CrossRef]
- Naga, N.; Kihara, Y.; Miyanaga, T.; Furukawa, H. A photo hydrosilylation reaction. Macromolecules 2009, 42, 3454–3462. [Google Scholar] [CrossRef]
- Moussa, K.; Decker, C. Semi-interpenetrating polymer networks synthesis. J. Polym. Sci. A Polym. Chem. 1993, 31, 2633–2642. [Google Scholar] [CrossRef]
- Pescosolido, L.; Schuurman, W.; Malda, J.; Matricardi, P.; Alhaique, F.; Coviello, P.; van Weeren, R.; Dhert, W.J.A.; Hennink, W.E.; Vermonden, T. Hyaluronic acid and dextran-based semi-IPN hydrogels as biomaterials for bioprinting. Biomacromolecules 2011, 12, 1831–1838. [Google Scholar] [CrossRef] [PubMed]
- Decker, C.; Decker, D.; Nguyen Thi Viet, T.; Le Xuan, H. Photoinitiated cationic polymerisation of multifunctional systems. Macromol. Symposia 1996, 102, 63–71. [Google Scholar] [CrossRef]
- Yang, T.; Malkoch, M.; Hult, A. Sequential interpenetrating poly(ethylene glycol) hydrogels prepared by UV-initiated thiol–ene coupling chemistry. J. Polym. Sci. A Polym. Chem. 2013, 51, 363–371. [Google Scholar] [CrossRef]
- Guymon, J.; Clapper, D.; Guymon, C.A. Compatibilization of immiscible polymer networks through photopolymerization in a lyotropic liquid crystal. Adv. Mater. 2006, 18, 1575–1580. [Google Scholar] [CrossRef]
- Ortiz, R.A.; Urbina, B.A.P.; Valdez, L.V.C.; Duarte, L.B.; Santos, R.G.; Valdez, A.I.G.; Santos, R.G.; Valdez, A.E.G.; Soucek, M.D. Effect of introducing a cationic system into a thiol–ene photopolymerizable formulation. J. Polym. Sci. A Polym. Chem. 2007, 45, 4829–4843. [Google Scholar] [CrossRef]
- Wei, H.; Li, Q.; Ojelade, M.; Madbouly, S.; Otaigbe, J.U.; Hoyle, C.E. Thiol−ene free-radical and vinyl ether cationic hybrid photopolymerization. Macromolecules 2007, 40, 8788–8793. [Google Scholar] [CrossRef]
- He, Y.; Xiao, M.; Wu, F.; Nie, J. Photopolymerization kinetics of cycloaliphatic epoxide–acrylate hybrid monomer. Polym. Int. 2007, 56, 1292–1297. [Google Scholar] [CrossRef]
- Xiao, M.; Shi, S.; Nie, J.; He, Y. Photopolymerization kinetics study of epoxide/acrylate hybrid oligomer. PMSE Prepr. 2006, 94, 408–409. [Google Scholar]
- Crivello, J.V. Synergistic effects in hybrid free radical/cationic photopolymerizations. J. Polym. Sci. A Polym. Chem. 2007, 45, 3759–3769. [Google Scholar] [CrossRef]
- Crivello, J.V. Hybrid free radical/cationic frontal photopolymerizations. J. Polym. A Polym. Chem. 2007, 45, 4331–4340. [Google Scholar] [CrossRef]
- Cai, Y.; Jessop, J.L.P. Decreased oxygen inhibition in photopolymerized acrylate/epoxide hybrid polymer coatings as demonstrated by Raman spectroscopy. Polymer 2006, 47, 6560–6566. [Google Scholar] [CrossRef]
- Crivello, J.V. Redox initiated cationic polymerization: The unique behavior of alkyl glycidyl ethers. J. Polym. Sci. A Polym. Chem. 2011, 49, 2147–2154. [Google Scholar] [CrossRef]
- Jian, Y.; He, Y.; Sun, Y.; Yang, H.; Yang, W.; Nie, J. Thiol–epoxy/thiol–acrylate hybrid materials synthesized by photopolymerization. J. Mater. Chem. C 2013, 1, 4481–4489. [Google Scholar] [CrossRef]
- De Ruiter, B.; El Ghayoury, A.; Hofmeir, H.; Schubert, U.S.; Manea, M. Two-step curing processes for coating application. Prog. Org. Coat. 2006, 55, 154–159. [Google Scholar] [CrossRef]
- Bourcier, S.; Vancaeyzeele, C.; Vidal, F.; Fichet, O. Microemulsion as the template for synthesis of interpenetrating polymer networks with predefined structure. Polymer 2013, 54, 4436–4445. [Google Scholar] [CrossRef]
