
Dilute Semiflexible Polymers with Attraction: Collapse, Folding and Aggregation


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Off-Lattice Polymer Models with Attractive Interaction
3. Monte Carlo Simulation and Analysis Methods
3.1. Generalized-Ensemble Methods: Flat Histogram
3.2. Generalized-Ensemble Methods: Locally-Confined Histograms
3.3. Reweighting from Generalized Ensembles
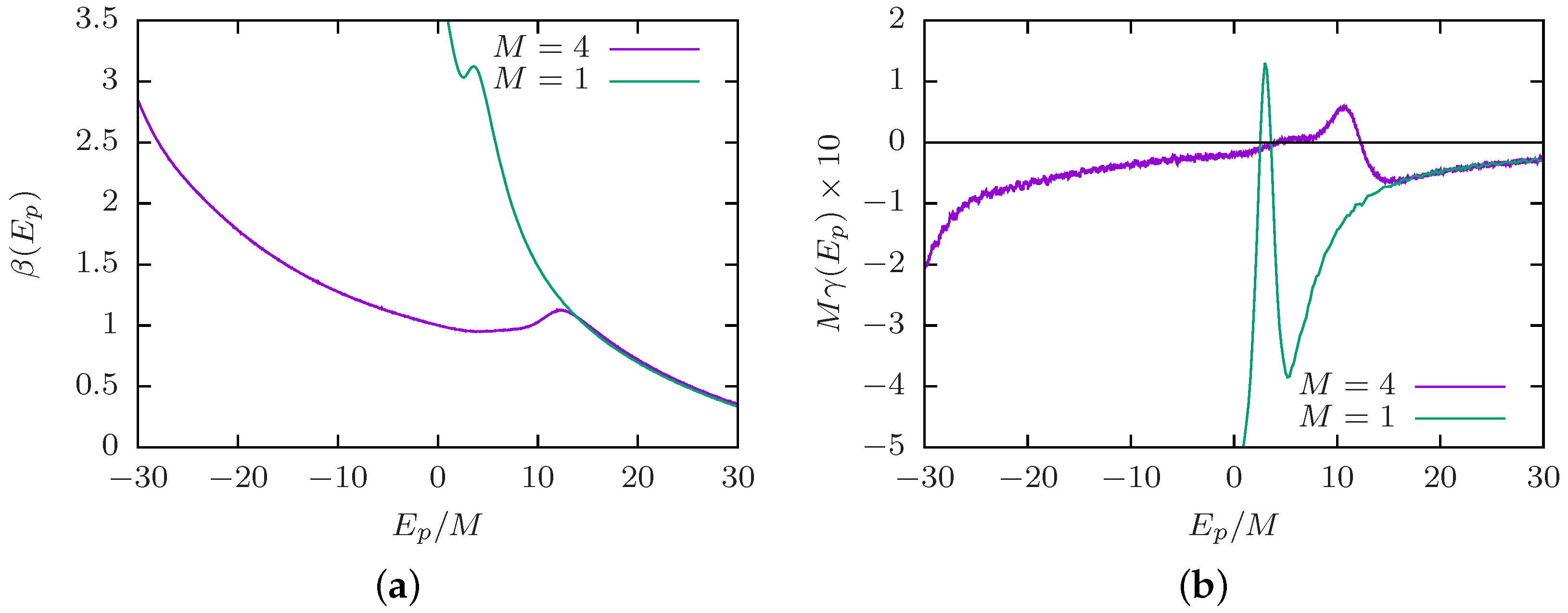
3.4. Canonical and Microcanonical Analysis
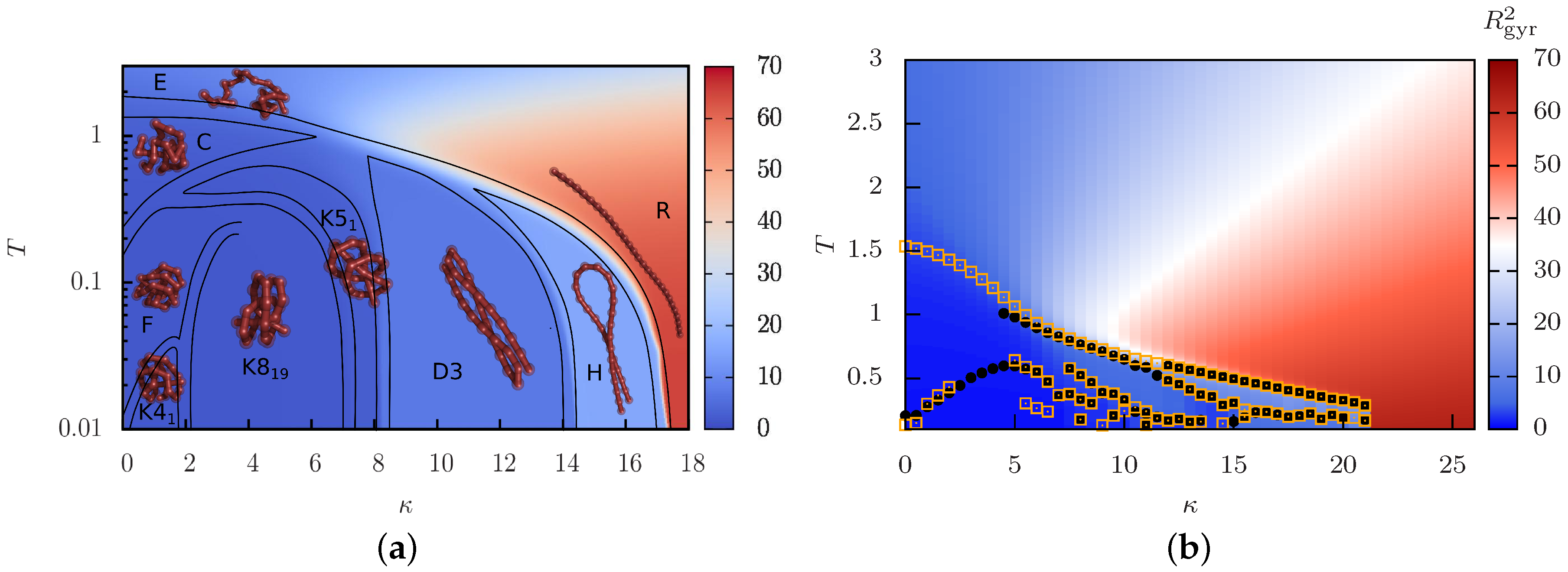
4. Phase Behavior of Isolated Semiflexible Polymers
4.1. Structural Phase Diagram
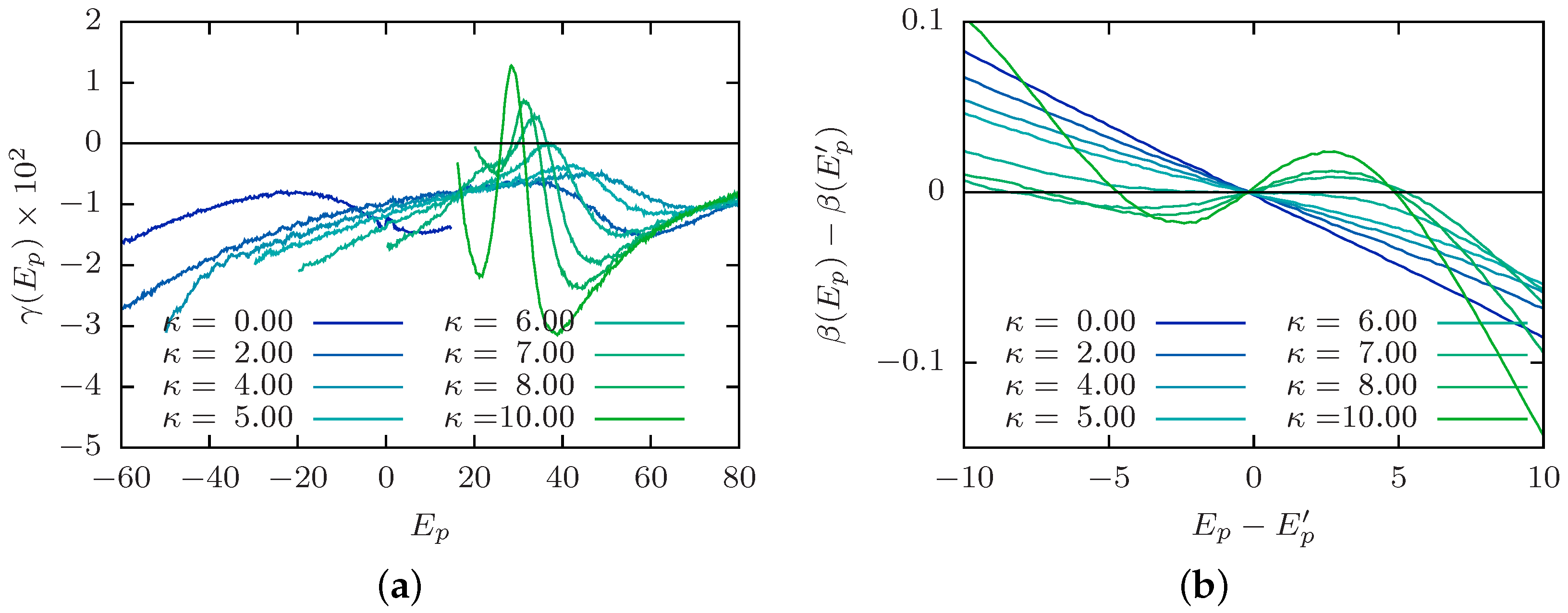
4.2. Order of the Collapse Transition Line
4.3. Knots as Stable Phase
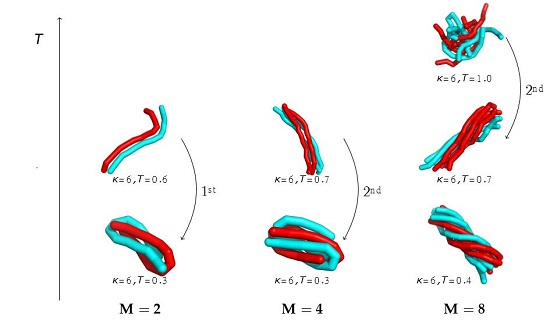
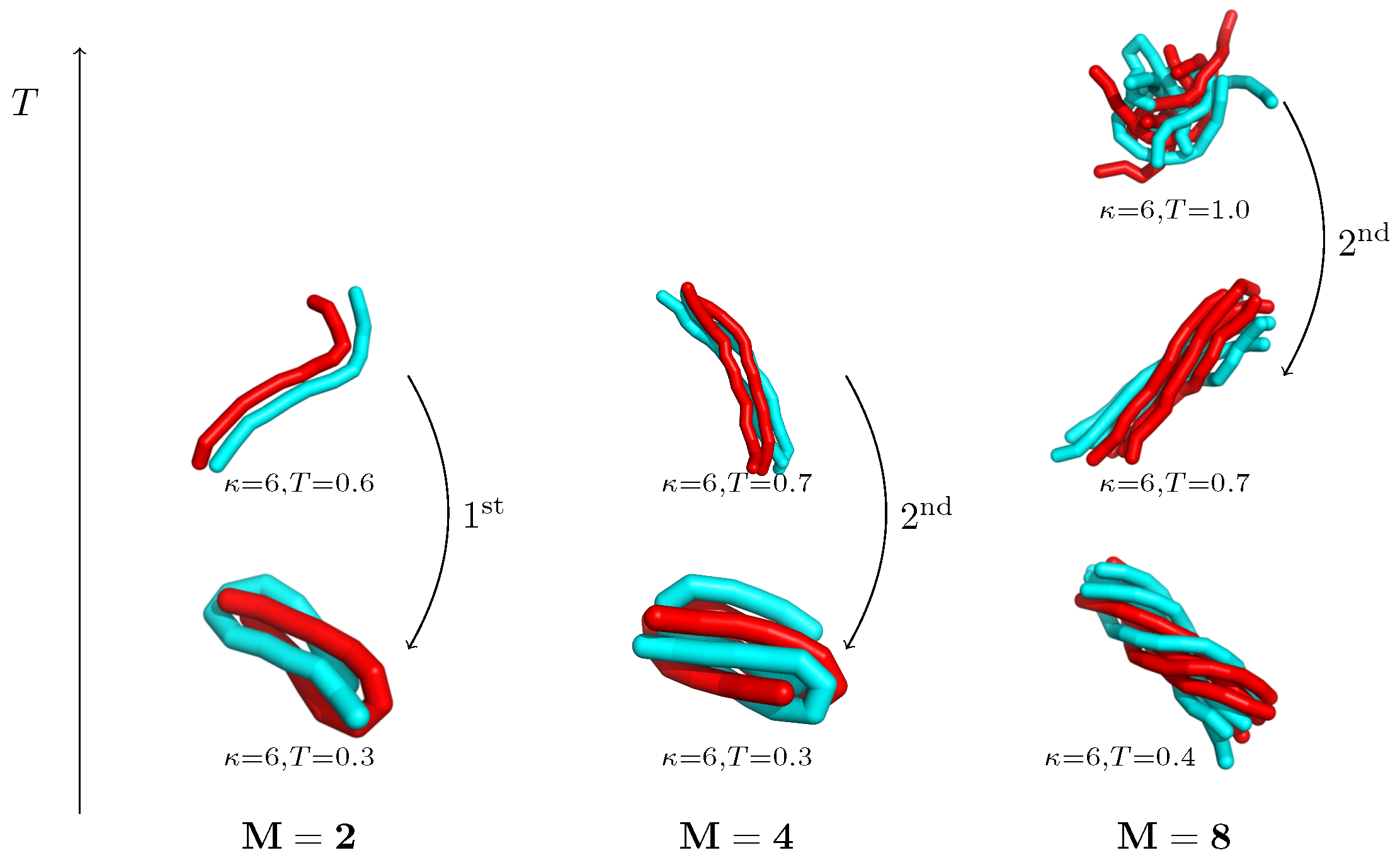
5. Aggregation of Dilute Semiflexible Polymers
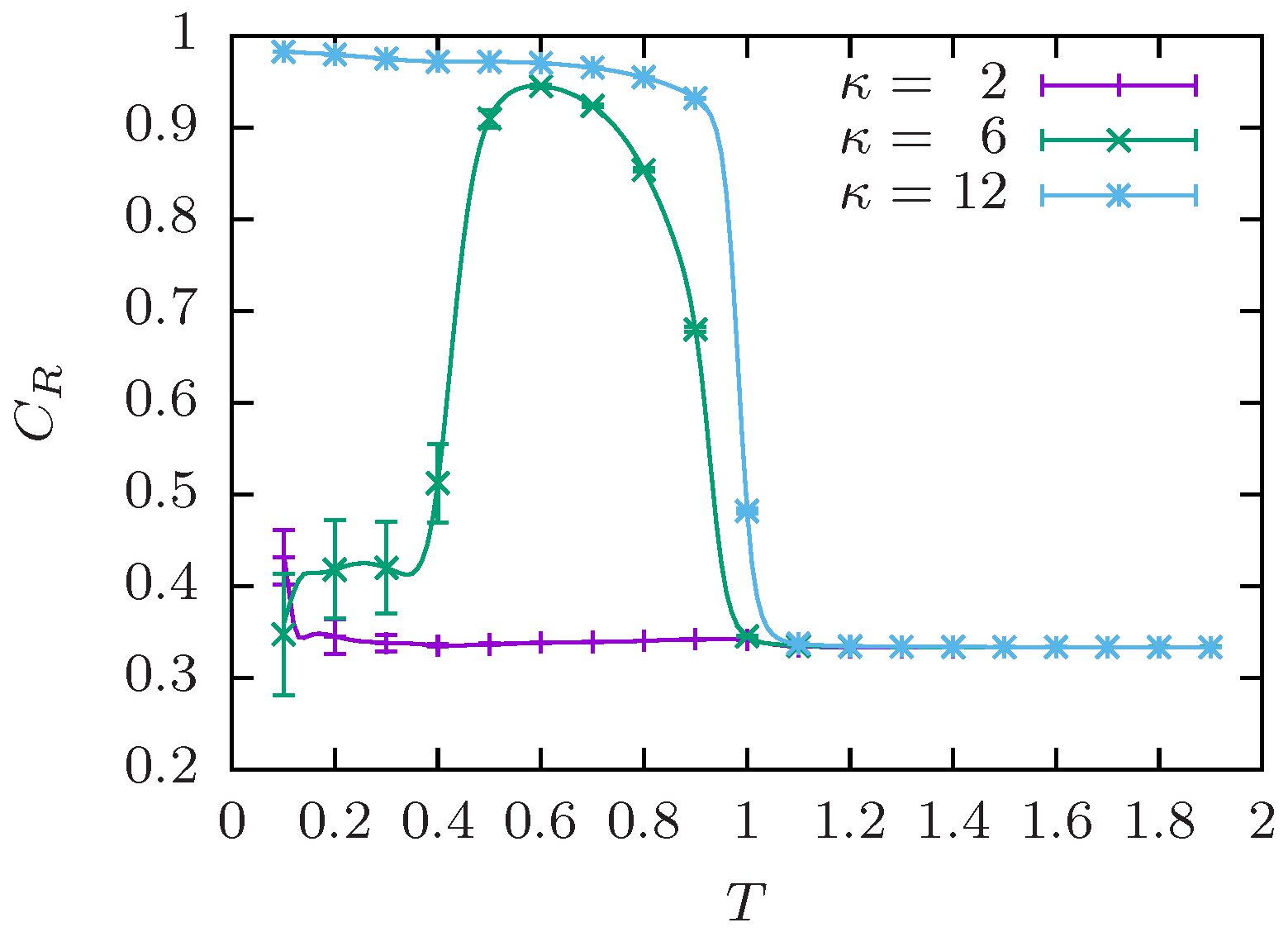
5.1. End-to-End Order Parameter
5.2. Structural Motifs Induced by Semiflexibility
5.3. Competition between Single-Chain Collapse and Many-Chain Aggregation
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- De Gennes, P.G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon Press: Oxford, UK, 1986. [Google Scholar]
- Des Cloizeaux, J.; Jannink, G. Polymers in Solution; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Clarendon Press: Oxford, UK, 1987. [Google Scholar]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications, 2nd ed.; Academic Press: New York, NY, USA, 2001. [Google Scholar]
- Rapaport, D.C. The Art of Molecular Dynamics Simulations, 2nd ed.; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Newman, M.E.J.; Barkema, G.T. Monte Carlo Methods in Statistical Physics; Clarendon Press: Oxford, UK, 1999. [Google Scholar]
- Landau, D.P.; Binder, K. Monte Carlo Simulations in Statistical Physics; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Berg, B.A. Markov Chain Monte Carlo Simulations and Their Statistical Analysis; World Scientific: Singapore, 2004. [Google Scholar]
- Janke, W. Monte Carlo simulations in statistical physics—From basic principles to advanced applications. In Order, Disorder and Criticality: Advanced Problems of Phase Transition Theory; Holovatch, Y., Ed.; World Scientific: Singapore, 2012; Volume 3, pp. 93–166. [Google Scholar]
