
What Happens When Threading is Suppressed in Blends of Ring and Linear Polymers?

Abstract
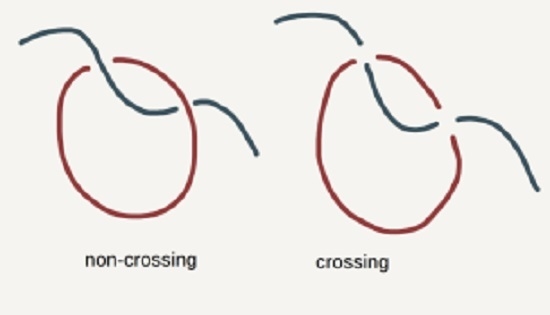
:
1. Introduction
2. Materials and Methods
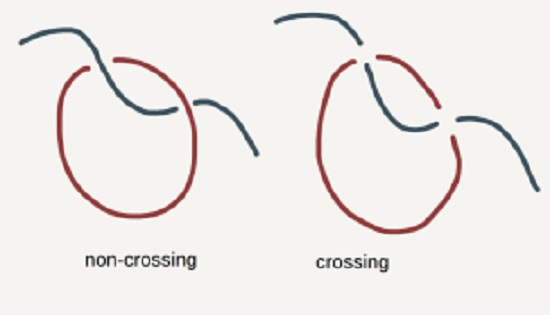
2.1. Bond-Fluctuation Model
- Excluded Volume—the target lattice site is empty and available,
- Finite Extensibility—none of the bond-lengths stretch beyond , and
- Chain Uncrossability—mid-points of bonds do not intersect.
2.2. Self-Diffusion Coefficient
2.3. Systems Studied
- In symmetric blends, = = 300 was held fixed, while the linear fraction = was varied between 0 and 1. Note that for = 0 (pure rings) and =1 (pure linears), the crossing simulations and the non-crossing simulations were identical, since there are no ring-linear interactions to suppress in these pure systems.
- In asymmetric blends, the concentration of the matrix LPs , and the number of RP monomers , were held fixed, while the number of LP monomers was varied between . To avoid ring-ring interactions in these probe systems, the concentration of the ring polymers was kept about 10 times lower than the overlap concentration [34].
3. Results
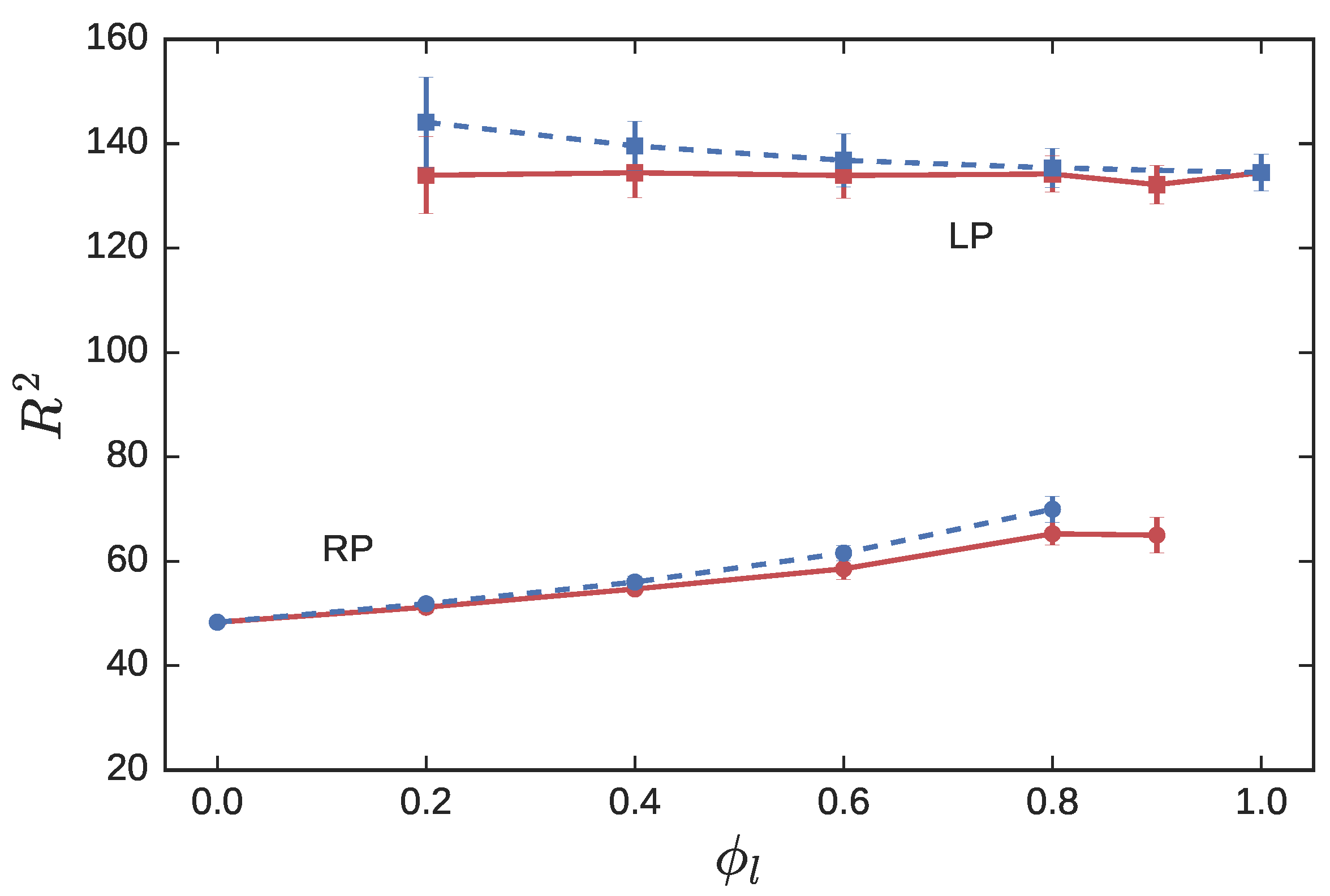
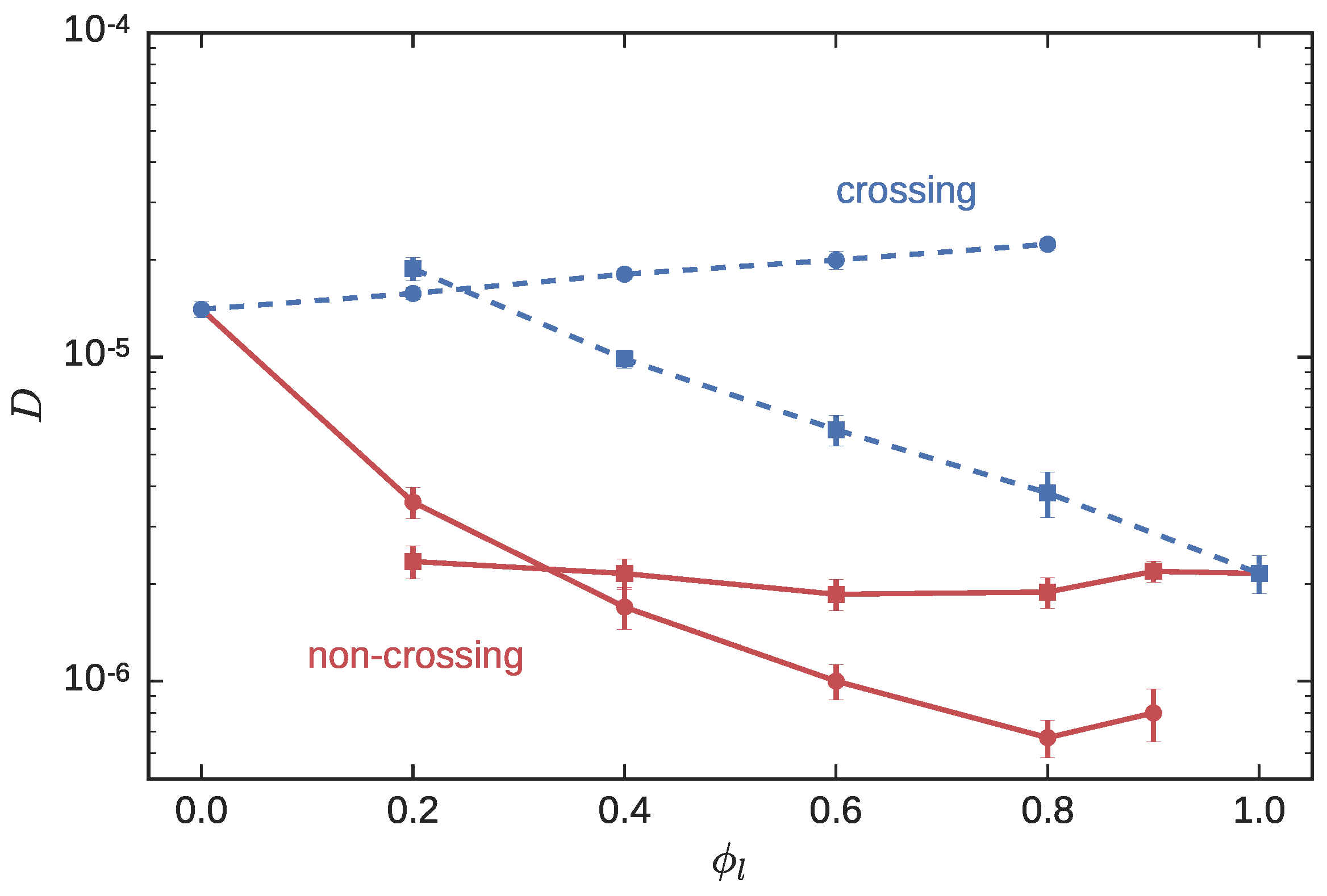
3.1. Symmetric Blends
3.1.1. Statics
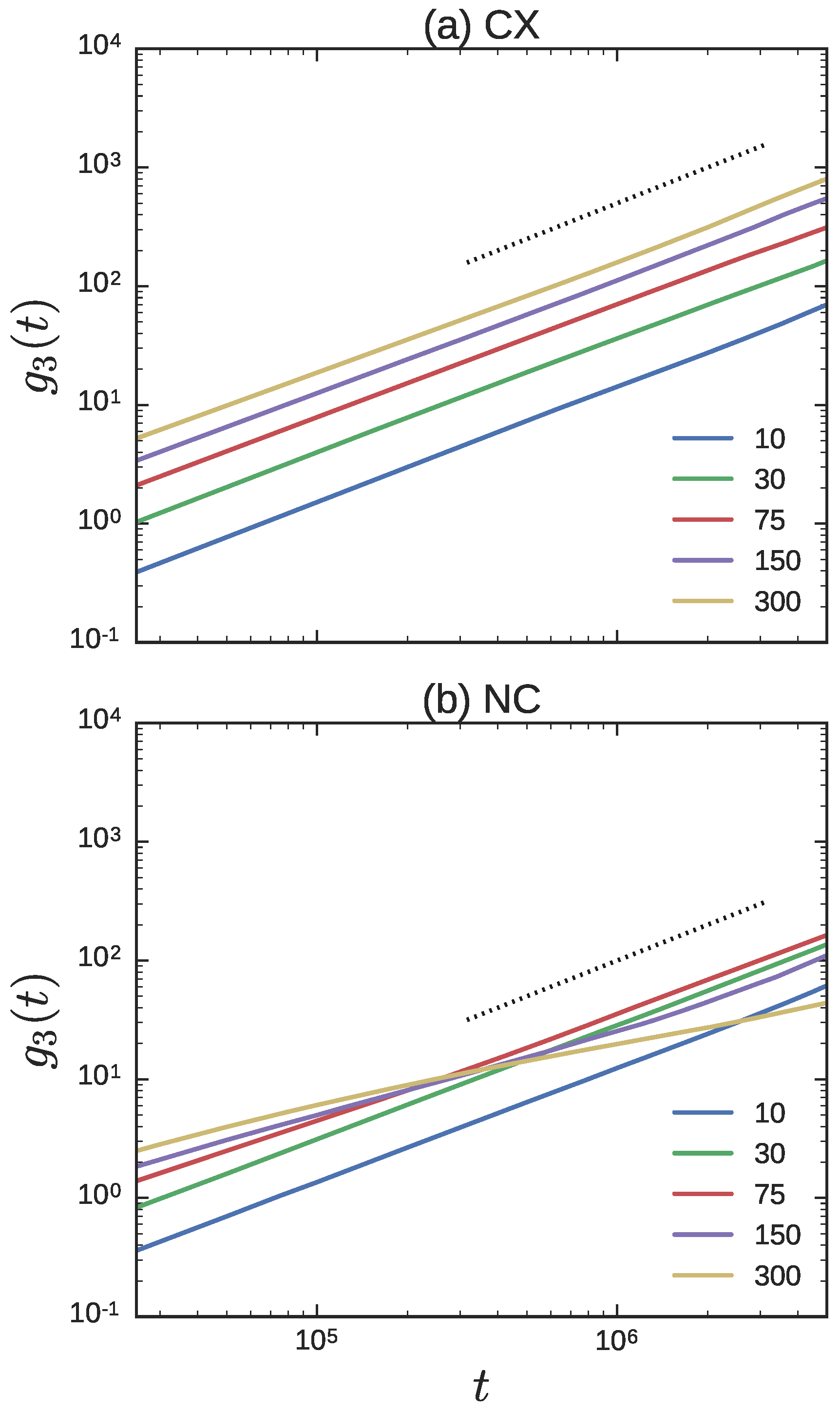
3.1.2. Dynamics
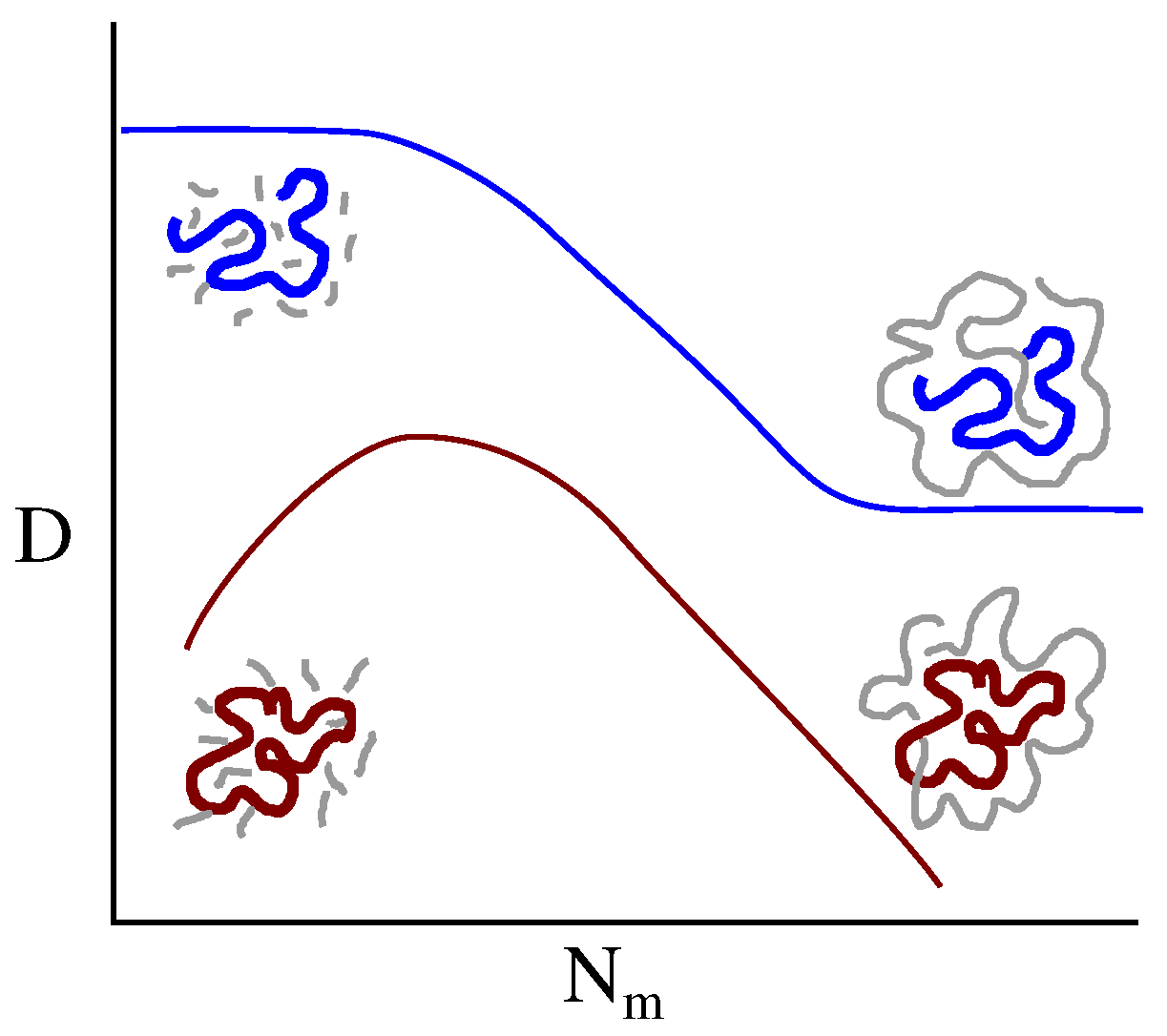
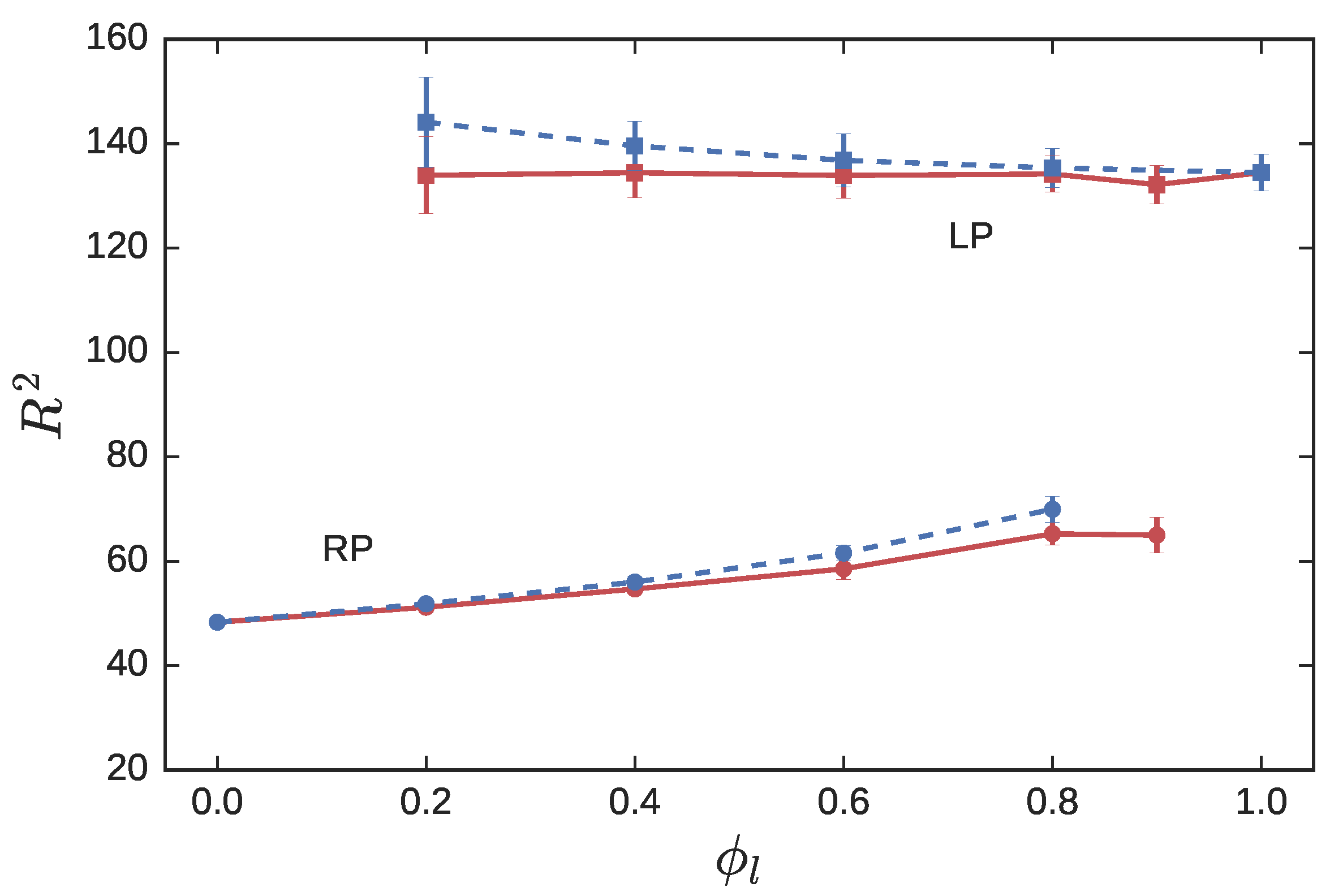
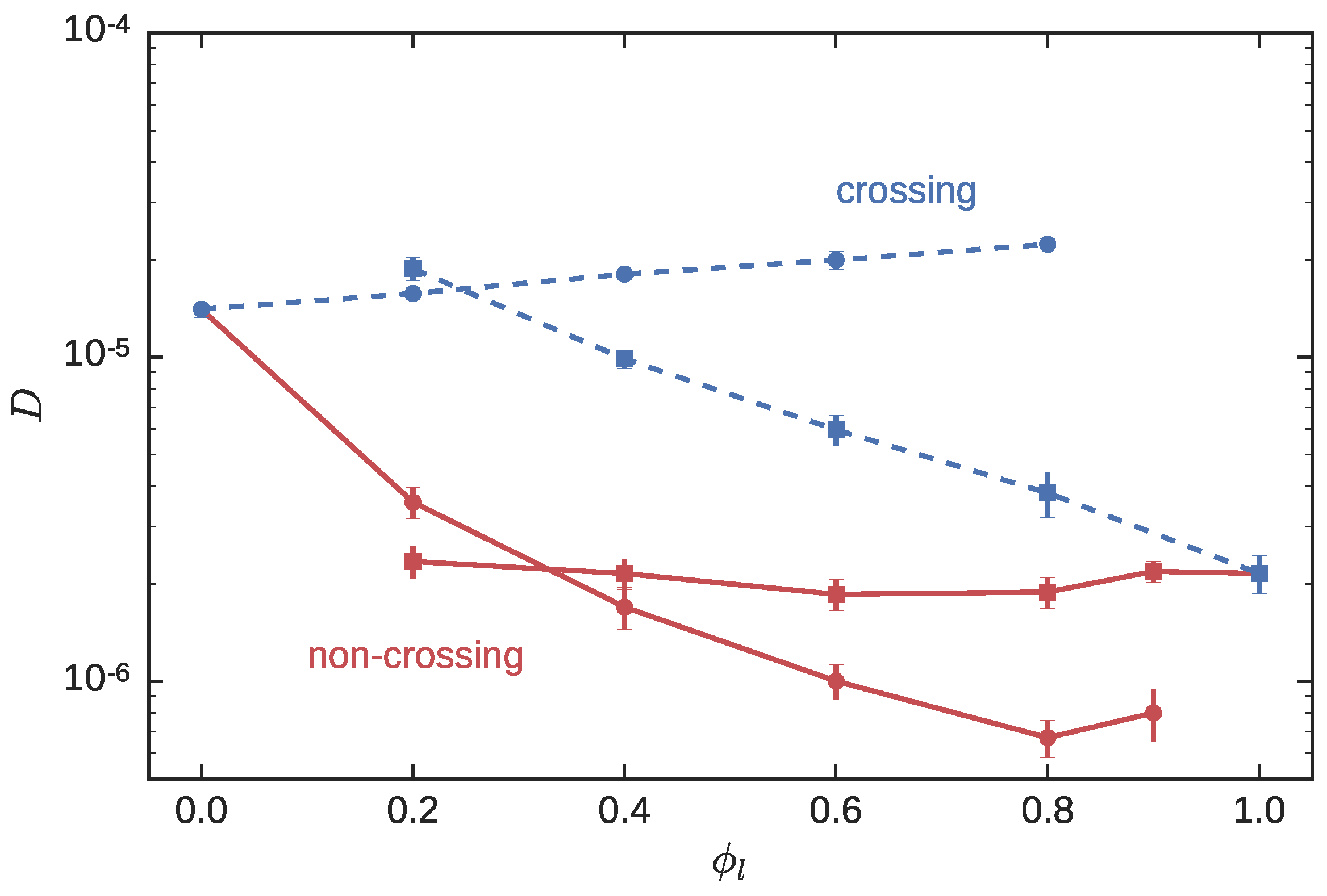
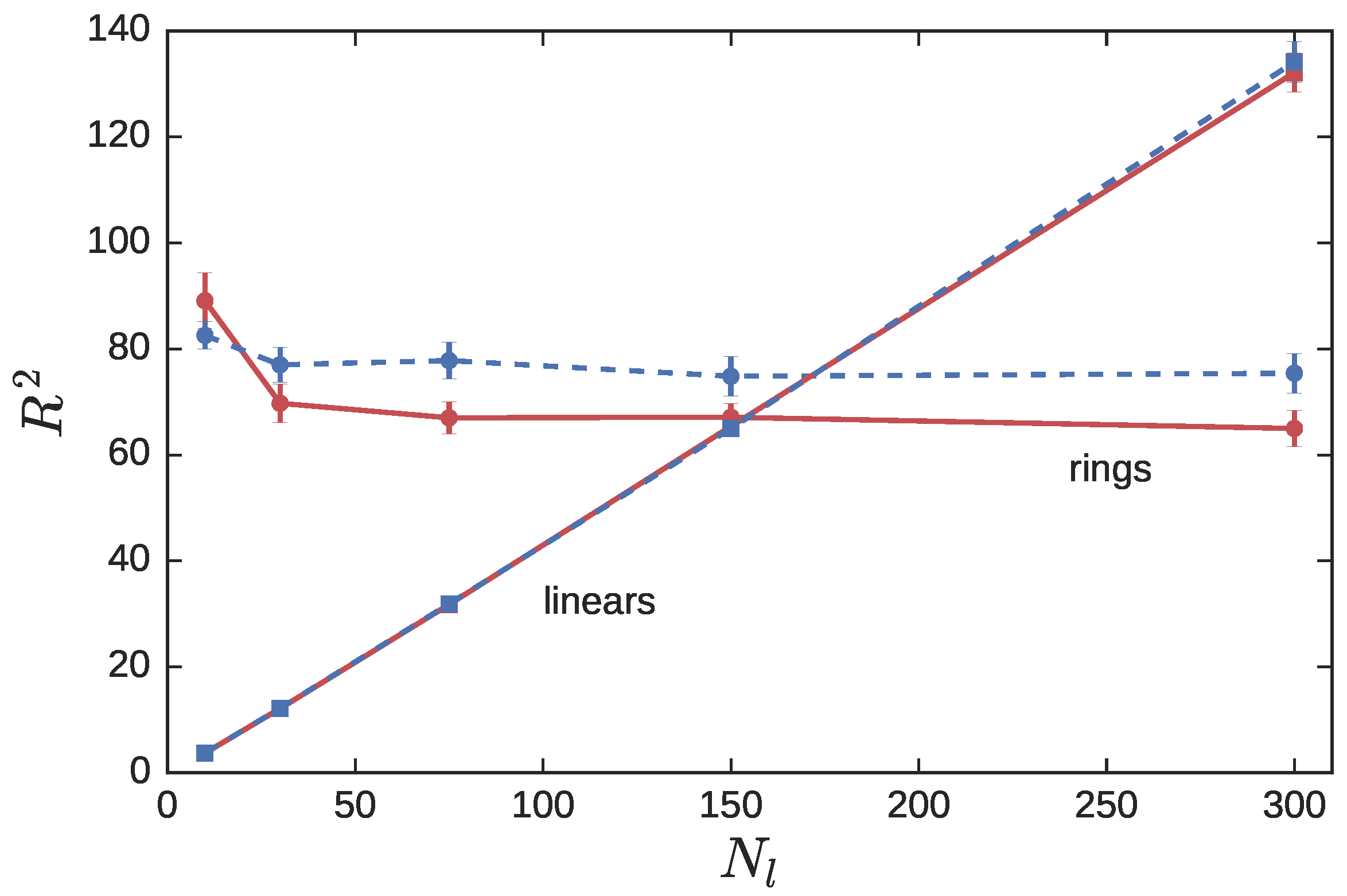
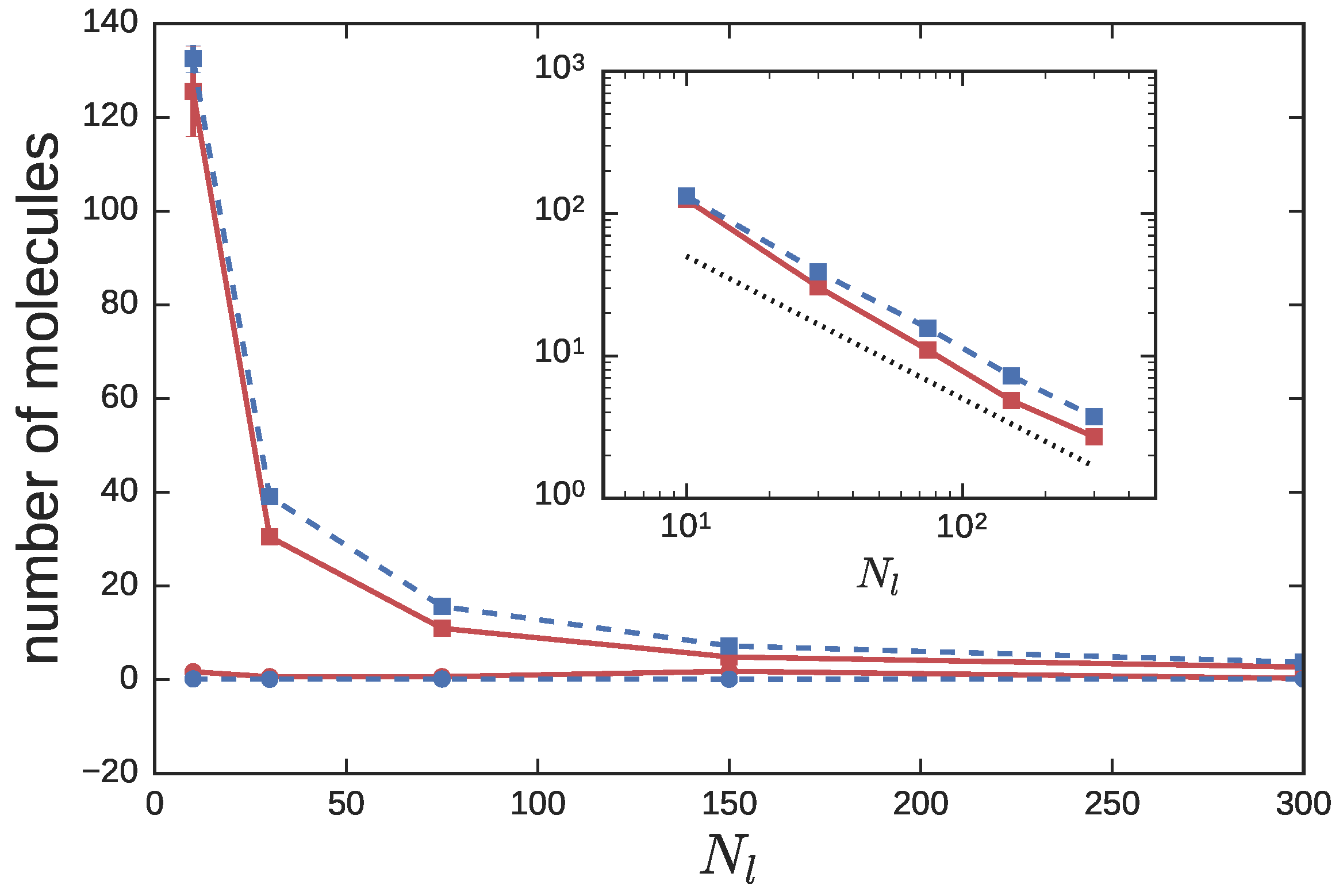
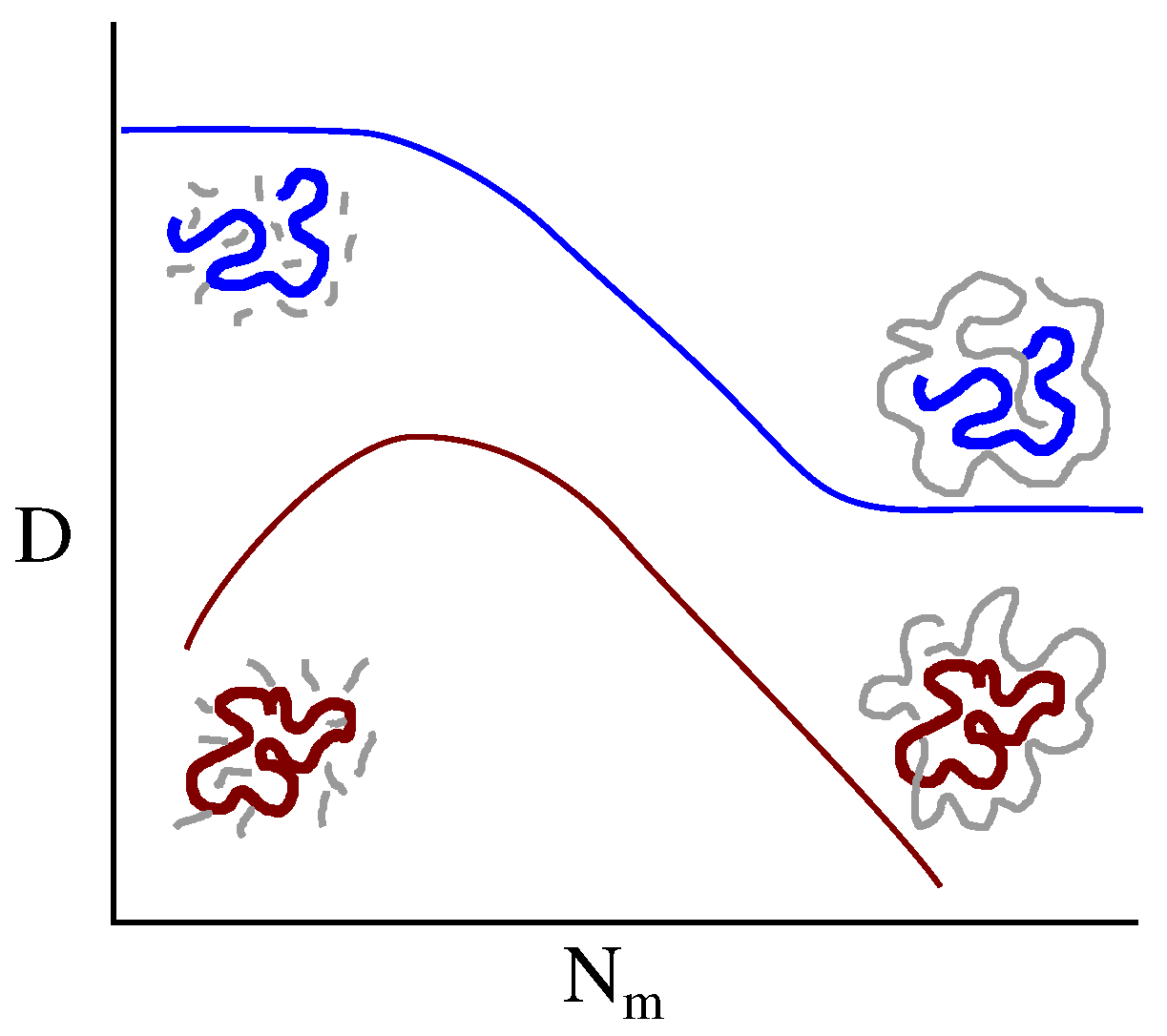
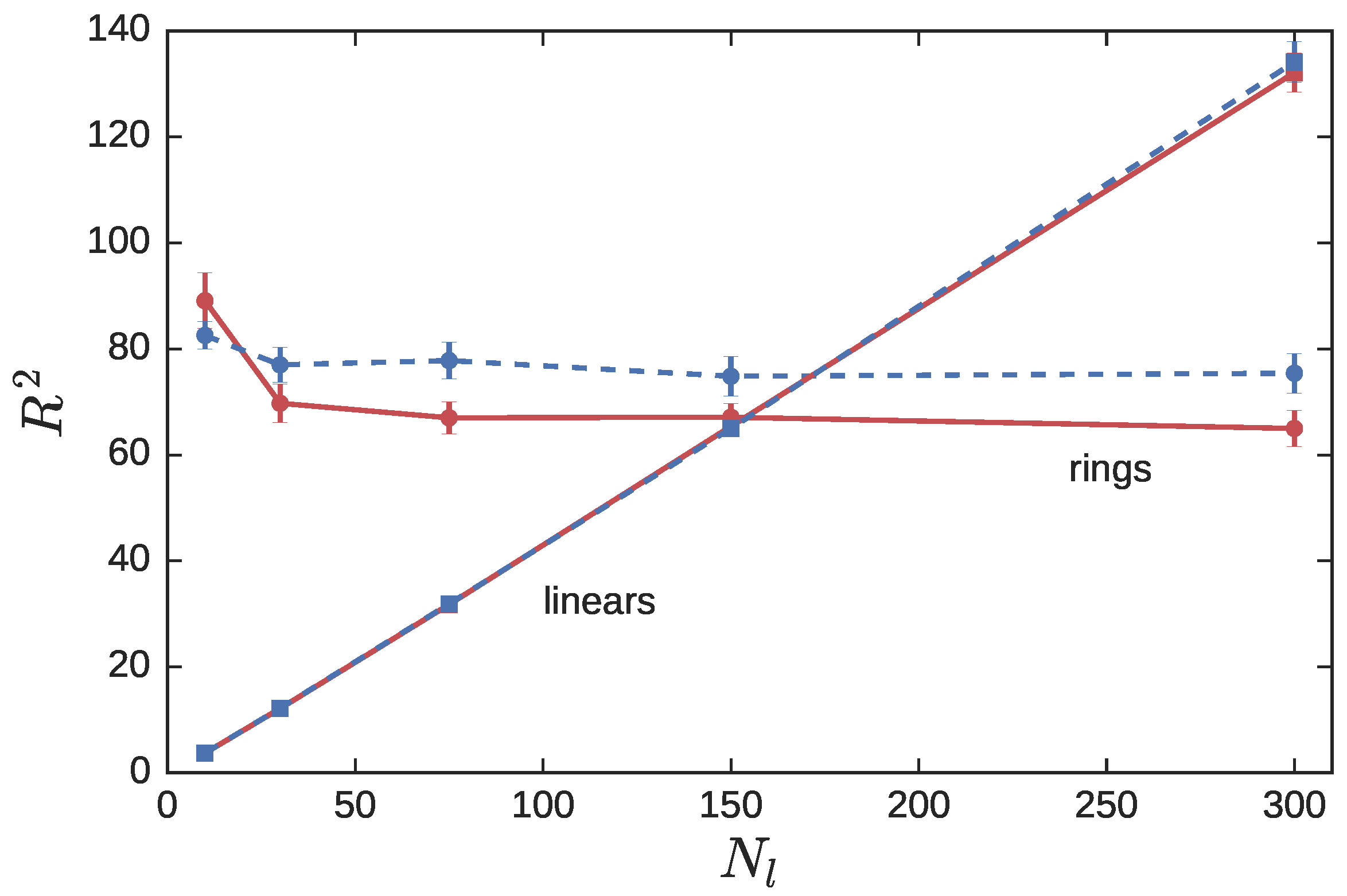
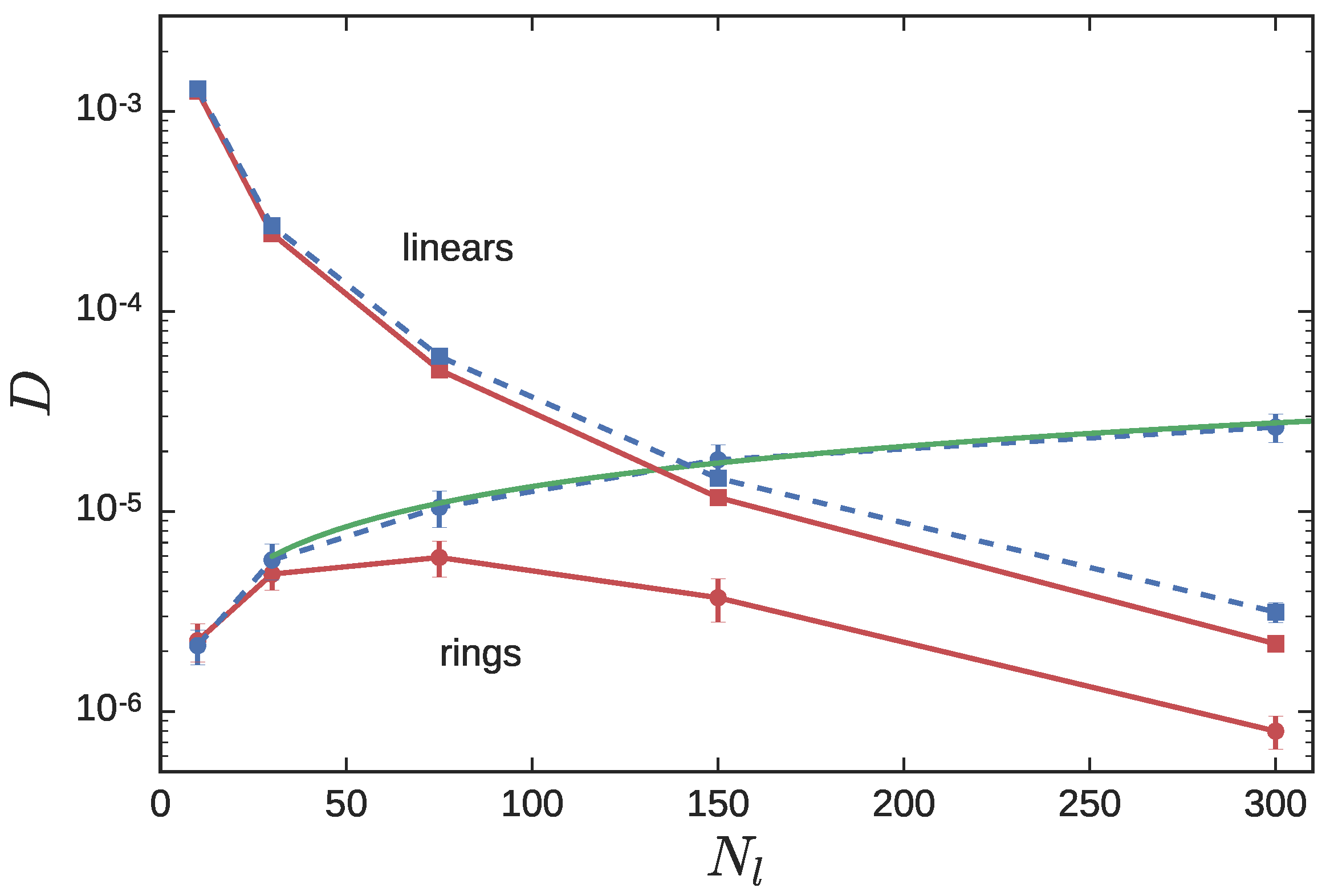
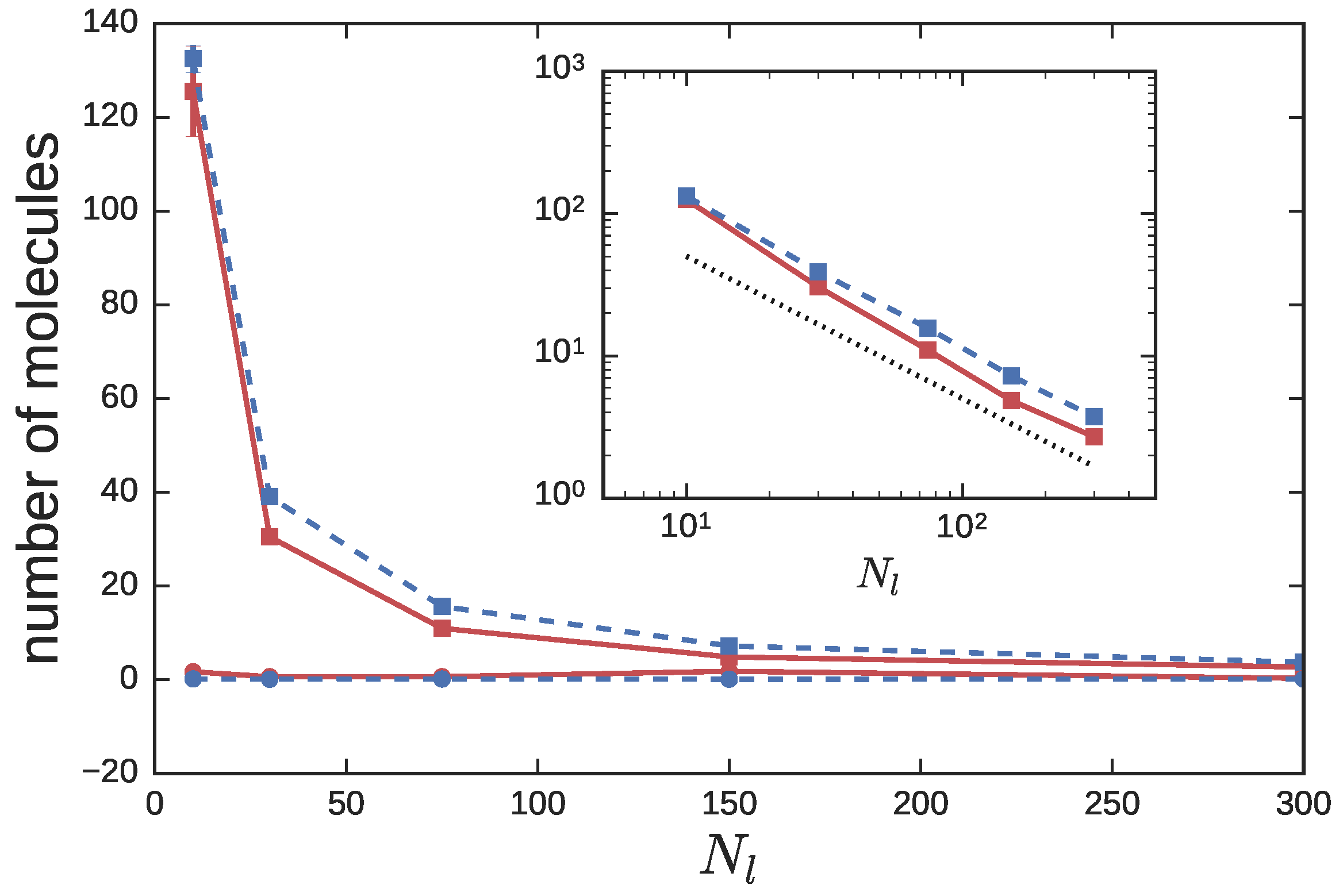
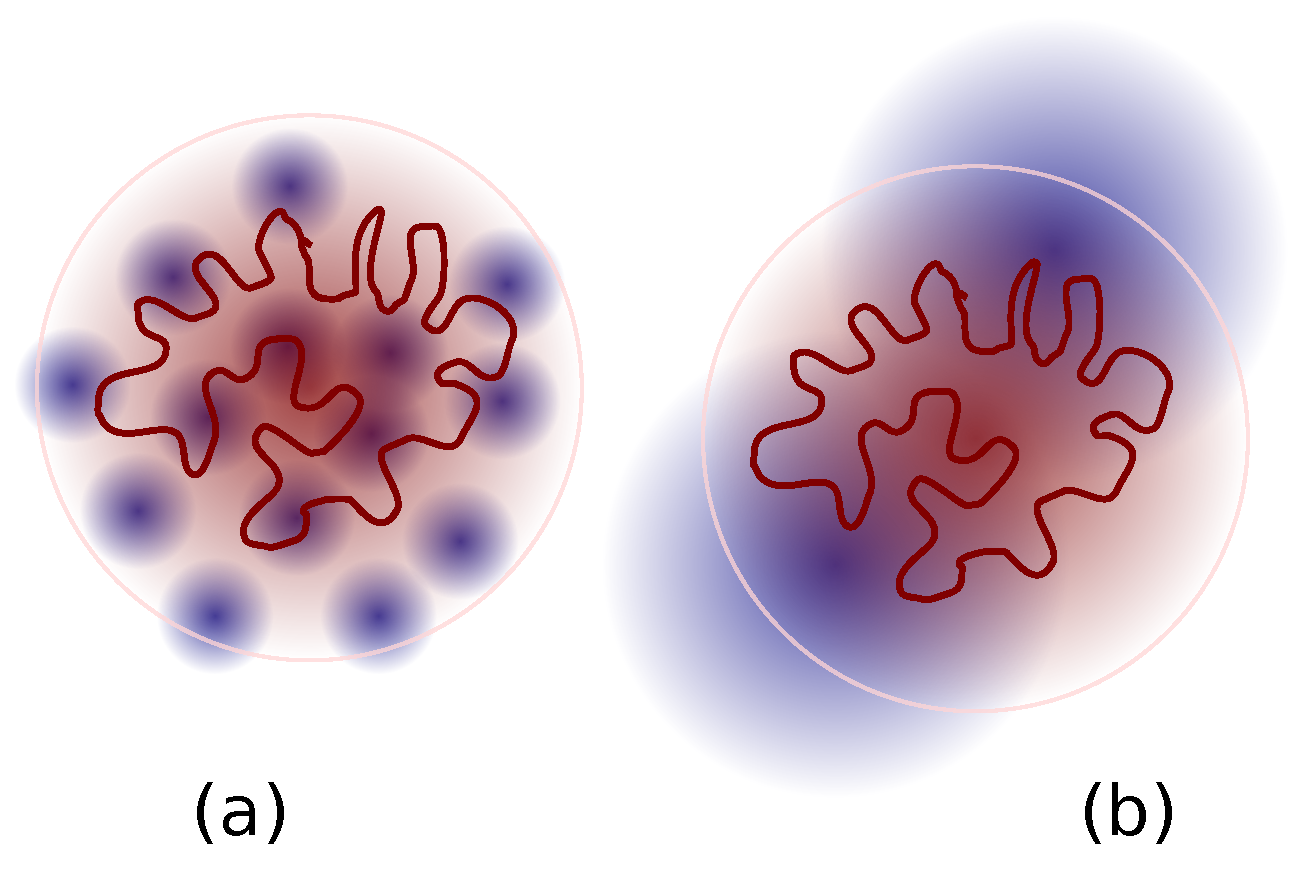
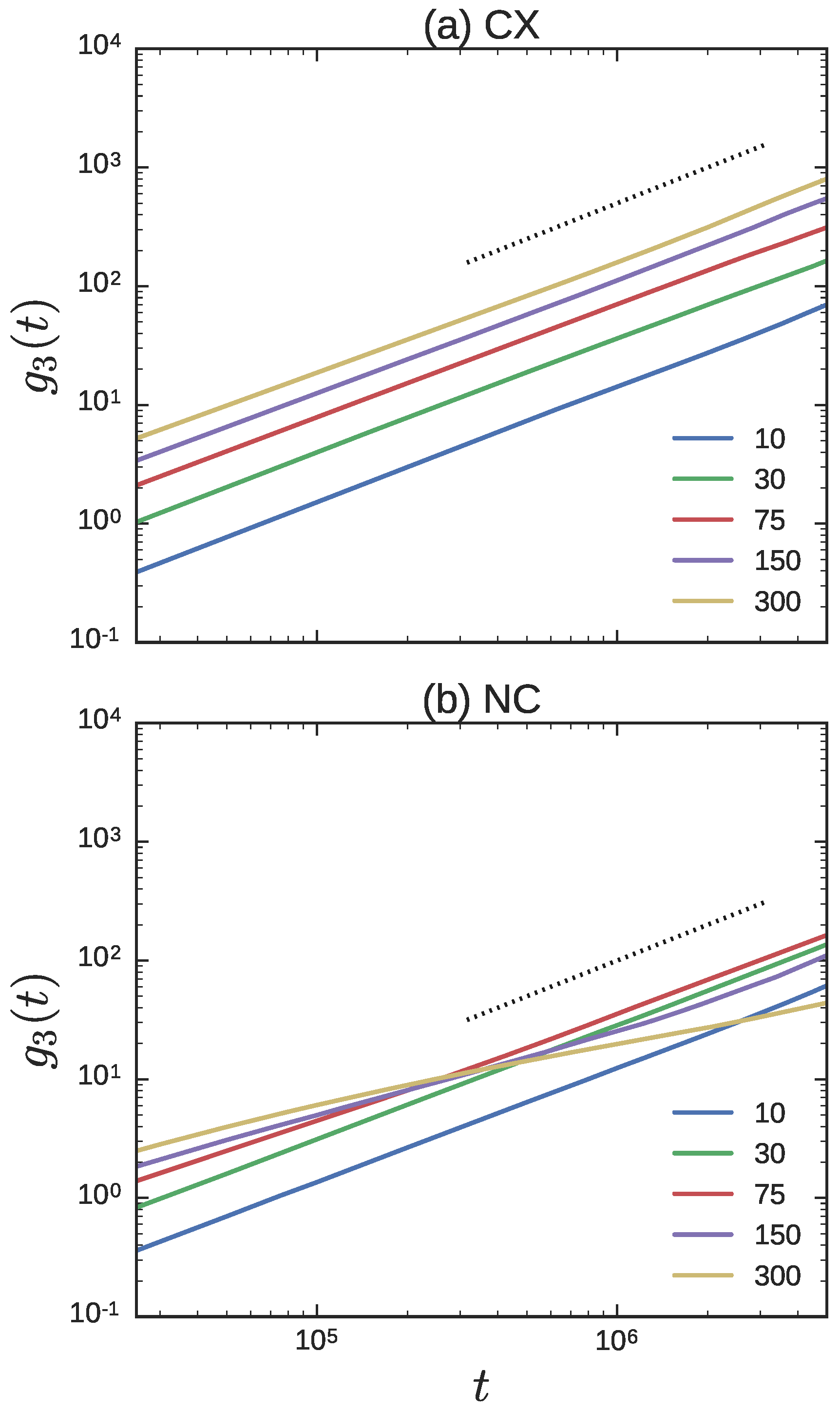
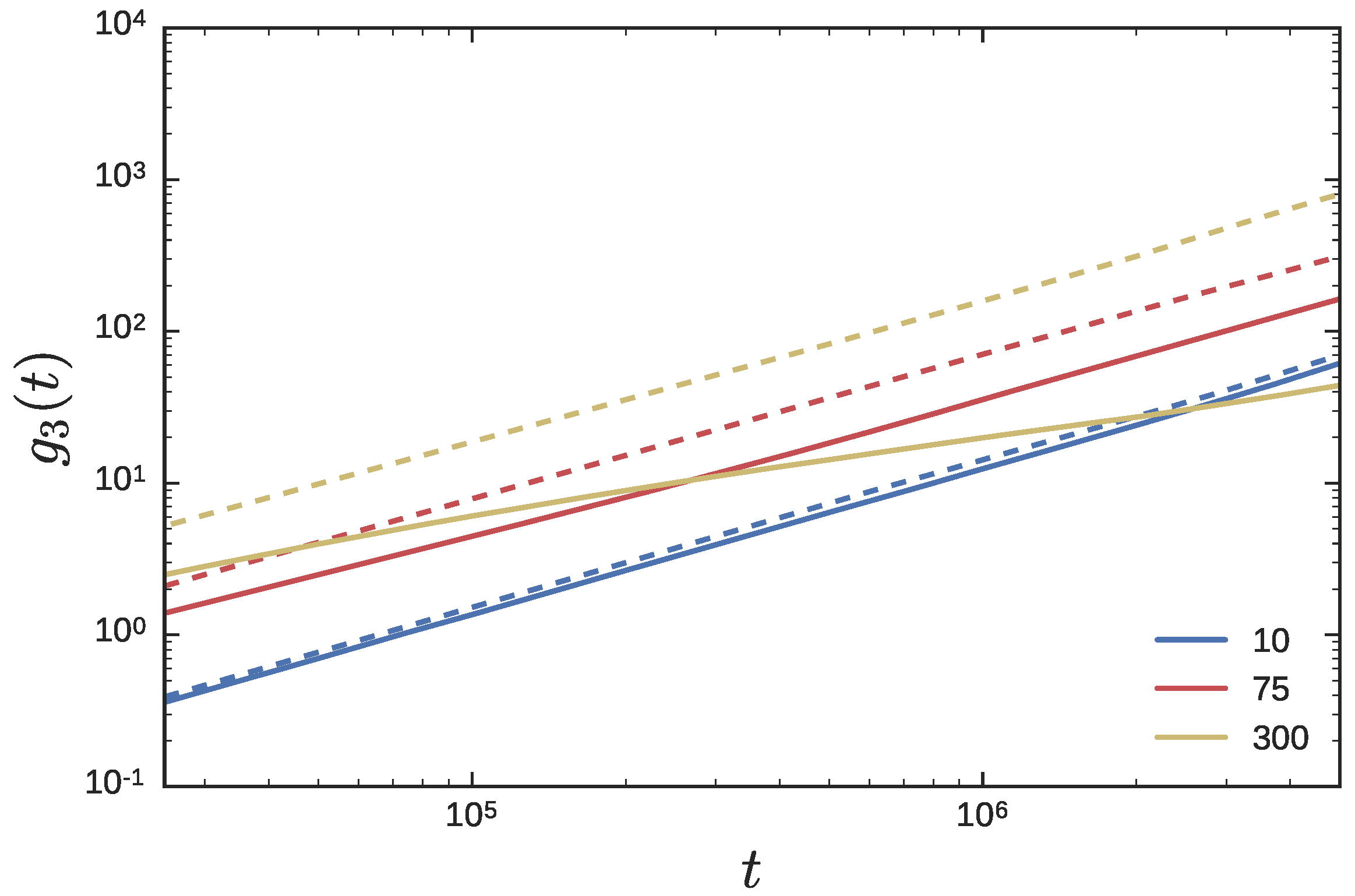
3.2. Ring Probes in Linear Matrix
3.2.1. Statics
3.2.2. Dynamics
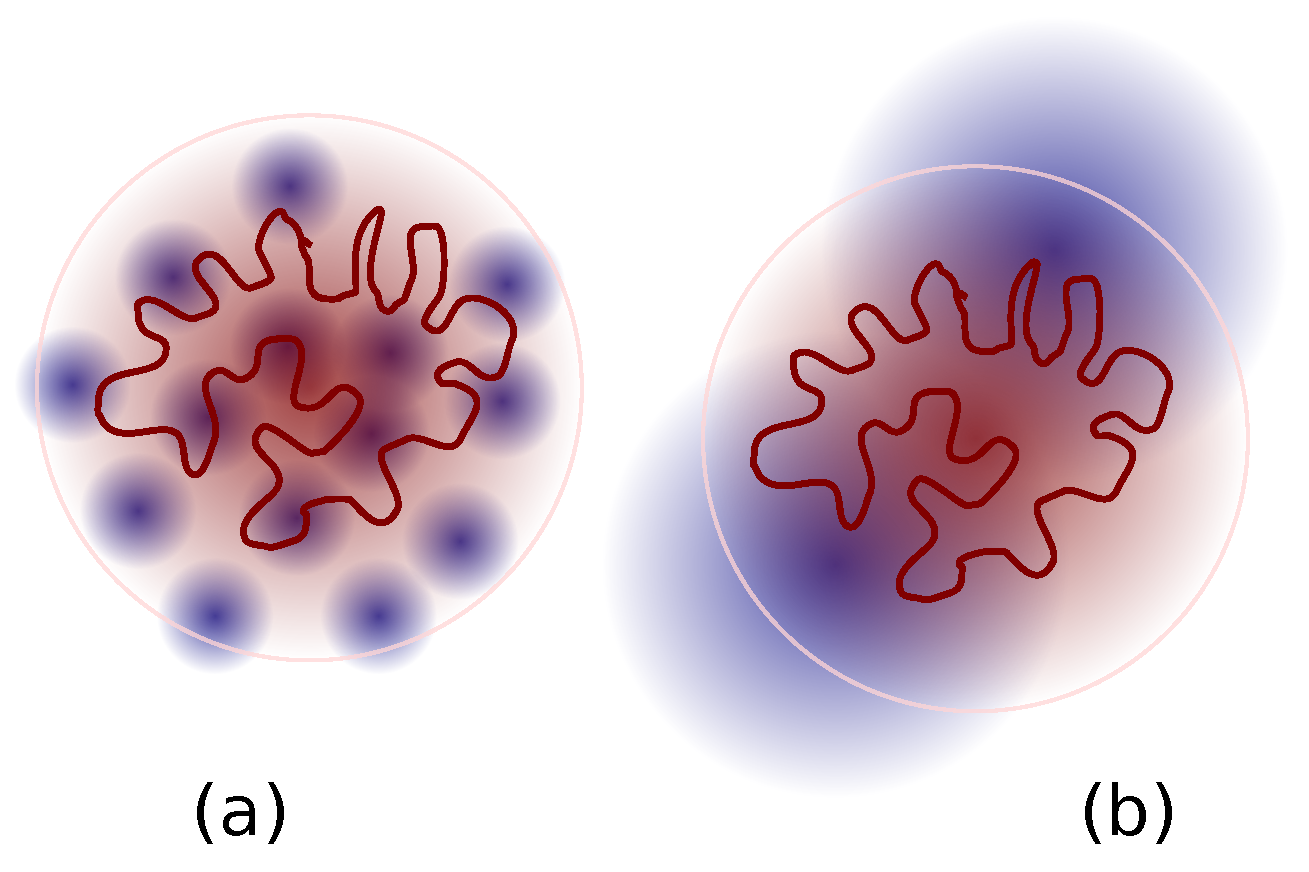
4. Discussion
Proposed Experiments and Simulations
5. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| RP | Ring Polymer |
| LP | Linear Polymer |
| RLB | Ring-Linear Blend |
| NC | Non-Crossing |
| CX | Crossing |
References
- Roovers, J.; Toporowski, P.M. Synthesis and characterization of ring polybutadienes. J. Polym. Sci. B 1988, 26, 1251–1259. [Google Scholar] [CrossRef]
- Bielawski, C.W.; Benitez, D.; Grubbs, R.H. An “endless” route to cyclic polymers. Science 2002, 297, 2041–2044. [Google Scholar] [CrossRef] [PubMed]
- Endo, K. Synthesis and properties of cyclic polymers. In Advances in Polymer Science: New Frontiers in Polymer Synthesis; Kobayashi, S., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 121–183. [Google Scholar]
- Tezuka, Y. (Ed.) Topological Polymer Chemistry: Progress of Cyclic Polymers in Syntheses, Properties and Functions; World Scientific: Singapore, 2013.
- Laib, S.; Robertson, R.M.; Smith, D.E. Preparation and characterization of a set of linear DNA molecules for polymer physics and rheology studies. Macromolecules 2006, 39, 4115–4119. [Google Scholar] [CrossRef]
- Lee, W.; Lee, H.; Lee, H.C.; Cho, D.; Chang, T.; Gorbunov, A.A.; Roovers, J. Retention behavior of linear and ring polystyrene at the chromatographic critical condition. Macromolecules 2002, 35, 529–538. [Google Scholar] [CrossRef]
- Takano, A.; Kushida, Y.; Aoki, K.; Masuoka, K.; Hayashida, K.; Cho, D.; Kawaguchi, D.; Matsushita, Y. HPLC characterization of cyclization reaction product obtained by end-to-end ring closure reaction of a telechelic polystyrene. Macromolecules 2007, 40, 679–681. [Google Scholar] [CrossRef]
- Ohta, Y.; Kushida, Y.; Matsushita, Y.; Takano, A. SEC–MALS characterization of cyclization reaction products: Formation of knotted ring polymer. Polymer 2009, 50, 1297–1299. [Google Scholar] [CrossRef]
- Ohta, Y.; Nakamura, M.; Matsushita, Y.; Takano, A. Synthesis, separation and characterization of knotted ring polymers. Polymer 2012, 53, 466–470. [Google Scholar] [CrossRef]
