Computational Study of a Heterostructural Model of Type I Collagen and Implementation of an Amino Acid Potential Method Applicable to Large Proteins


Abstract
:1. Introduction
2. Electronic Structure of a Model of 7-2 Heterostructural Type I Collagen
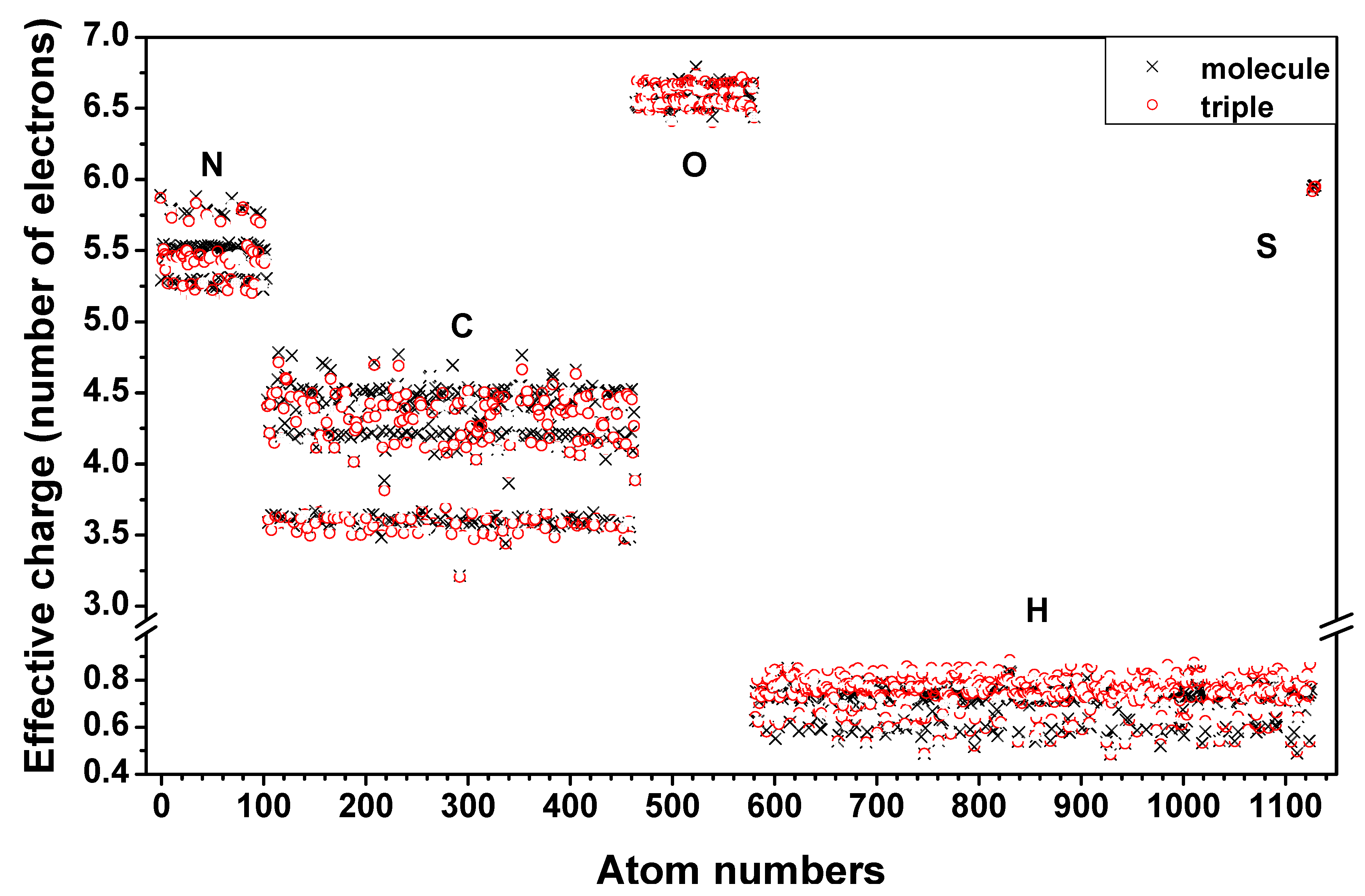
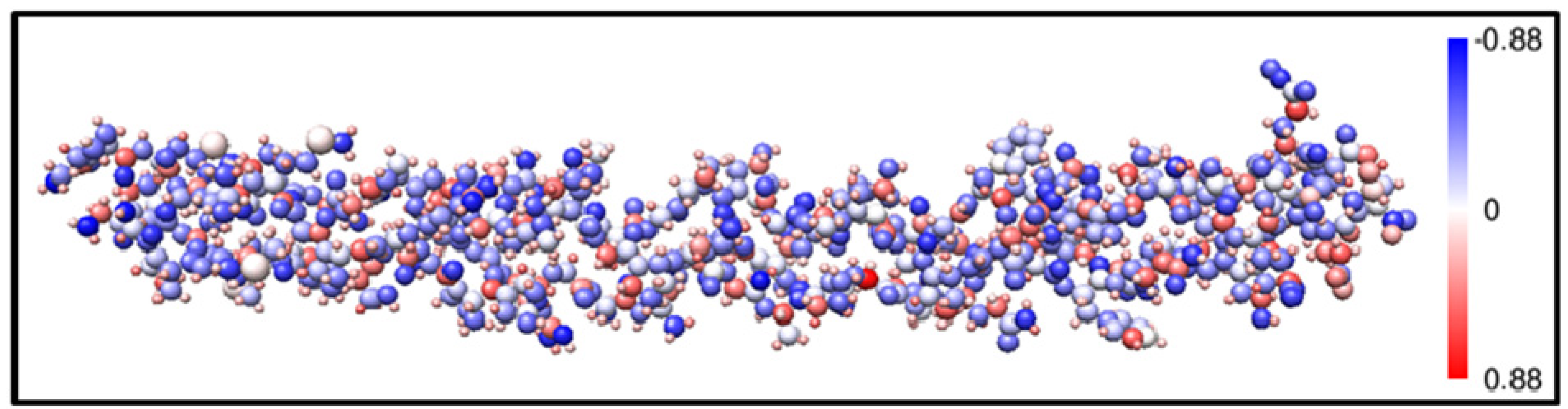
2.1. Description of the 7-2 Heterostructural Model

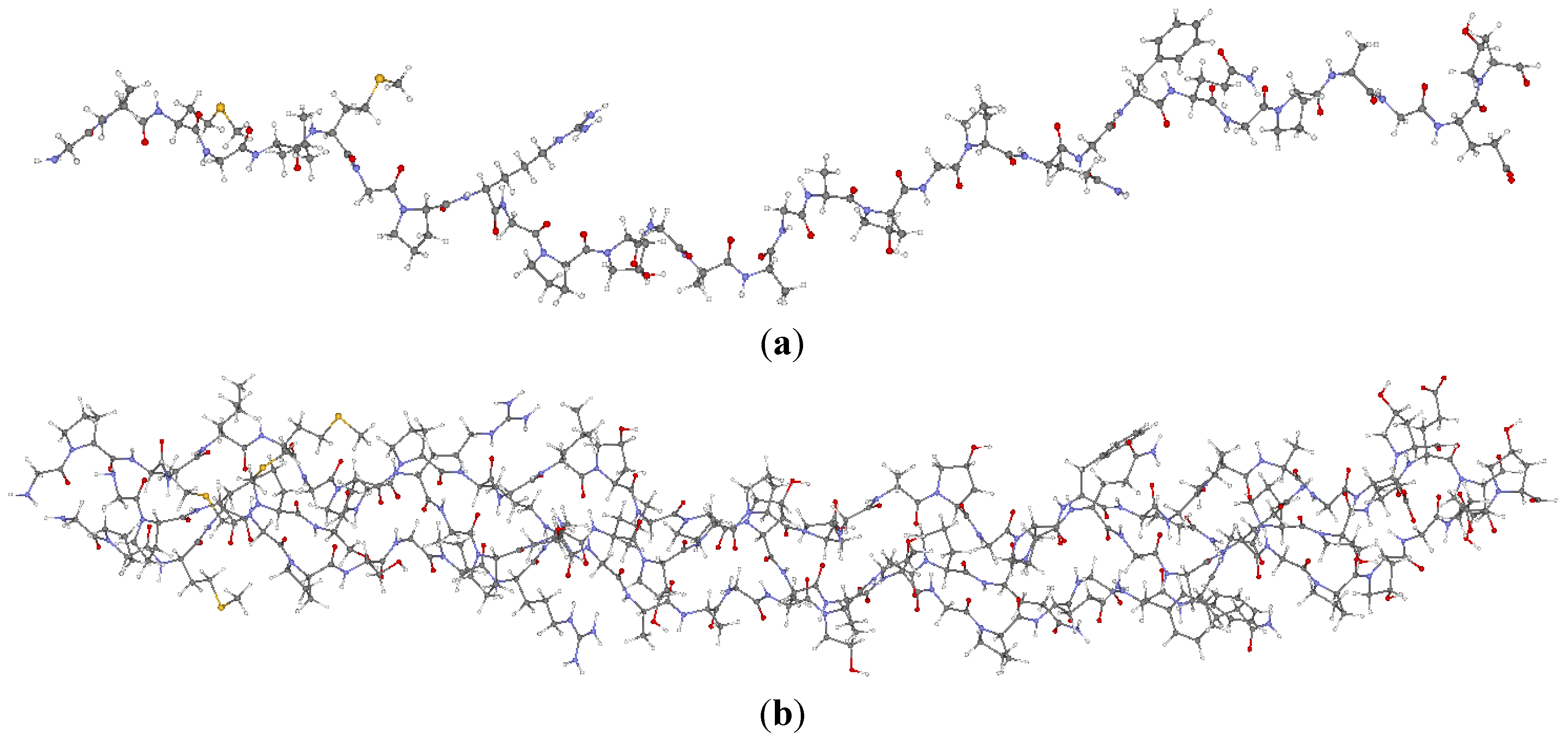{kind=link}
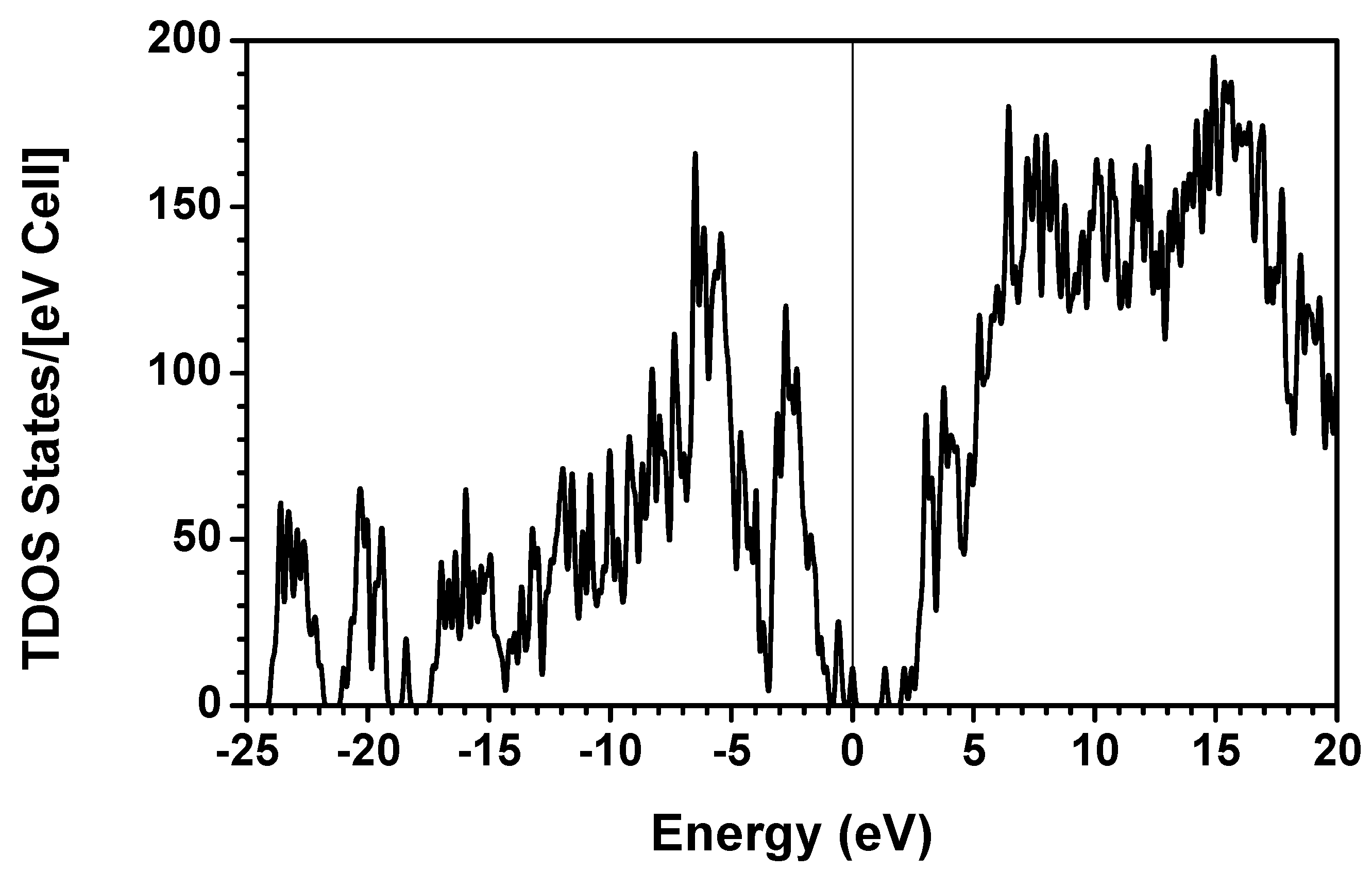{kind=link}
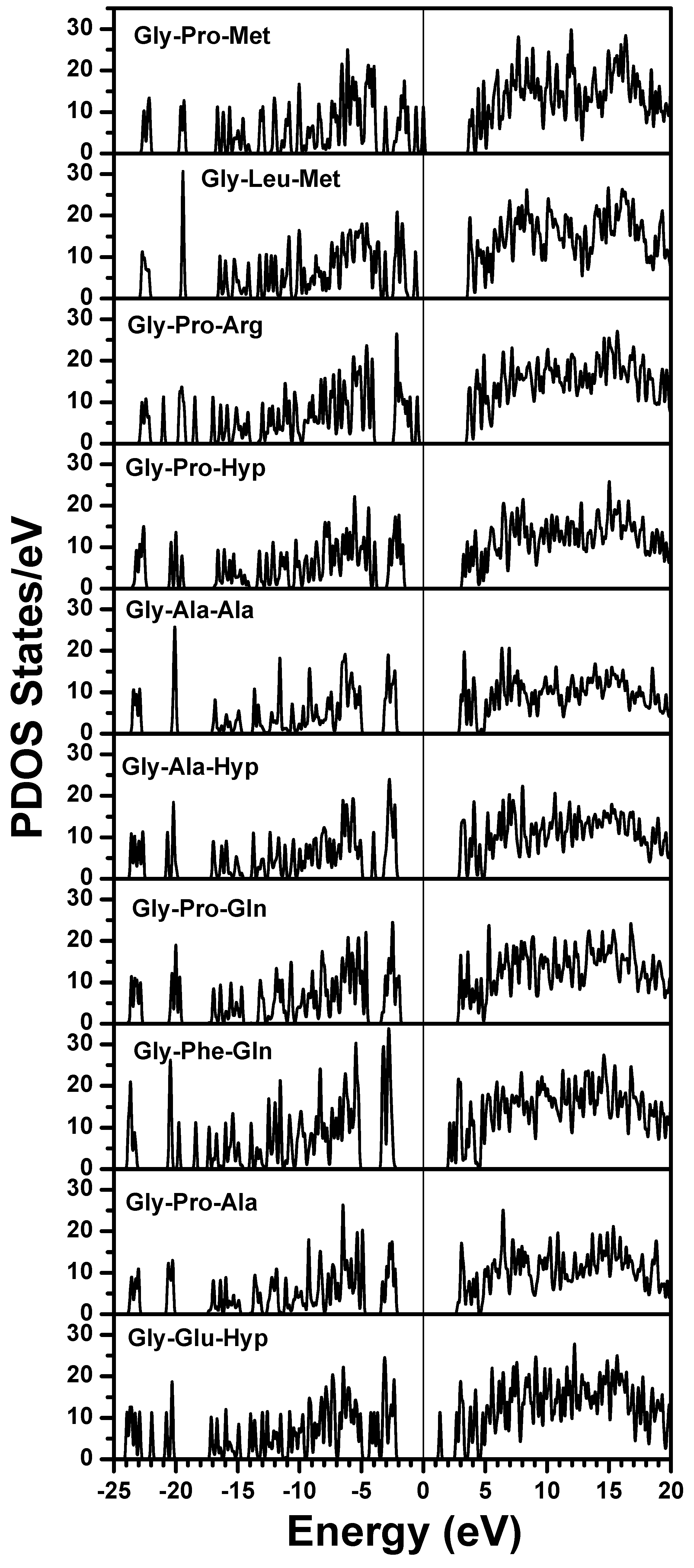{kind=link}
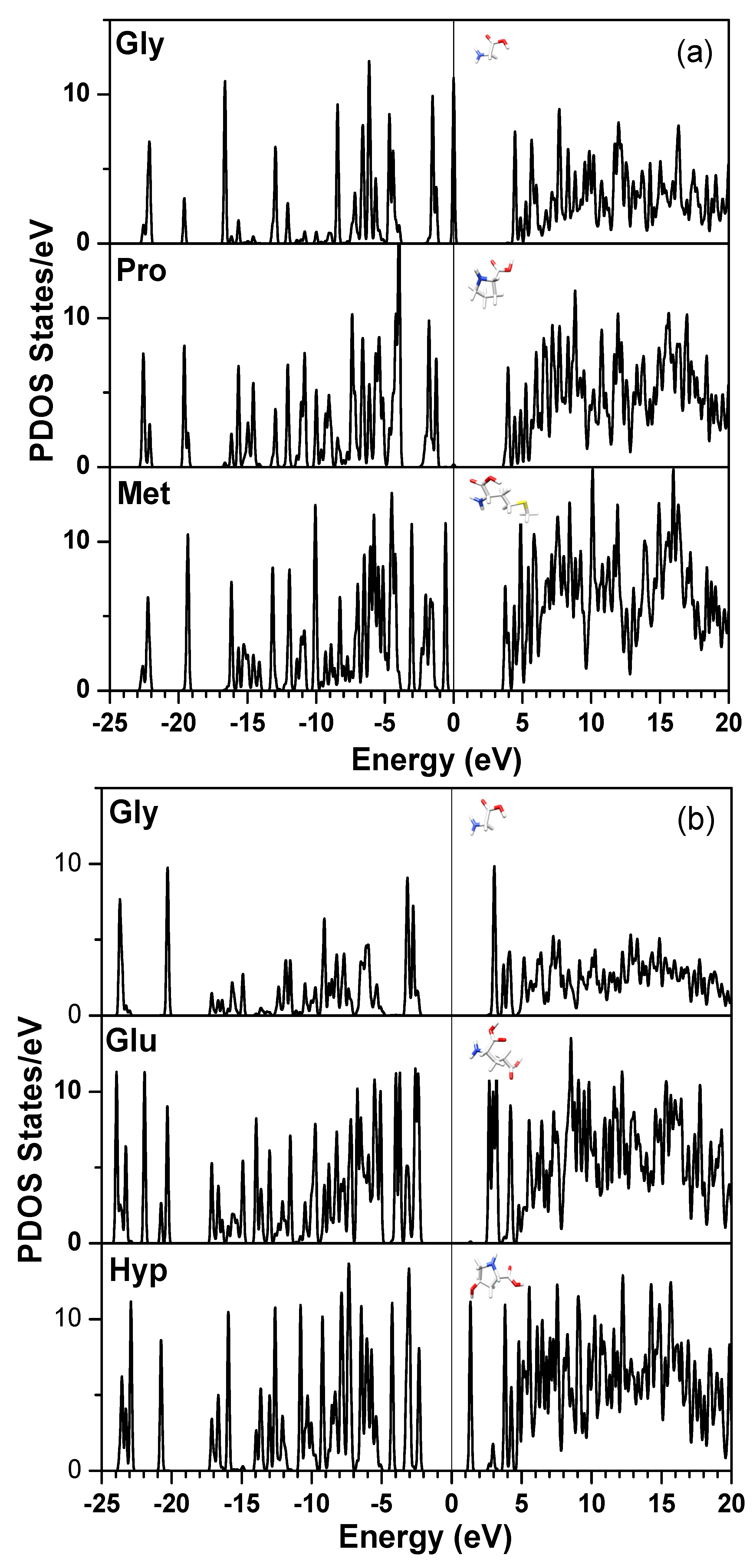{kind=link}