- Cook, W.D. Photopolymerization kinetics of oligo(ethylene oxide) and oligo(methylene) oxide dimethacrylates. J. Polym. Sci. A Polym. Chem. 1993, 31, 1053–1067. [Google Scholar] [CrossRef]
- Chen, F.; Cook, W.D. Hybrid UV-cured organic–inorganic IPNs. Eur. Polym. J. 2008, 44, 1796–1813. [Google Scholar] [CrossRef]
- De Brito, M.; Allonas, X.; Croutxé-Barghorn, C.; Palmieri, M.; Dietlin, C.; Agarwal, S.; Lellinger, D.; Alig, I. Kinetic study of photoinduced quasi-simultaneous interpenetrating polymer networks. Prog. Org. Coat. 2012, 73, 186–193. [Google Scholar] [CrossRef]
- Nowers, J.R.; Costanzo, J.A.; Narasimhan, B. Structure–property relationships in acrylate/epoxy interpenetrating polymer networks: Effects of the reaction sequence and composition. J. Appl. Polym. Sci. 2007, 104, 891–901. [Google Scholar] [CrossRef]
- Lee, S.; Jeong, Y.C.; Heo, Y.; Kim, S., II; Choi, Y.-S.; Park, J.-K. Holographic photopolymers of organic/inorganic hybrid interpenetrating networks for reduced volume shrinkage. J. Mater. Chem. 2009, 19, 1105–1114. [Google Scholar] [CrossRef]
- Carioscia, J.A.; Stansbury, J.W.; Bowman, C.N. Evaluation and control of thiol-ene/thiol-epoxy hybrid networks. Polymer 2007, 48, 1526–1532. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, F. Synthesis and application of ion-imprinted interpenetrating polymer network gel for selective solid phase extraction of Cd2+. Chem. Eng. J. 2014, 242, 117–126. [Google Scholar] [CrossRef]
- Wang, J.; Liu, F. UV-radiation curing of simultaneous interpenetrating polymer network hydrogels for enhanced heavy metal ion removal. Mater. Sci. Eng. B 2012, 177, 1633–1640. [Google Scholar] [CrossRef]
- Nakanishi, H.; Namikawa, N.; Norisuye, T.; Tran-Cong-Miyata, Q. Interpenetrating polymer networks with spatially graded morphology controllable by UV-radiation curing. Macromol. Symposia 2006, 242, 157–164. [Google Scholar] [CrossRef]
- Daniele, M.D.; Adams, A.A.; Naciri, J.; North, S.H.; Ligler, F.S. Interpenetrating networks based on gelatin methacrylamide and PEG formed using concurrent thiol click photopolymerization for hydrogel tissue engineering scaffolds. Biomaterials 2014, 35, 1845–1856. [Google Scholar] [CrossRef] [PubMed]
- Kundu, J.; Poole-Warren, L.A.; Martens, P.; Kundu, S.C. Silk fibroin/poly(vinyl alcohol) photocrosslinked hydrogels for delivery of macromolecular drugs. Acta Biomater. 2012, 8, 1720–1729. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; He, J.; Nichol, J.W.; Wang, L.; Hutson, C.B.; Wang, B.; Du, Y.; Fan, H.; Khademhosseini, A. Synthesis and characterization of photocrosslinkable gelatin and silk fibroin interpenetrating polymer network hydrogels. Acta Biomater. 2011, 7, 2384–2393. [Google Scholar] [CrossRef] [PubMed]
- Matricardi, P.; di Meo, C.; Coviello, T.; Hennink, W.E.; Alhaique, F. Interpenetrating polymer networks polysaccharide hydrogels for drug delivery and tissue engineering. Adv. Drug Deliver. Rev. 2013, 65, 1172–1187. [Google Scholar] [CrossRef]