- Robertson, R.M.; Laib, S.; Smith, D.E. Diffusion of isolated DNA molecules: Dependence on length and topology. Proc. Natl. Acad. Sci. USA 2006, 103, 7310–7314. [Google Scholar] [CrossRef] [PubMed]
- Habuchi, S.; Satoh, N.; Yamamoto, T.; Tezuka, Y.; Vacha, M. Multimode diffusion of ring polymer molecules revealed by a single-molecule study. Angew. Chem. Int. Ed. 2010, 49, 1418–1421. [Google Scholar] [CrossRef] [PubMed]
- Habuchi, S.; Fujiwara, S.; Yamamoto, T.; Vacha, M.; Tezuka, Y. Single-molecule study on polymer diffusion in a melt state: Effect of chain topology. Anal. Chem. 2013, 85, 7369–7376. [Google Scholar] [CrossRef] [PubMed]
- Regan, K.; Ricketts, S.; Robertson-Anderson, R.M. DNA as a model for probing polymer entanglements: Circular polymers and non-classical dynamics. Polymers 2016, 8, 336. [Google Scholar] [CrossRef]
- Fetters, L.J.; Lohse, D.J.; Graessley, W.W. Chain dimensions and entanglement spacings in dense macromolecular systems. J. Polym. Sci. Polym. Phys. Ed. 1999, 37, 1023–1033. [Google Scholar] [CrossRef]
- Kapnistos, M.; Lang, M.; Vlassopoulos, D.; Pyckhout-Hintzen, W.; Richter, D.; Cho, D.; Chang, T.; Rubinstein, M. Unexpected power-law stress relaxation of entangled ring polymers. Nat. Mater. 2008, 7, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.M.; Smith, D.E. Strong effects of molecular topology on diffusion of entangled DNA molecules. Proc. Natl. Acad. Sci. USA 2007, 104, 4824–4827. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.M.; Smith, D.E. Self-diffusion of entangled linear and circular DNA molecules: Dependence on length and concentration. Macromolecules 2007, 40, 3373–3377. [Google Scholar] [CrossRef]
- Chapman, C.D.; Shanbhag, S.; Smith, D.E.; Robertson-Anderson, R.M. Complex effects of molecular topology on diffusion in entangled biopolymer blends. Soft Matter 2012, 8, 9177–9182. [Google Scholar] [CrossRef]