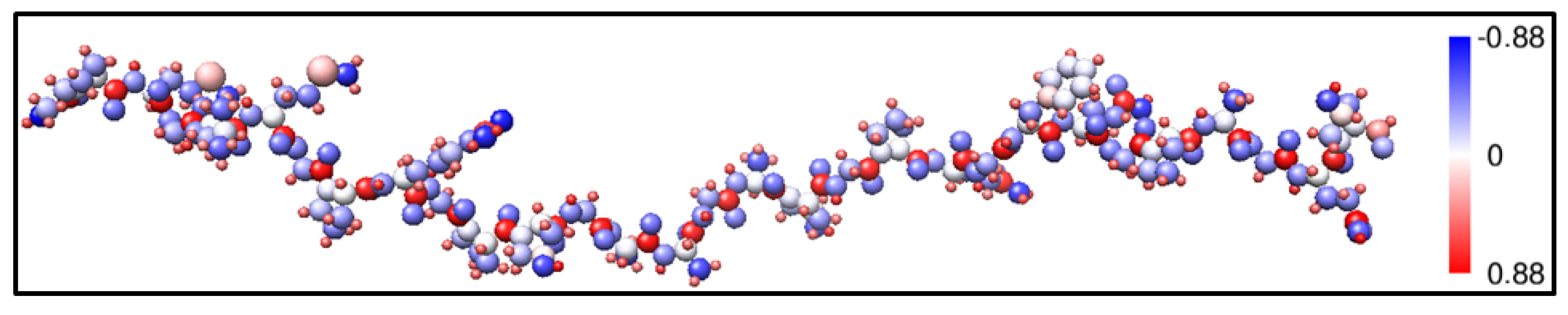{kind=link}
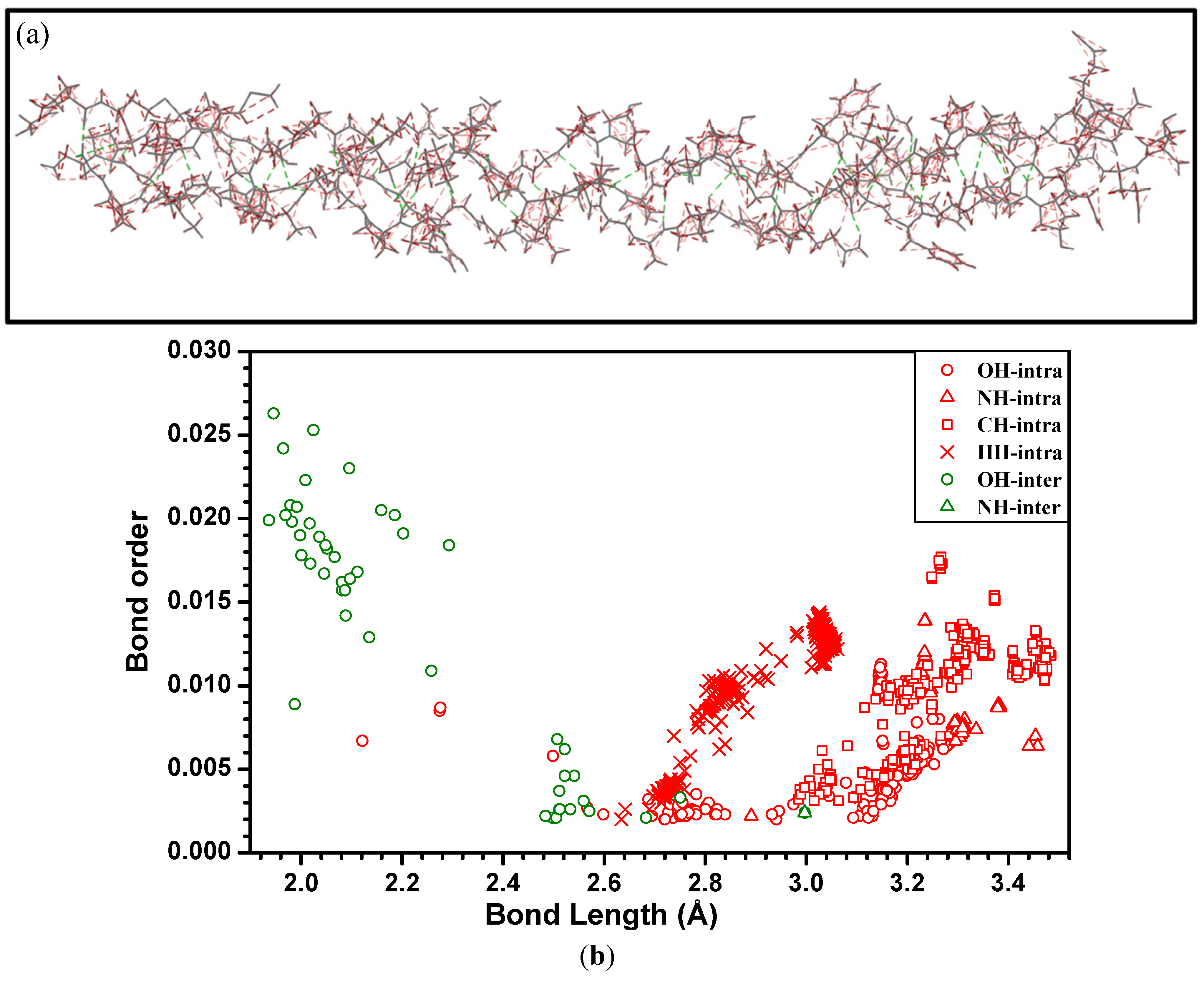{kind=link}
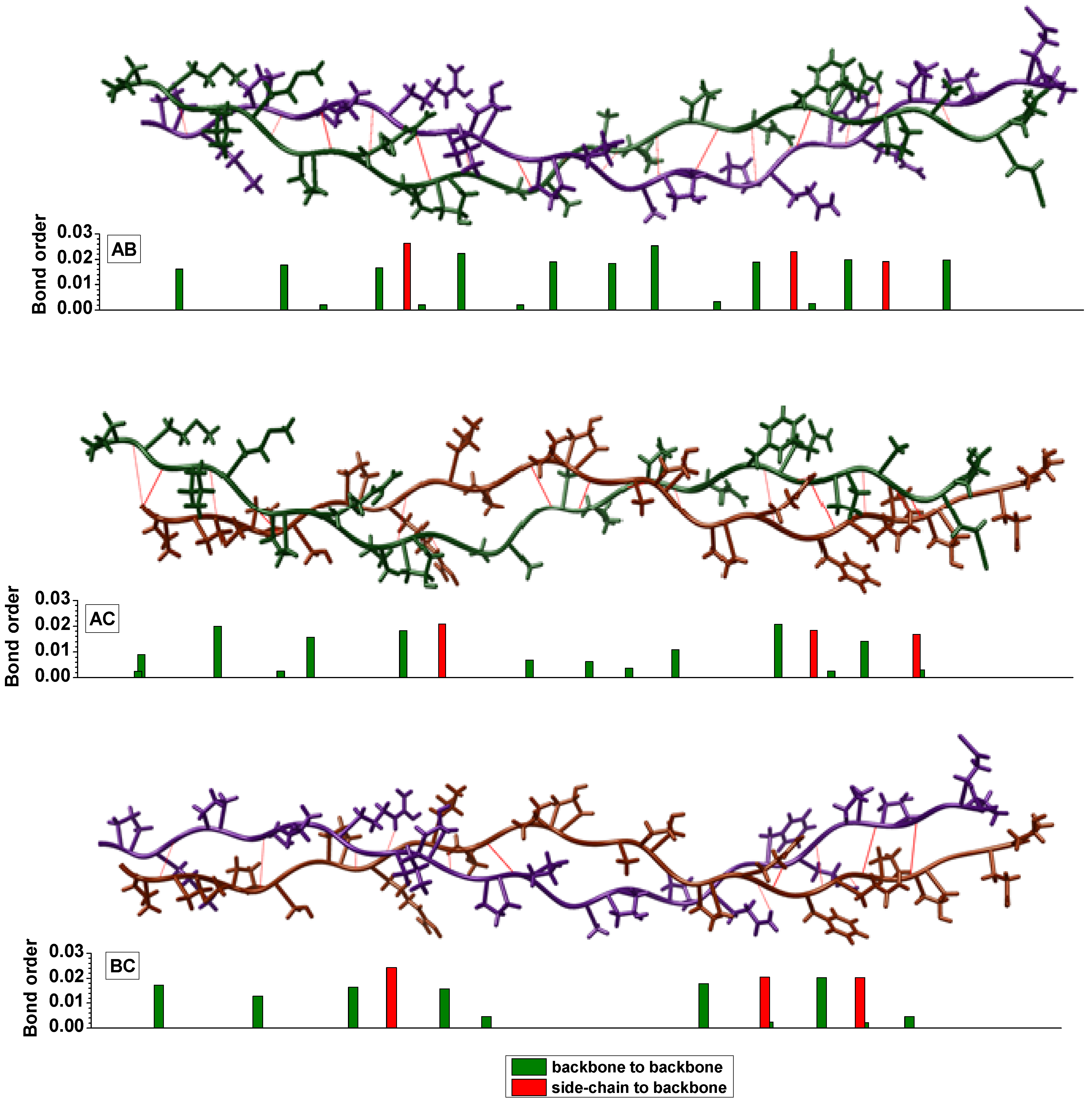{kind=link}
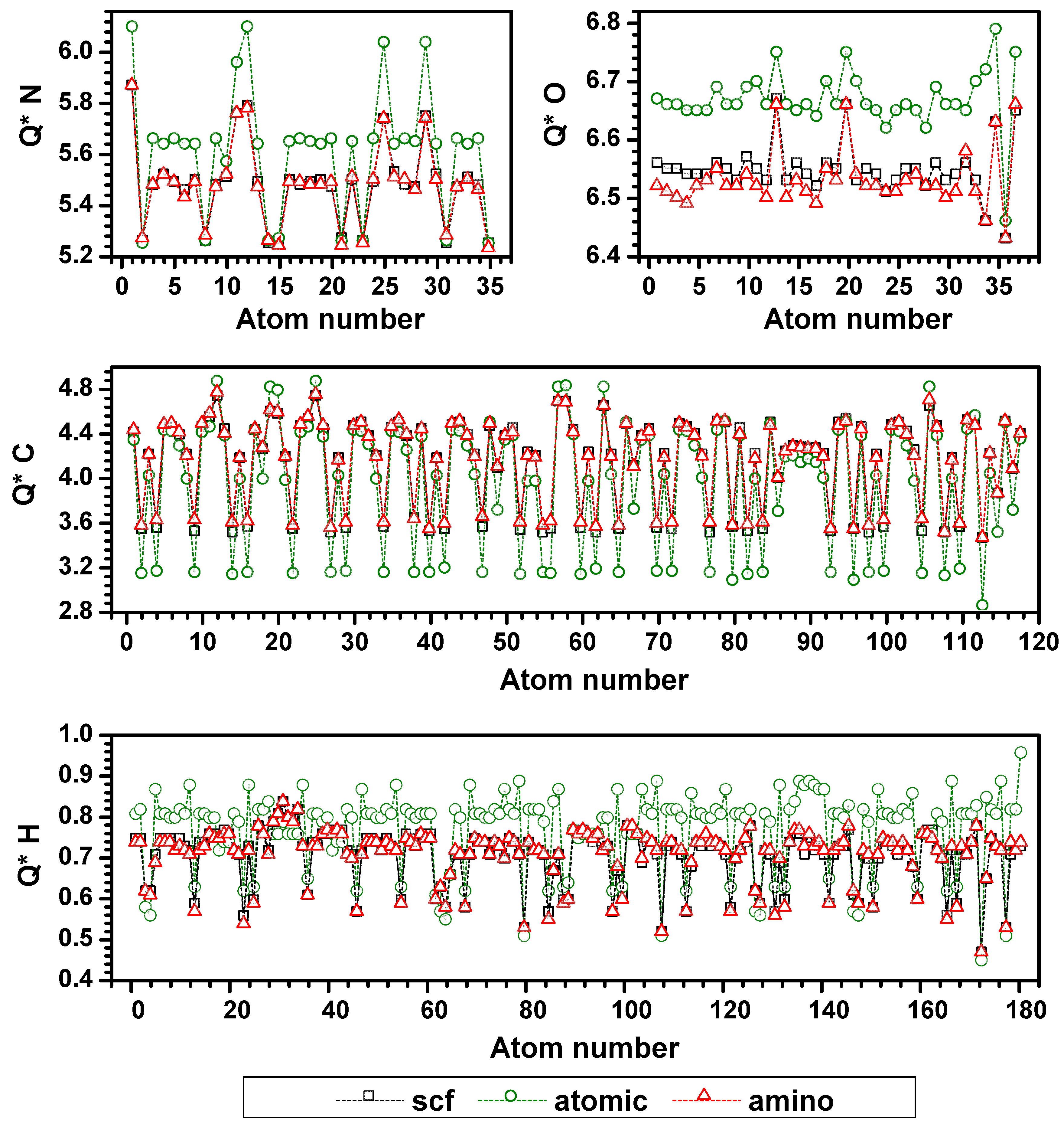{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chain 1: α2[I] | Chain 2: α1[I] | Chain 3: α1[I] |
|---|---|---|
| Gly-Pro-Met | Gly-Pro-Met | Gly-Pro-Met |
| Gly-Leu-Met | Gly-Pro-Ser | Gly-Pro-Ser |
| Gly-Pro-Arg | Gly-Pro-Arg | Gly-Pro-Arg |
| Gly-Pro-Hyp | Gly-Leu-Hyp | Gly-Leu-Hyp |
| Gly-Ala-Ala | Gly-Pro-Hyp | Gly-Pro-Hyp |
| Gly-Ala-Hyp | Gly-Ala-Hyp | Gly-Ala-Hyp |
| Gly-Pro-Gln | Gly-Pro-Gln | Gly-Pro-Gln |
| Gly-Phe-Gln | Gly-Phe-Gln | Gly-Phe-Gln |
| Gly-Pro-Ala | Gly-Pro-Hyp | Gly-Pro-Hyp |
| Gly-Glu-Hyp | Gly-Glu-Hyp | Gly-Glu-Hyp |

2.2. Method of Calculation
 is expanded in terms of atomic orbitals uiα centered at the atomic site Rα. The uiα are expanded as products of Gaussian-type-orbitals (GTOs) and spherical harmonics [51].
is expanded in terms of atomic orbitals uiα centered at the atomic site Rα. The uiα are expanded as products of Gaussian-type-orbitals (GTOs) and spherical harmonics [51].
