
Activated Charge-Reversal Polymeric Nano-System: The Promising Strategy in Drug Delivery for Cancer Therapy

Abstract
:
1. Introduction
2. Role of Surface Charge on Cellular Uptake
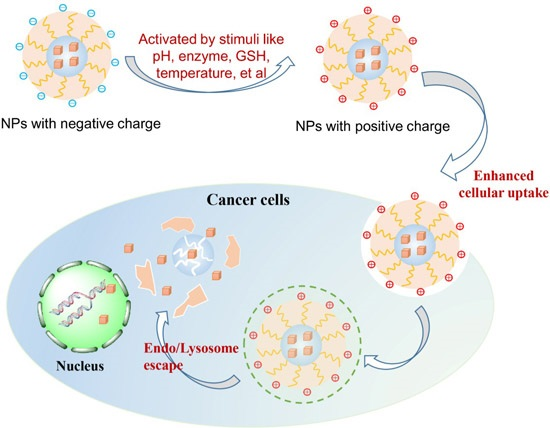
3. Activated Approaches of Surface Charge Reversal
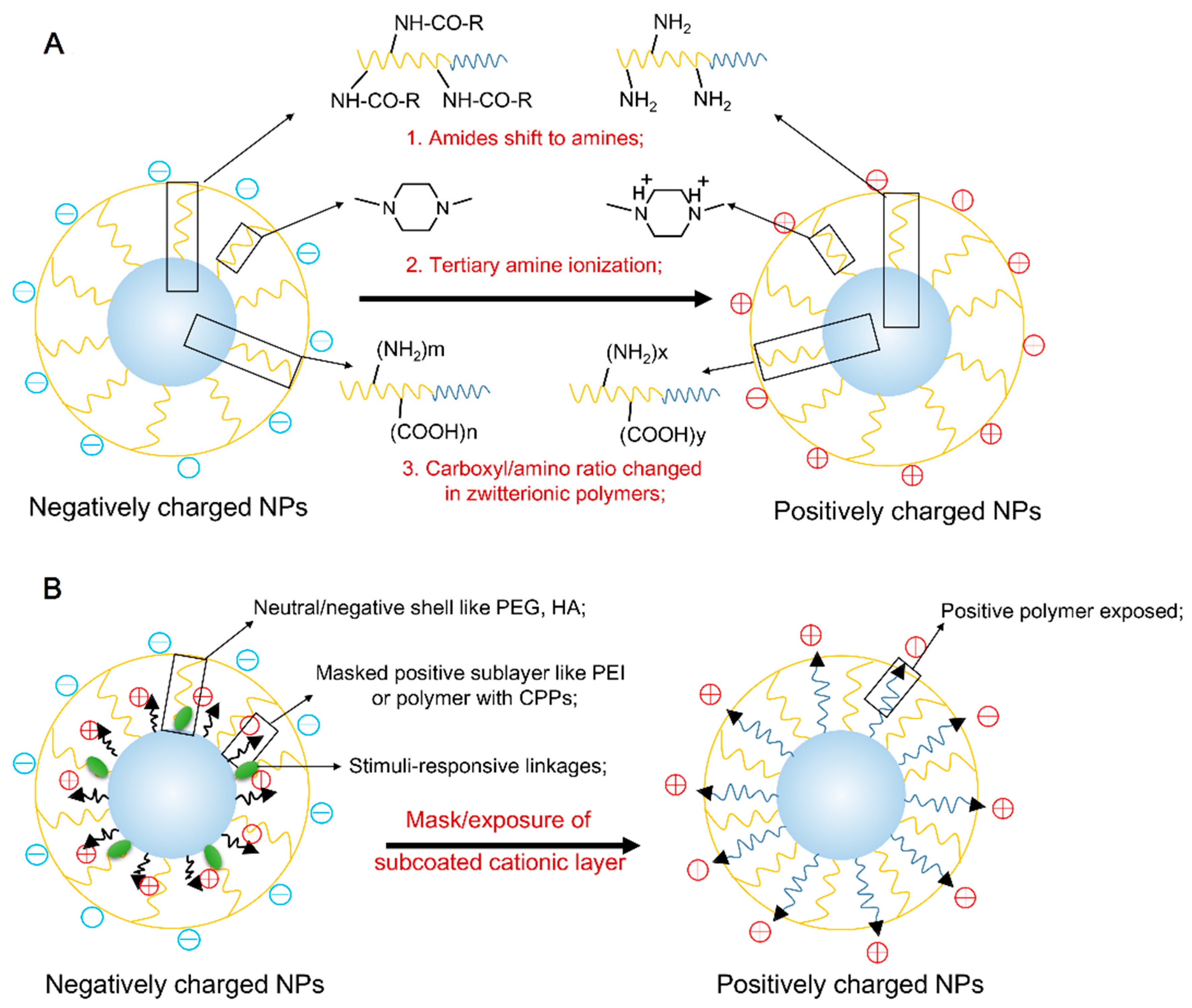
3.1. The Stimuli-Responsive Protonation/Deprotonation of Polymer
3.2. The Stimuli-Responsive Mask/Exposure of Subcoated Cationic Nano-Layer
4. Activated Surface Charge Reversal NPs for Cancer Treatment
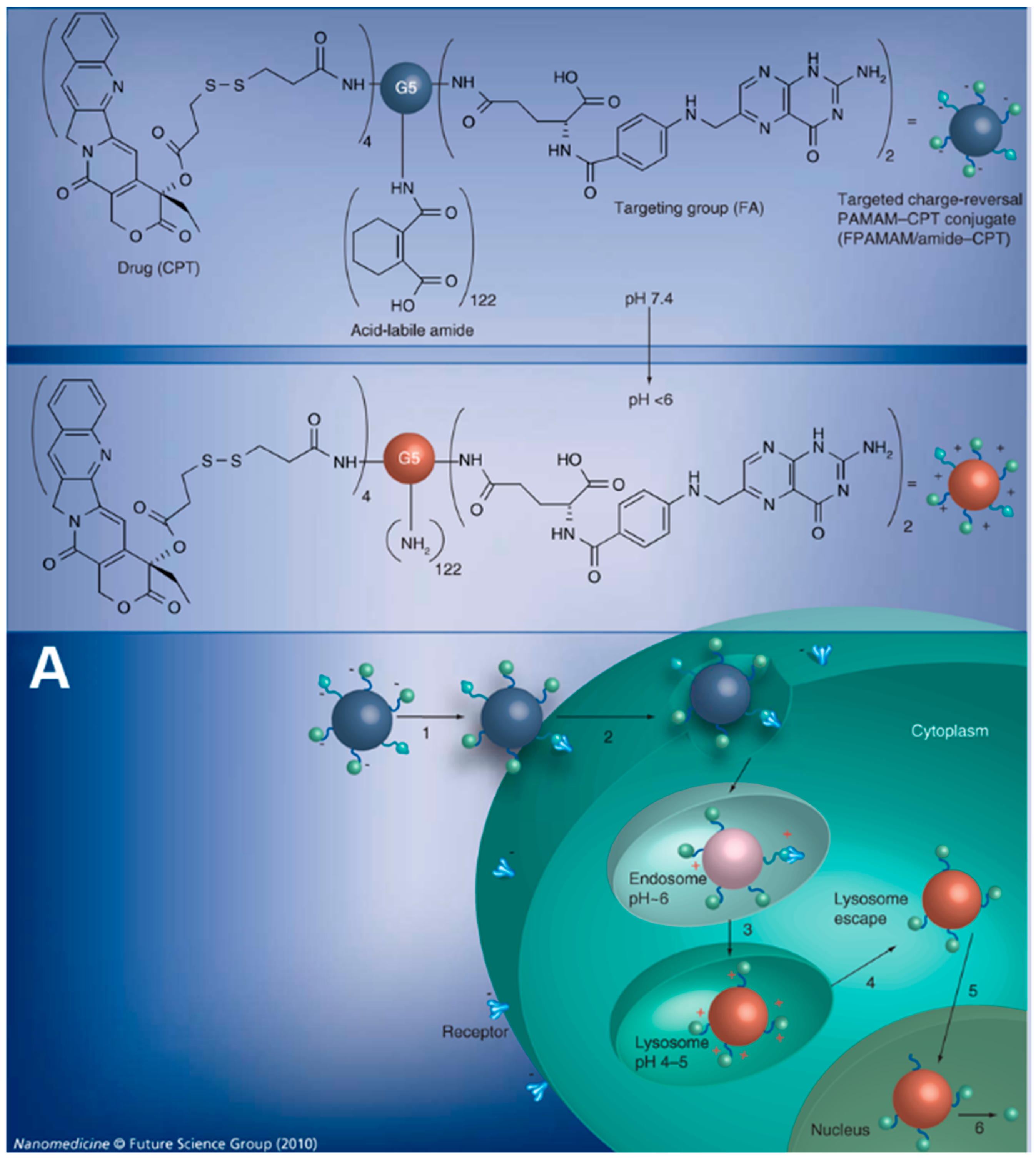
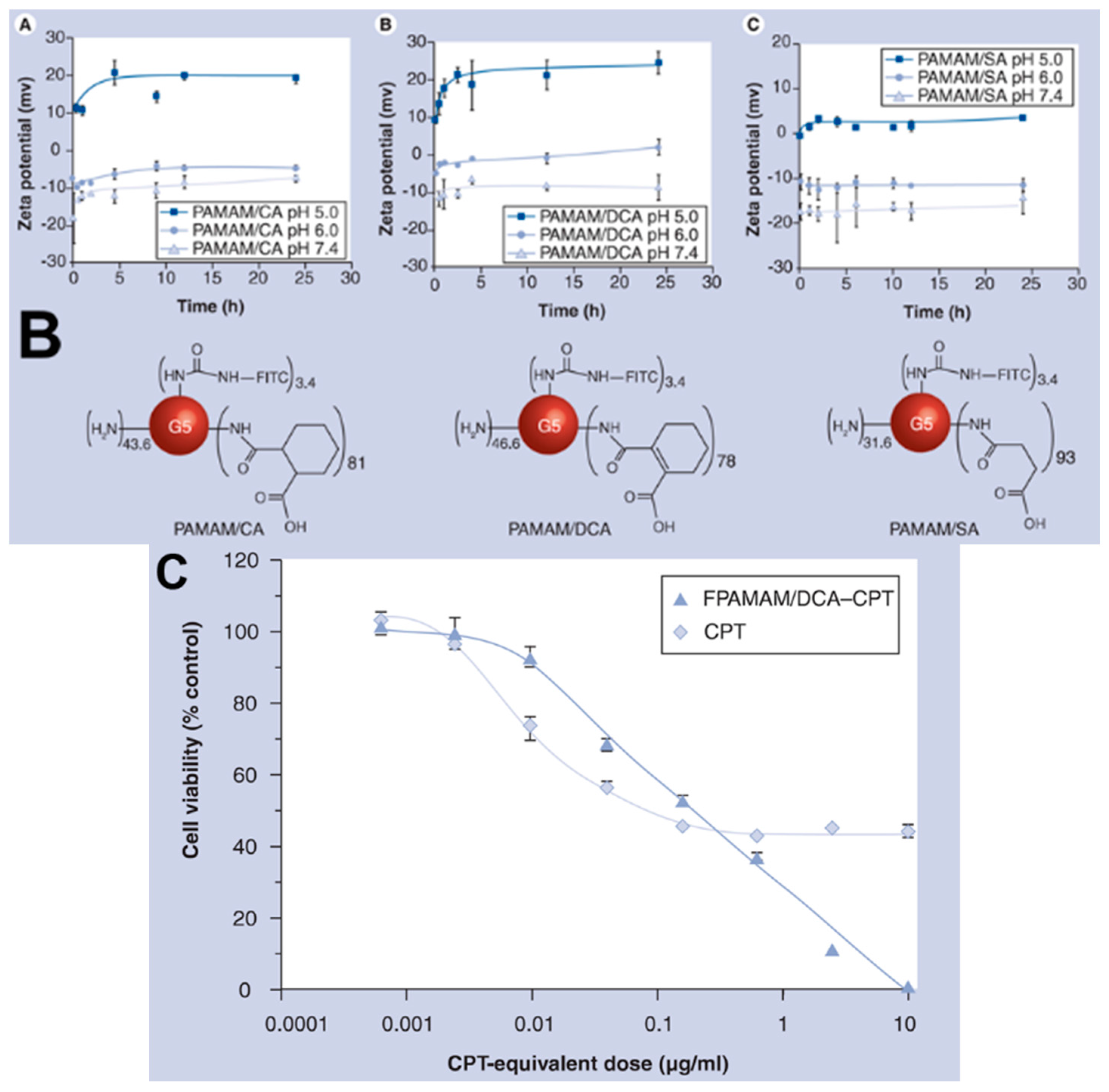
4.1. Enhanced Cellular Uptake and Intracellular Delivery
4.2. Enhanced Drug Delivery to Cancer Stem Cell
4.3. Enhanced Multidrug Resistance Reversal
5. Conclusions and Future Prospects
Acknowledgments
Author Contributions
Conflicts of Interest
References
- WHO. Cancer. In World Cancer Report; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- Liu, M.; Kurosaki, T.; Suzuki, M.; Enomoto, Y.; Nishimatsu, H.; Arai, T.; Sawabe, M.; Hosoi, T.; Homma, Y.; Kitamura, T. Significance of common variants on human chromosome 8q24 in relation to the risk of prostate cancer in native Japanese men. BMC Genet. 2009, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Suzuki, M.; Arai, T.; Sawabe, M.; Enomoto, Y.; Nishimatsu, H.; Kume, H.; Homma, Y.; Kitamura, T. A replication study examining three common single-nucleotide polymorphisms and the risk of prostate cancer in a Japanese population. Prostate 2011, 71, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotech. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Shin, D.M. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat. Rev. Drug Discov. 2008, 7, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D.A.; Bristol, M.L.; Yalowich, J.C. Toxicity issues in cancer drug development. Curr. Opin. Investig. Drug 2010, 11, 612–614. [Google Scholar]
- Hermanson, T.; Norris, L.B.; Bian, J.; Sartor, O.; Bennett, C.L. Toxicity and costs of toxicity associated with new cancer drugs: International implications. J. Clin. Oncol. 2014, 32, 3591–3592. [Google Scholar] [CrossRef] [PubMed]
- Zahreddine, H.; Borden, K.L. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer. 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Billone, P.S.; Mullett, W.M. Nanomedicine in action: An overview of cancer nanomedicine on the market and in clinical trials. J. Nanomater. 2013, 2013, 1–12. [Google Scholar] [CrossRef]
- Sandhiya, S.; Dkhar, S.A.; Surendiran, A. Emerging trends of nanomedicine—An overview. Fund Clin. Pharmacol. 2009, 23, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Rink, J.S.; Plebanek, M.P.; Tripathy, S.; Thaxton, C.S. Update on current and potential nanoparticle cancer therapies. Curr. Opin. Oncol. 2013, 25, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M. Frontiers in cancer nanomedicine: Directing mass transport through biological barriers. Trends Biotechnol. 2010, 28, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Xiao, Z.; Valencia, P.M.; Radovic-Moreno, A.F.; Farokhzad, O.C. Targeted polymeric therapeutic nanoparticles: Design, development and clinical translation. Chem. Soc. Rev. 2012, 41, 2971–3010. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gu, F.; Chan, J.; Wang, A.; Langer, R.; Farokhzad, O. Nanoparticles in medicine: Therapeutic applications and developments. Clin. Pharmacol. Ther. 2008, 83, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y.C. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves abraxane for metastatic pancreatic cancer. Available online: http://journals.lww.com/oncology-times/blog/onlinefirst/pages/post.aspx?PostID=812 (accessed on 6 September 2013).