- Gooßen, S.; Krutyeva, M.; Sharp, M.; Feoktystov, A.; Allgaier, J.; Pyckhout-Hintzen, W.; Wischnewski, A.; Richter, D. Sensing polymer chain dynamics through ring topology: A neutron spin echo study. Phys. Rev. Lett. 2015, 115, 148302. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, G.D.; Tsalikis, D.G.; Mavrantzas, V.G. Microscopic dynamics and topology of polymer rings immersed in a host matrix of longer linear polymers: Results from a detailed molecular dynamics simulation study and comparison with experimental data. Polymers 2016, 8, 283. [Google Scholar] [CrossRef]
- McKenna, G.B.; Plazek, D.J. The viscosity of blends of linear and cyclic molecules of similar molecular mass. Polym. Commun. 1986, 27, 304–306. [Google Scholar]
- Tead, S.F.; Kramer, E.J.; Hadziioannou, G.; Antonietti, M.; Sillescu, H.; Lutz, P.; Strazielle, C. Polymer topology and diffusion—A comparison of diffusion in linear and cyclic macromolecules. Macromolecules 1992, 25, 3942–3947. [Google Scholar] [CrossRef]
- Mills, P.J.; Mayer, J.W.; Kramer, E.J.; Hadziioannou, G.; Lutz, P.; Strazielle, C.; Rempp, P.; Kovacs, A.J. Diffusion of polymer rings in linear polymer matrices. Macromolecules 1987, 20, 513–518. [Google Scholar] [CrossRef]
- Vasquez, R.; Shanbhag, S. Percolation of trace amounts of linear polymers in melts of cyclic polymers. Macromol. Theory Simul. 2011, 20, 205–211. [Google Scholar] [CrossRef]
- Halverson, J.D.; Lee, W.B.; Grest, G.S.; Grosberg, A.Y.; Kremer, K. Molecular dynamics simulation study of nonconcatenated ring polymers in a melt. II. Dynamics. J. Chem. Phys. 2011, 134, 204904. [Google Scholar] [CrossRef] [PubMed]
- Halverson, J.D.; Grest, G.S.; Grosberg, A.Y.; Kremer, K. Rheology of ring polymer melts: From linear contaminants to ring-linear blends. Phys. Rev. Lett. 2012, 108. [Google Scholar] [CrossRef] [PubMed]
- Tsalikis, D.G.; Mavrantzas, V.G. Threading of ring poly(ethylene oxide) molecules by linear chains in the melt. ACS Macro Lett. 2014, 3, 763–766. [Google Scholar] [CrossRef]