- Rennerfeldt, D.A.; Renth, A.N.; Talata, Z.; Gehrke, S.H.; Detamore, M.S. Tuning mechanical performance of poly(ethylene glycol) and agarose interpenetrating network hydrogels for cartilage tissue engineering. Biomaterials 2013, 34, 8241–8257. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chan-Park, M.B. Hydrogel based on interpenetrating polymer networks of dextran and gelatin for vascular tissue engineering. Biomaterials 2009, 30, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Myung, D.; Koh, W.; Ko, J.; Hu, Y.; Carrasco, M.; Noolandi, J.; Ta, C.N.; Frank, C.W. Biomimetic strain hardening in interpenetrating polymer network hydrogels. Polymer 2007, 48, 5376–5387. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, D.N.; Choi, D.; Lee, W.; Park, J.; Koh, W.G. Preparation of interpenetrating polymer network composed of poly(ethylene glycol) and poly(acrylamide) hydrogels as a support of enzyme immobilization. Polym. Adv. Technol. 2008, 19, 852–858. [Google Scholar] [CrossRef]
- Bae, K.H.; Wang, L.-S.; Kurisawa, M. Injectable biodegradable IPN hydrogels: Progress and challenges. J. Mater. Chem. B 2013, 1, 5371–5388. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Lü, J.; Shao, Y.-H. Preparation and characterization of poly(n-isopropylacrylamide)-modified poly(2-hydroxyethyl acrylate) hydrogels by interpenetrating polymer networks for sustained drug release. Macromol. Biosci. 2006, 6, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.-P.; Ma, D.; Zhang, L.-M. New semi-interpenetrating network hydrogels: Synthesis, characterization and properties. Macromol. Biosci. 2006, 6, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Dragan, E.S. Design and applications of interpenetrating polymer network hydrogels. A review. Chem. Eng. J. 2014, 243, 572–590. [Google Scholar] [CrossRef]
- Cha, C.; Kim, S.R.; Jin, Y.-S.; Kong, H. Tuning structural durability of yeast-encapsulating alginate gel beads with interpenetrating networks for sustained bioethanol production. Biotechnol. Bioeng. 2012, 109, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Elisseeff, J.; McIntosh, W.; Anseth, K.; Riley, S.; Ragan, P.; Langer, R. Photoencapsulation of chondrocytes in poly(ethylene oxide)-based semi-interpenetrating networks. J. Biomed. Mater. Res. 2000, 51, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhao, N.; Rudenja, S. Surface interpenetrating networks of poly(ethylene terephthalate) and polyamides for effective biocidal properties. Macromol. Chem. Phys. 2010, 211, 286–296. [Google Scholar] [CrossRef]
- Li, J.; Lin, F.; Li, L.; Li, J.; Liu, S. Surface engineering of poly(ethylene terephthalate) for durable hemocompatibility via a surface interpenetrating network technique. Macromol. Chem. Phys. 2012, 213, 2120–2129. [Google Scholar] [CrossRef]
- Zhang, S.; Feng, Y.; Zhang, L.; Sun, J.; Xu, X.; Xu, Y. Novel interpenetrating networks with shape-memory properties. J. Polym. Sci. A Polym. Chem. 2007, 45, 768–775. [Google Scholar] [CrossRef]
- Myung, D.; Farooqui, N.; Zheng, L.L.; Koh, W.; Gupta, S.; Bakri, A.; Noolandi, J.; Cochran, J.R.; Frank, C.W.; Ta, C.N. Bioactive interpenetrating polymer network hydrogels that support corneal epithelial wound healing. J. Biomed. Mater. Res. A 2009, 90A, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Akpalo, E.; Bidault, L.; Boissière, M.; Vancaeyzeele, C.; Fichet, O.; Larreta-Garde, V. Fibrin–polyethylene oxide interpenetrating polymer networks: New self-supported biomaterials combining the properties of both protein gel and synthetic polymer. Acta Biomater. 2011, 7, 2418–2427. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.L.; Weng, J.-B.; Huang, Q.-M.; Hu, B.-H.; Qiao, T.; Deng, P. Fabrication of a stable poly(vinylpyrrolidone)/poly(urushiol) multilayer ultrathin film through layer-by-layer assembly and photo-induced polymerization. Colloids Surf. A Physicochem. Eng. Asp. 2009, 337, 15–20. [Google Scholar] [CrossRef]