- Ryan, S.M.; Brayden, D.J. Progress in the delivery of nanoparticle constructs: Towards clinical translation. Curr. Opin. Pharmacol. 2014, 18, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Schütz, C.A.; Juillerat-Jeanneret, L.; Mueller, H.; Lynch, I.; Riediker, M. Therapeutic nanoparticles in clinics and under clinical evaluation. Nanomedicine 2013, 8, 449–467. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Stylianopoulos, T. Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Onconl. 2010, 7, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, H.; Akita, H.; Harashima, H. A multifunctional envelope type nano device (MEND) for gene delivery to tumours based on the EPR effect: A strategy for overcoming the PEG dilemma. Adv. Drug Deliv. Rev. 2011, 63, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, H.; Akita, H.; Harashima, H. The polyethyleneglycol dilemma: Advantage and disadvantage of PEGylation of liposomes for systemic genes and nucleic acids delivery to tumors. Biol. Pharm. Bull. 2013, 36, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, Q.; Peng, F.; Liu, L.; Gong, C. Strategies of polymeric nanoparticles for enhanced internalization in cancer therapy. Colloid Surf. B 2015, 135, 56–72. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Mohanty, C.; Sahoo, S.K. Ligand-based targeted therapy for cancer tissue. Expert Opin. Drug Deliv. 2009, 6, 285–304. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, Q.; Qiu, L. Smart ligand: Aptamer-mediated targeted delivery of chemotherapeutic drugs and siRNA for cancer therapy. J. Control. Release 2013, 171, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Trapani, G.; Denora, N.; Trapani, A.; Laquintana, V. Recent advances in ligand targeted therapy. J. Drug Target 2012, 20, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. BBA Rev. Cancer 2010, 1805, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Azzi, S.; Hebda, J.K.; Gavard, J. Vascular permeability and drug delivery in cancers. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.G.; Wei, W.; Lv, P.P.; Yue, H.; Wang, L.Y.; Su, Z.G.; Ma, G.H. Surface charge affects cellular uptake and intracellular trafficking of chitosan-based nanoparticles. Biomacromolecules 2011, 12, 2440–2446. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Stellacci, F. Effect of surface properties on nanoparticle—Cell interactions. Small 2010, 6, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Foged, C.; Brodin, B.; Frokjaer, S.; Sundblad, A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int. J. Pharm. 2005, 298, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Kovochich, M.; Liong, M.; Meng, H.; Kabehie, S.; George, S.; Zink, J.I.; Nel, A.E. Polyethyleneimine coating enhances the cellular uptake of mesoporous silica nanoparticles and allows safe delivery of siRNA and DNA constructs. ACS Nano 2009, 3, 3273–3286. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Mccrate, J.M.; Lee, J.C.; Li, H. The role of surface charge on the uptake and biocompatibility of hydroxyapatite nanoparticles with osteoblast cells. Nanotechnology 2011, 22. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Shi, W.; Freund, L.B. Mechanics of receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 2005, 102, 9469–9474. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.; Davies, P. Mechanisms of endocytosis and exocytosis. Symp. Soc. Exp. Biol. 1974, 28, 419–446. [Google Scholar] [PubMed]
- Sahay, G.; Alakhova, D.Y.; Kabanov, A.V. Endocytosis of nanomedicines. J. Control. Release 2010, 145, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Leroueil, P.R.; Hong, S.; Mecke, A.; Baker, J.R., Jr.; Orr, B.G.; Banaszak Holl, M.M. Nanoparticle interaction with biological membranes: Does nanotechnology present a Janus face? Acc. Chem. Res. 2007, 40, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.C.; Tang, C.H.; Fang, T.Y. The role of dextran conjugation in transfection mediated by dextran-grafted polyethylenimine. J. Gene Med. 2004, 6, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Arvizo, R.R.; Miranda, O.R.; Thompson, M.A.; Pabelick, C.M.; Bhattacharya, R.; Robertson, J.D.; Rotello, V.M.; Prakash, Y.; Mukherjee, P. Effect of nanoparticle surface charge at the plasma membrane and beyond. Nano Lett. 2010, 10, 2543–2548. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Zhao, Y.; Liu, Y.; Chang, X.; Chen, C.; Zhao, Y. Cellular uptake, intracellular trafficking, and cytotoxicity of nanomaterials. Small 2011, 7, 1322–1337. [Google Scholar] [CrossRef] [PubMed]
