
Intrinsic Delocalization during the Decay of Excitons in Polymeric Solar Cells


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
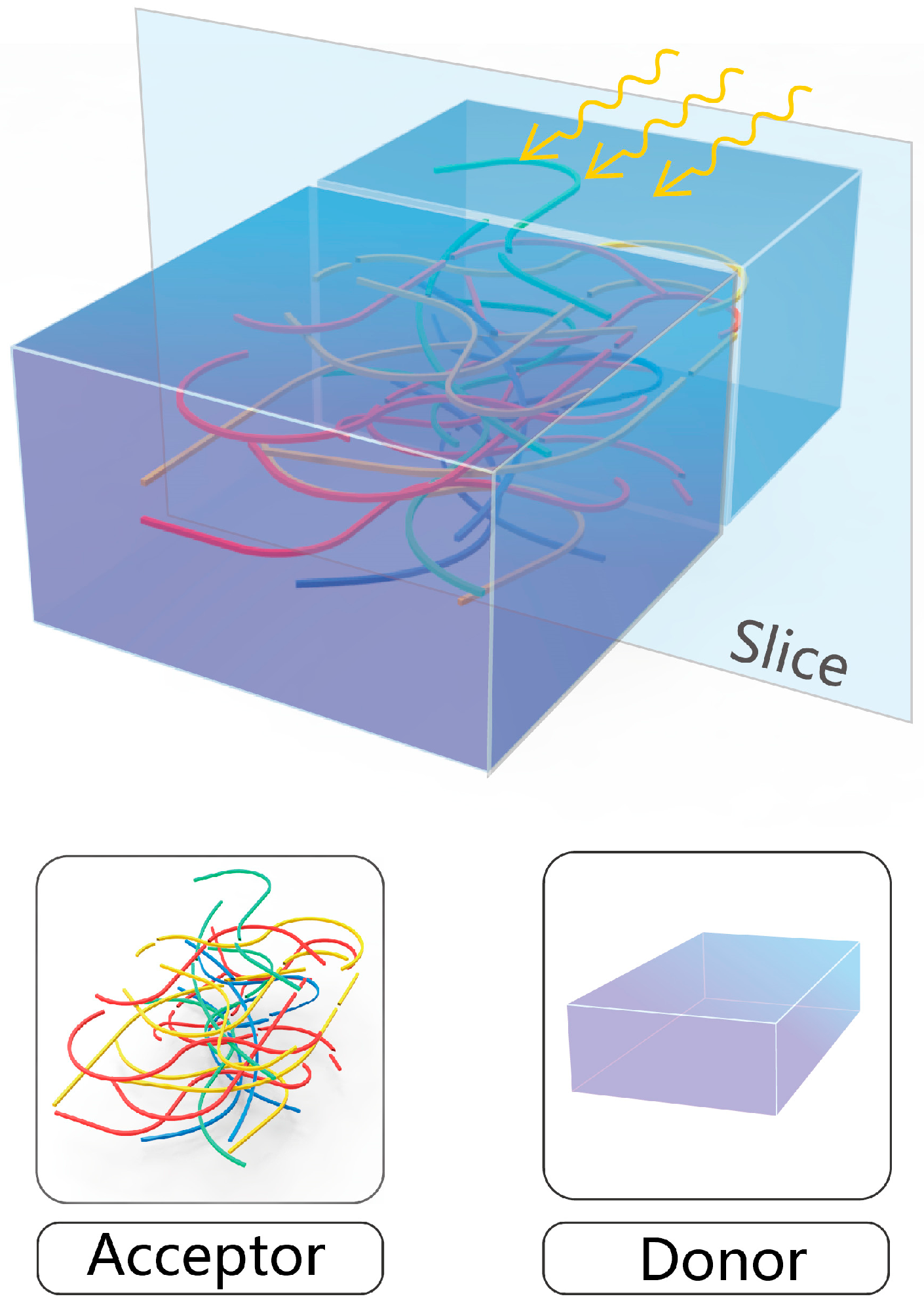
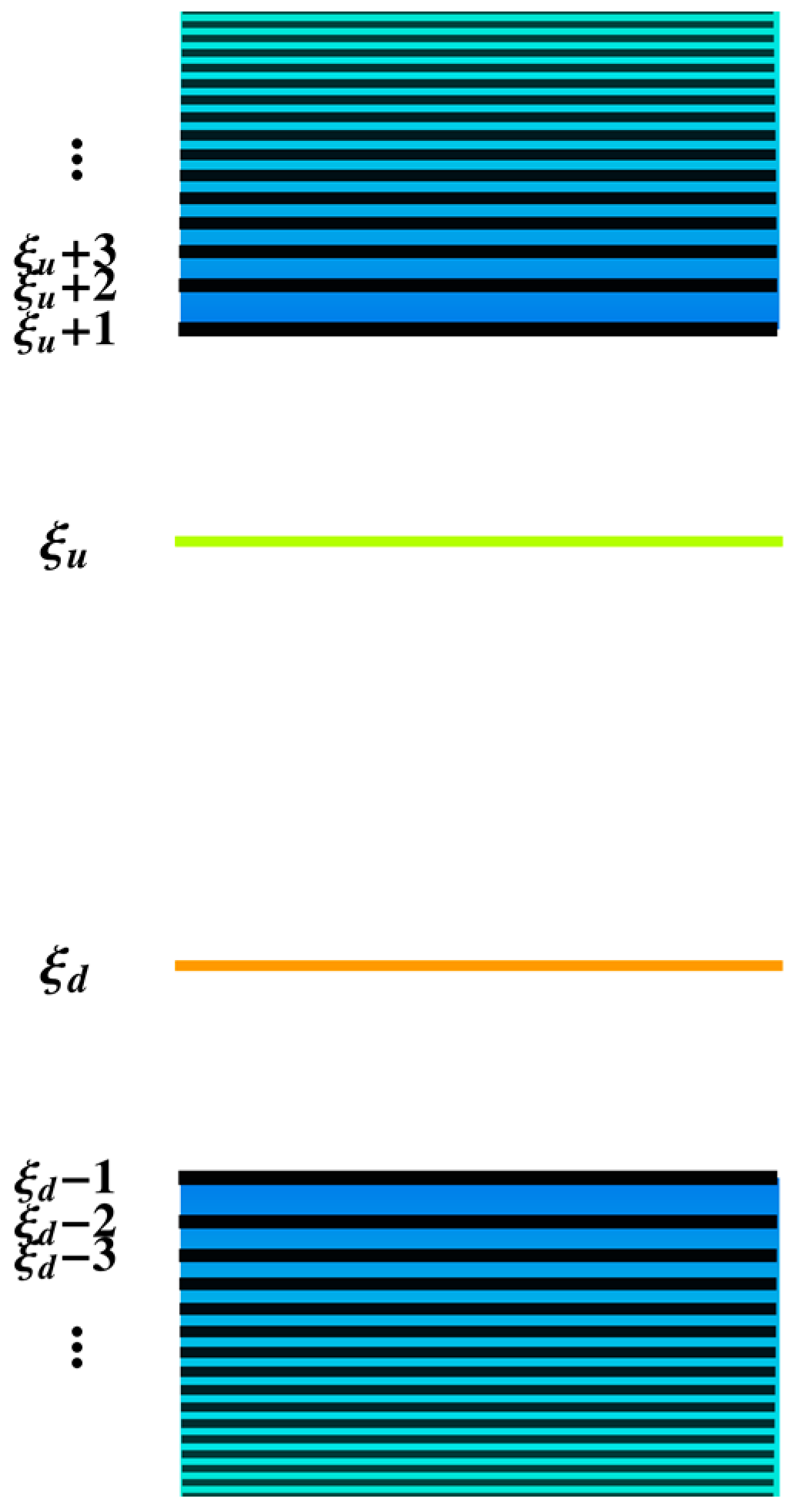
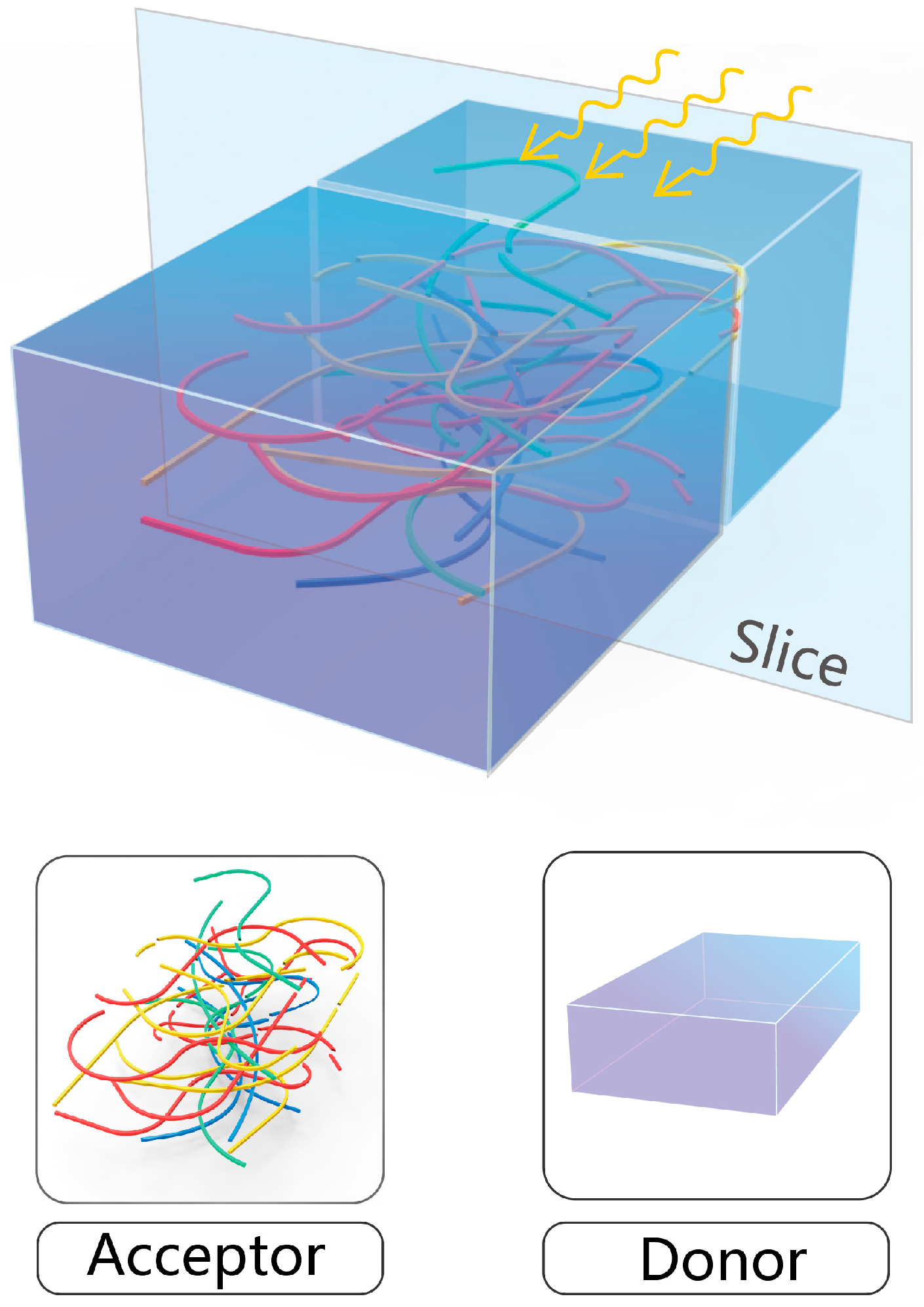
2. Methodology
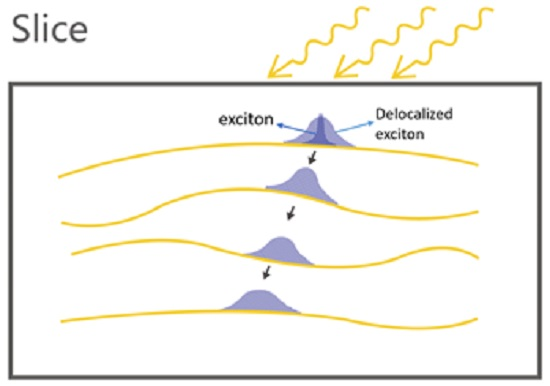
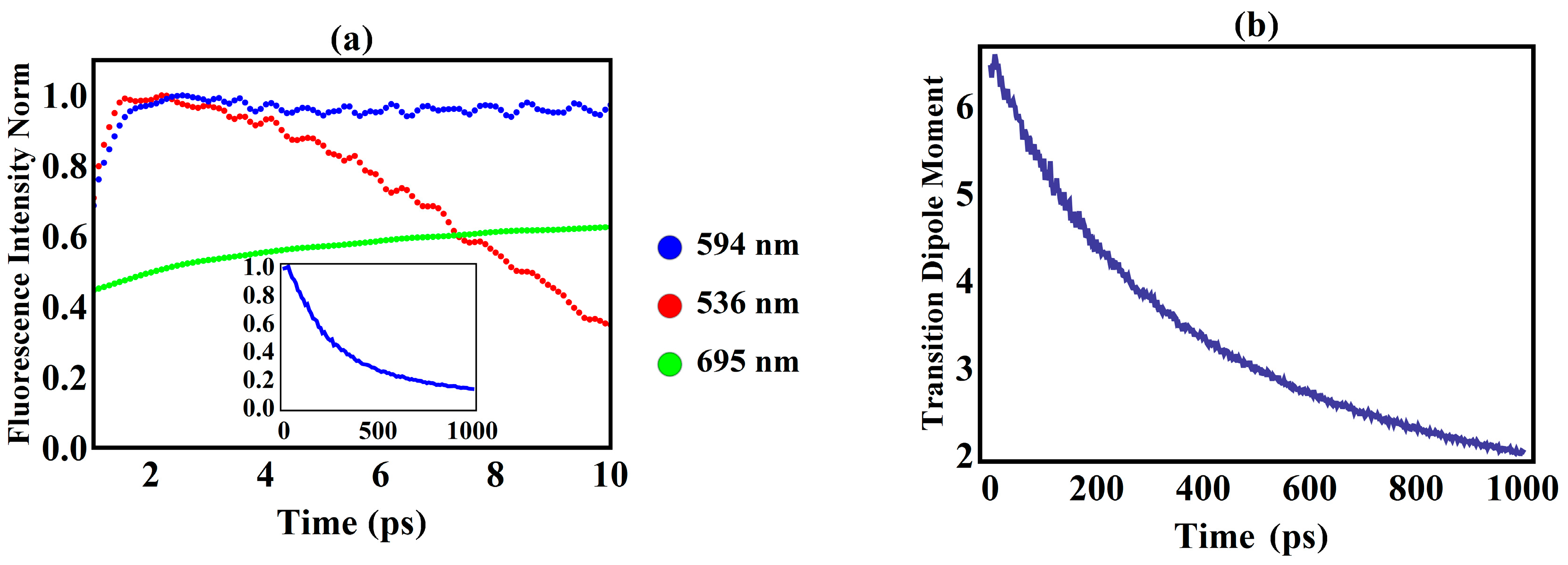
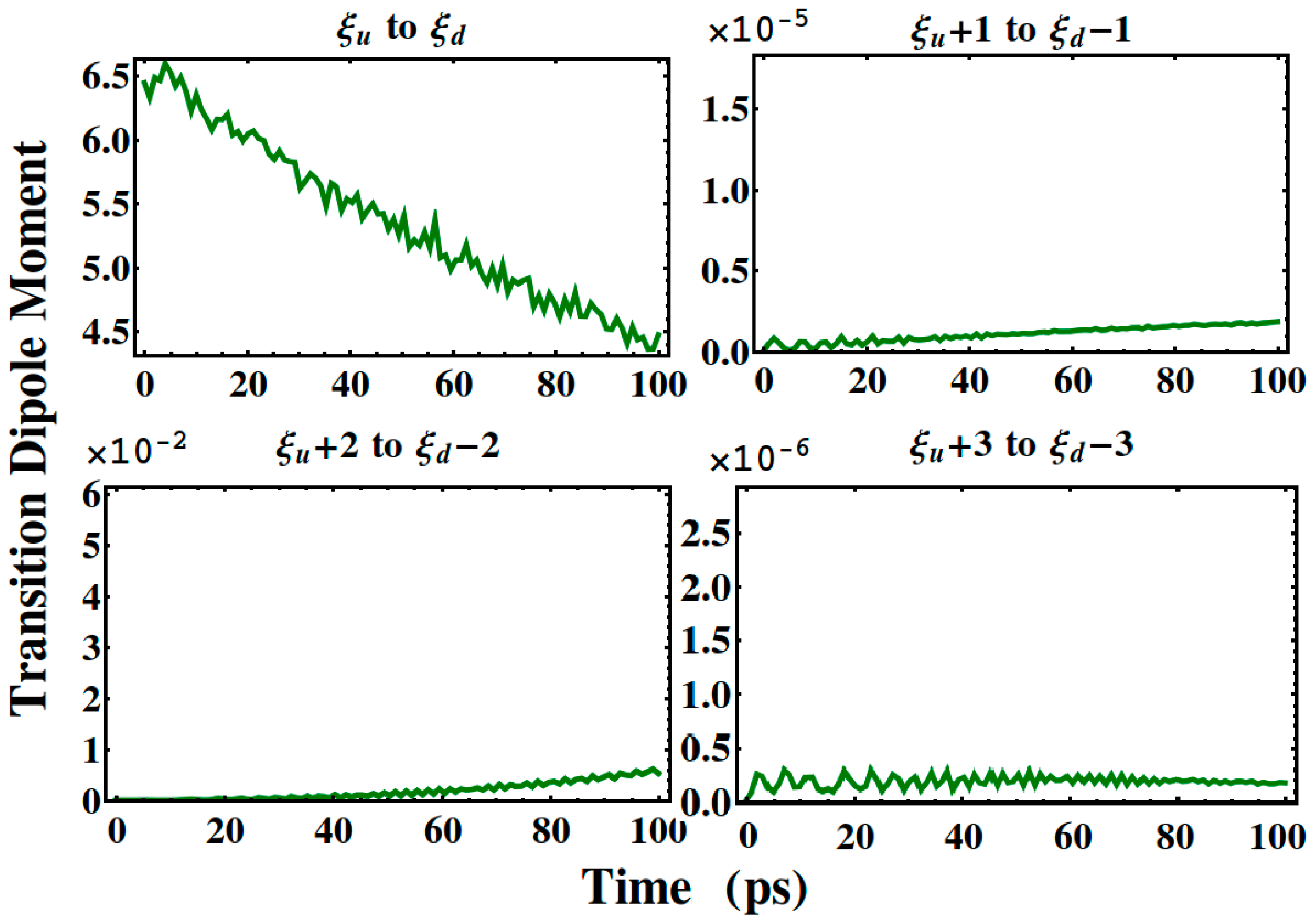
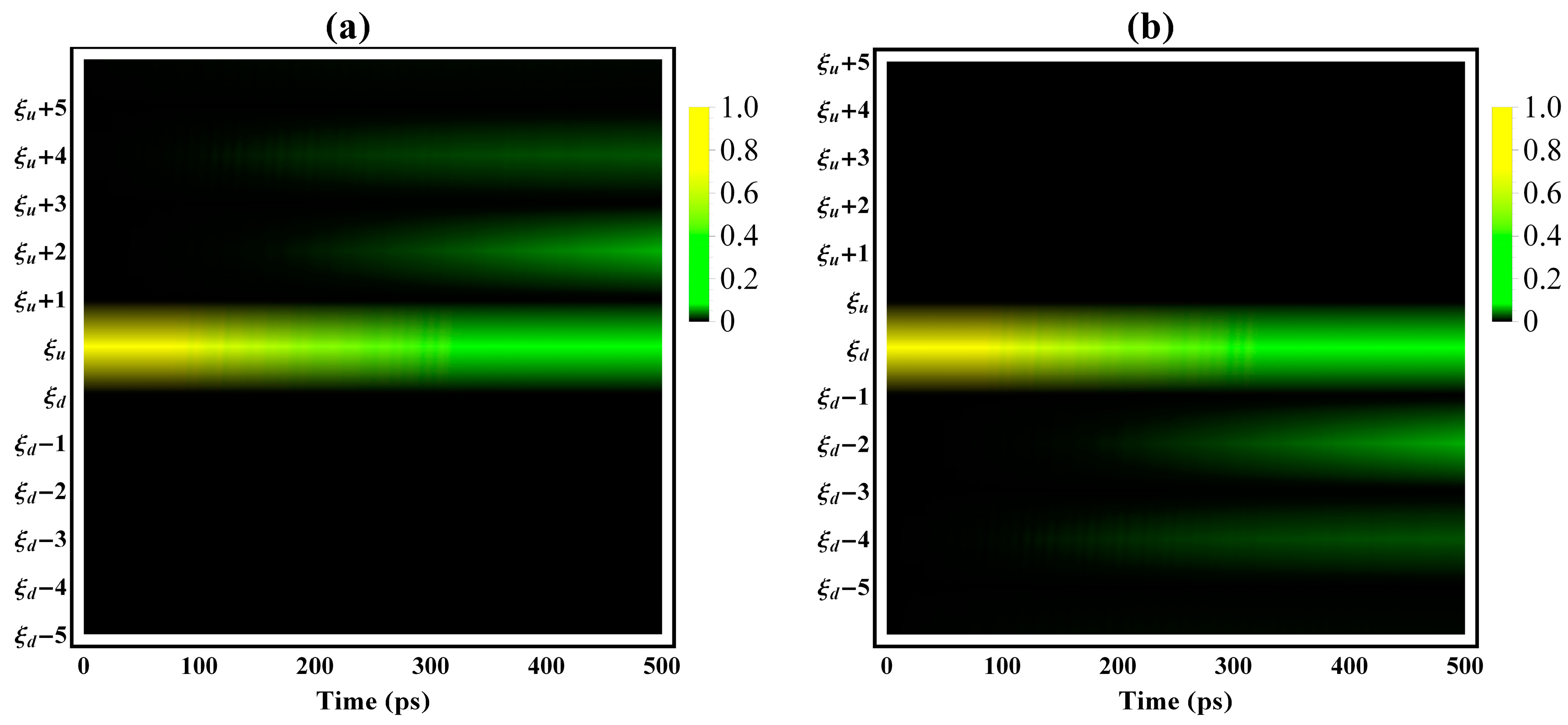
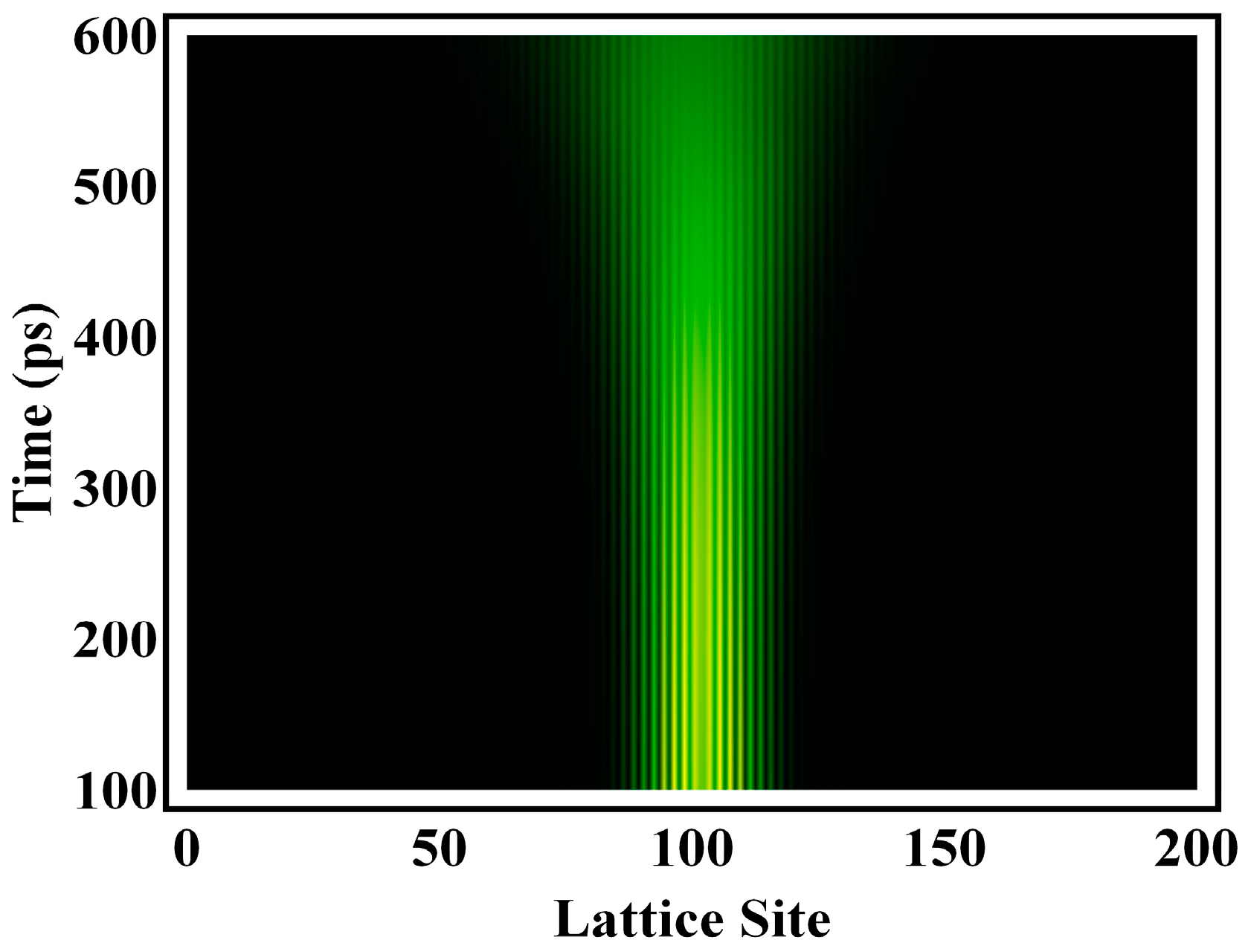
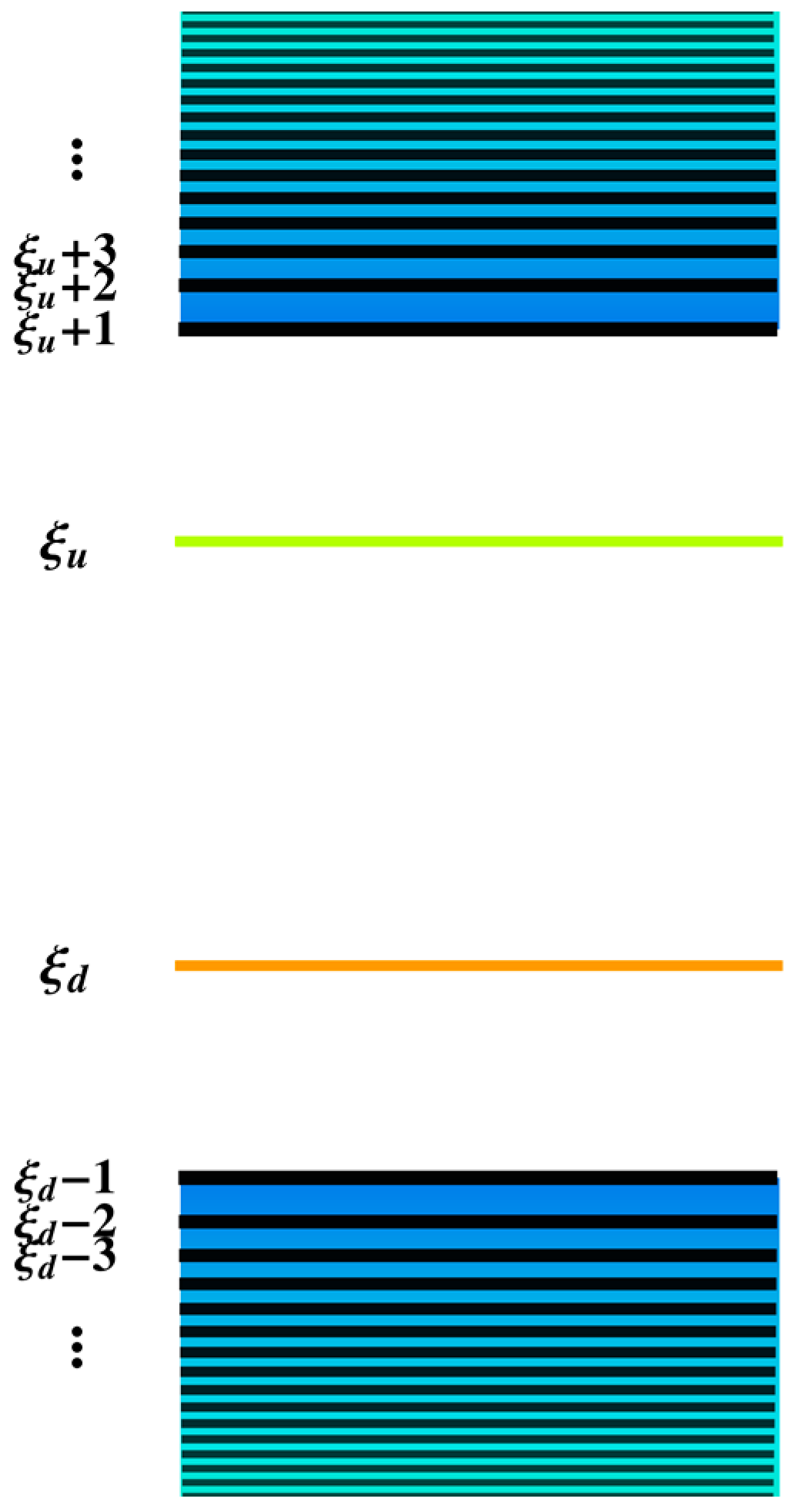
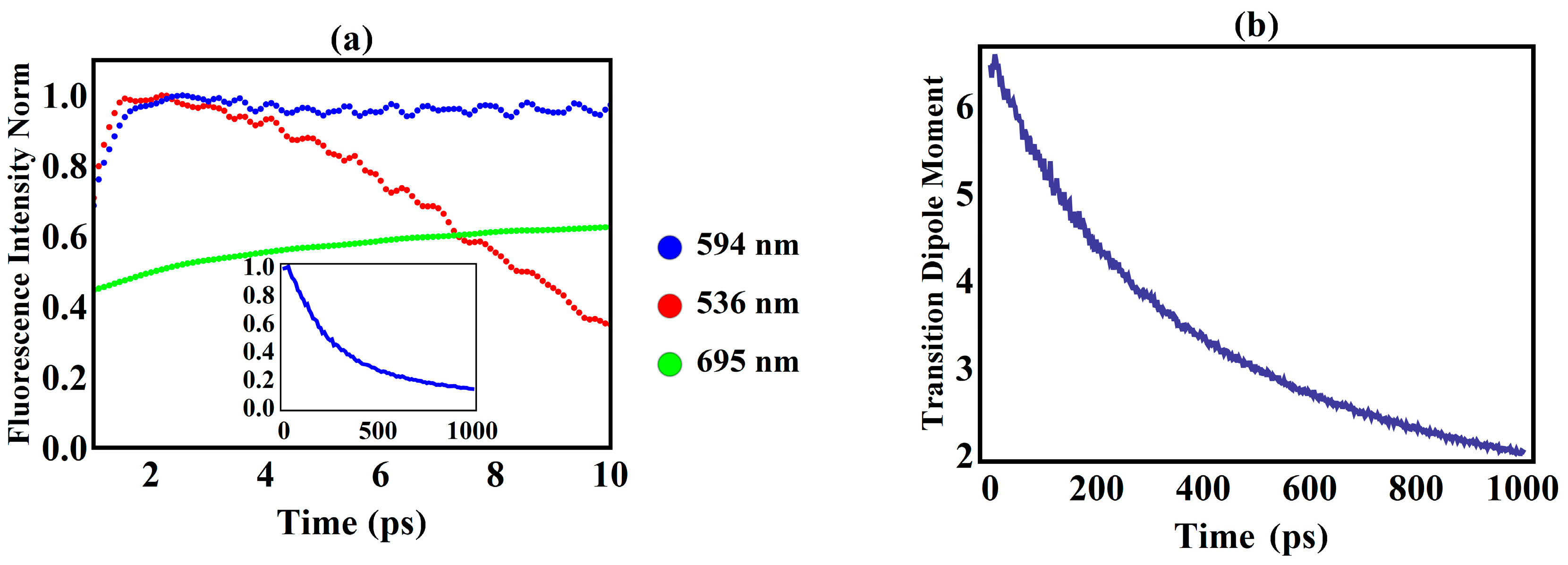
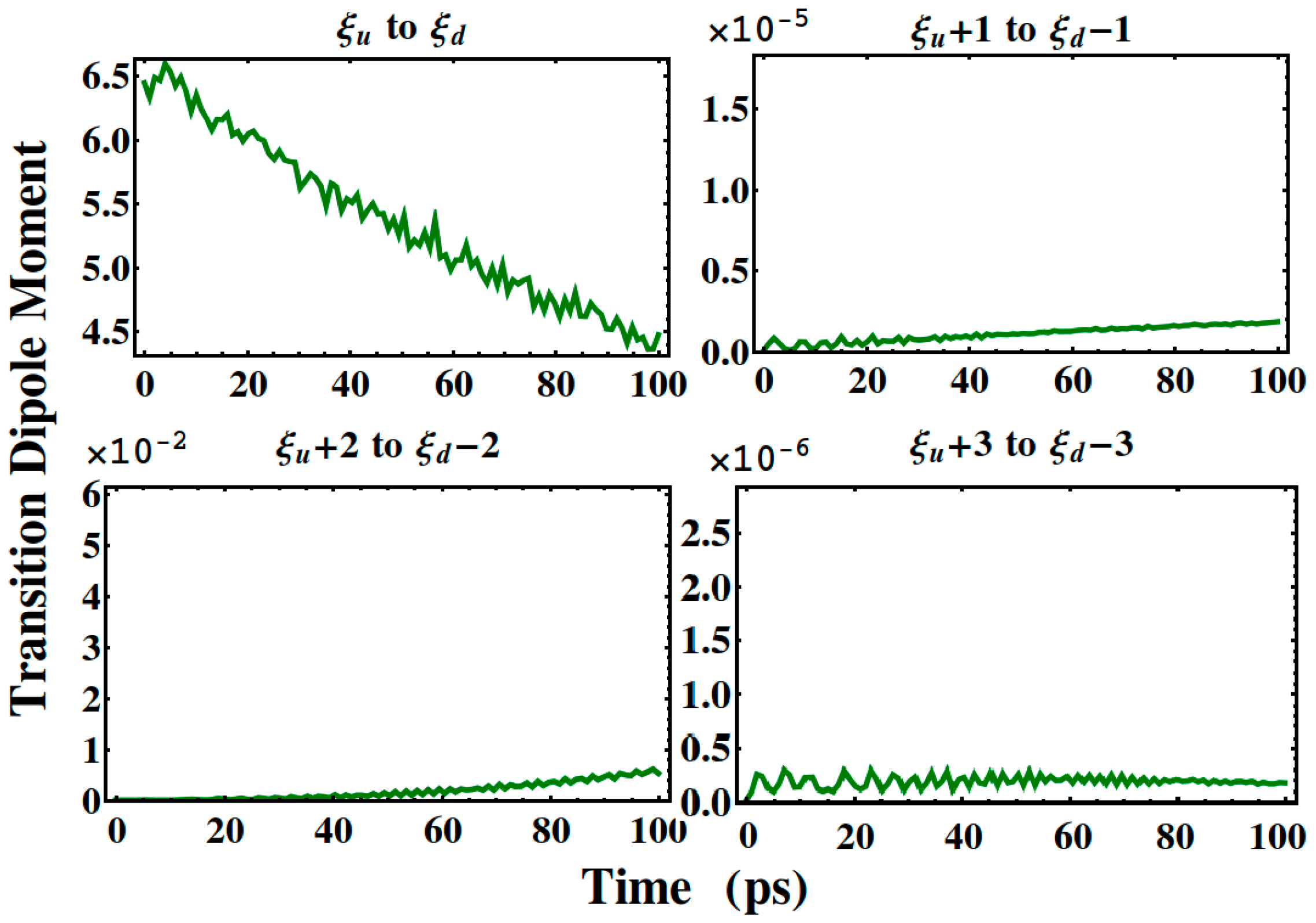
3. Results and Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chamberlain, G.A. Organic solar cells: A review. Sol. Cells 1983, 8, 47–83. [Google Scholar] [CrossRef]
- Wouhrle, D.; Meissner, D. Organic solar cells. Adv. Mater. 1991, 3, 129–138. [Google Scholar] [CrossRef]
- Brabec, C.J.; Sariciftci, N.S.; Hummelen, J.C. Plastic solar cells. Adv. Funct. Mater. 2001, 11, 15–26. [Google Scholar] [CrossRef]