- Karabanova, L.V.; Mikhalovsky, S.V.; Lloyd, A.W. Gradient semi-interpenetrating polymer networks based on polyurethane and poly(2-hydroxyethyl methacrylate) for biomedical applications. J. Mater. Chem. 2012, 22, 7919–7928. [Google Scholar] [CrossRef]
- Jain, S.H.; Murata, K.; Anazawa, T. Nanostructures developed from semi-interpenetrating polymer network structures. Macromol. Chem. Phys. 2003, 204, 893–902. [Google Scholar] [CrossRef]
- Forney, B.S.; Baguenard, C.; Guymon, C.A. Improved stimuli-response and mechanical properties of nanostructured poly(N-isopropylacrylamide-co-dimethylsiloxane) hydrogels generated through photopolymerization in lyotropic liquid crystal templates. Soft Matter 2013, 9, 7458–7467. [Google Scholar] [CrossRef]
- Lav, T.X.; Tran-Van, F.; Vidal, F.; Péralta, S.; Chevrot, C.; Teyssié, D.; Grazulevicius, J.V.; Getautis, V.; Derbal, H.; Nunzi, J.-M.; et al. Synthesis and characterization of p and n dopable interpenetrating polymer networks for organic photovoltaic devices. Thin Solid Films 2008, 516, 7223–7229. [Google Scholar] [CrossRef]
- Park, S.; Bearinger, J.P.; Lautenschlager, E.P.; Castner, D.G.; Healy, K.E. Surface modification of poly(ethylene terephthalate) angioplasty balloons with a hydrophilic poly(acrylamide-co-ethylene glycol) interpenetrating polymer network coatings. J. Biomed. Mater. Res. 2000, 53, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Wippich, C.C.; Wu, C.J.; Sivasankar, P.M.; Schmidt, G. Robust and semi-interpenetrating hydrogels from poly(ethylene glycol) and collagen for elastomeric tissue scaffolds. Macromol. Biosci. 2012, 12, 1490–1501. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, N. Contribution of polymer chemistry to dentistry: Development of an impermeable interpenetrating polymer network to protect teeth from acid demineralization. Polym. Int. 2008, 57, 159–162. [Google Scholar] [CrossRef]
- Garoushi, S.; Vallittu, P.K.; Lassila, L.V.J. Short glass fiber reinforced restorative composite resin with semi-inter penetrating polymer network matrix. Dent. Mater. 2007, 23, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Fouassier, J.P.; Lalevée, J. Photoinitiators for Polymer Synthesis-Scope, Reactivity, and Efficiency; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Dietliker, K. A Compilation of Photoinitiators Commercially Available for UV Today; Sita Technology Ltd.: London, UK, 2002. [Google Scholar]
- Crivello, J.V. Photoinitiators for Free Radical, Cationic and Anionic Photopolymerization, 2nd ed.; John Wiley & Sons: Chichester, UK, 1998. [Google Scholar]
- Green, W.A. Industrial Photoinitiators; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Vabrik, R.; Czajlik, I.; Túry, G.; Rusznák, I.; Ille, A.; Víg, A.; Hult, A. Study of epoxy resin–acrylated polyurethane semi-interpenetrating polymer networks. J. Appl. Polym. Sci. 1998, 68, 111–119. [Google Scholar] [CrossRef]