- Baschnagel, J.; Meyer, H.; Wittmer, J.; Kulić, I.; Mohrbach, H.; Ziebert, F.; Lee, N.-K.; Nam, G.-M.; Johner, A. Semiflexible chains at surfaces: Worm-like chains and beyond. Polymers 2016, 8, 286. [Google Scholar] [CrossRef]
- Broedersz, C.P.; MacKintosh, F.C. Modeling semiflexible polymer networks. Rev. Mod. Phys. 2014, 86, 995–1036. [Google Scholar] [CrossRef]
- Karatrantos, A.; Clarke, N.; Kröger, M. Modeling of polymer structure and conformations in polymer nanocomposites from atomistic to mesoscale: A Review. Polym. Rev. 2016, 56, 385–428. [Google Scholar] [CrossRef]
- Vanderzande, C. Lattice Models of Polymers; Cambridge Lecture Notes in Physics; Cambridge University Press: Cambridge, UK, 1998; Volume 11. [Google Scholar]
- Carmesin, I.; Kremer, K. The bond fluctuation method: A new effective algorithm for the dynamics of polymers in all spatial dimensions. Macromolecules 1988, 21, 2819–2923. [Google Scholar] [CrossRef]
- Kremer, K.; Binder, K. Monte Carlo simulation of lattice models for macromolecules. Comp. Phys. Rep. 1988, 7, 259–310. [Google Scholar] [CrossRef]
- Kratky, O.; Porod, G. Diffuse small-angle scattering of X-rays in colloid systems. J. Colloid Sci. 1949, 4, 35–70. [Google Scholar] [CrossRef]
- Milchev, A.; Paul, W.; Binder, K. Off-lattice Monte Carlo simulation of dilute and concentrated polymer solutions under theta conditions. J. Chem. Phys. 1993, 99, 4786–4798. [Google Scholar] [CrossRef]
- Milchev, A.; Bhattacharya, A.; Binder, K. Formation of block copolymer micelles in solution: A Monte Carlo study of chain length dependence. Macromolecules 2001, 34, 1881–1893. [Google Scholar] [CrossRef]
- Kremer, K.; Grest, G.S. Dynamics of entangled linear polymer melts: A molecular-dynamics simulation. J. Chem. Phys. 1990, 92, 5057–5086. [Google Scholar] [CrossRef]
- Schnabel, S.; Bachmann, M.; Janke, W. Elastic Lennard–Jones polymers meet clusters: Differences and similarities. J. Chem. Phys. 2009, 131, 124904. [Google Scholar] [CrossRef] [PubMed]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- Swendsen, R.H.; Wang, J.-S. Replica Monte Carlo simulation of spin-glasses. Phys. Rev. Lett. 1986, 57, 2607–2609. [Google Scholar] [CrossRef] [PubMed]