- Nelson, J. Organic photovoltaic films. Curr. Opin. Solid State Mater. Sci. 2002, 6, 87–95. [Google Scholar] [CrossRef]
- Li, Y.; Zou, Y. Conjugated polymer photovoltaic materials with broad absorption band and high charge carrier mobility. Adv. Mater. 2008, 20, 2952–2958. [Google Scholar] [CrossRef]
- Dennler, G.; Scharber, M.C.; Brabec, C.J. Polymer-Fullerene bulk-heterojunction solar cells. Adv. Mater. 2009, 21, 1323–1338. [Google Scholar] [CrossRef]
- Li, C.; Liu, M.; Pschirer, N.G.; Baumgarten, M.; Mullen, K. Polyphenylene-based materials for organic photovoltaics. Chem. Rev. 2010, 110, 6817–6855. [Google Scholar] [CrossRef] [PubMed]
- Facchetti, A. π-Conjugated polymers for organic electronics and photovoltaic applications. Chem. Mater. 2011, 23, 733–758. [Google Scholar] [CrossRef]
- Piliego, C.; Loi, M.A. Charge transfer state in highly efficient polymer–fullerene bulk heterojunction solar cells. J. Mater. Chem. 2012, 22, 4141–4150. [Google Scholar] [CrossRef]