- Dean, K.; Cook, W.D. Effect of curing sequence on the photopolymerization and thermal curing kinetics of dimethacrylate/epoxy interpenetrating polymer networks. Macromolecules 2002, 35, 7942–7954. [Google Scholar] [CrossRef]
- Podsiadły, R.; Podemska, K.; Szymczak, A.M. Novel visible photoinitiators systems for free-radical/cationic hybrid photopolymerization. Dyes Pigment. 2011, 91, 422–426. [Google Scholar] [CrossRef]
- Lalevee, J.; Dumur, F.; Gigmes, D.; Graff, B.; Xiao, P.; Fouassier, J.P.; Hong, W.; Li, Y. Green light sensitive diketopyrrolopyrrole derivatives used in versatile photoinitiating systems for photopolymerizations. Polym. Chem. 2014, 5, 2293–2300. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Thirion, D.; Fagour, S.; Vacher, A.; Sallenave, X.; Morlet-Savary, F.; Graff, B.; Fouassier, J.P.; Gigmes, D.; et al. Multicolor photoinitiators for radical and cationic polymerization: Mono vs. poly functional thiophene derivatives. Macromolecules 2013, 46, 6786–6793. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Graff, B.; Morlet-Savary, F.; Vidal, L.; Gigmes, D.; Fouassier, J.P.; Lalevee, J. Indanedione skeleton for the design of low intensity 300–500 nm light sensitive initiators. Macromolecules 2014, 47, 26–34. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Graff, B.; Morlet-Savary, F.; Gigmes, D.; Fouassier, J.P.; Lalevee, J. Tunable organophotocatalysts for polymerization reactions. Macromolecules 2014, 47, 973–978. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevee, J. Blue light sensitive naphthalic anhydride derivatives. Macromolecules 2014, 47, 601–608. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevee, J. Cationic and thiol–ene photopolymerization upon red lights using anthraquinone derivatives as photoinitiators. Macromolecules 2013, 46, 6744–6750. [Google Scholar] [CrossRef]
- Tehfe, M.A.; Dumur, F.; Xiao, F.; Delgove, M.; Graff, B.; Fouassier, J.P.; Gigmes, D.; Lalevee, J. Chalcone derivatives as highly versatile photoinitiators for radical, cationic, thiol–ene and IPN polymerization reactions upon visible lights. Polym. Chem. 2014, 5, 382–390. [Google Scholar] [CrossRef]
- Mokbel, H.; Dumur, F.; Telitel, S.; Vidal, L.; Xiao, P.; Versace, D.L.; Tehfe, M.A.; Morlet-Savary, F.; Graff, B.; Fouassier, J.P.; et al. Photoinitiating systems of polymerization and in-situ incorporation of silver nanoparticles in polymer matrixes upon visible lights: Push-pull malonate and malonitrile based dyes. Polym. Chem. 2013, 4, 5679–5687. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Frigoli, M.; Tehfe, M.A.; Graff, B.; Fouassier, J.P.; Gigmes, D.; Lalevee, J. Naphthalimide based methacrylated photoinitiators in radical and cationic photopolymerization under visible light. Polym. Chem. 2013, 4, 5440–5448. [Google Scholar] [CrossRef]