- Geyer, C.J. Markov chain Monte Carlo maximum likelihood. In Computing Science and Statistics, Proceedings of the 23rd Symposium on the Interface, Seattle, WA, USA, 21–24 April 1991; Keramidas, E.M., Ed.; Interface Foundation: Fairfax Station, VA, USA, 1991; pp. 156–163. [Google Scholar]
- Hukushima, K.; Nemoto, K. Exchange Monte Carlo method and application to spin glass simulations. J. Phys. Soc. Jpn. 1996, 65, 1604–1608. [Google Scholar] [CrossRef]
- Hansmann, U.H.E. Parallel tempering algorithm for conformational studies of biological molecules. Chem. Phys. Lett. 1997, 281, 140–150. [Google Scholar] [CrossRef]
- Hansmann, U.H.E.; Okamoto, Y.; Eisenmenger, F. Molecular dynamics, Langevin and hydrid Monte Carlo simulations in a multicanonical ensemble. Chem. Phys. Lett. 1996, 259, 321–330. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Straub, J.E.; Keyes, T. Statistical-temperature Monte Carlo and molecular dynamics algorithms. Phys. Rev. Lett. 2006, 97, 050601. [Google Scholar] [CrossRef] [PubMed]
- Junghans, C.; Perez, D.; Vogel, T. Molecular dynamics in the multicanonical ensemble: Equivalence of Wang-Landau sampling, statistical temperature molecular dynamics, and metadynamics. J. Chem. Theory Comput. 2014, 10, 1843–1847. [Google Scholar] [CrossRef] [PubMed]
- Lal, M. Monte Carlo computer simulations of chain molecules. I. Mol. Phys. 1969, 17, 57–64. [Google Scholar] [CrossRef]
- Madras, N.; Sokal, A.D. The pivot algorithm: A highly efficient Monte Carlo method for the self-avoiding walk. J. Stat. Phys. 1988, 50, 109–186. [Google Scholar] [CrossRef]
- Bachmann, M.; Arkın, H.; Janke, W. Multicanonical study of coarse-grained off-lattice models for folding heteropolymers. Phys. Rev. E 2005, 71, 031906. [Google Scholar] [CrossRef] [PubMed]
- Baschnagel, J.; Wittmer, J.P.; Meyer, H. Monte Carlo simulation of polymers: Coarse-grained models. In Computational Soft Matter: From Synthetic Polymers to Proteins, Lecture notes of the Winter School, Bonn, Germany, 29 February–6 March 2004; Attig, N., Binder, K., Grubmüller, H., Kremer, K., Eds.; 2004; Volume 23, pp. 83–140. [Google Scholar]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comp. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Berg, B.A.; Neuhaus, T. Multicanonical algorithms for first order phase transitions. Phys. Lett. B 1991, 267, 249–253. [Google Scholar] [CrossRef]