- Merhi, M.; Dombu, C.Y.; Brient, A.; Chang, J.; Platel, A.; Le Curieux, F.; Marzin, D.; Nesslany, F.; Betbeder, D. Study of serum interaction with a cationic nanoparticle: Implications for in vitro endocytosis, cytotoxicity and genotoxicity. Int. J. Pharm. 2012, 423, 37–44. [Google Scholar] [CrossRef] [PubMed]
- He, W.Y.; Zheng, X.; Zhao, Q.; Duan, L.J.; Lv, Q.; Gao, G.H.; Yu, S.J. pH-triggered charge-reversal polyurethane micelles for controlled release of doxorubicin. Macromol.Biosci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Van Kirk, E.A.; Zhan, Y.; Murdoch, W.J.; Radosz, M.; Shen, Y. targeted charge-reversal nanoparticles for nuclear drug delivery. Angew. Chem. Int. Ed. 2007, 46, 4999–5002. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Sun, T.; Song, W.; Wu, J.; Wang, J. A tumor acidity activated charge conversional nanogel as an intelligent vehicle for promoted tumoral cell uptake and drug delivery. Angew. Chem. Int. Ed. 2010, 122, 3703–3708. [Google Scholar] [CrossRef]
- Keeney, M.; Ong, S.G.; Padilla, A.; Yao, Z.; Goodman, S.; Wu, J.C.; Yang, F. Development of poly(β-amino ester)-based biodegradable nanoparticles for nonviral delivery of minicircle DNA. ACS Nano 2013, 7, 7241–7250. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Tang, H.; Zhan, Y.; van Kirk, E.A.; Murdoch, W.J. Degradable poly(β-amino ester) nanoparticles for cancer cytoplasmic drug delivery. Nanomed. Nanotechnol. 2009, 5, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhang, J.; Wang, L.; Wang, Y.; Chen, M. Tumor pHe-triggered charge-reversal and redox-responsive nanoparticles for docetaxel delivery in hepatocellular carcinoma treatment. Nanoscale 2015, 7, 15763–15779. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, T.; Wu, D.; Zhang, X.; Yan, J.; Du, S.; Guo, Y.; Wang, J.; Zhang, A. stimuli-responsive zwitterionic block copolypeptides: Poly(N-isopropylacrylamide)-block-poly(lysine-co-glutamic acid). Biomacromolecules 2008, 9, 2670–2676. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Tang, Z.; Zhang, X.; Yu, H.; Sun, H.; Pang, X.; Chen, X. pH-triggered charge-reversal polypeptide nanoparticles for cisplatin delivery: Preparation and in vitro evaluation. Biomacromolecules 2013, 14, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Fawell, S.; Seery, J.; Daikh, Y.; Moore, C.; Chen, L.L.; Pepinsky, B.; Barsoum, J. Tat-mediated delivery of heterologous proteins into cells. Proc. Natl. Acad. Sci. USA 1994, 91, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, C.; Plank, C.; Lausier, J.; Schillinger, U.; Müller, R.H.; Rosenecker, J. Oligomers of the arginine-rich motif of the HIV-1 TAT protein are capable of transferring plasmid DNA into cells. J. Biol. Chem. 2003, 278, 11411–11418. [Google Scholar] [CrossRef] [PubMed]
- Sethuraman, V.A.; Bae, Y.H. TAT peptide-based micelle system for potential active targeting of anti-cancer agents to acidic solid tumors. J. Control. Release 2007, 118, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Gao, Z.; Kim, D.; Park, K.; Kwon, I.C.; Bae, Y.H. Super pH-sensitive multifunctional polymeric micelle for tumor pHe specific TAT exposure and multidrug resistance. J. Control. Release 2008, 129, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gao, S.; Zhang, F.; Yang, K.; Ma, Q.; Zhu, L. Activatable hyaluronic acid nanoparticle as a theranostic agent for optical/photoacoustic image-guided photothermal therapy. ACS Nano 2014, 8, 12250–12258. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Jambhrunkar, S.; Thorn, P.; Chen, J.; Gu, W.; Yu, C. Hyaluronic acid modified mesoporous silica nanoparticles for targeted drug delivery to CD44-overexpressing cancer cells. Nanoscale 2013, 5, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; Min, K.H.; Na, J.H.; Choi, K.; Kim, K.; Park, J.H.; Kwon, I.C.; Jeong, S.Y. Self-assembled hyaluronic acid nanoparticles as a potential drug carrier for cancer therapy: Synthesis, characterization, and in vivo biodistribution. J. Mater. Chem. 2009, 19, 4102–4107. [Google Scholar] [CrossRef]