- Winder, C.; Sariciftci, N.S. Low bandgap polymers for photon harvesting in bulk heterojunction solar cells. J. Mater. Chem. 2004, 14, 1077–1086. [Google Scholar] [CrossRef]
- Morel, D.L.; Gosh, A.K.; Feng, T.; Stogryn, E.L.; Purwin, P.E.; Shaw, R.F.; Fishman, C. High-efficiency organic solar cells. Appl. Phys. Lett. 1978, 32, 495–497. [Google Scholar] [CrossRef]
- Gosh, A.K.; Feng, T. Merocyanine organic solar cells. J. Appl. Phys. 1978, 49, 5982–5989. [Google Scholar] [CrossRef]
- Bakulin, A.A.; Rao, A.; Pavelyev, V.G.; van Loosdrecht, P.H.M.; Pshenichnikov, M.S.; Niedzialek, D.; Cornil, J.; Beljonne, D.; Freiend, R.H. The role of driving energy and delocalized states for charge separation in organic semiconductors. Science 2012, 335, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- Jailaubekov, A.E.; Willard, A.P.; Tritsch, J.R.; Chan, W.L.; Sai, N.; Gearba, R.; Kaake, L.G.; Williams, K.J.; Leung, K.; Rossky, P.J.; et al. Hot charge-transfer excitons set the time limit for charge separation at donor/acceptor interfaces in organic photovoltaics. Nat. Mater. 2013, 12, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chan, W.L. Dynamical localization limiting the coherent transport range of excitons in organic crystals. J. Phys. Chem. Lett. 2014, 5, 1812–1818. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Kwok, W.M.; Ma, C.; Chan, C.T.L.; Zhu, M.X.; Che, C.M. Organic triplet excited states of gold(I) complexes with oligo(o-or m-phenyleneethynylene) ligands: Conjunction of steady-state and time-resolved spectroscopic studies on exciton delocalization and emission pathways. J. Am. Chem. Soc. 2011, 133, 14120–14135. [Google Scholar] [CrossRef] [PubMed]
- Paraecattil, A.A.; Banerji, N. Charge separation pathways in a highly efficient polymer: Fullerene solar cell material. J. Am. Chem. Soc. 2014, 136, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Grancini, G.; Maiuri, M.; Fazzi, D.; Petrozza, A.; Egelhaaf, H.J.; Brida, D.; Cerullo, G. Hot exciton dissociation in polymer solar cells. Nat. Mater. 2013, 12, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, T.; Yamada, Y.; Kanemitsu, Y. Dynamics of exciton-hole recombination in hole-doped single-walled carbon nanotubes. Phys. Rev. B 2012, 86, 075449. [Google Scholar] [CrossRef]
- Mouri, S.; Miyauchi, Y.; Iwamura, M.; Matsuda, K. Temperature dependence of photoluminescence spectra in hole-doped single-walled carbon nanotubes: Implications of trion localization. Phys. Rev. B 2013, 87, 045408. [Google Scholar] [CrossRef]
- Chen, S.; Yoshita, M.; Ishikawa, A.; Mochizuki, T.; Maruyama, S.; Akiyama, H.; Yuhei, H.; Pfeiffer, L.N.; West, K.W. Intrinsic radiative lifetime derived via absorption cross section of one-dimensional excitons. Sci. Rep. 2013, 3, 1941. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, P.; Kambhampati, P. Independent control of electron and hole localization in core/barrier/shell nanostructures. J. Phys. Chem. C 2012, 116, 8154–8160. [Google Scholar] [CrossRef]
- Piris, J.; Dykstra, T.E.; Bakulin, A.A.; Loosdrecht, P.H.M.V.; Knulst, W.; Trinh, M.T.; Schins, J.M.; Siebbeles, L.D.A. Photogeneration and ultrafast dynamics of excitons and charges in P3HT/PCBM blends. J. Phys. Chem. C 2009, 113, 14500–14506. [Google Scholar] [CrossRef]
- Banerji, N.; Cowan, S.; Leclerc, M.; Vauthey, E.; Heeger, A.J. Exciton formation, relaxation, and decay in PCDTBT. J. Am. Chem. Soc. 2010, 132, 17459–17470. [Google Scholar] [CrossRef] [PubMed]
- Banerji, N.; Cowan, S.; Vauthey, E.; Heeger, A.J. Ultrafast relaxation of the poly (3-hexylthiophene) emission spectrum. J. Phys. Chem. C 2011, 115, 9726–9739. [Google Scholar] [CrossRef]
- Banerji, N.; Seifter, J.; Wang, M.; Vauthey, E.; Wudl, F.; Heeger, A.J. Ultrafast spectroscopic investigation of a fullerene poly (3-hexylthiophene) dyad. Phys. Rev. B 2011, 84, 075206. [Google Scholar] [CrossRef]