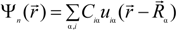
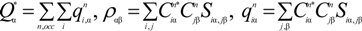
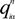 is the fractional charge of the ith orbital in the αth atom of the nth state which contains two electrons (one if the spin quantum number is included in i) and Siα,jβ are the overlap integrals. The calculation of Qα* and BO values enables us to quantitatively assess the charge transfer and bond strength in various parts of the system. The self-consistent potential for the collagen model was obtained by iteratively solving the Kohn-Sham equation with a full basis (FB) set (1s, 2s, 3s, 2p, 3p atomic orbitals for C, O, and N and 1s, 2s, 2p orbitals for H). A minimal basis (MB) set (1s, 2s, 2p for C, O, and N and 1s orbitals for H) was used for effective charge and BO calculations since Mulliken scheme works only for the more localized basis set.
is the fractional charge of the ith orbital in the αth atom of the nth state which contains two electrons (one if the spin quantum number is included in i) and Siα,jβ are the overlap integrals. The calculation of Qα* and BO values enables us to quantitatively assess the charge transfer and bond strength in various parts of the system. The self-consistent potential for the collagen model was obtained by iteratively solving the Kohn-Sham equation with a full basis (FB) set (1s, 2s, 3s, 2p, 3p atomic orbitals for C, O, and N and 1s, 2s, 2p orbitals for H). A minimal basis (MB) set (1s, 2s, 2p for C, O, and N and 1s orbitals for H) was used for effective charge and BO calculations since Mulliken scheme works only for the more localized basis set. and total charge density
and total charge density  :
:
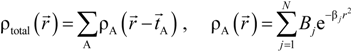
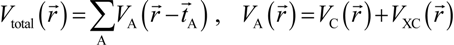
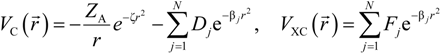
 . Because the accuracy of the calculation depends largely on how accurately the true charge density can be represented by Equations (3)–(5), the optimal choice of the fitting set {βj} is extremely important. When carefully constructed, these site-specific atom-centered potential functions are transferable. Thus, it is possible to obtain a self-consistent potential from the calculation of a simpler system (say quartz, α-SiO2) and then use it in the calculation of a similar but larger and more complicated system (amorphous SiO2 glass) without needing to perform self-consistent calculations. It should be noted that although the atom-centered functions
. Because the accuracy of the calculation depends largely on how accurately the true charge density can be represented by Equations (3)–(5), the optimal choice of the fitting set {βj} is extremely important. When carefully constructed, these site-specific atom-centered potential functions are transferable. Thus, it is possible to obtain a self-consistent potential from the calculation of a simpler system (say quartz, α-SiO2) and then use it in the calculation of a similar but larger and more complicated system (amorphous SiO2 glass) without needing to perform self-consistent calculations. It should be noted that although the atom-centered functions  and
and  consist of spherical Gaussians around each atom, the superposition of them is non-spherical in real space and can accurately represent different types of bonding in different types of structural configurations without shape approximation [51]. For large biomolecules with more than several thousand of atoms, the self-consistent procedure described above is still not feasible but we can make use of the accurately calculated and obtained from smaller subunits and apply them in the calculation of the larger system. We will return to this part later when we present the simplified method for large proteins using the self-consistent charge densities and potentials obtained from individual amino acids. We list the important parameters that enter into the OLCAO calculation for the 7-2 heterostructural model of collagen as follows: (1) number of chains 3; (2) number of trimmers 30; (3) number of amino acids 90; (4) number of atoms 1135; (5) number of valence electrons 3246; (6) size of total potential column data 12,706; (7) size of secular equation (FB) 7456.
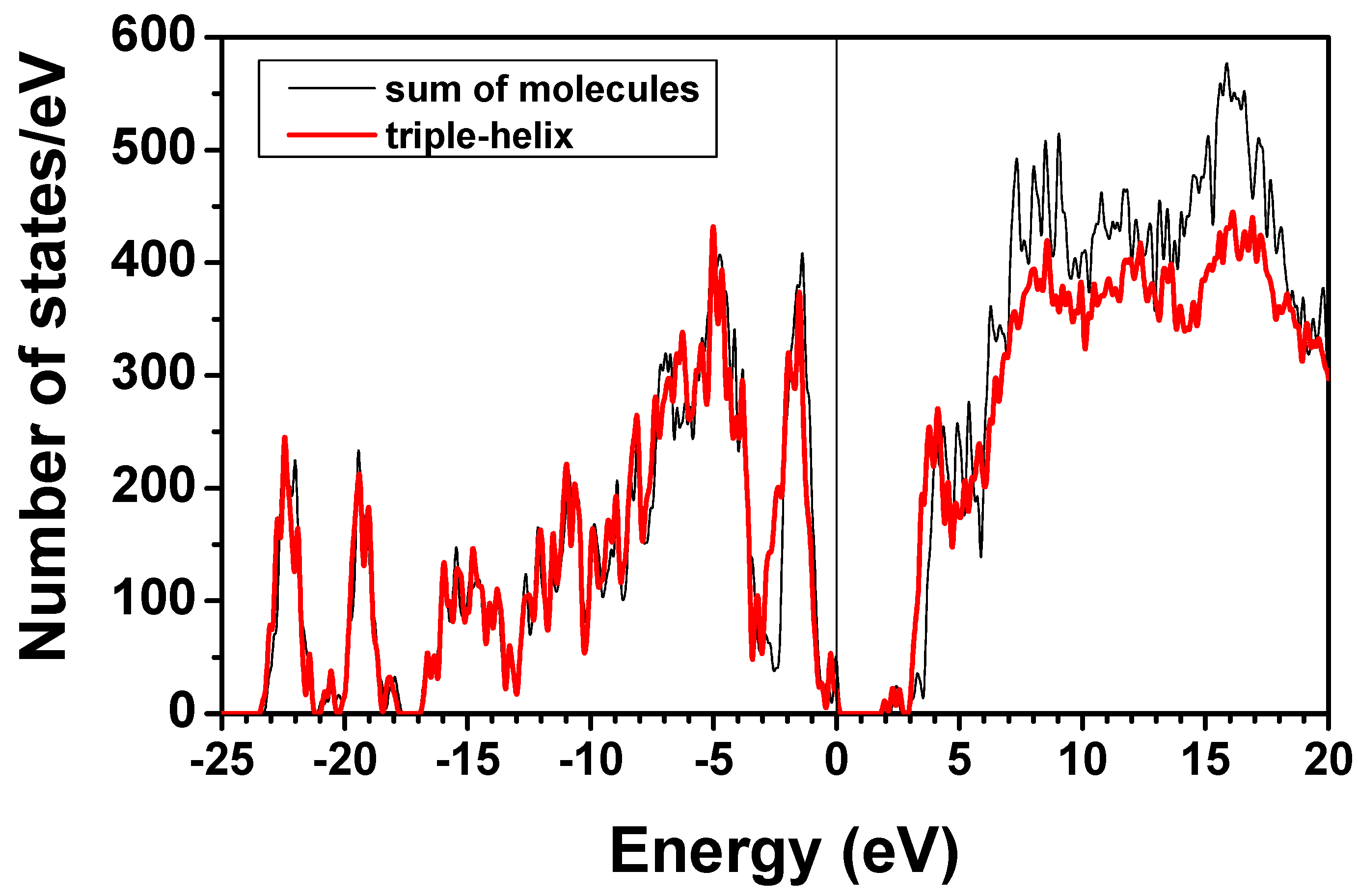