- He, M.; Zhao, Z.; Yin, L.; Tang, C.; Yin, C. Hyaluronic acid coated poly(butyl cyanoacrylate) nanoparticles as anticancer drug carriers. Int. J. Pharm. 2009, 373, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Lin, L.; Chen, J.; Chen, X.; Park, T.G.; Maruyama, A. RGD targeting hyaluronic acid coating system for PEI-PBLG polycation gene carriers. J. Control. Release 2011, 155, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.M.; Drevs, J.; Esser, N.; Hamada, F.M.; Badary, O.A.; Unger, C.; Fichtner, I.; Kratz, F. A new approach for the treatment of malignant melanoma: Enhanced antitumor efficacy of an albumin-binding doxorubicin prodrug that is cleaved by matrix metalloproteinase 2. Cancer Res. 2003, 63, 4062–4066. [Google Scholar] [PubMed]
- Chen, W.H.; Luo, G.F.; Lei, Q.; Jia, H.Z.; Hong, S.; Wang, Q.R.; Zhuo, R.X.; Zhang, X.Z. MMP-2 responsive polymeric micelles for cancer-targeted intracellular drug delivery. Chem. Commun. 2015, 51, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gemeinhart, R.A. Improving matrix metalloproteinase-2 specific response of a hydrogel system using electrophoresis. Int. J. Pharm. 2012, 429, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Kate, P.; Torchilin, V.P. Matrix metalloprotease 2-responsive multifunctional liposomal nanocarrier for enhanced tumor targeting. ACS Nano 2012, 6, 3491–3498. [Google Scholar] [CrossRef] [PubMed]
- Kuai, R.; Yuan, W.; Qin, Y.; Chen, H.; Tang, J.; Yuan, M.; Zhang, Z.; He, Q. Efficient delivery of payload into tumor cells in a controlled manner by TAT and thiolytic cleavable PEG co-modified liposomes. Mol. Pharm. 2010, 7, 1816–1826. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Lee, Y.; Kim, J.S.; Jeong, J.H.; Park, T.G. Thermally triggered cellular uptake of quantum dots immobilized with poly(N-isopropylacrylamide) and cell penetrating peptide. Langmuir 2010, 26, 14965–14969. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, Y.; Xie, X.; Cai, X.; Zhang, H.; Gong, W.; Wang, Z.; Mei, X. PEGylated liposomes with NGR ligand and heat-activable cell-penetrating peptide–doxorubicin conjugate for tumor-specific therapy. Biomaterials 2014, 35, 4368–4381. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.B.; van Gaal, E.; Minten, I.; Storm, G.; van Hest, J.C.; Löwik, D.W. Constrained and UV-activatable cell-penetrating peptides for intracellular delivery of liposomes. J. Control. Release 2012, 164, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wang, J.; Lin, Z.; Li, X.; Li, J. Tumor-acidity activated surface charge-conversion of polymeric nanocarriers for enhanced cell adhesion and targeted drug release. Macromol. Rapid Commun. 2014, 35, 1679–1684. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Li, Z.; Zhu, J.; Han, K.; Zeng, Z.; Hong, W.; Li, W.; Jia, H.; Liu, Y.; Zhuo, R. Dual-pH sensitive charge-reversal polypeptide micelles for tumor-triggered targeting uptake and nuclear drug delivery. Small 2015, 11, 2543–2554. [Google Scholar] [CrossRef] [PubMed]
- Mok, H.; Park, J.W.; Park, T.G. Enhanced intracellular delivery of quantum dot and adenovirus nanoparticles triggered by acidic pH via surface charge reversal. Bioconj. Chem. 2008, 19, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.Y.; Mao, C.Q.; Du, X.J.; Du, J.Z.; Wang, F.; Wang, J. Surface charge switchable nanoparticles based on zwitterionic polymer for enhanced drug delivery to tumor. Adv. Mater. 2012, 24, 5476–5480. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Du, X.; Mao, C.; Wang, J. Tailor-made dual pH-sensitive polymer–doxorubicin nanoparticles for efficient anticancer drug delivery. J. Am. Chem. Soc. 2011, 133, 17560–17563. [Google Scholar] [CrossRef] [PubMed]
