Living Radical Polymerization via Organic Superbase Catalysis

Abstract
:1. Introduction


2. Experimental Section
2.1. Materials
2.2. GPC Measurements
2.3. NMR Measurement
2.4. Radical Trap Experiments
2.5. Polymerizations
3. Results and Discussion
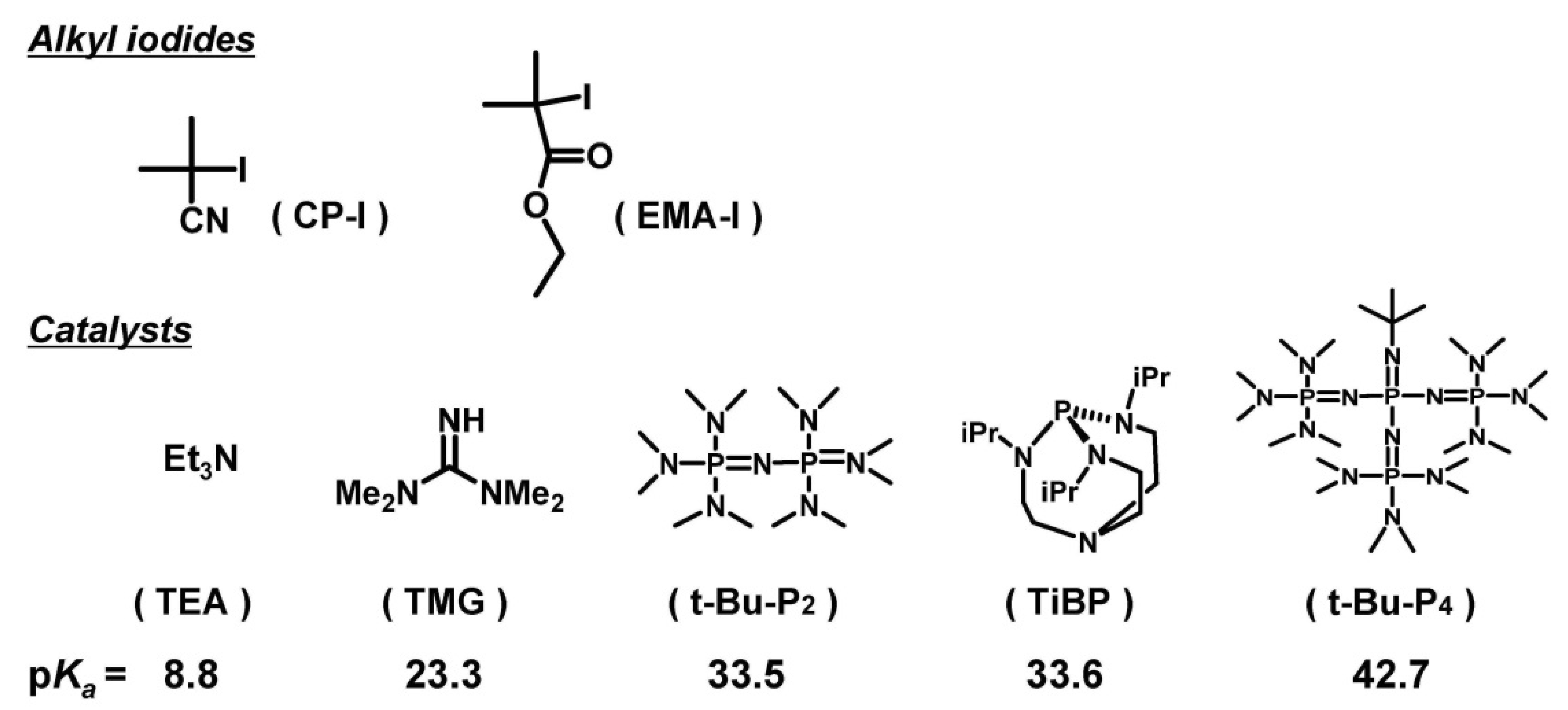
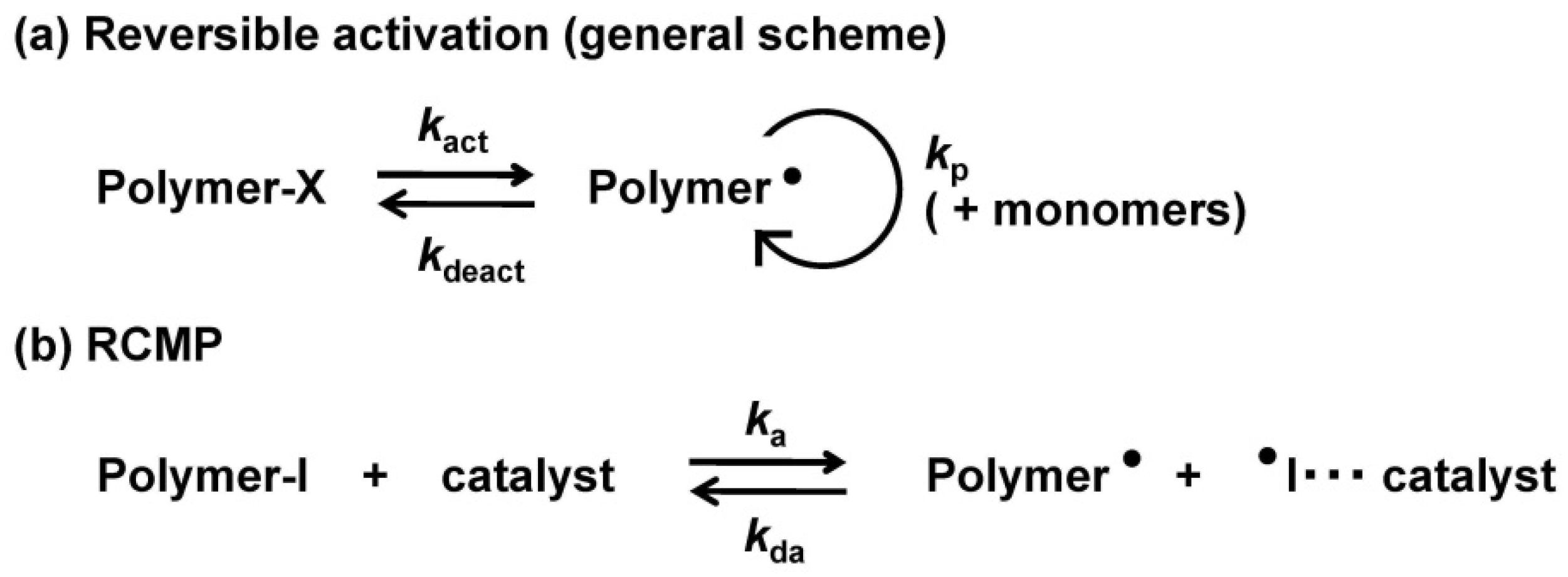
3.1. Experimental Proof for Generation of R• from R–I with Superbase Catalysts

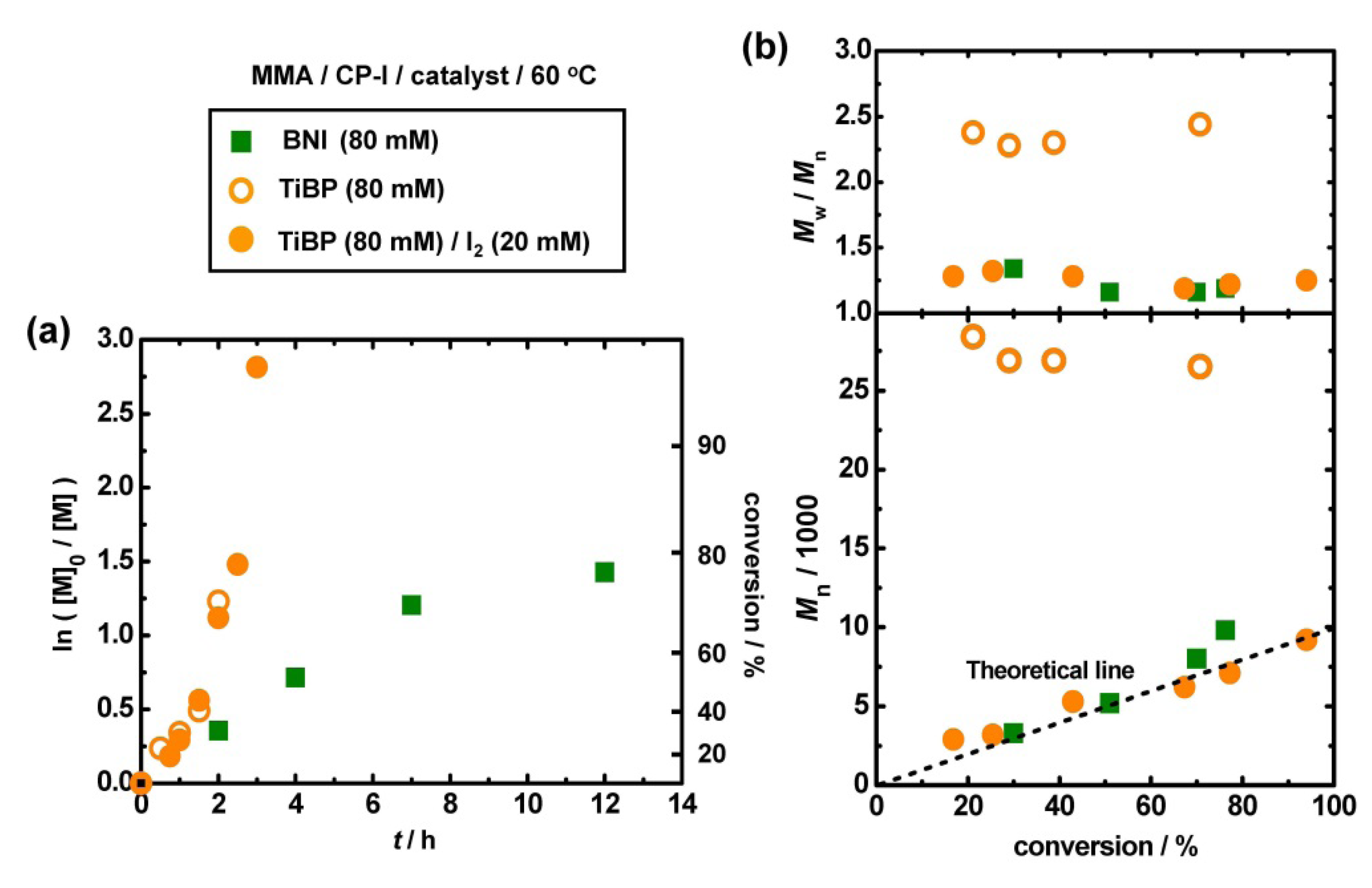
3.2. Polymerization of MMA with TiBP


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Target DP | Catalyst | [CP–I]0/[catalyst]0/[I2]0 (mM) | Solvent | t (h) | T (°C) | Conv (%) | Mn (Mn,theo) | PDI |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 100 | BNI | 80/80 | bulk | 7 | 60 | 70 | 8,000 (7,000) | 1.16 |
| 2 | 100 | TiBP | 80/80 | bulk | 2 | 60 | 71 | 27,000 (7,100) | 2.44 |
| 3 | 100 | TiBP | 80/80/20 | bulk | 3 | 60 | 94 | 9,200 (9,400) | 1.25 |
| 4 | 100 | TiBP | 80/40/5 | bulk | 4 | 60 | 74 | 10,000 (7,400) | 1.29 |
| 5 | 100 | TMG | 80/40/5 | bulk | 12 | 60 | 44 | 6,000 (4,400) | 1.13 |
| 6 | 100 | TMG | 80/80/0 | bulk | 10 | 60 | 63 | 9,000 (6,300) | 1.33 |
| 7 | 100 | t-Bu-P2 | 80/40/5 | bulk | 12 | 60 | 76 | 17,000 (7,600) | 1.23 |
| 8 | 100 | t-Bu-P2 | 80/40/0 | bulk | 6 | 60 | 64 | 14,000 (6,400) | 1.16 |
| 9 | 100 | t-Bu-P4 | 80/40/5 | bulk | 8 | 60 | 82 | 14,000 (8,200) | 1.33 |
| 10 | 100 | t-Bu-P4 | 80/40/0 | bulk | 6 | 60 | 65 | 14,000 (7,000) | 1.40 |
| 11 | 100 | TEA | 80/40/5 | bulk | 10 | 60 | 45 | 6,300 (4,500) | 1.29 |