consist of spherical Gaussians around each atom, the superposition of them is non-spherical in real space and can accurately represent different types of bonding in different types of structural configurations without shape approximation [51]. For large biomolecules with more than several thousand of atoms, the self-consistent procedure described above is still not feasible but we can make use of the accurately calculated and obtained from smaller subunits and apply them in the calculation of the larger system. We will return to this part later when we present the simplified method for large proteins using the self-consistent charge densities and potentials obtained from individual amino acids. We list the important parameters that enter into the OLCAO calculation for the 7-2 heterostructural model of collagen as follows: (1) number of chains 3; (2) number of trimmers 30; (3) number of amino acids 90; (4) number of atoms 1135; (5) number of valence electrons 3246; (6) size of total potential column data 12,706; (7) size of secular equation (FB) 7456.2.3. Results and Discussions on the Collagen Model
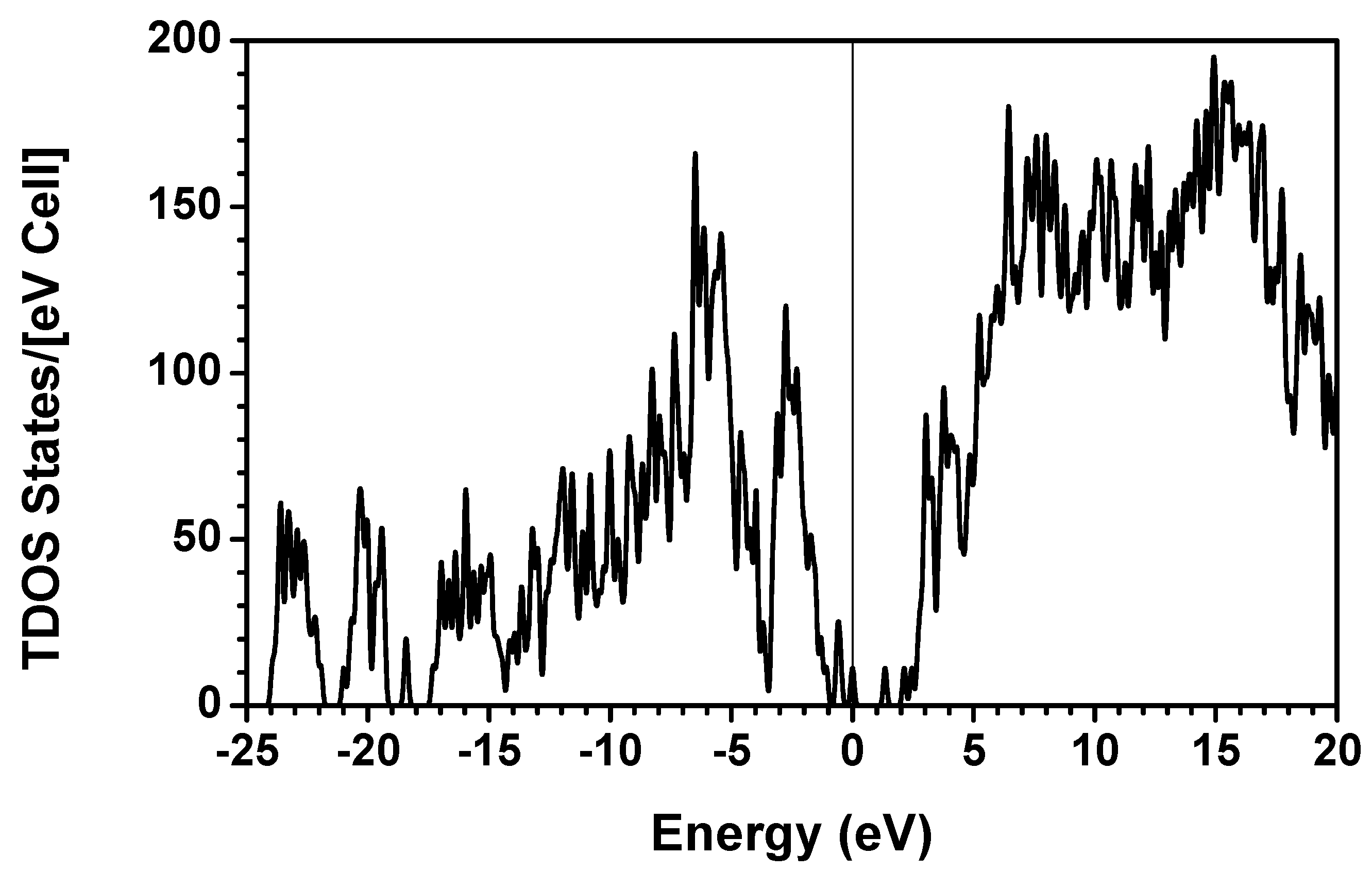
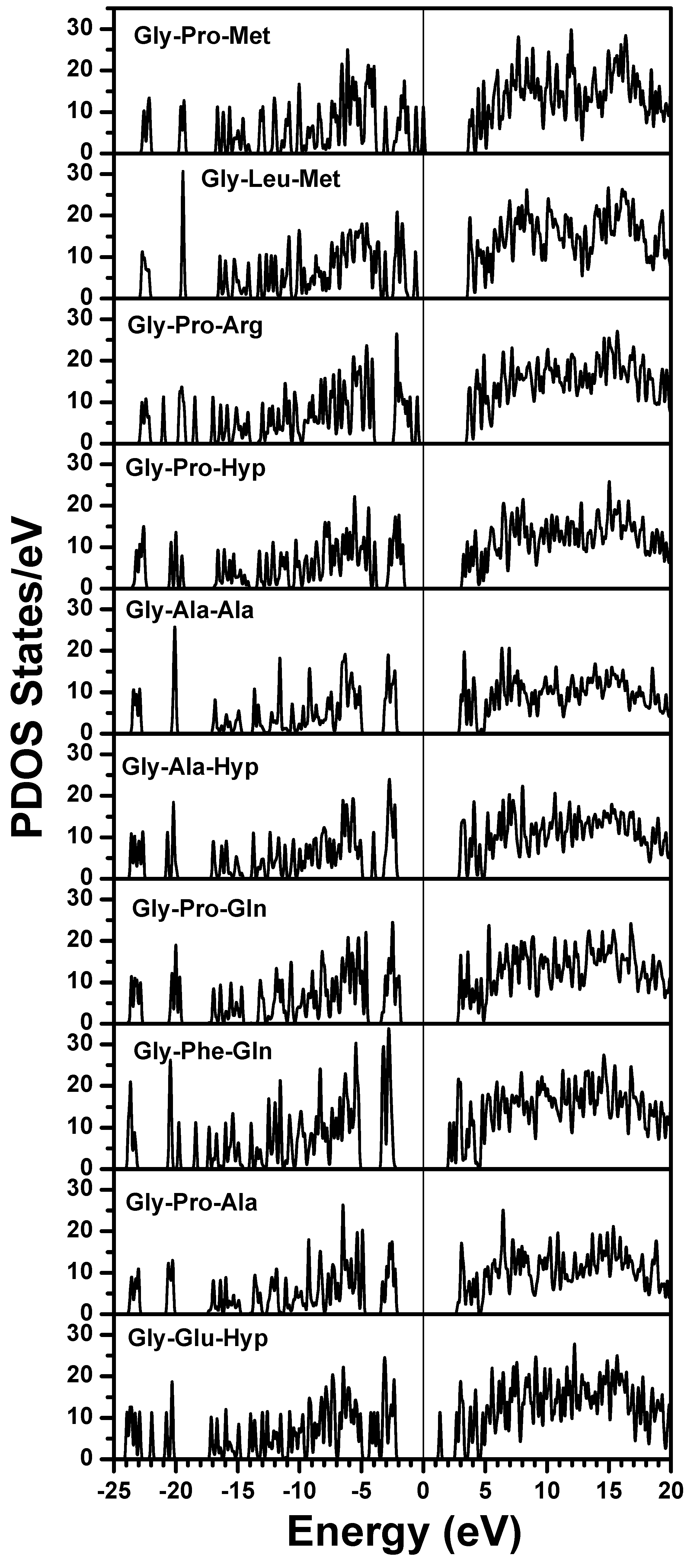

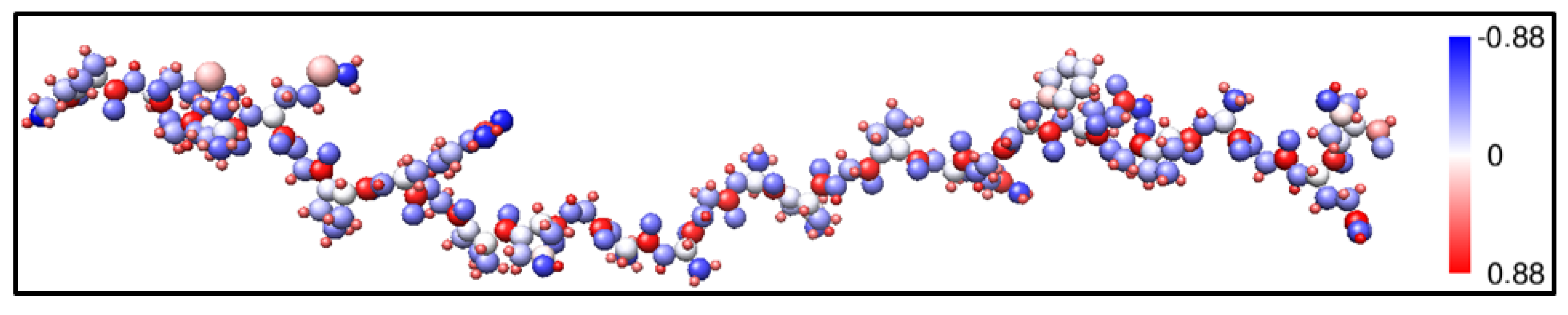
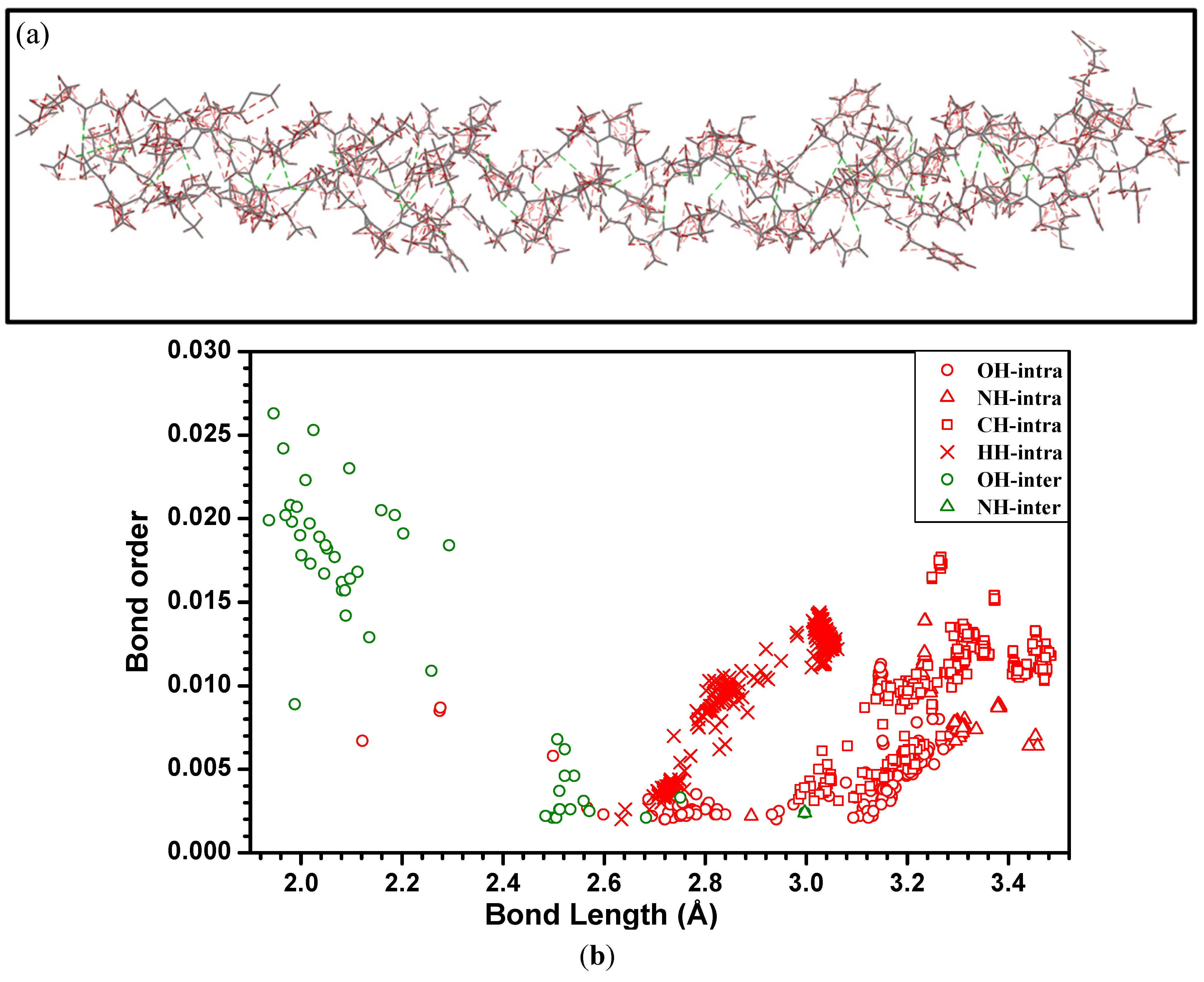
3. Amino-Acid Potential Method for Large Proteins
3.1. Current Trends in Computational Biophysical Research
3.2. Amino-Acid Potential Method for Large Proteins
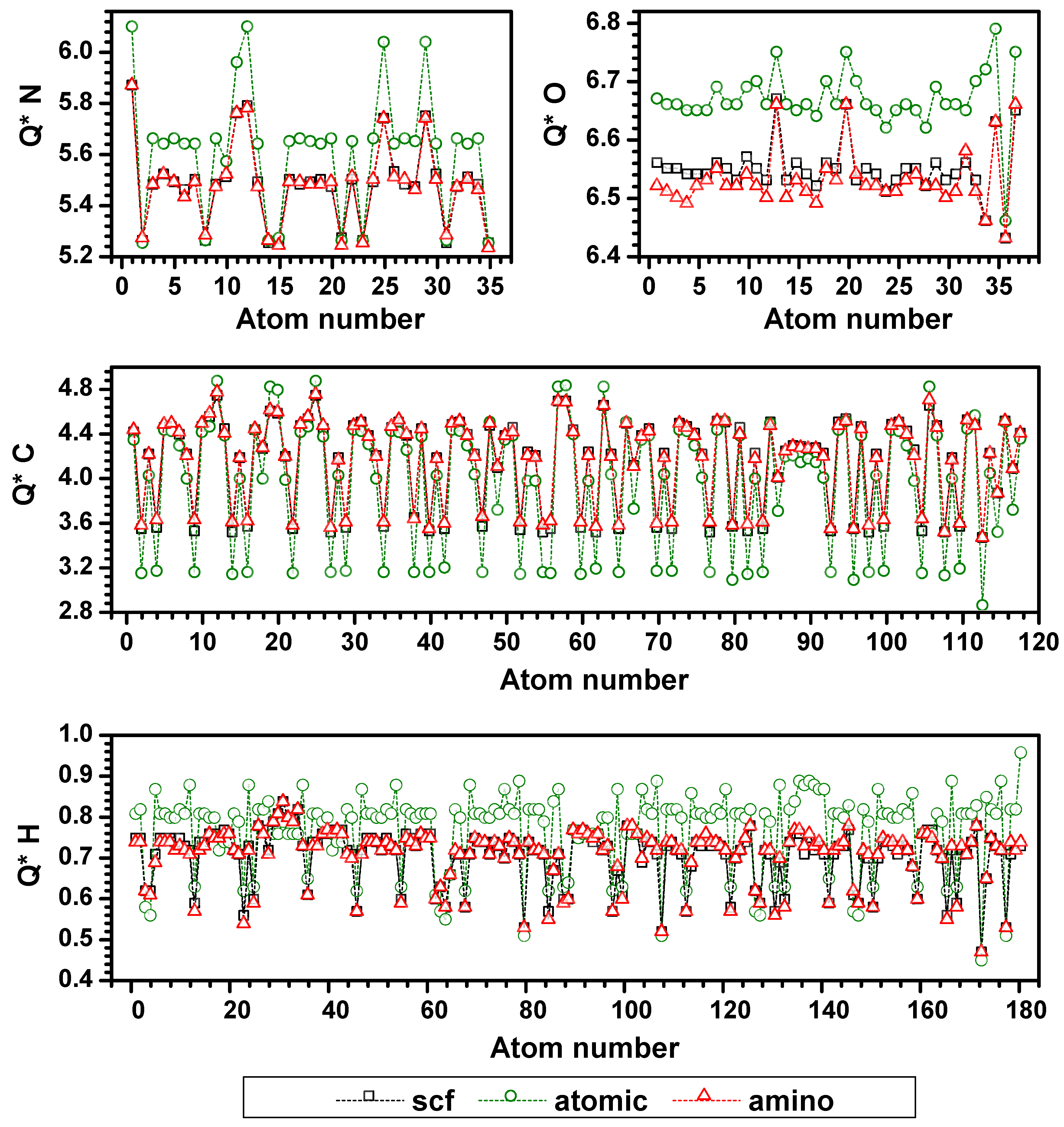
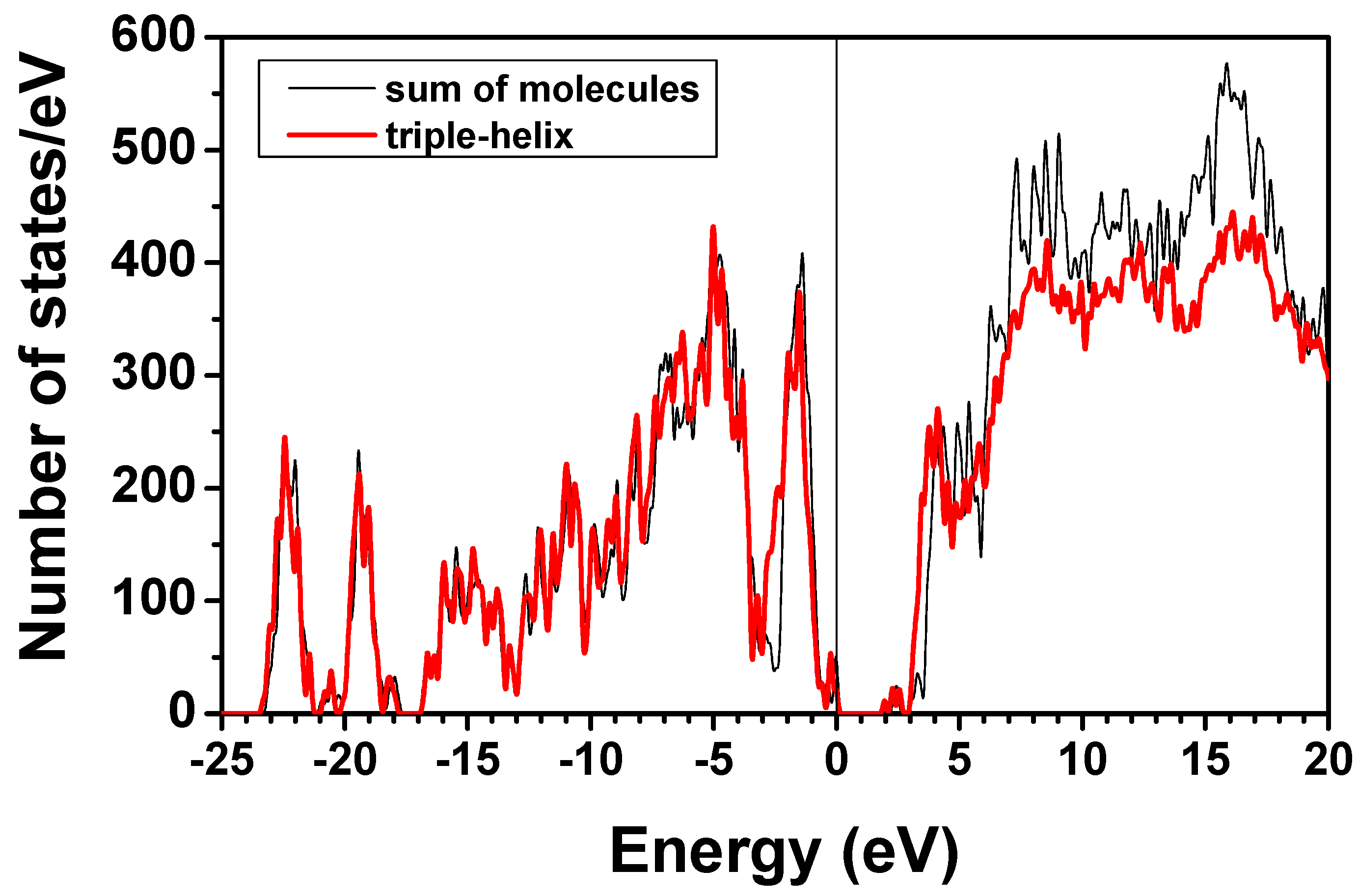
3.3. Validation of the AAPM
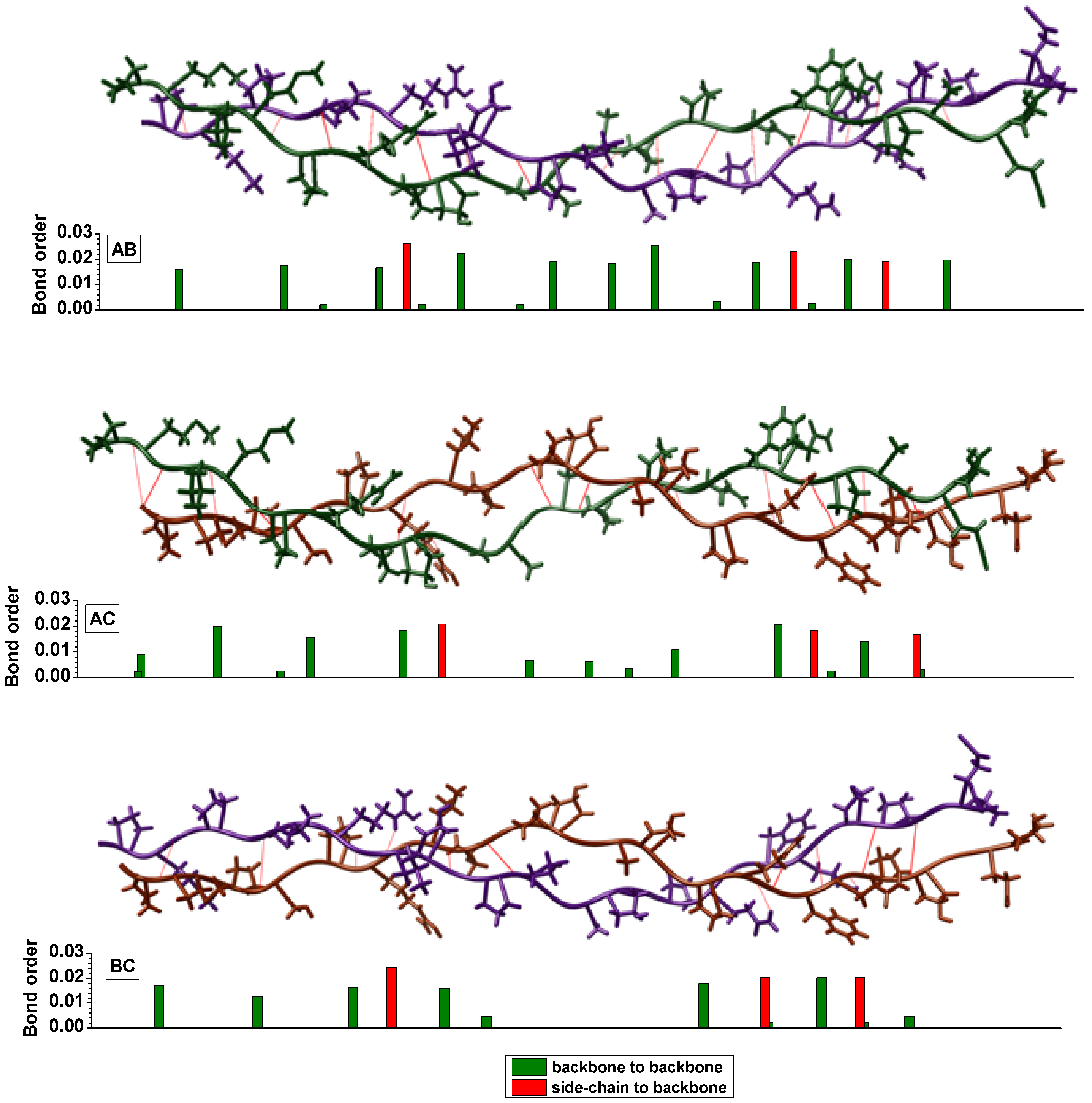
3.4. Possible Application of the AAPM Approach

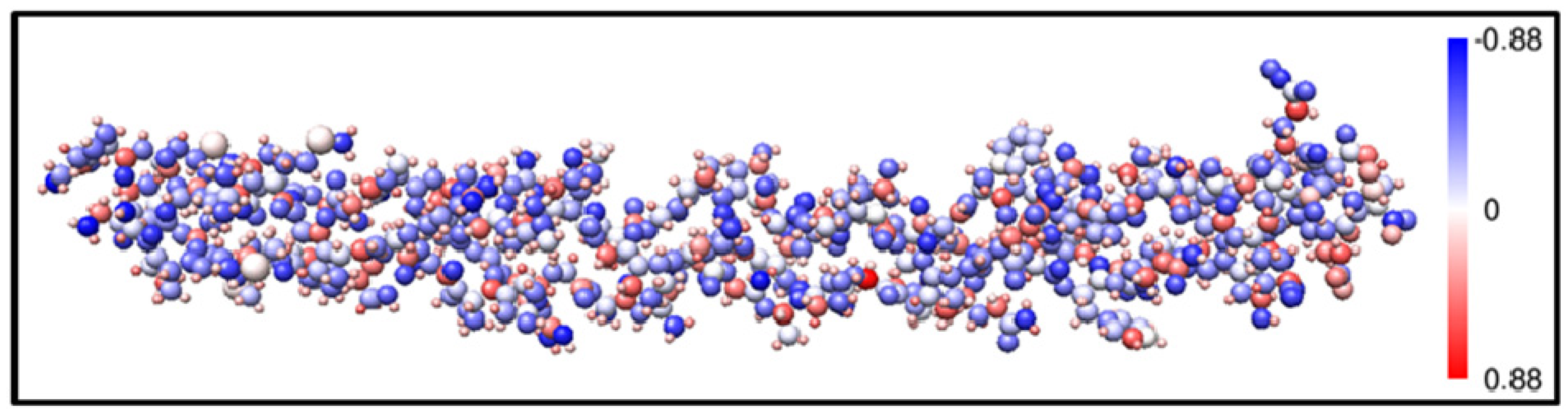
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Fratzl, P. Collagen: Structure and Mechanics; Springer: New York, NY, USA, 2008; Volume 1. [Google Scholar]
- Maynes, R.; Burgeson, R. Structure and Function of Collagen Types; Academic Press: London, UK, 1987. [Google Scholar]
- Bateman, J.F.; Lamande, S.; Ramshaw, J.A.M. Collagen Superfamily; Comper, W.D., Ed.; Harwood Academic Publishers: Amsterdam, The Netherlands, 1996; Volume 2, pp. 22–67. [Google Scholar]
- Ottani, V.; Raspanti, M.; Ruggeri, A. Collagen structure and functional implications. Micron 2001, 32, 251–260. [Google Scholar]
- Brodsky, B.; Perishov, A.V. Molecular structure of the collagen triple helix. Adv. Protein Chem. 2005, 70, 301–339. [Google Scholar] [CrossRef]
- Rich, A.; Crick, F.H.C. The structure of collagen. Nature 1955, 176, 915–916. [Google Scholar] [CrossRef]
- Wess, T.J.; Hammersley, A.P.; Wess, L.; Miller, A. A consensus model for molecular packing of type I collagen. J. Struct. Biol. 1998, 122, 92–100. [Google Scholar]
- Ramshaw, J.A.M.; Shah, N.K.; Brodsky, B. Gly-X-Y tripeptide frequencies in collagen: A context for host-guest triple-helical properties. J. Struct. Biol. 1998, 122, 86–91. [Google Scholar]
- Kramer, R.Z.; Vitagliano, L.; Bella, J.; Berisio, R.; Mazzarelaa, L.; Brodsky, B.; Zagari, A.; Berman, H.M. X-ray crystallographic determination of a collagen-like peptide with repeating sequence (Por-Por-Gly). J. Mol. Biol. 1998, 280, 623–638. [Google Scholar] [CrossRef]
- Kramer, R.Z.; Venugopal, M.G.; Bella, J.; Mayville, P.; Brodsky, B.; Berman, H.M. Staggered molecular packing in crystals of a collagen-like peptide with a single charge pair. J. Mol. Biol. 2000, 301, 1191–1205. [Google Scholar]
- Kramer, R.Z.; Bella, J.; Mayville, P.; Brodsky, B.; Berman, H.M. The crystal and molecular structure of a collagen-like peptide with a biologically related sequence. J. Mol. Biol. 2001, 311, 131–147. [Google Scholar]
- Berisio, R.; Vitagliano, L.; Mazzarella, L.; Zagari, A. Crystal structure of a collagen-like polypeptide with repeating sequence Pro-Hyp-Gly at 1.4 Å resolution: Implication for collagen hydration. Biopolymers 2001, 56, 8–13. [Google Scholar]
- Berisio, R.; Vitagliano, L.; Mazzarella, L.; Zagari, A. Crystal structure of the collagen triple helix model [(Pro-Pro-Gly)10]3. Protein Sci. 2002, 11, 262–270. [Google Scholar]
- Ottani, V.; Martini, D.; Franchi, M.; Ruggeri, A.; Raspanti, M. Hierarchical structures in fibrillar collagens. Micron 2002, 33, 587–596. [Google Scholar]
- Christiansen, D.L.; Huang, E.K.; Silver, F.H. Assembly of type I collagen: Fusion of fibril subunits and the influence of fibril diameter on mechanical properties. Matrix Biol. 2000, 19, 409–420. [Google Scholar]
- Gautieri, A.; Vesentini, S.; Redealli, A.; Buehler, M.J. Hierarchical structure and nanomechanics of collagen microfibrils from the atomistic scale up. Nano Lett. 2011, 11, 757–766. [Google Scholar]
- Brodsky, B.; Ramshaw, J.A.M. The collagen triple-helix structure. Matrix Biol. 1997, 15, 545–554. [Google Scholar] [CrossRef]
- Persikov, A.V.; Ramshaw, J.A.M.; Kirkpatrick, A.; Brodsky, B. The collagen triple-helix structure. Biochemistry 2000, 39, 14960–14967. [Google Scholar] [CrossRef]
- Persikov, A.; Ramshaw, J.A.M.; Brodksy, B. Collagen model peptides: Sequence dependence of triple helix stability. Biopolymers 2000, 55, 436–450. [Google Scholar] [CrossRef]
- Almora-Barrios, N.; de Leeuw, N.H. Molecular dynamics simulation of the early stages of nucleation of hydroxyapatite at a collagen template. Cryst. Growth Des. 2012, 12, 756–763. [Google Scholar] [CrossRef]