- Berg, B.A.; Neuhaus, T. Multicanonical ensemble: A new approach to simulate first-order phase transitions. Phys. Rev. Lett. 1992, 68, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Janke, W. Multicanonical simulation of the two-dimensional 7-state potts model. Int. J. Mod. Phys. C 1992, 3, 1137–1146. [Google Scholar] [CrossRef]
- Janke, W. Multicanonical Monte Carlo simulations. Physica A 1998, 254, 164–178. [Google Scholar] [CrossRef]
- Wang, F.; Landau, D.P. Efficient, multiple-range random walk algorithm to calculate the density of states. Phys. Rev. Lett. 2001, 86, 2050–2053. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Landau, D.P. Determining the density of states for classical statistical models: A random walk algorithm to produce a flat histogram. Phys. Rev. E 2001, 64, 056101. [Google Scholar] [CrossRef] [PubMed]
- Liang, F. A theory on flat histogram Monte Carlo algorithms. J. Stat. Phys. 2006, 122, 511–529. [Google Scholar] [CrossRef]
- Liang, F.; Liu, C.; Carroll, R.J. Stochastic approximation in Monte Carlo computation. J. Am. Stat. Assoc. 2007, 102, 305–320. [Google Scholar] [CrossRef]
- Belardinelli, R.E.; Pereyra, V.D. Fast algorithm to calculate density of states. Phys. Rev. E 2007, 75, 046701. [Google Scholar] [CrossRef] [PubMed]
- Janke, W.; Paul, W. Thermodynamics and structure of macromolecules from flat-histogram Monte Carlo simulations. Soft Matter 2016, 12, 642–657. [Google Scholar] [CrossRef] [PubMed]
- Zierenberg, J.; Marenz, M.; Janke, W. Scaling properties of a parallel implementation of the multicanonical algorithm. Comput. Phys. Commun. 2013, 184, 1155–1160. [Google Scholar] [CrossRef]
- Vogel, T.; Li, Y.W.; Wüst, T.; Landau, D.P. A generic, hierarchical framework for massively parallel Wang-Landau sampling. Phys. Rev. Lett. 2013, 110, 210603. [Google Scholar] [CrossRef] [PubMed]
- Belardinelli, R.E.; Pereyra, D.V. Nonconvergence of the Wang-Landau algorithms with multiple random walkers. Phys. Rev. E 2016, 93, 053306. [Google Scholar] [CrossRef] [PubMed]
- Martin-Mayor, V. Microcanonical approach to the simulation of first-order phase transitions. Phys. Rev. Lett. 2007, 98, 137207. [Google Scholar] [CrossRef] [PubMed]