| 12 | 400 | TiBP | 20/40/10 | Toluene a | 24 | 60 | 91 | 45,000 (36,000) | 1.45 |
| 13 | 400 | TiBP | 20/80/25 | Toluene a | 14 | 60 | 82 | 38,000 (33,000) | 1.20 |
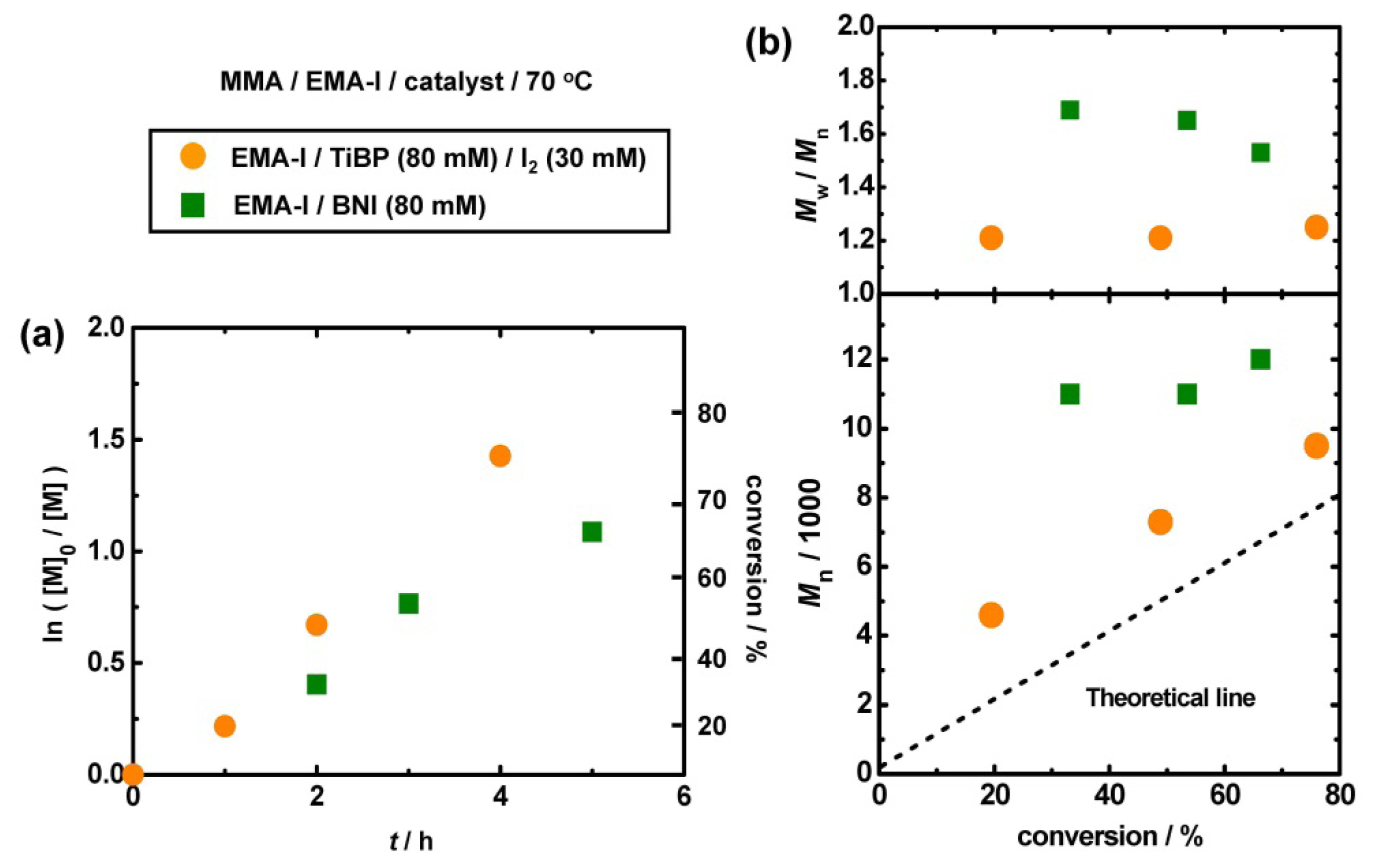
3.3. Polymerization from EMA-I

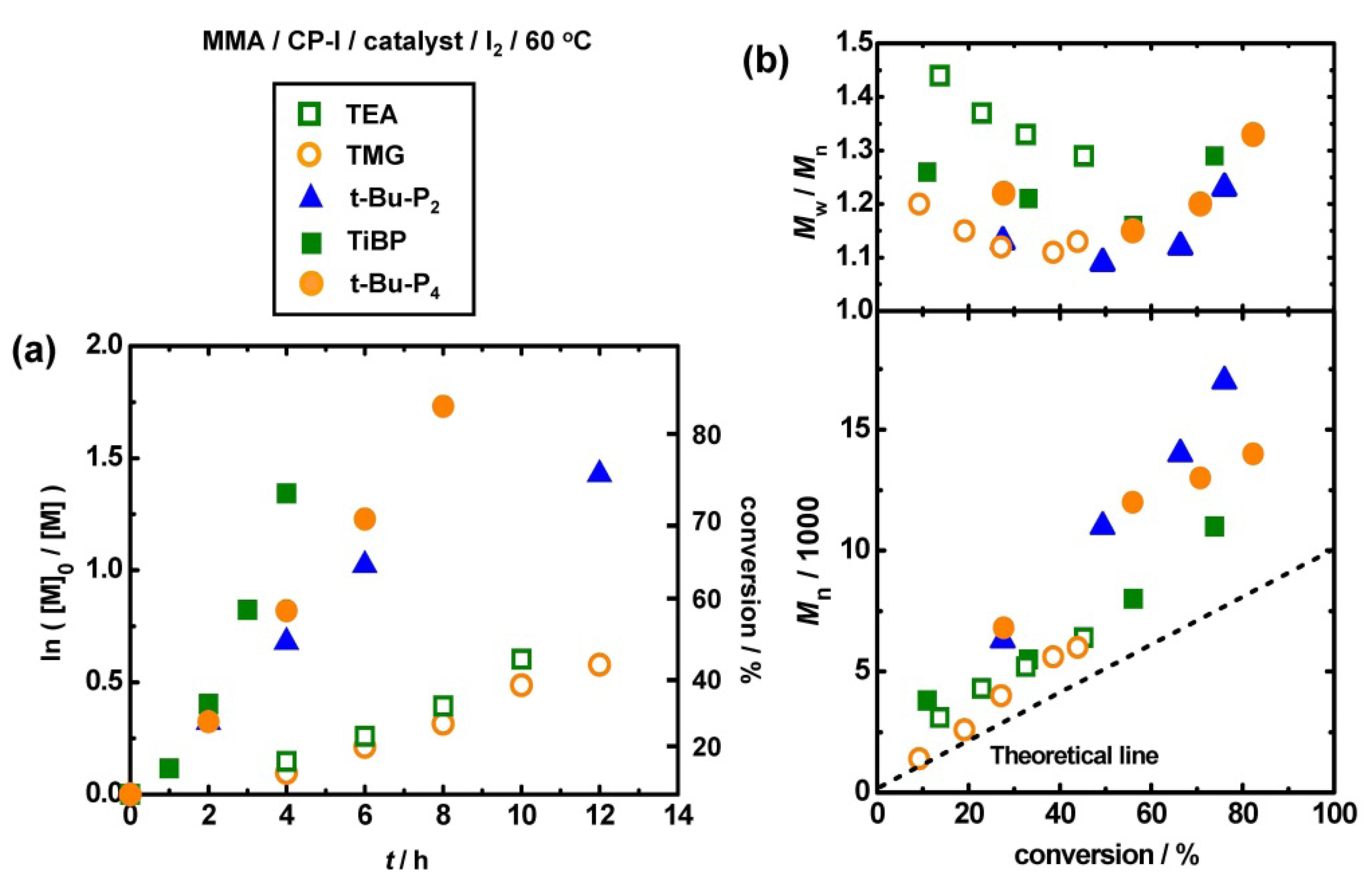
3.4. Other Superbase Catalysts

3.5. Higher Molecular Weights Polymers and Some Functional Methacrylates
| Entry | Monomer | Target DP | Catalyst | [CP–I]0/[catalyst]0/[I2]0 (mM) | T (oC) | t (h) | Conv (%) | Mn(Mn,theo) | PDI |
|---|---|---|---|---|---|---|---|---|---|
| 1 | BzMA | 100 | TiBP | 80/40/10 | 60 | 8 | 73 | 12,000 (13,000) | 1.39 |
| 2 | BzMA | 100 | TMG | 80/80/2 | 60 | 30 | 67 | 15,000 (12,000) | 1.37 |
| 3 | GMA | 100 | TiBP | 80/10/7 | 60 | 9 | 72 | 7,500 (10,000) | 1.27 |
| 4 | PEGMA a | 100 | TiBP | 80/40/10 | 60 | 6 | 100 | 19,000 (30,000) | 1.36 |
| 5 | PEGMA a | 100 | TMG | 80/40/2 | 60 | 6 | 100 | 16,000 (30,000) | 1.40 |

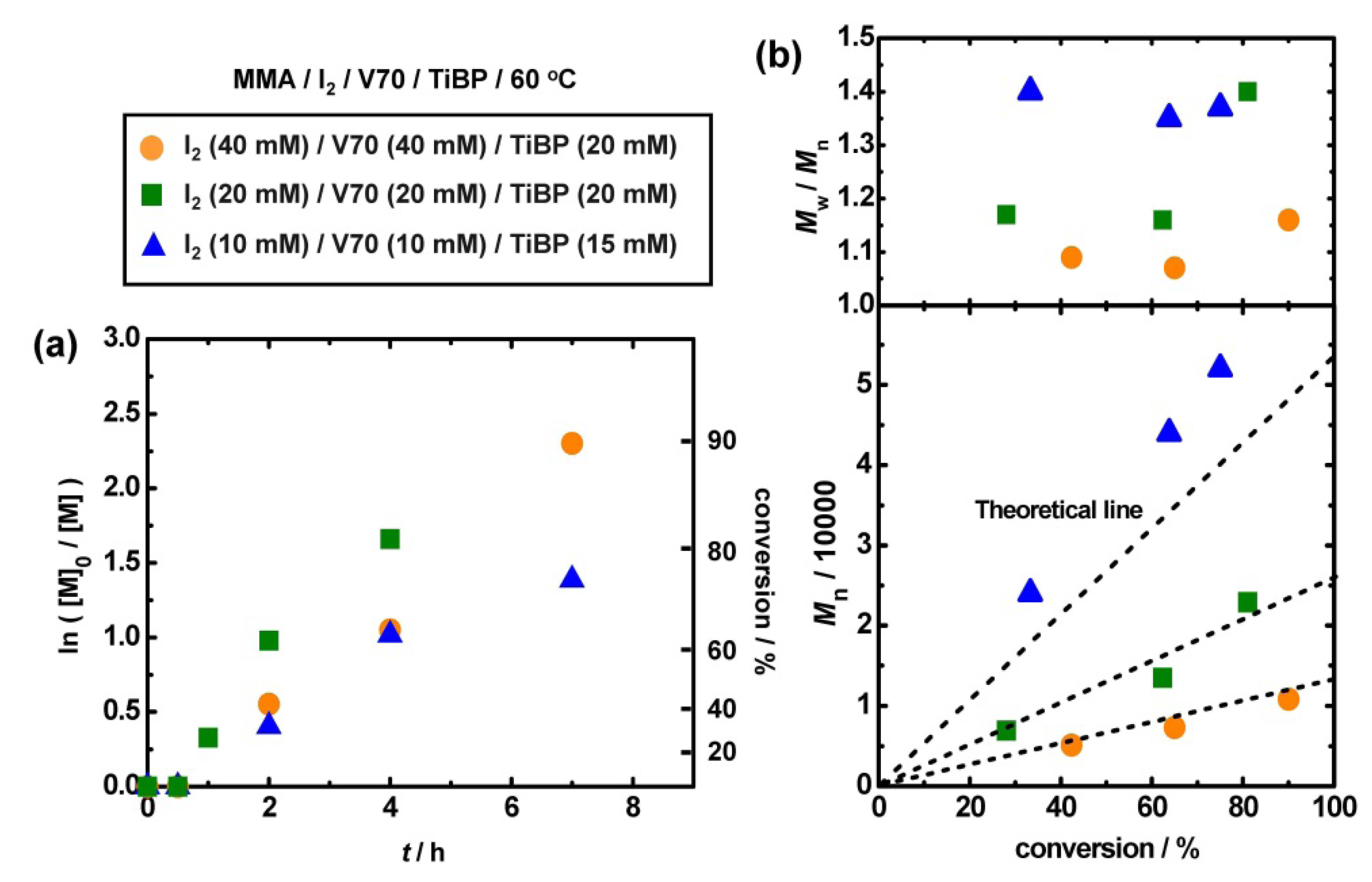
3.6. Use of Alkyl iodide Formed in Situ

| Entry | Monomer | Target DP | Catalyst | [monomer]0/[I2]0 /[V70]0/[catalyst]0 (mM) | Solvent | T (°C) | t (h) | Conv (%) | Mn (Mn,theo) | PDI |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | MMA | 130 | TiBP | 8000/40/40/20 | Bulk | 60 | 7 | 95 | 11,000 (12,000) | 1.16 |
| 2 | MMA | 270 | TiBP | 8000/20/20/20 | Bulk | 60 | 4 | 81 | 23,000 (22,000) | 1.40 |
| 3 | MMA | 530 | TiBP | 8000/10/10/15 a | Toluene b | 60 | 23 | 74 | 52,000 (40,000) | 1.36 |