- Kuibaniemi, H.; Tormp, G.; Prockop, D. Mutations in collagen genes: Causes of rare and some common diseases in humans. FASEB J. 1991, 5, 2052–2060. [Google Scholar]
- Scheuler-Furman, O.; Wang, C.; Bradeley, P.; Misura, K.; Baker, D. Progress in modeling of protein structures and interactions. Science 2005, 310, 638–642. [Google Scholar] [CrossRef]
- Butter, D.; Docherty, R.; Swart, R.M. The application of computational chemistry to the study of chemistry of collagen. J. Am. Leather Chem. Assoc. 1997, 92, 185–199. [Google Scholar]
- Klien, T.E.; Huang, C.C. Computational investigations of structure changes resulting from point mutation in a collegen-like peptide. Biopolymers 1999, 49, 167–183. [Google Scholar] [CrossRef]
- Mooney, S.D.; Huang, C.C.; Kollman, P.A.; Klein, T.E. Computed free energy differences between point mutations in a collagen-like peptide. Biopolymers 2001, 58, 347–353. [Google Scholar] [CrossRef]
- Vaidyanathan, J.; Vaidyanathan, T.K.; Yadav, P.; Linarus, C.E. Collagen-ligand interaction in dentinal adhesion: Computer visualization and analysis. Biomaterials 2001, 22, 2911–2920. [Google Scholar] [CrossRef]
- Madhan, B.; Thanikaivelan, P.; Subramanian, V.; Raghava, J.R.; Balachandran, U.N.; Ramasami, T. Molecular mechanics and dynamics on the interaction of gallic acid with collagen-like peptides. Chem. Phys. Lett. 2001, 346, 334–340. [Google Scholar] [CrossRef]
- Improta, R.; Freanca, M.; Crescenzi, O.; Benzi, C.; Barone, V. Understanding the role of stereoelectronic effects in determining collagen stability: 2. A quantum mechanical/molecular mechanical study of (Proline-Proline-Glycine)(n) polypeptides. J. Am. Chem. Soc. 2002, 124, 7857–7865. [Google Scholar] [CrossRef]
- Lorenzo, A.C.; Caffarena, W.R. Elastic properties, Young’s modulus determination and structural stability of the tropocollagen molecule: A computational study by steered molecular dynamics. J. Biomech. 2005, 38, 1527–1533. [Google Scholar] [CrossRef]
- Vesentini, S.; Fitie, C.F.C.; Montevecchi, F.M.; Redaelli, A. Molecular assessment of the elastic properties of collagen-like homotrimer sequences. Biomech. Model Mechan. 2005, 3, 224–234. [Google Scholar]
- Gautieri, A.; Vesentini, S.; Montevecchi, F.M.; Redaelli, A. Mechanical properties of physiological and pathological models of collagen peptides investigated via steered molecular dynamics simulations. J. Biomech. 2008, 41, 3073–3077. [Google Scholar] [CrossRef]
- Gautieri, A.; Uzel, S.; Vesentini, S.; Redaelli, A.; Buehler, M.J. Molecular and mesoscale mechanisms of osteogenesis imperfecta disease in collagen fibrils. Biophys. J. 2009, 97, 857–865. [Google Scholar] [CrossRef]
- Gautieri, A.; Russo, A.; Vesentini, S.; Redaelli, A.; Buehler, M.J. Coarse-grained model of collagen molecules using an extended MARTINI force field. J. Chem. Theory Comput. 2010, 6, 1210–1218. [Google Scholar]
- Rainey, J.K.; Goh, M.C. A statistically derived parameterization for the collagen triple-helix. Protein Sci. 2002, 11, 2748–2754. [Google Scholar] [CrossRef]
- Buehler, M.J. Nature designs tough collagen: Explaining the nanostructure of collagen fibrils. P Natl. Acad. Sci. 2006, 103, 12285–12290. [Google Scholar] [CrossRef]
- Gautieri, A.; Pate, M.I.; Vesentini, S.; Redaelli, A.; Buehler, M.J. Hydration and distance dependence of intermolecular shearing between collagen molecules in a model microfibril. J. Biomech. 2012, 45, 2079–2083. [Google Scholar]
- Rhys, N.H.; Dougan, L. The emerging role of hydrogen bond interactions in polyglutamine structure, stability and association. Soft Matter 2013, 9, 2359–2364. [Google Scholar] [CrossRef]
- Gautieri, A.; Vesentini, S.; Redaelli, A.; Buehler, M. Intermolecular slip mechanism in tropocollagen nanofibrils. Int. J. Mater. Res. 2009, 7, 921–925. [Google Scholar]
- Qin, Z.; Buehler, M.J. Molecular dynamics simulation of the α-helix to β-sheet transition in coiled protein filaments: Evidence for a critical filament length scale. Phys. Rev. Lett. 2010, 104, 198304:1–198304:4. [Google Scholar]
- Grant, C.A.; Phillips, M.A.; Thomson, N.H. Dynamic mechanical analysis of collagen fibrils at the nanoscale. J. Mech. Behav. Biomed. 2012, 5, 165–170. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R. The amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Parthasarathi, R.; Madhan, B.; Subramanian, V.; Ramasami, T. Ab initio and density functional theory based studies on collagen triplets. Theor. Chem. Acc. 2003, 110, 19–27. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry: III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1988, 45, 13244–13249. [Google Scholar] [CrossRef]