- Xiao, P.; Simonet-Jegat, C.; Dumur, F.; Schrodj, G.; Tehfe, M.A.; Fouassier, J.P.; Gigmes, D.; Lalevee, J. Silicon polyoxomolybdate ([SiMo12O40]4−) as photoinitiator in radical and cationic photopolymerization: Application to the fabrication of polyoxometalate/polymer hybrid materials. Polym. Chem. 2013, 4, 4526–4530. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, P.; Tehfe, M.A.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevee, J. Effect of substituents on the photoinitiating abilities of acridinediones. Macromol. Chem. Phys. 2013, 214, 2276–2282. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Tehfe, M.A.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevee, J. Difunctional acridinediones as photoinitiators of polymerization under UV and visible lights: Structural effects. Polymer 2013, 54, 3458–3466. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Tehfe, M.A.; Graff, B.; Fouassier, J.P.; Gigmes, D.; Lalevee, J. Keggin-type polyoxometalate ion ([PMo12O40]3−) in radical initiating systems: Application to radical and cationic photopolymerization. Macromol. Chem. Phys. 2013, 214, 1749–1755. [Google Scholar] [CrossRef]
- Tehfe, M.A.; Dumur, F.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevee, J. Blue-to-red light sensitive push-pull structured photoinitiators: Indandione derivatives for radical and cationic photopolymerization reactions. Macromolecules 2013, 46, 3332–3341. [Google Scholar] [CrossRef]
- Telitel, S.; Ouhib, F.; Fouassier, J.P.; Jerome, G.; Detrembleur, C.; Lalevee, J. Thiophene derivatives with donor–π–acceptor structures for enhanced light-absorption properties and efficient cationic polymerization upon green-light irradiation. Macromol. Chem. Phys. 2014, 215, 1514–1524. [Google Scholar] [CrossRef]
- Tehfe, M.A.; Dumur, F.; Xiao, P.; Graff, B.; Morlet-Savary, F.; Fouassier, J.P.; Gigmes, D.; Lalevee, J. New chromone based photoinitiators for polymerization reactions upon visible lights. Polym. Chem. 2013, 4, 4234–4244. [Google Scholar]
- Tehfe, M.A.; Dumur, F.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevee, J. Green light induced cationic ring opening polymerization reactions: Perylene bis-dicarboximides as efficient photosensitizers. Macromol. Chem. Phys. 2013, 214, 1052–1060. [Google Scholar] [CrossRef]
- Tehfe, M.A.; Dumur, F.; Contal, E.; Graff, B.; Morlet-Savary, F.; Gigmes, D.; Fouassier, J.P.; Lalevee, J. New insights in radical and cationic polymerization upon visible light exposure: Role of novel photoinitiator systems based on the pyrene chromophore. Polym. Chem. 2013, 4, 1625–1634. [Google Scholar] [CrossRef]
- Tehfe, M.A.; Dumur, F.; Telitel, S.; Gigmes, D.; Contal, E.; Bertin, D.; Morlet-Savary, F.; Graff, B.; Fouassier, J.P.; Lalevee, J. Metal based photoinitiators: A new progress using zinc complexes. Eur. Polym. J. 2013, 49, 1040–1049. [Google Scholar] [CrossRef]
- Tehfe, M.A.; Zein-Fakih, A.; Lalevee, J.; Dumur, F.; Gigmes, D.; Graff, B.; Morlet-Savary, F.; Hamied, T.; Fouassier, J.P. Pyridinium salts: New systems for photopolymerization reactions upon visible light exposure. Eur. Polym. J. 2013, 49, 567–574. [Google Scholar] [CrossRef]
- Telitel, S.; Lalevee, J.; Blanchard, N.; Kavalli, T.; Tehfe, M.A.; Schweitzer, S.; Morlet-Savary, F.; Graff, B.; Fouassier, J.P. Photopolymerization of cationic monomers and acrylate/divinylether blends under visible lights using pyrromethene dyes. Macromolecules 2012, 45, 6864–6868. [Google Scholar] [CrossRef]
- Tehfe, M.A.; Dumur, F.; Graff, B.; Morlet-Savary, F.; Fouassier, J.P.; Gigmes, D.; Lalevee, J. Trifunctional photoinitiators based on a triazine skeleton for visible light sources and UV LED induced polymerizations. Macromolecules 2012, 45, 8639–8647. [Google Scholar]