| 4 | St | 100 | TiBP | 8000/40/55/20 | Bulk | 80 | 7 | 74 | 11,000 (7,700) | 1.4 |
| 5 | St | 100 | TMG | 8000/40/50/40 | Bulk | 80 | 10 | 100 | 12,000 (11,000) | 1.4 |
| 6 | St | 100 | t-Bu-P4 | 8000/40/60/5 | Bulk | 80 | 9 | 81 | 11,000 (8,500) | 1.38 |
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric Enamine Catalysis. Chem. Rev. 2007, 107, 5471–5569. [Google Scholar] [CrossRef]
- Hegedus, L.S. Organocatalysts in Organic Synthesis. J. Am. Chem. Soc. 2009, 131, 17995–17997. [Google Scholar] [CrossRef]
- Zhong, C.; Shi, X. When Organocatalysis Meets Transition-Metal Catalysis. Eur. J. Org. Chem. 2010, 2010, 2999–3025. [Google Scholar] [CrossRef]
- Grasa, G.A.; Singh, R.; Nolan, S.P. Transesterification/Acylation Reactions Catalyzed by Molecular Catalysts. Synthesis 2004, 2004, 971–985. [Google Scholar]
- Sohtome, Y.; Hashimoto, Y.; Nagasawa, K. Guanidine-Thiourea Bifunctional Organocatalyst for the Asymmetric Henry (Nitroaldol) Reaction. Adv. Synth. Catal. 2005, 347, 1643–1648. [Google Scholar] [CrossRef]
- Ishikawa, T.; Kumamoto, T. Guanidines in Organic Synthesis. Synthesis 2006, 2006, 737–752. [Google Scholar] [CrossRef]
- Melchiorre, P.; Marigo, M.; Carlone, A.; Bartoli, G. Asymmetric Aminocatalysis—Gold Rush in Organic Chemistry. Angew. Chem. Int. Ed. 2008, 47, 6138–6171. [Google Scholar] [CrossRef]