- Oh, N.M.; Kwag, D.S.; Oh, K.T.; Youn, Y.S.; Lee, E.S. Electrostatic charge conversion processes in engineered tumor-identifying polypeptides for targeted chemotherapy. Biomaterials 2012, 33, 1884–1893. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.T.; Kim, D.; You, H.H.; Ahn, Y.S.; Lee, E.S. pH-Sensitive properties of surface charge-switched multifunctional polymeric micelle. Int. J. Pharm. 2009, 376, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Mo, R.; Sun, Q.; Xue, J.; Li, N.; Li, W.; Zhang, C.; Ping, Q. Multistage pH-responsive liposomes for mitochondrial-targeted anticancer drug delivery. Adv. Mater. 2012, 24, 3659–3665. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Zhang, G.; Liu, S. pH-Disintegrable Polyelectrolyte multilayer-coated mesoporous silica nanoparticles exhibiting triggered co-release of cisplatin and model drug molecules. Macromol. Rapid Commun. 2011, 32, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Guan, X.; Li, J.; Pei, Q.; Liu, M.; Xie, Z.; Jing, X. Hybrid polymer micelles capable of cRGD targeting and pH-triggered surface charge conversion for tumor selective accumulation and promoted uptake. Chem. Commun. 2014, 50, 9188–9191. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Gu, J.; Qu, X.; Yang, Z. Preparation of multifunctional drug carrier for tumor-specific uptake and enhanced intracellular delivery through the conjugation of weak acid labile linker. Bioconj. Chem. 2009, 20, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Cheng, W.P.; Liu, J.; Lo, S.Y.; Smith, D.; Qu, X.; Yang, Z. pH-triggered reversible “stealth” polycationic micelles. Biomacromolecules 2007, 9, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Zhao, X.; Liu, J.; Deng, L.; Zhang, J.; Liu, J.; Dong, A. Reactive oxygen species (ROS) responsive PEG-PCL nanoparticles with pH-controlled negative-to-positive charge reversal for intracellular delivery of doxorubicin. J. Mater. Chem. B 2015, 3, 9397–9408. [Google Scholar] [CrossRef]
- Hu, J.; Miura, S.; Na, K.; Bae, Y.H. pH-responsive and charge shielded cationic micelle of poly(l-histidine)-block-short branched PEI for acidic cancer treatment. J. Control. Release 2013, 172, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Sethuraman, V.A.; Lee, M.C.; Bae, Y.H. A biodegradable pH-sensitive micelle system for targeting acidic solid tumors. Pharm. Res. 2008, 25, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Koren, E.; Apte, A.; Jani, A.; Torchilin, V.P. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J. Control. Release 2012, 160, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Sun, W.; Zhong, J.; Yang, Q.; Zhu, X.; Zhou, Z.; Zhang, Z.; Huang, Y. multistage nanovehicle delivery system based on stepwise size reduction and charge reversal for programmed nuclear targeting of systemically administered anticancer drugs. Adv. Funct. Mater. 2015, 25, 4101–4113. [Google Scholar] [CrossRef]
- Ito, T.; Iida-Tanaka, N.; Koyama, Y. Efficient in vivo gene transfection by stable DNA/PEI complexes coated by hyaluronic acid. J. Drug Target 2008, 16, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Zhang, Z.; Zhang, Y.; Lv, H.; Zhou, J.; Li, C.; Hou, L.; Zhang, Q. Dual-functional liposomes based on pH-responsive cell-penetrating peptide and hyaluronic acid for tumor-targeted anticancer drug delivery. Biomaterials 2012, 33, 9246–9258. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.J.; von Maltzahn, G.; Lord, M.E.; Park, J.H.; Agrawal, A.; Min, D.H.; Sailor, M.J.; Bhatia, S.N. Protease-triggered unveiling of bioactive nanoparticles. Small 2008, 4, 1307–1312. [Google Scholar] [CrossRef] [PubMed]