- Schierz, P.; Zierenberg, J.; Janke, W. Molecular dynamics and Monte Carlo simulations in the microcanonical ensemble: Quantitative comparison and reweighting techniques. J. Chem. Phys. 2015, 143, 134114. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, T.; Hager, J.S. Free-energy calculations with multiple Gaussian modified ensembles. Phys. Rev. E 2006, 74, 036702. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Keyes, T.; Straub, J.E. Generalized replica exchange method. J. Chem. Phys. 2010, 132, 224107. [Google Scholar] [CrossRef] [PubMed]
- Schierz, P.; Zierenberg, J.; Janke, W. First-order phase transitions in the real microcanonical ensemble. Phys. Rev. E 2016, 94, 021301. [Google Scholar] [CrossRef]
- Ferrenberg, A.M.; Swendsen, R.H. Optimized Monte Carlo data analysis. Phys. Rev. Lett. 1989, 63, 1195–1198. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Kim, J.; Keyes, T.; Straub, J.E. Communication: Iteration-free, weighted histogram analysis method in terms of intensive variables. J. Chem. Phys. 2011, 135, 061103. [Google Scholar] [CrossRef] [PubMed]
- Efron, B. The Jackknife, the Bootstrap and Other Resampling Plans; Society for Industrial and Applied Mathematics: Philadelphia, PA, USA, 1982. [Google Scholar]
- Efron, B.; Tibshirani, R.J. An Introduction to the Bootstrap; Springer Science+Business Media: Dordrecht, The Netherlands, 1994. [Google Scholar]
- Gross, D.H.E. Microcanonical Thermodynamics; World Scientific: Singapore, 2001. [Google Scholar]
- Janke, W. Canonical versus microcanonical analysis of first-order phase transitions. Nucl. Phys. B (Proc. Suppl.) 1998, 63, 631–633. [Google Scholar] [CrossRef]
- Junghans, C.; Bachmann, M.; Janke, W. Microcanonical analyses of peptide aggregation processes. Phys. Rev. Lett. 2006, 97, 218103. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, S.; Seaton, D.T.; Landau, D.P.; Bachmann, M. Microcanonical entropy inflection points: Key to systematic understanding of transitions in finite systems. Phys. Rev. E 2011, 84, 011127. [Google Scholar] [CrossRef] [PubMed]
- Zierenberg, J.; Schierz, P.; Janke, W. Canonical free-energy barrier of droplet formation. Available online: http://arxiv.org/abs/1607.08355 (accessed on 19 August 2016).