- Lohmeijer, B.G.G.; Pratt, R.C.; Leibfarth, F.; Logan, J.W.; Long, D.A.; Dove, A.P.; Nederberg, F.; Choi, J.; Wade, C.; Waymouth, R.M.; Hedrick, J.L. Guanidine and Amidine Organocatalysts for Ring-Opening Polymerization of Cyclic Esters. Macromolecules 2006, 39, 8574–8583. [Google Scholar] [CrossRef]
- Kakuchi, T.; Chen, Y.; Kitakado, J.; Mori, K.; Fuchise, K.; Satoh, T. Organic Superbase as an Efficient Catalyst for Group Transfer Polymerization of Methyl Methacrylate. Macromolecules 2011, 44, 4641–4647. [Google Scholar] [CrossRef]
- Tsarevsky, N.V.; Sumerlin, B.S. Fundamentals of Controlled/Living Radical Polymerization, 1st ed.; Royal Society of Chemistry: London, UK, 2013. [Google Scholar]
- Matyjaszewski, K.; Möller, M. Polymer Science: A Comprehensive Reference, 1st ed.; Elsevier: Amsterdam, the Netherlands, 2012. [Google Scholar]
- Moad, G.; Solomon, D.H. The Chemistry of Radical Polymerization, 2nd ed.; Elsevier: Amsterdam, the Netherlands, 2006. [Google Scholar]
- Nicolas, J.; Guillaneuf, Y.; Lefay, C.; Bertin, D.; Gigmes, D.; Charleux, B. Nitroxide-mediated polymerization. Prog. Polym. Sci. 2013, 38, 63–235. [Google Scholar]
- Matyjaszewski, K. Atom transfer radical polymerization (ATRP): Current status and future perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Ouchi, M.; Terashima, T.; Sawamoto, M. Transition metal-catalyzed living radical polymerization: Toward perfection in catalysis and precision polymer synthesis. Chem. Rev. 2009, 109, 4963–5050. [Google Scholar] [CrossRef]