- Mok, H.; Bae, K.H.; Ahn, C.H.; Park, T.G. PEGylated and MMP-2 specifically dePEGylated quantum dots: Comparative evaluation of cellular uptake. Langmuir 2008, 25, 1645–1650. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Fan, X.; Li, F.; Suo, R.; Li, H.; Yang, Z.; Zhang, W.; Bai, Y.; Tian, W. Thermo and pH dual-controlled charge reversal amphiphilic graft copolymer micelles for overcoming drug resistance in cancer cells. J. Mater. Chem. B 2015, 3, 4585–4596. [Google Scholar] [CrossRef]
- Lammers, T.; Kiessling, F.; Hennink, W.E.; Storm, G. Drug targeting to tumors: Principles, pitfalls and (pre-) clinical progress. J. Control. Release 2012, 161, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Ciani, L.; Ristori, S.; Bonechi, C.; Rossi, C.; Martini, G. Effect of the preparation procedure on the structural properties of oligonucleotide/cationic liposome complexes (lipoplexes) studied by electron spin resonance and Zeta potential. Biophys. Chem. 2007, 131, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Bernfield, M.; Götte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, C.; Billotey, C.; Roger, J.; Pons, J.; Bacri, J.C.; Gazeau, F. Intracellular uptake of anionic superparamagnetic nanoparticles as a function of their surface coating. Biomaterials 2003, 24, 1001–1011. [Google Scholar] [CrossRef]
- Shen, Y.; Zhou, Z.; Sui, M.; Tang, J.; Xu, P.; Kirk, E.A.V.; Murdoch, W.J.; Fan, M.; Radosz, M. Charge-reversal polyamidoamine dendrimer for cascade nuclear drug delivery. Nanomedicine 2010, 5, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.Q.; Chen, K.G.; Yu, X.Y.; Zhao, G.; Shen, S.; Cao, Z.T.; Luo, Y.L.; Wang, Y.C.; Wang, J. Promoting tumor penetration of nanoparticles for cancer stem cell therapy by TGF-β signaling pathway inhibition. Biomaterials 2016, 82, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.; Ng, V.W.; Gao, S.; Tan, M.H.; Hedrick, J.L.; Yang, Y.Y. Codelivery of dual drugs from polymeric micelles for simultaneous targeting of both cancer cells and cancer stem cells. Nanomedicine 2015, 10, 2819–2832. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhao, G.; Liu, J.; Ma, N.; Chivukula, P.; Perelman, L.; Okada, K.; Chen, Z.; Gough, D.; Yu, L. Novel biodegradable lipid nano complex for siRNA delivery significantly improving the chemosensitivity of human colon cancer stem cells to paclitaxel. J. Control. Release 2009, 140, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Issels, R.D. Hyperthermia adds to chemotherapy. Eur. J. Cancer 2008, 44, 2546–2554. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, L.R.; Stafford, R.; Bankson, J.; Sershen, S.; Rivera, B.; Price, R.; Hazle, J.; Halas, N.; West, J. Nanoshell-mediated near-infrared thermal therapy of tumors under magnetic resonance guidance. Proc. Natl. Acad. Sci. USA 2003, 100, 13549–13554. [Google Scholar] [CrossRef] [PubMed]
- Du, J.Z.; Mao, C.Q.; Yuan, Y.Y.; Yang, X.Z.; Wang, J. Tumor extracellular acidity-activated nanoparticles as drug delivery systems for enhanced cancer therapy. Biotechnol. Adv. 2014, 32, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.H.; Wu, S.H.; Yao, M.; Lu, C.W.; Lin, Y.S.; Hung, Y.; Mou, C.Y.; Chen, Y.C.; Huang, D.M. The effect of surface charge on the uptake and biological function of mesoporous silica nanoparticles in 3T3-L1 cells and human mesenchymal stem cells. Biomaterials 2007, 28, 2959–2966. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Shen, J.; Zhang, Z.; Yu, H.; Li, Y. Reversal of multidrug resistance by stimuli-responsive drug delivery systems for therapy of tumor. Adv. Drug Deliv. Rev. 2013, 65, 1699–1715. [Google Scholar] [CrossRef] [PubMed]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Palakurthi, S.; Yellepeddi, V.K.; Vangara, K.K. Recent trends in cancer drug resistance reversal strategies using nanoparticles. Expert Opin. Drug Deliv. 2012, 9, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, D.; List, A.F. Targeting the multidrug resistance-1 transporter in AML: Molecular regulation and therapeutic strategies. Blood 2004, 104, 1940–1951. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.R.; Pattni, B.S.; Abouzeid, A.H.; Torchilin, V.P. Nanopreparations to overcome multidrug resistance in cancer. Adv. Drug Deliv. Rev. 2013, 65, 1748–1762. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.K. Recent advances in nanooncology. Technol. Cancer Res. T 2008, 7, 1–13. [Google Scholar] [CrossRef]