- Jalkanen, K.J.; Olsen, K.; Knapp-Mohammady, M.; Bohr, J. First-principles structures for the close-packed and the 7/2 motif of collagen. Europhys. Lett. 2012, 100, 28003:1–28003:4. [Google Scholar] [CrossRef]
- Bohr, J.; Olsen, K. The close-packed triple helix as a possible new structural motif for collagen. Theor. Chem. Acc. 2011, 130, 1095–103. [Google Scholar] [CrossRef]
- Ching, W.Y. Theoretical studies of electronic properties of ceramic materials. J. Am. Ceram. Soc. 1990, 71, 3135–3160. [Google Scholar] [CrossRef]
- Ching, W.Y.; Rulis, P. Electronic Structure Methods for Complex Materials: The Orthogonalized Linear Combination of Atomic Orbitals; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Okuyama, K.; Takayanagi, M.; Ashida, T.; Kakudo, M. A new structural model for collagen. Polym. J. 1977, 9, 341–343. [Google Scholar] [CrossRef]
- Rich, A.; Crick, F.H.C. The molecular structure of collagen. J. Mol. Biol. 1961, 3, 483–506. [Google Scholar] [CrossRef]
- Bella, J. A new method for describing the helical conformation of collagen: Dependence of the triple helical twist on amino acid sequence. J. Struct. Biol. 2010, 170, 377–391. [Google Scholar] [CrossRef]
- Okuyama, K.; Kawaguchi, T.; Shimura, M.; Noguchi, K.; Mizuno, K.; Bachinger, H.P. Crystal structure of the collagen model peptide (Pro-Pro-Gly) 4-Hyp-Asp-Gly-(Pro-Pro-Gly) 4 at 1.0 Å resolution. Biopolymers 2013, 99, 436–447. [Google Scholar] [CrossRef]
- Okuyama, K.; Miyama, K.; Mizuno, K.; Bachinger, H.P. Crystal structure of (Gly-Pro-Hyp) 9: Implications for the collagen molecular model. Biopolymers 2012, 97, 607–616. [Google Scholar] [CrossRef]
- Okuyama, K.; Xu, X.; Iguchi, M.; Noguchi, K. Revision of collagen molecular structure. Peptide Sci. 2006, 84, 181–191. [Google Scholar] [CrossRef]
- Vesentini, S.; Redaelli, A.; Montevecchi, F.M. Estimation of the binding force of the collagen molecule-decorin core protein complex in collagen fibril. J. Biomech. 2005, 38, 433–443. [Google Scholar] [CrossRef]
- Ching, W.Y.; Xu, Y.N.; Lou, Y.; Jean, Y.C. Electronic states and Fermi surfaces in the organic superconductor k-(ET)2CuN(CN)2Br. Phys. Rev. B 1997, 55, 2780–2783. [Google Scholar] [CrossRef]
- Caruso, A.N.; Pokhodyna, K.I.; Shum, W.W.; Ching, W.Y.; Anderson, B.; Bremer, M.T.; Vescovo, E.; Rulis, P.; Epstein, A.J.; Miller, J.S. Spin polarization from organic-based magnet [FeII(TCNE)(NCMe)2][FeIIICl4]. Phys. Rev. B 2009, 79, 195202–1. [Google Scholar]
- Ouyang, L.; Randaccio, L.; Rulis, P.; Kurmaev, E.Z.; Moewes, A.; Ching, W.Y. Electronic structure and bonding in Vitamin B12 cyanocobalamin. J. Mol. Struct. (Theo. Chem.) 2003, 622, 221–227. [Google Scholar] [CrossRef]
- Ouyang, L.; Rulis, P.; Ching, W.Y.; Nadin, G.; Randaccio, L. Electronic structure and bonding in adenosylcobalamin. Inorg. Chem. 2004, 43, 1235–1241. [Google Scholar]
- MacNaughton, J.B.; Moewes, A.; Lee, J.S.; Wetting, S.D.; Kraatz, H.-B.; Ouyang, L.; Ching, W.Y.; Kurmaev, E.Z. Dependence of the DNA electronic structure on environmental and structural variations. J. Phys. Chem. 2006, 110, 15742–15748. [Google Scholar]
- Liang, L.; Rulis, P.; Kahr, B.; Ching, W.Y. Theoretical study of the large linear dichroism of herapathite. Phys. Rev. B 2009, 80, 235132:1–235132:5. [Google Scholar]
- Ching, W.Y.; Rulis, P.; Misra, A. Ab initio elastic properties and tensile strength in hydroxyapatite crystal. Acta Biomater. 2009, 5, 3067–3075. [Google Scholar] [CrossRef]
- Liang, L.; Rulis, P.; Ching, W.Y. Mechanical properties, electronic structure and bonding of α- and β-tri-calcium phosphates with surface characterization. Acta Biomater. 2010, 6, 3763–3771. [Google Scholar]
- Liang, L.; Rulis, P.; Ouyang, L.; Ching, W.Y. Ab initio investigation of hydrogen bonding and network structure in a supercooled model of water. Phys. Rev. B 2011, 83, 024201:1–024201:7. [Google Scholar]
- Mulliken, R.S. Electron population analysis on LCAO-MO molecular wave functions I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Pearson, R.G. Recent advances in the concept of hard and soft acids and bases. J. Chem. Educ. 1987, 64, 561–567. [Google Scholar] [CrossRef]
- Parr, R.G.; Chattaraj, P.K. Principle of maximum hardness. J. Am. Chem. Soc. 1991, 113, 1854–1855. [Google Scholar] [CrossRef]
- De Proft, F.; Geerling, P. Conceptual and computational DFT in the study of aromaticity. Chem. Rev. 2001, 101, 1451–1464. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Fuentealba, P.; Jaque, P.; Toro-Labbe, A. Validity of the minimal polarizability principle in molecular vibrations and internal rotations: An ab initio SCF study. J. Phys. Chem. A 1999, 103, 9307–9312. [Google Scholar]