- Tsalikis, D.G.; Koukoulas, T.; Mavrantzas, V.G. Dynamic, conformational and topological properties of ring–linear poly(ethylene oxide) blends from molecular dynamics simulations. React. Funct. Polym. 2014, 80, 61–70. [Google Scholar] [CrossRef]
- Lee, E.; Kim, S.; Jung, Y. Slowing down of ring polymer diffusion caused by inter-ring threading. Macromol. Rapid Commun. 2015, 36, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Michieletto, D.; Marenduzzo, D.; Orlandini, E.; Alexander, G.P.; Turner, M.S. Threading dynamics of ring polymers in a gel. ACS Macro Lett. 2014, 3, 255–259. [Google Scholar] [CrossRef]
- Tsalikis, D.G.; Mavrantzas, V.G.; Vlassopoulos, D. Analysis of slow modes in ring polymers: Threading of rings controls long-time relaxation. ACS Macro Lett. 2016, 5, 755–760. [Google Scholar] [CrossRef]
- Yang, Y.B.; Sun, Z.Y.; Fu, C.L.; An, L.J.; Wang, Z.G. Monte Carlo simulation of a single ring among linear chains: Structural and dynamic heterogeneity. J. Chem. Phys. 2010, 133. [Google Scholar] [CrossRef] [PubMed]
- Henke, S.F.; Shanbhag, S. Self-diffusion in asymmetric ring-linear blends. React. Funct. Polym. 2014, 80, 57–60. [Google Scholar] [CrossRef]
- Shanbhag, S. Unusual dynamics of ring probes in linear matrices. J. Polym. Sci. B Polym. Phys. 2016. [Google Scholar] [CrossRef]
- Graessley, W.W. Entangled linear, branched and network polymer systems—Molecular theories. Adv. Polym. Sci. 1982, 47, 67–117. [Google Scholar]
- Klein, J. Dynamics of entangled linear, branched, and cyclic polymers. Macromolecules 1986, 19, 105–118. [Google Scholar] [CrossRef]
- Hess, W. Tracer diffusion in polymeric mixtures. Macromolecules 1987, 20, 2587–2599. [Google Scholar] [CrossRef]
- Kolinski, A.; Skolnick, J.; Yaris, R. Monte Carlo studies on the long time dynamic properties of dense cubic lattice multichain systems. II. Probe polymer in a matrix of different degrees of polymerization. J. Chem. Phys. 1987, 86, 7174–7180. [Google Scholar] [CrossRef]