- Lena, F.; Matyjaszewski, K. Transition metal catalysts for controlled radical polymerization. Prog. Polym. Sci. 2010, 35, 959–1021. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the RAFT process—A second update. Aust. J. Chem. 2009, 62, 1402–1472. [Google Scholar] [CrossRef]
- Keddie, D.J.; Moad, G.; Rizzado, E.; Thang, S.H. RAFT agent design and synthesis. Macromolecules 2012, 45, 5321–5342. [Google Scholar] [CrossRef]
- David, G.; Boyer, C.; Tonnar, J.; Ameduri, B.; Lacroix-Desmazes, P.; Boutevin, B. Use of iodocompounds in radical polymerization. Chem. Rev. 2006, 106, 3936–3962. [Google Scholar] [CrossRef]
- Yamago, S. Precision polymer synthesis by degenerative transfer controlled/living radical polymerization using organotellurium, organostibine and organobismuthine chain-transfer agents. Chem. Rev. 2009, 109, 5051–5068. [Google Scholar] [CrossRef]
- Fukuda, T. Fundamental kinetic aspects of living radical polymerization and the use of gel permeation chromatography to shed light on them. J. Polym. Sci. A Polym. Chem. 2004, 42, 4743–4755. [Google Scholar] [CrossRef]
- Fischer, H. The persistent radical effect: A principle for selective radical reactions and living radical polymerizations. Chem. Rev. 2001, 101, 3581–3618. [Google Scholar] [CrossRef]
- Goto, A.; Fukuda, T. Kinetics of living radical polymerization. Prog. Polym. Sci. 2004, 29, 329–385. [Google Scholar] [CrossRef]
- Fukuda, T.; Goto, A. Controlled and Living Radical Polymerization—Principles and Fundamentals. In Polymer Science: A Comprehensive Reference, 1st ed.; Matyjaszewski, K., Möller, M., Eds.; Elsevier: Amsterdam, the Netherlands, 2012; pp. 120–157. [Google Scholar]