- Jeffrey, G.A. An Introduction to H-Bonding (Topics in Physical Chemistry); Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Klamt, A.; Reinisch, J.; Eckert, F.; Graton, J.; le Questel, J. Interpretation of experimental hydrogen-bond enthalpies and entropies from COSMO polarisation charge densities. Phys. Chem. Chem. Phys. 2013, 15, 7147–7154. [Google Scholar]
- Devereux, M.; Popelier, P.; McLay, I. A refined model for prediction of hydrogen bond acidity and basicity parameters from quantum chemical molecular descriptors. Phys. Chem. Chem. Phys. 2009, 11, 1595–1603. [Google Scholar] [CrossRef]
- Kenny, P. Hydrogen bonding, electrostatic potential, and molecular design. J. Chem. Inf. Model 2009, 49, 1234–1244. [Google Scholar] [CrossRef]
- Nocker, M.; Handschuh, S.; Tautermann, C.; Liedl, K. Theoretical prediction of hydrogen bond strength for use in molecular modeling. J. Chem. Inf. Model 2009, 49, 2067–2076. [Google Scholar] [CrossRef]
- Foster, M.E.; Sohlberg, K. Empirically corrected DFT and semi-empirical methods for non-bonding interactions. Phys. Chem. Chem. Phys. 2010, 12, 307–322. [Google Scholar] [CrossRef]
- Brodsky, B. Hydrogen bonding in the triple-helix. Proc. Indian Acad. Sci. 1999, 111, 13–8. [Google Scholar]
- French, R.H.; Parsegian, V.A.; Podgornik, R.; Rajter, R.F.; Jagota, A.; Luo, J.; Asthagiri, D.; Chaudhury, M.K.; Chiang, Y.; Granick, S.; et al. Long range interactions in nanoscale science. Rev. Mod. Phys. 2010, 82, 1887–1944. [Google Scholar] [CrossRef]
- Tajkhorshi, E.; Aksimentiev, A.; Balabin, I.; Gao, M.; Isralewitz, B.; Phillips, J.C.; Zhu, F.; Schulten, K. Large scale simulation of protein mechanics and function. Adv. Protein Chem. 2003, 66, 195–247. [Google Scholar] [CrossRef]
- Enkovaara, J.; Rostgaard, C.; Mortensen, J.J.; Chen, J.; Dulak, M.; Ferrighi, L.; Gavnholt, J.; Glinsvad, C.; Haikola, V; Hansen, H.A.; et al. Electronic structure calculations with GPAW: A real-space implementation of the projector augmented-wave method. J. Phys. Condens. Matter 2010, 22, 253202:1–253202:24. [Google Scholar]
- Chaban, G.M.; Pizzarello, S. Ab initio calculations of 6- and 7-carbon meteoritic amino acids and their diastereomers. Meteor. Planet Sci. 2010, 45, 1053–1060. [Google Scholar] [CrossRef]
- Szalay, P.G.; Watson, T.; Perera, A.; Lotrich, V.F.; Bartlett, R.J. Benchmark studies on the building blocks of DNA: 1. Superiority of coupled cluster methods in describing the excited states of nucleobases in the Franck-Condon region. J. Chem. Phys. A 2012, 116, 6702–6710. [Google Scholar]
- Berisio, R.; DeSimone, A.; Ruggiero, A.; Importa, R.; Vitagliano, L. Role of side chains in collagen triple helix stabilization and partner recognition. J. Pept. Sci. 2009, 15, 131–140. [Google Scholar] [CrossRef]
- Yildiz, I.; Tvestkova, I.; Wev, A.W.; Shukla, S.; Masarapu, H.; Dragnea, B.; Steinmetz, N.F. Engineering brome mosaic virus for biomedical applications. RSC Adv. 2012, 2, 3670–3677. [Google Scholar]
- Al jabali, A.A.A.; Steinmetz, N.F.; Barclay, J.E.; Lomonossoff, G.P.; Evans, D.J. Controlled immobilisation of active enzymes on the cowpea mosaic virus capsid. Nanoscale 2012, 4, 4560–4564. [Google Scholar]
- Senn, H.; Thiel, W. QM/MM methods for biomolecular systems. Angew. Chem. Int. Ed. 2009, 48, 1198–1229. [Google Scholar]
- Menikarachchi, L.; Gascon, J. QM/MM approaches in medicinal chemistry research. Curr. Topics Med. Chem. 2010, 10, 46–54. [Google Scholar] [CrossRef]
- Floris, F.M.; Tomasi, J.; Ahuir, J.L.P. Dispersion and repulsion contributions to the solvation energy: refinements to a simple computational model in the continuum approximation. J. Comput. Chem. 1991, 12, 784–791. [Google Scholar] [CrossRef]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Ratjer, F.; French, R.H.; Ching, W.Y.; Podgornik, R.; Parsegian, A. Chirality-dependent properties of carbon nanotube: Electronic structure, optical dispersion properties, Hamaker coefficients and van der Waals–London dispersion interactions. RSC Adv. 2013, 824, 823–842. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Eifler, J.; Rulis, P.; Tai, R.; Ching, W.-Y. Computational Study of a Heterostructural Model of Type I Collagen and Implementation of an Amino Acid Potential Method Applicable to Large Proteins. Polymers 2014, 6, 491-514. https://doi.org/10.3390/polym6020491
Eifler J, Rulis P, Tai R, Ching W-Y. Computational Study of a Heterostructural Model of Type I Collagen and Implementation of an Amino Acid Potential Method Applicable to Large Proteins. Polymers. 2014; 6(2):491-514. https://doi.org/10.3390/polym6020491
Chicago/Turabian StyleEifler, Jay, Paul Rulis, Rex Tai, and Wai-Yim Ching. 2014. "Computational Study of a Heterostructural Model of Type I Collagen and Implementation of an Amino Acid Potential Method Applicable to Large Proteins" Polymers 6, no. 2: 491-514. https://doi.org/10.3390/polym6020491