- Maritan, A.; Micheletti, C.; Trovato, A.; Banavar, J.R. Optimal shapes of compact strings. Nature 2000, 406, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Banavar, J.R.; Maritan, A. Colloquium: Geometrical approach to protein folding: A tube picture. Rev. Mod. Phys. 2003, 75, 23–34. [Google Scholar] [CrossRef]
- Auer, S.; Miller, M.A.; Krivov, S.V.; Dobson, C.M.; Karplus, M.; Vendruscolo, M. Importance of metastable states in the free energy landscapes of polypeptide chains. Phys. Rev. Lett. 2007, 99, 178104. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, O.; Maddocks, J.H. Global curvature, thickness, and the ideal shapes of knots. Proc. Natl. Acad. Sci. USA 1999, 96, 4769–4773. [Google Scholar] [CrossRef] [PubMed]
- Vogel, T.; Neuhaus, T.; Bachmann, M.; Janke, W. Thickness-dependent secondary structure formation of tubelike polymers. Europhys. Lett. 2009, 85, 10003. [Google Scholar] [CrossRef]
- Vogel, T.; Neuhaus, T.; Bachmann, M.; Janke, W. Thermodynamics of tubelike flexible polymers. Phys. Rev. E 2009, 80, 011802. [Google Scholar] [CrossRef] [PubMed]
- Vogel, T.; Neuhaus, T.; Bachmann, M.; Janke, W. Ground-state properties of tubelike flexible polymers. Eur. Phys. J. E 2009, 30, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, E.; Varilly, P.; Knowles, T.P.J.; Frenkel, D.; Abeln, S. Consistent treatment of hydrophobicity in protein lattice models accounts for cold denaturation. Phys. Rev. Lett. 2016, 116, 078101. [Google Scholar] [CrossRef] [PubMed]
- Koniaris, K.; Muthukumar, M. Knottedness in ring polymers. Phys. Rev. Lett. 1991, 66, 2211–2214. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, T.; Tsurusaki, K. Universality of random knotting. Phys. Rev. E 1997, 55, 6245–6248. [Google Scholar] [CrossRef]
- Virnau, P.; Kantor, Y.; Kardar, M. Knots in globule and coil phases of a model polyethylene. J. Am. Chem. Soc. 2005, 127, 15102–15106. [Google Scholar] [CrossRef] [PubMed]
- Lua, R.; Borovinskiy, A.L.; Grosberg, A.Y. Fractal and statistical properties of large compact polymers: A computational study. Polymer 2004, 45, 717–731. [Google Scholar] [CrossRef]
- Frank-Kamenetskii, M.; Lukashin, A.; Vologodskii, A. Statistical mechanics and topology of polymer chains. Nature 1975, 258, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, M.L. Are there knots in proteins? Nat. Struct. Biol. 1994, 1, 213–214. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.R. A deeply knotted protein structure and how it might fold. Nature 2000, 406, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Lua, R.C.; Grosberg, A.Y. Statistics of knots, geometry of conformations, and evolution of proteins. PLoS Comput. Biol. 2006, 2, e45. [Google Scholar] [CrossRef] [PubMed]
- Virnau, P.; Mirny, L.A.; Kardar, M. Intricate knots in proteins: Function and evolution. PLoS Comput. Biol. 2006, 2, e122. [Google Scholar] [CrossRef] [PubMed]
- Jamroz, M.; Niemyska, W.; Rawdon, E.J.; Stasiak, A.; Millett, K.C.; Sułkowski, P.; Sulkowska, J.I. KnotProt: A database of proteins with knots and slipknots. Nucleic Acids Res. 2015, 43, D306. [Google Scholar] [CrossRef] [PubMed]
- Wüst, T.; Reith, D.; Virnau, P. Sequence determines degree of knottedness in a coarse-grained protein model. Phys. Rev. Lett. 2015, 114, 028102. [Google Scholar] [CrossRef] [PubMed]
- Arsuaga, J.; Vázquez, M.; Trigueros, S.; Sumners, D.W.; Roca, J. Knotting probability of DNA molecules confined in restricted volumes: DNA knotting in phage capsids. Proc. Natl. Acad. Sci. USA 2002, 99, 5373–5377. [Google Scholar] [CrossRef] [PubMed]
- Arsuaga, J.; Vázquez, M.; McGuirk, P.; Trigueros, S.; Sumners, D.W.; Roca, J. DNA knots reveal a chiral organization of DNA in phage capsids. Proc. Natl. Acad. Sci. USA 2005, 102, 9165–9169. [Google Scholar] [CrossRef] [PubMed]
- Reith, D.; Cifra, P.; Stasiak, A.; Virnau, P. Effective stiffening of DNA due to nematic ordering causes DNA molecules packed in phage capsids to preferentially form torus knots. Nucleic Acids Res. 2012, 40, 5129–5137. [Google Scholar] [CrossRef] [PubMed]
- Virnau, P.; Rieger, F.C.; Reith, D. Influence of chain stiffness on knottedness in single polymers. Biochem. Soc. Trans. 2013, 41, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Trefz, B.; Siebert, J.; Virnau, P. How molecular knots can pass through each other. Proc. Natl. Acad. Sci. USA. 2014, 111, 7948–7951. [Google Scholar] [CrossRef] [PubMed]
- Doniach, S.; Garel, T.; Orland, H. Phase diagram of a semiflexible polymer chain in a θ solvent: Application to protein folding. J. Chem. Phys. 1996, 105, 1601–1608. [Google Scholar] [CrossRef]
- Kolinski, A.; Skolnick, J.; Yaris, R. The collapse transition of semiflexible polymers. A Monte Carlo simulation of a model system. J. Chem. Phys. 1986, 85, 3585–3597. [Google Scholar] [CrossRef]
- Bastolla, U.; Grassberger, P. Phase transitions of single semistiff polymer chains. J. Stat. Phys. 1997, 89, 1061–1078. [Google Scholar] [CrossRef]