- Tehfe, M.A.; Ma, L.; Graff, B.; Morlet-Savary, F.; Fouassier, J.P.; Zhao, J.; Lalevee, J. Cyclometallated Pt(II) complexes in visible light photoinitiating systems. Macromol. Chem. Phys. 2012, 213, 2282–2286. [Google Scholar] [CrossRef]
- Tehfe, M.A.; Lalevee, J.; Telitel, S.; Contal, E.; Dumur, F.; Gigmes, D.; Bertin, D.; Nechab, M.; Graff, B.; Morlet-Savary, F.; et al. Polyaromatic structures as organophotocatalysts for efficient dual radical/cationic photopolymerizations under visible lights. Macromolecules 2012, 45, 4454–4460. [Google Scholar] [CrossRef]
- Lalevee, J.; Blanchard, N.; Tehfe, M.A.; Peter, M.; Morlet-Savary, F.; Gigmes, D.; Fouassier, J.P. Efficient dual radical/cationic photoinitiator under visible lights: A new concept. Polym. Chem. 2011, 2, 1986–1991. [Google Scholar] [CrossRef]
- Tehfe, M.A.; Lalevee, J.; Morlet-Savary, F.; Graff, B.; Blanchard, N.; Fouassier, J.P. Polymerization reactions under visible lights. Macromolecules 2012, 45, 1746–1752. [Google Scholar] [CrossRef]
- Xiao, P.; Dumur, F.; Graff, B.; Gigmes, D.; Lalevee, J. Panchromatic photopolymerizable cationic films using indoline and squaraine dye based photoinitiating systems. Macromolecules 2013, 46, 7661–7667. [Google Scholar] [CrossRef]
- Decker, C.; Bendaikha, T. Interpenetrating polymer networks. II. Sunlight induced polymerization. J. Appl. Polym. Sci. 1998, 70, 2269–2282. [Google Scholar] [CrossRef]
- Decker, C.; Nguyen Thi Viet, T.; Le Xuan, H. New systems for photopolymerization reactions upon visible light exposure. Eur. Polym. J. 1996, 32, 1319–1326. [Google Scholar] [CrossRef]
- Esposito Corcione, C.; Striani, R.; Frigione, M. UV-cured methacrylic-silica hybrids: Effect of oxygen inhibition on photo-curing kinetics. Thermochim. Acta 2014, 576, 47–55. [Google Scholar] [CrossRef]
- Esposito Corcione, C. Development and characterization of novel photopolymerizable formulations for stereolithography. J. Polym. Eng. 2014, 34, 85–93. [Google Scholar]
- Esposito Corcione, C.; Greco, A.; Maffezzoli, A. Photopolymerization kinetics of an epoxy based resin for stereolithography. J. Therm. Anal. Calorim. 2003, 72, 687–693. [Google Scholar] [CrossRef]
- Esposito Corcione, C.; Greco, A.; Maffezzoli, A. Photopolymerization kinetics of an epoxy-based resin for stereolithography. J. Appl. Polym. Sci. 2004, 92, 3484–3491. [Google Scholar] [CrossRef]
- Esposito Corcione, C.; Greco, A.; Maffezzoli, A. Time–temperature and time-irradiation intensity superposition for photopolymerization of an epoxy based resin. Polymer 2005, 46, 8018–8027. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fouassier, J.P.; Lalevée, J. Photochemical Production of Interpenetrating Polymer Networks; Simultaneous Initiation of Radical and Cationic Polymerization Reactions. Polymers 2014, 6, 2588-2610. https://doi.org/10.3390/polym6102588
Fouassier JP, Lalevée J. Photochemical Production of Interpenetrating Polymer Networks; Simultaneous Initiation of Radical and Cationic Polymerization Reactions. Polymers. 2014; 6(10):2588-2610. https://doi.org/10.3390/polym6102588
Chicago/Turabian StyleFouassier, Jean Pierre, and Jacques Lalevée. 2014. "Photochemical Production of Interpenetrating Polymer Networks; Simultaneous Initiation of Radical and Cationic Polymerization Reactions" Polymers 6, no. 10: 2588-2610. https://doi.org/10.3390/polym6102588