- Barsky, S. Molecular dynamics study of diffusion in bidisperse polymer melts. J. Chem. Phys. 2000, 112, 3450–3456. [Google Scholar] [CrossRef]
- Lin, H.; Mattice, W.L.; von Meerwall, E.D. Chain dynamics of bidisperse polyethylene melts: A Monte Carlo study on a high-coordination lattice. Macromolecules 2007, 40, 959–966. [Google Scholar] [CrossRef]
- Picu, R.C.; Rakshit, A. Coarse grained model of diffusion in entangled bidisperse polymer melts. J. Chem. Phys. 2007, 127. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Larson, R.G. Constraint release in entangled binary blends of linear polymers: A molecular dynamics study. Macromolecules 2008, 41, 4945–4960. [Google Scholar] [CrossRef]
- Green, P.F.; Mills, P.J.; Palmstrøm, C.J.; Mayer, J.W.; Kramer, E.J. Limits of reptation in polymer melts. Phys. Rev. Lett. 1984, 53, 2145–2148. [Google Scholar] [CrossRef]
- Green, P.F.; Kramer, E.J. Matrix effects on the diffusion of long polymer chains. Macromolecules 1986, 19, 1108–1114. [Google Scholar] [CrossRef]
- Antonietti, M.; Coutandin, J.; Sillescu, H. Diffusion of linear polystyrene molecules in matrixes of different molecular weights. Macromolecules 1986, 19, 793–798. [Google Scholar] [CrossRef]
- Seggern, J.V.; Klotz, S.; Cantow, H.J. Reptation and constraint release in linear polymer melts: An experimental study. Macromolecules 1991, 24, 3300–3303. [Google Scholar] [CrossRef]
- Patteson, A.E.; Gopinath, A.; Purohit, P.K.; Arratia, P.E. Particle diffusion in active fluids is non-monotonic in size. Soft Matter 2016, 12, 2365–2372. [Google Scholar] [CrossRef] [PubMed]
- Kasyap, T.V.; Koch, D.L.; Wu, M. Hydrodynamic tracer diffusion in suspensions of swimming bacteria. Phys. Fluids 2014, 26. [Google Scholar] [CrossRef]
- Weber, S.N.; Weber, C.A.; Frey, E. Binary mixtures of particles with different diffusivities demix. Phys. Rev. Lett. 2016, 116. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, J.S. Effects of chain topology on polymer dynamics—Bulk melts. J. Chem. Phys. 1994, 101, 4205–4213. [Google Scholar] [CrossRef]