- Goto, A.; Zushi, H.; Hirai, N.; Wakada, T.; Tsujii, Y.; Fukuda, T. Living radical polymerizations with germanium, tin and phosphorus catalysts—Reversible chain transfer catalyzed polymerizations (RTCPs). J. Am. Chem. Soc. 2007, 129, 13347–13354. [Google Scholar] [CrossRef]
- Goto, A.; Hirai, N.; Wakada, T.; Nagasawa, K.; Tsujii, Y.; Fukuda, T. Living radical polymerization with nitrogen catalyst: Reversible chain transfer catalyzed polymerization with N-iodosuccinimide. Macromolecules 2008, 41, 6261–6264. [Google Scholar] [CrossRef]
- Goto, A.; Hirai, N.; Nagasawa, K.; Tsujii, Y.; Fukuda, T.; Kaji, H. Phenols and carbon compounds as efficient organic catalysts for reversible chain transfer catalyzed living radical polymerization (RTCP). Macromolecules 2010, 43, 7971–7978. [Google Scholar] [CrossRef]
- Vana, P.; Goto, A. Kinetic simulations of reversible chain transfer catalyzed polymerization (RTCP): Guidelines to optimum molecular weight control. Macromol. Theory Simul. 2010, 19, 24–35. [Google Scholar]
- Yorizane, M.; Nagasuga, T.; Kitayama, Y.; Tanaka, A.; Minami, H.; Goto, A.; Fukuda, T.; Okubo, M. Reversible chain transfer catalyzed polymerization (RTCP) of methyl methacrylate with nitrogen catalyst in an aqueous microsuspension system. Macromolecules 2010, 43, 8703–8705. [Google Scholar] [CrossRef]
- Goto, A.; Tsujii, Y.; Fukuda, T. Reversible chain transfer catalyzed polymerization (RTCP): A new class of living radical polymerization. Polymer 2008, 49, 5177–5185. [Google Scholar] [CrossRef]