- Heeger, A.J.; Kivelson, S.; Schrieffer, J.R.; Su, W.P. Solitons in conducting polymers. Mod. Phys. 1988, 60, 781. [Google Scholar] [CrossRef]
- Wang, C.; Zhuang, L.Q.; Chen, R.A.; Li, S.; George, T.F. Localization and relaxation of singlet exciton formation in conjugated polymers under photoexcitation. J. Phys. Chem. B 2013, 117, 3258–3263. [Google Scholar] [CrossRef] [PubMed]
- Devizis, A.; Serbenta, A.; Meerholz, K.; Hertel, D.; Gulbinas, V. Excited state relaxation in poly (spirobifluorene-co-benzothiadiazole) films. J. Chem. Phys. 2009, 131, 104902. [Google Scholar] [CrossRef]
- Marciniak, H.; Fiebig, M.; Huth, M.; Schiefer, S.; Nickel, B.; Selmaier, F.; Lochbrunner, S. Ultrafast exciton relaxation in microcrystalline pentacene films. Phys. Rev. Lett. 2007, 99, 176402. [Google Scholar] [CrossRef] [PubMed]
- Synooka, O.; Kretschmer, F.; Hager, M.D.; Himmerlich, M.; Krischok, S.; Gehrig, D.; Laquai, F.; Schubert, U.S.; Gobsch, G.; Hoppe, H. Modification of the active layer/PEDOT: PSS interface by solvent additives resulting in improvement of the performance of organic solar cells. ACS Appl. Mater. Interfaces 2014, 6, 11068–11081. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.; Jiang, D.; Chen, R.; Li, S.; George, T.F. Intrinsic Delocalization during the Decay of Excitons in Polymeric Solar Cells. Polymers 2016, 8, 414. https://doi.org/10.3390/polym8120414
Chen W, Jiang D, Chen R, Li S, George TF. Intrinsic Delocalization during the Decay of Excitons in Polymeric Solar Cells. Polymers. 2016; 8(12):414. https://doi.org/10.3390/polym8120414
Chicago/Turabian StyleChen, Weikang, Deyao Jiang, Renai Chen, Sheng Li, and Thomas F. George. 2016. "Intrinsic Delocalization during the Decay of Excitons in Polymeric Solar Cells" Polymers 8, no. 12: 414. https://doi.org/10.3390/polym8120414