- Hauck, M.L.; LaRue, S.M.; Petros, W.P.; Poulson, J.M.; Yu, D.; Spasojevic, I.; Pruitt, A.F.; Klein, A.; Case, B.; Thrall, D.E. Phase I trial of doxorubicin-containing low temperature sensitive liposomes in spontaneous canine tumors. Clin. Cancer Res. 2006, 12, 4004–4010. [Google Scholar] [CrossRef] [PubMed]
- Vives, E.; Richard, J.; Rispal, C.; Lebleu, B. TAT peptide internalization: Seeking the mechanism of entry. Curr. Protein Pept. Sci. 2003, 4, 125–132. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
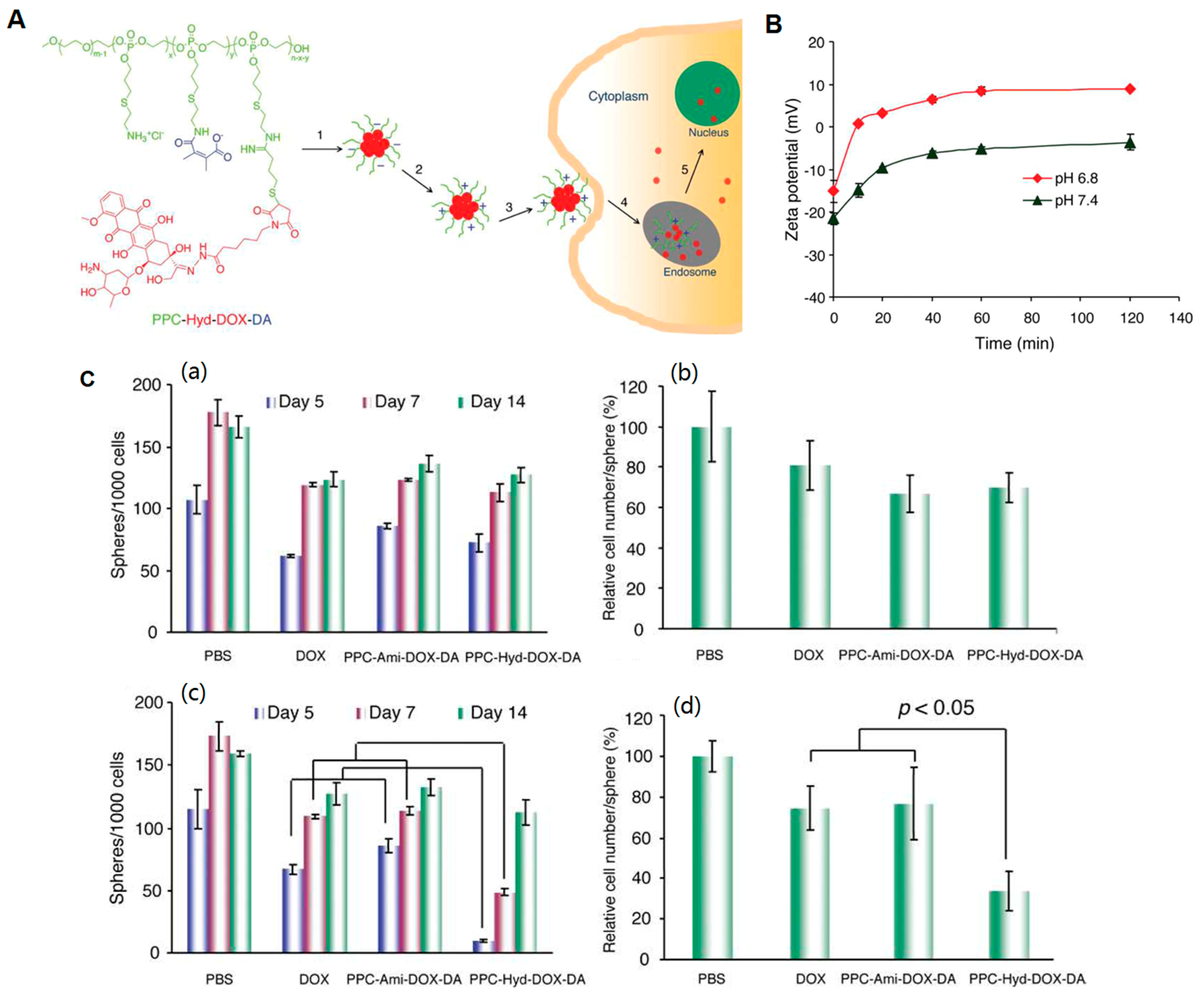{kind=link}
| Activation mechanism | Polymers | Reversal approach | Nano-carriers | Cargo | Test cells | References |
|---|---|---|---|---|---|---|
| pH-sensitive | polycaprolactone-block-PEI/amide-folic acid | 20% of its primary and secondary amines in PEI converted into their amides | NPs | Doxorubicin | SKOV3 | [48] |
| d-α-tocopheryl polyethylene glycol 1000-poly-(β-amino ester) block copolymer containing disulfide linkages | Tertiary amine activated in acidic environment | NPs | Docetaxel | HepG2 and SMCC 7721 | [52] | |
| poly(l-glutamic acid-co-l-lysine) | Manipulating the feed ratio of l-glutamic acid/l-lysine ratio | NPs | Cisplatin | HeLa | [54] | |
| poly(d,l-lactide)-block-poly(2-aminoethyl methacrylate)/DMMA | β-carboxylic amide produced by the reaction between amino on PAEMA and DMMA | Micelles | Doxorubicin | Hela | [72] | |
| poly(l-leucine)-block-Poly(l-lysine)/DMMA-TAT/succinyl chloride | β-carboxylic amide produced by the reaction between lysine amino and DMMA; TAT peptide masked by succinyl chloride | Micelles | Doxorubicin | Hela | [73] | |
| poly(2-aminoethyl methacrylate hydrochloride)-DMMA | Amides produced by the reaction between amino groups and DMMA | Nanogel | Doxorubicin | MDA-MB-435s | [49] | |
| biotin-poly(ethylene glycol) grafted poly(l-lysine) | Primary amine groups in the PLL backbone postmodified by citraconic anhydride | NPs | Quantum dot; Adenovirus | _ | [74] | |
| poly(ε-caprolactone)-block-poly(allyl ethylene phosphate)-graft-2-(mercaptoethyl) trimethylammonium chloride/DMMA | pH-triggered amide bond | NPs | Doxorubicin | MDA-MB-231 | [75] | |
| poly(ethylene glycol)-b-poly(allyl ethylene phosphate)/cysteamine-Hyd-DOX/DMMA | pH-triggered amide bond | Polymer-drug conjugate NPs | Doxorubicin | MDA-MB-231 | [76] | |
| poly(l-aspartic acid)/DMMA | pH-triggered amide bond | Polypeptide NPs | Chlorin e6 | B16F10, KB, MCF-7, A549, HeLa | [77] | |
| poly(l-lactic acid)-b-poly(ethylene glycol)-b-poly(l-lysine-Nε-(2,3-dimethyl maleic acid)) | pH-triggered amide bond | Micelles | Doxorubicin | _ | [78] | |
| 1,5-dioctadecyl-l-glutamy-l,2-histidyl-hexahydrobenzoic acid | hexahydrobenzoic amide in HHG2C18 | Liposomes | Temsirolimus | A498 | [79] | |
| P(DMA-co-TPAMA)/PAH polyelectrolyte multilayers | The mixture of cationic and negatively charged polyelectrolyte | Polyelectrolyte -coated mesoporous silica NPs | Cisplatin; Rhodamine B | _ | [80] | |
| c(RGDfK)-PEG-b-PLA/N-(3-aminopropyl)-imidazole -PEG-b-PLA | Imidazole (pKa = ~ 6.8) could achieve the transformation of protonation-deprotonation | Mixed micelles | Doxorubicin | EMT6 | [81] | |
| PEG-b-C18 | pH-dependent hydrolysis of benzoic-imine bond | Micelles | Doxorubicin | HepG2 | [82] | |