- Goto, A.; Tsujii, Y.; Kaji, H. Living Radical Polymerizations with Organic Catalysts. In Fundamentals of Controlled/Living Radical Polymerization, 1st ed.; Tsarevsky, N.V., Sumerlin, B.S., Eds.; Royal Society of Chemistry: London, UK, 2013; pp. 250–286. [Google Scholar]
- Goto, A.; Suzuki, T.; Ohfuji, H.; Tanishima, M.; Fukuda, T.; Tsujii, Y.; Kaji, H. Reversible Complexation Mediated Living Radical Polymerization (RCMP) using organic catalysts. Macromolecules 2011, 44, 8709–8715. [Google Scholar] [CrossRef]
- Goto, A.; Tsujii, Y.; Kaji, H. Reversible Complexation Mediated Polymerization (RCMP) of Methyl Methacrylate. ACS Symp. Ser. 2012, 1100, 305–315. [Google Scholar] [CrossRef]
- Ohtsuki, A.; Goto, A.; Kaji, H. Visible-Light-Induced reversible complexation mediated living radical polymerization of methacrylates with organic catalysts. Macromolecules 2013, 46, 96–102. [Google Scholar] [CrossRef]
- Goto, A.; Ohtsuki, A.; Ohfuji, H.; Tanishima, M.; Kaji, H. Reversible generation of a carbon-centered radical from alkyl iodide using organic salts and their application as organic catalysts in living radical polymerization. J. Am. Chem. Soc. 2013, 135, 11131–11139. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E. Alkoxyamine-Initiated Living Radical Polymerization: Factors Affecting Alkoxyamine Homolysis Rate. Macromolecules 1995, 28, 8722–8728. [Google Scholar] [CrossRef]