- Krawczyk, J.; Owczarek, A.L.; Prellberg, T. A semi-flexible attracting segment model of two-dimensional polymer collapse. Physica A 2010, 389, 1619–1624. [Google Scholar] [CrossRef]
- Noguchi, H.; Yoshikawa, K. Morphological variation in a collapsed single homopolymer chain. J. Chem. Phys. 1998, 109, 5070–5077. [Google Scholar] [CrossRef]
- Ivanov, V.A.; Paul, W.; Binder, K. Finite chain length effects on the coil-globule transition of stiff-chain macromolecules: A Monte Carlo simulation. J. Chem. Phys. 1998, 109, 5659–5669. [Google Scholar] [CrossRef]
- Stukan, M.R.; Ivanov, V.A.; Grosberg, A.Y.; Paul, W.; Binder, K. Chain length dependence of the state diagram of a single stiff-chain macromolecule: Theory and Monte Carlo simulation. J. Chem. Phys. 2003, 118, 3392–3400. [Google Scholar] [CrossRef]
- Martemyanova, J.A.; Stukan, M.R.; Ivanov, V.A.; Müller, M.; Paul, W.; Binder, K. Dense orientationally ordered states of a single semiflexible macromolecule: An expanded ensemble Monte Carlo simulation. J. Chem. Phys. 2005, 122, 174907. [Google Scholar] [CrossRef] [PubMed]
- Seaton, D.T.; Schnabel, S.; Landau, D.P.; Bachmann, M. From flexible to stiff: Systematic analysis of structural phases for single semiflexible polymers. Phys. Rev. Lett. 2013, 110, 028103. [Google Scholar] [CrossRef] [PubMed]
- Marenz, M.; Janke, W. Knots as a topological order parameter for semiflexible polymers. Phys. Rev. Lett. 2016, 116, 128301. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Huang, M.; Lei, Q.; Larson, R.G. A simple analytical model for predicting the collapsed state of self-attractive semiflexible polymers. Polymers 2016, 8, 264. [Google Scholar] [CrossRef]
- Maurstad, G.; Stokke, B.T. Metastable and stable states of xanthan polyelectrolyte complexes studied by atomic force microscopy. Biopolymers 2004, 74, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.P.; Paul, W.; Binder, K. Phase transitions of a single polymer chain: A Wang-Landau simulation study. J. Chem. Phys. 2009, 131, 114907. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.P.; Paul, W.; Binder, K. All-or-none proteinlike folding transition of a flexible homopolymer chain. Phys. Rev. E 2009, 79, 050801. [Google Scholar] [CrossRef] [PubMed]
- Gross, J.; Neuhaus, T.; Vogel, T.; Bachmann, M. Effects of the interaction range on structural phases of flexible polymers. J. Chem. Phys. 2013, 138, 074905. [Google Scholar] [CrossRef] [PubMed]
- Koci, T.; Bachmann, M. Confinement effects upon the separation of structural transitions in linear systems with restricted bond fluctuation ranges. Phys. Rev. E 2015, 92, 042142. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.J.; Bachmann, M. Stabilization of helical macromolecular phases by confined bending. Phys. Rev. Lett. 2015, 115, 048301. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.J.; Bachmann, M. Significance of bending restraints for the stability of helical polymer conformations. Phys. Rev. E 2016, 93, 062501. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.-P.; Paul, W.; Binder, K. Standard definitions of persistence length do not describe the local “intrinsic” stiffness of real polymer chains. Macromolecules 2010, 43, 3094–3102. [Google Scholar] [CrossRef]
- Schnabel, S.; Vogel, T.; Bachmann, M.; Janke, W. Surface effects in the crystallization process of elastic flexible polymers. Chem. Phys. Lett. 2009, 476, 201–204. [Google Scholar] [CrossRef]
- Seaton, D.T.; Wüst, T.; Landau, D.P. Collapse transitions in a flexible homopolymer chain: Application of the Wang-Landau algorithm. Phys. Rev. E 2010, 81, 011802. [Google Scholar] [CrossRef] [PubMed]
- Zierenberg, J.; Janke, W. From amorphous aggregates to polymer bundles: The role of stiffness on structural phases in polymer aggregation. Europhys. Lett. 2015, 109, 28002. [Google Scholar] [CrossRef]
- Kauffman, L.H. Knots and Physics, 2nd ed.; World Scientific: Singapore, 1991. [Google Scholar]
- Virnau, P. Detection and visualization of physical knots in macromolecules. Phys. Procedia 2010, 6, 117–125. [Google Scholar] [CrossRef]
- Janke, W. Accurate first-order transition points from finite-size data without power-law corrections. Phys. Rev. B 1993, 47, 14757–14770. [Google Scholar] [CrossRef]
- Janke, W. First-order phase transitions. In Computer Simulations of Surfaces and Interfaces; Dünweg, B., Landau, D.P., Milchev, A.I., Eds.; Kluwer: Dordrecht, The Netherlands, 2003; Volume 114, pp. 111–135. [Google Scholar]
- Irbäck, A.; Mohanty, S. All-atom Monte Carlo simulations of protein folding and aggregation. In Computational Methods to Study the Structure and Dynamics of Biomolecules and Biomolecular Processes; Liwo, A., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 433–444. [Google Scholar]
- Junghans, C.; Bachmann, M.; Janke, W. Thermodynamics of peptide aggregation processes: An analysis from perspectives of three statistical ensembles. J. Chem. Phys. 2008, 128, 085103. [Google Scholar] [CrossRef] [PubMed]