| PEG-benzoic imine-poly(l-lysine)-cholic acid | pH-dependent hydrolysis of benzoic-imine bond | Micelles | _ | Caco-2 | [83] | |
| mPEG-ros-P(CL-co-DCL) | ROS sensitive thioether linkage; acid-labile β-carboxylic amides | NPs | Doxorubicin | HepG2 and L02 cell | [84] | |
| mPEG2k-b-polysulfadimethoxine4k/poly(l-histidine)3.7k-polyethyleneimine1.8k | charge shielding/deshielding | Mixed micelles | Paclitaxel | MCF-7;SKOV-3 | [85] | |
| TAT-PEG-PLLA/poly(methacryloyl sulfadimethoxine) (PSD)-b-PEG | shielding/deshielding transition of TAT peptide | Mixed micelles | Doxorubicin | MCF-7 | [57] | |
| poly(l-cystine bisamide-g-sulfadiazine)(PCBS)-b-PEG-TAT | shielding/deshielding transition of TAT peptide | Micelles | Doxorubicin | MCF-7 | [86] | |
| mAb 2C5-PEG3.4k-PE/PEG2k-Hz-PE/TAT-PEG1k-PE | Deshielded TAT peptide by the clearance of pH-sensitive bond | Liposome | Doxorubicin | B16-F10, HeLa, MCF-7 | [87] | |
| PLA3k-b-PEG2k-b-poly His2k-TAT/polyHis5k-b-PEG3.4k | pH-triggered “pop-up” of TAT | Micelles | Doxorubicin | A2780/Dox R | [58] | |
| DMA-N-(2-hydroxypropyl) methacrylamide/N-(2-hydroxypropyl) methacrylamide-DoxR8 peptide | CPP R8 exposed by the clearance of DMA in acid environment | Polymer-drug conjugate NPs | Doxorubicin | Hela | [88] | |
| Enzyme-sensitive | RGD-Hyaluronic acid coating polyethylenimine-poly(γ-benzyl l-glutamate) | Hyaluronic acid is used to shield the positive charges of PEI | Multiple layer NPs | DNA | Hela | [63] |
| Polyethylenimine/Hyaluronic acid/Plasmid DNA | Hyaluronic acid is used to shield the positive charges of PEI | Ternary nano-complexes | Plasmid DNA | B16 | [89] | |
| Hyaluronic acid coating R6H4 liposome | Hyaluronic acid is used to shield the positive charges of R6H4 | Multiple layer liposome | Paclitaxel | HepG2 | [90] | |
| Long-chain PEG-MMP2 substrate peptide-dextran-coated iron oxide | MMP2-responsive PEG | Multiple layer NPs | _ | HT-1080 | [91] | |
| PEG-MMP2 substrate peptide-QD | MMP2-responsive peptide linker | Multiple layer NPs | QDs | MDA-MB-435 | [92] | |
| mAb 2C5-PEG3.4k-MMP2 peptide-DOPE/TAT-PEG2k-DSPE | MMP2-responsive PEG layer | Liposomes | _ | 4T1, H9C2 | [67] | |
| Redox-sensitive | DOPE-S-S-mPEG5k/DOPE-PEG1.6k-TAT | Shielded TAT could be exposed in higher GSH concentration | Liposomes | Calcein | HepG2 | [68] |
| Thermo-sensitive | poly (N-iso-propylacrylamide) (PNIPAAm) | thermosensitive characteristics of PNIPAAm chain length to activate CPPs | Liposome | QDs | MDA-MB-435 | [69] |
| CPP-Dox/NGR-TSL | CPPs would be activated via heat stimulus | Liposome | Doxorubicin | HT-1080, MCF-7 | [70] | |
| UV-sensitive | 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimido(polyethylene glycol)-2000/1,2-distearoyl-SN-glycero-3-phospho-ethanolamine-N-[methoxy(polyethylene glycol)-2000 | Reactivation of the peptide can be accomplished by releasing the constrain via UV-irradiation | Liposome | _ | Hela | [71] |
| Thermo and pH dual-sensitive | poly(styrene-co-maleic anhydride)-graft-poly(2-(N,N-dimethylamino)ethyl methacrylate | (+) at 25 °C and pH 7.4; → (−) at 37 °C and pH 7.4; → (+) at 37 C and pH 6.8 | Micelles | Doxorubicin | A2780/Dox R | [93] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Y.; Gong, X.; Zhang, J.; Chen, F.; Fu, C.; Li, P.; Zou, L.; Zhao, G. Activated Charge-Reversal Polymeric Nano-System: The Promising Strategy in Drug Delivery for Cancer Therapy. Polymers 2016, 8, 99. https://doi.org/10.3390/polym8040099
Hu Y, Gong X, Zhang J, Chen F, Fu C, Li P, Zou L, Zhao G. Activated Charge-Reversal Polymeric Nano-System: The Promising Strategy in Drug Delivery for Cancer Therapy. Polymers. 2016; 8(4):99. https://doi.org/10.3390/polym8040099
Chicago/Turabian StyleHu, Yichen, Xiao Gong, Jinming Zhang, Fengqian Chen, Chaomei Fu, Peng Li, Liang Zou, and Gang Zhao. 2016. "Activated Charge-Reversal Polymeric Nano-System: The Promising Strategy in Drug Delivery for Cancer Therapy" Polymers 8, no. 4: 99. https://doi.org/10.3390/polym8040099