- Goto, A.; Fukuda, T. Determination of the activation rate constants of alkyl halide initiators for atom transfer radical polymerization. Macromol. Rapid Commun. 1999, 20, 633–636. [Google Scholar] [CrossRef]
- Schulte, T.; Studer, A. New Seven- and Eight-Membered Cyclic Alkoxyamines for the Living Free Radical Polymerization. Macromolecules 2003, 36, 3078–3084. [Google Scholar] [CrossRef]
- Ishikawa, T. Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts; John Wiley & Sons: London, UK, 2009. [Google Scholar]
- Lacroix-Desmazes, P.; Severac, R.; Boutevin, B. Reverse iodine transfer polymerization of methyl acrylate and n-butyl acrylate. Macromolecules 2005, 38, 6299–6309. [Google Scholar] [CrossRef]
- Tonnar, P.; Lacroix-Desmazes, P. Use of sodium iodide as the precursor to the control agent in ab initio emulsion polymerization. Angew. Chem. Int. Ed. 2008, 47, 1294–1297. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lei, L.; Tanishima, M.; Goto, A.; Kaji, H. Living Radical Polymerization via Organic Superbase Catalysis. Polymers 2014, 6, 860-872. https://doi.org/10.3390/polym6030860
Lei L, Tanishima M, Goto A, Kaji H. Living Radical Polymerization via Organic Superbase Catalysis. Polymers. 2014; 6(3):860-872. https://doi.org/10.3390/polym6030860
Chicago/Turabian StyleLei, Lin, Miho Tanishima, Atsushi Goto, and Hironori Kaji. 2014. "Living Radical Polymerization via Organic Superbase Catalysis" Polymers 6, no. 3: 860-872. https://doi.org/10.3390/polym6030860