- Auer, S.; Kashchiev, D. Phase diagram of α-helical and β-sheet forming peptides. Phys. Rev. Lett. 2010, 104, 168105. [Google Scholar] [CrossRef] [PubMed]
- Enciso, M.; Schütte, C.; Delle Site, L. Influence of pH and sequence in peptide aggregation via molecular simulation. J. Chem. Phys. 2015, 143, 243130. [Google Scholar] [CrossRef] [PubMed]
- Auer, S.; Dobson, C.M.; Vendruscolo, M.; Maritan, A. Self-templated nucleation in peptide and protein aggregation. Phys. Rev. Lett. 2008, 101, 258101. [Google Scholar] [CrossRef] [PubMed]
- Abeln, S.; Vendruscolo, M.; Dobson, C.M.; Frenkel, D. A simple lattice model that captures protein folding, aggregation and amyloid formation. PLoS ONE 2014, 9, e85185. [Google Scholar] [CrossRef] [PubMed]
- Irbäck, A.; Linnemann, N.; Linse, B.; Wallin, S. Aggregate geometry in amyloid fibril nucleation. Phys. Rev. Lett. 2013, 110, 058101. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, L.G.; Head, D.A.; Auer, S. Universality in the morphology and mechanics of coarsening amyloid fibril networks. Phys. Rev. Lett. 2015, 114, 078102. [Google Scholar] [CrossRef] [PubMed]
- Irbäck, A.; Wessén, J. Thermodynamics of amyloid formation and the role of intersheet interactions. J. Chem. Phys. 2015, 143, 105104. [Google Scholar] [CrossRef] [PubMed]
- Junghans, C.; Bachmann, M.; Janke, W. Statistical mechanics of aggregation and crystallization for semiflexible polymers. Europhys. Lett. 2009, 87, 40002. [Google Scholar] [CrossRef]
- Zierenberg, J.; Mueller, M.; Schierz, P.; Marenz, M.; Janke, W. Aggregation of theta-polymers in spherical confinement. J. Chem. Phys. 2014, 141, 114908. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.; Zierenberg, J.; Marenz, M.; Schierz, P.; Janke, W. Probing the effect of density on the aggregation temperature of semi-flexible polymers in spherical confinement. Phys. Procedia 2015, 68, 95–99. [Google Scholar] [CrossRef]
- Zierenberg, J.; Janke, W. Exploring different regimes in finite-size scaling of the droplet condensation-evaporation transition. Phys. Rev. E 2015, 92, 012134. [Google Scholar] [CrossRef] [PubMed]
- Giurleo, J.T.; He, X.; Talaga, D.S. β-lactoglobulin assembles into amyloid through sequential aggregated intermediates. J. Mol. Biol. 2008, 381, 1332–1348. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, R.J.; Edwards, L.; Johnston, D.; Becich, P.; Hirst, L.S. Designing highly tunable semiflexible filament networks. Phys. Rev. E 2014, 89, 062602. [Google Scholar] [CrossRef] [PubMed]
- Kouwer, P.H.J.; Koepf, M.; Le Sage, V.A.A.; Jaspers, M.; van Buul, A.M.; Eksteen-Akeroyd, Z.H.; Woltinge, T.; Schwartz, E.; Kitto, H.J.; Hoogenboom, R.; et al. Responsive biomimetic networks from polyisocyanopeptide hydrogels. Nature 2013, 493, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Kierfeld, J.; Lipowsky, R. Unbundling and desorption of semiflexible polymers. Europhys. Lett. 2003, 62, 285–291. [Google Scholar] [CrossRef]
- Kierfeld, J.; Kühne, T.; Lipowsky, R. Discontinuous unbinding transitions of filament bundles. Phys. Rev. Lett. 2005, 95, 038102. [Google Scholar] [CrossRef] [PubMed]
- Heussinger, C.; Schüller, F.; Frey, E. Statics and dynamics of the wormlike bundle model. Phys. Rev. E 2010, 81, 021904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grason, G.M.; Bruinsma, R.F. Chirality and equilibrium biopolymer bundles. Phys. Rev. Lett. 2007, 99, 098101. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.S.; Briehl, R.W.; Ferrone, F.A.; Josephs, R. Twisted protein aggregates and disease: The stability of sickle hemoglobin fibers. Phys. Rev. Lett. 2003, 90, 128103. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, Y.; Lin, Y.; Yagi, H.; Lee, Y.H.; Kitayama, H.; Sakurai, K.; So, M.; Ogi, H.; Naiki, H.; Goto, Y. Distinguishing crystal-like amyloid fibrils and glass-like amorphous aggregates from their kinetics of formation. Proc. Natl. Acad. Sci. USA 2012, 109, 14446–14451. [Google Scholar] [CrossRef] [PubMed]
- Ni, R.; Abeln, S.; Schor, M.; Stuart, M.A.C.; Bolhuis, P. Interplay between folding and assembly of fibril-forming polypeptides. Phys. Rev. Lett. 2013, 111, 058101. [Google Scholar] [CrossRef] [PubMed]







© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zierenberg, J.; Marenz, M.; Janke, W. Dilute Semiflexible Polymers with Attraction: Collapse, Folding and Aggregation. Polymers 2016, 8, 333. https://doi.org/10.3390/polym8090333
Zierenberg J, Marenz M, Janke W. Dilute Semiflexible Polymers with Attraction: Collapse, Folding and Aggregation. Polymers. 2016; 8(9):333. https://doi.org/10.3390/polym8090333
Chicago/Turabian StyleZierenberg, Johannes, Martin Marenz, and Wolfhard Janke. 2016. "Dilute Semiflexible Polymers with Attraction: Collapse, Folding and Aggregation" Polymers 8, no. 9: 333. https://doi.org/10.3390/polym8090333