- Iyer, B.V.S.; Lele, A.K.; Shanbhag, S. What is the size of a ring polymer in a ring-linear blend? Macromolecules 2007, 40, 5995–6000. [Google Scholar] [CrossRef]
- Iyer, B.V.S.; Shanbhag, S.; Juvekar, V.A.; Lele, A.K. Self-diffusion coefficient of ring polymers in semidilute solution. J. Polym. Sci. B Polym. Phys. 2008, 46, 2370–2379. [Google Scholar] [CrossRef]
- Subramanian, G.; Shanbhag, S. Conformational properties of blends of cyclic and linear polymer melts. Phys. Rev. E 2008, 77. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, G.; Shanbhag, S. Conformational free energy of melts of ring-linear polymer blends. Phys. Rev. E 2009, 80. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, G.; Shanbhag, S. Self-diffusion in binary blends of cyclic and linear polymers. Macromolecules 2008, 41, 7239–7242. [Google Scholar] [CrossRef]
- Brown, S.; Lenczycki, T.; Szamel, G. Influence of topological constraints on the statics and dynamics of ring polymers. Phys. Rev. E 2001, 63. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, S. Extraction of self-diffusivity in systems with nondiffusive short-time behavior. Phys. Rev. E 2013, 88. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, S. Estimating self-diffusion in polymer melts: How long is a long enough molecular simulation? Mol. Simul. 2016, 42, 162–172. [Google Scholar] [CrossRef]
- Shanbhag, S.; Larson, R.G. Chain retraction potential in a fixed entanglement network. Phys. Rev. Lett. 2005, 94. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, S.; Larson, R.G. Identification of topological constraints in entangled polymer melts using the bond-fluctuation model. Macromolecules 2006, 39, 2413–2417. [Google Scholar] [CrossRef]
- Uzcategui, A.V.; Shanbhag, S. Self-entanglement of a single polymer chain confined in a cubic box. J. Polym. Sci. B Polym. Phys. 2014, 52, 1283–1290. [Google Scholar] [CrossRef]
- Shaffer, J.S. Effects of chain topology on polymer dynamics—Configurational relaxation in polymer melts. J. Chem. Phys. 1995, 103, 761–772. [Google Scholar] [CrossRef]
- Tsolou, G.; Stratikis, N.; Baig, C.; Stephanou, P.S.; Mavrantzas, V.G. Melt structure and dynamics of unentangled polyethylene rings: Rouse theory, atomistic molecular dynamics simulation, and comparison with the linear analogues. Macromolecules 2010, 43, 10692–10713. [Google Scholar] [CrossRef]
- Kuriata, A.; Sikorski, A. Computer simulation of cyclic polymers in disordered media. Comput. Methods Sci. Technol. 2015, 21, 21–27. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (NC) | (CX) | |||||
|---|---|---|---|---|---|---|
| Symmetric blends | ||||||
| 300 | 300 | 360 | 0 | 0.0 | 30.0 | 30.0 |
| 300 | 300 | 288 | 72 | 0.2 | 70.0 | 20.0 |
| 300 | 300 | 216 | 144 | 0.4 | 70.0 | 20.0 |
| 300 | 300 | 144 | 216 | 0.6 | 80.0 | 20.0 |
| 300 | 300 | 72 | 288 | 0.8 | 80.0 | 20.0 |
| 300 | 300 | 36 | 324 | 0.9 | 100.0 | 15.0 |
| 300 | 300 | 0 | 360 | 1.0 | 40.0 | 40.0 |
| Asymmetric blends | ||||||
| 300 | 10 | 36 | 9,720 | 0.9 | 20.0 | 15.0 |
| 300 | 30 | 43 | 3,932 | 0.9 | 22.5 | 15.0 |
| 300 | 75 | 43 | 1,572 | 0.9 | 21.4 | 15.0 |
| 300 | 150 | 43 | 786 | 0.9 | 20.0 | 15.0 |
| 300 | 300 | 36 | 324 | 0.9 | 100.0 | 15.0 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crysup, B.; Shanbhag, S. What Happens When Threading is Suppressed in Blends of Ring and Linear Polymers? Polymers 2016, 8, 409. https://doi.org/10.3390/polym8120409
Crysup B, Shanbhag S. What Happens When Threading is Suppressed in Blends of Ring and Linear Polymers? Polymers. 2016; 8(12):409. https://doi.org/10.3390/polym8120409
Chicago/Turabian StyleCrysup, Benjamin, and Sachin Shanbhag. 2016. "What Happens When Threading is Suppressed in Blends of Ring and Linear Polymers?" Polymers 8, no. 12: 409. https://doi.org/10.3390/polym8120409