Supramolecular Interactions Induce Unexpectedly Strong Emissions from Triphenylamine-Functionalized Polytyrosine Blended with Poly(4-vinylpyridine)



Abstract
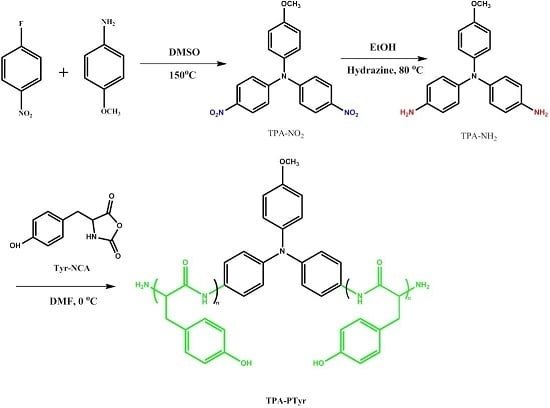
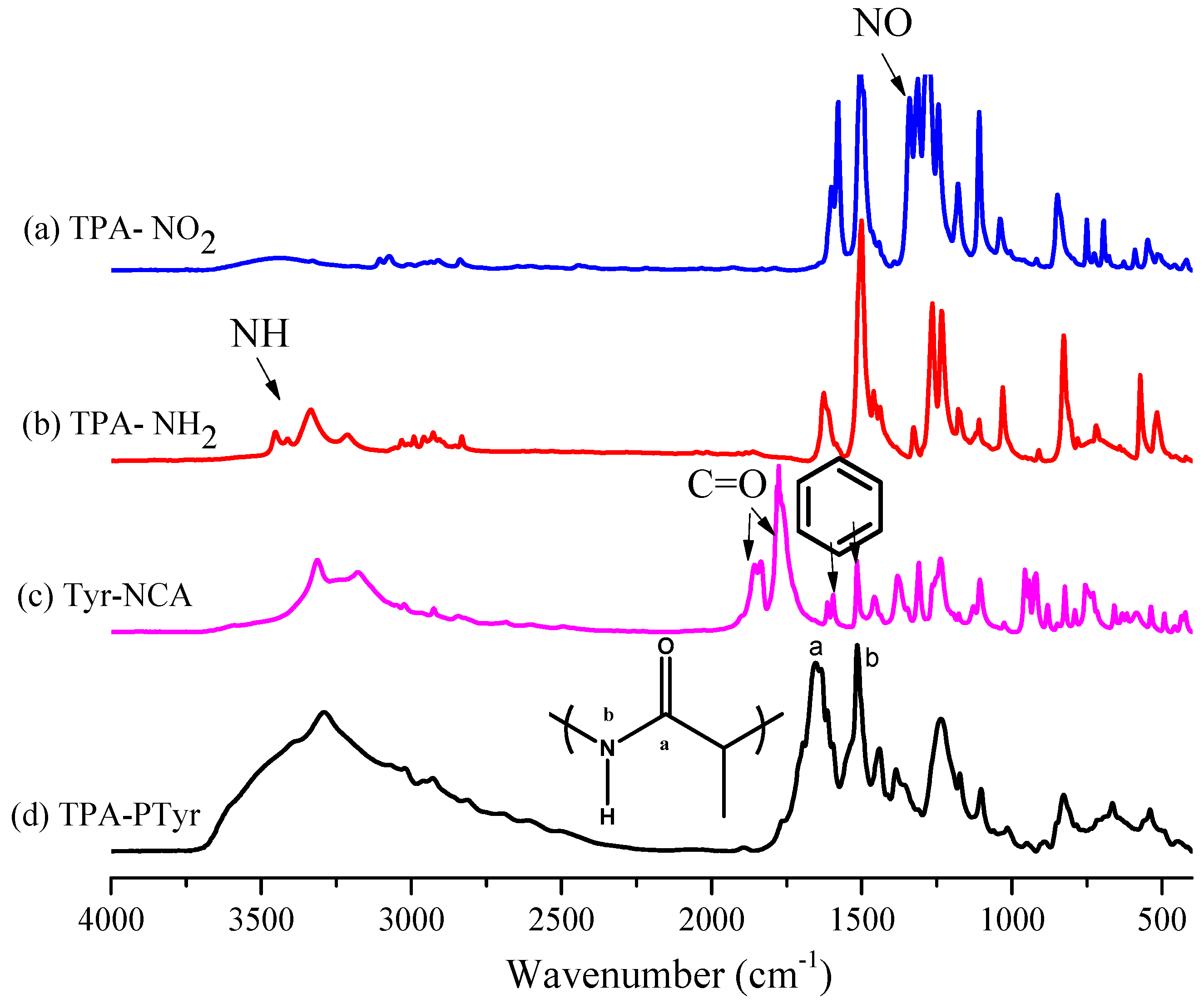
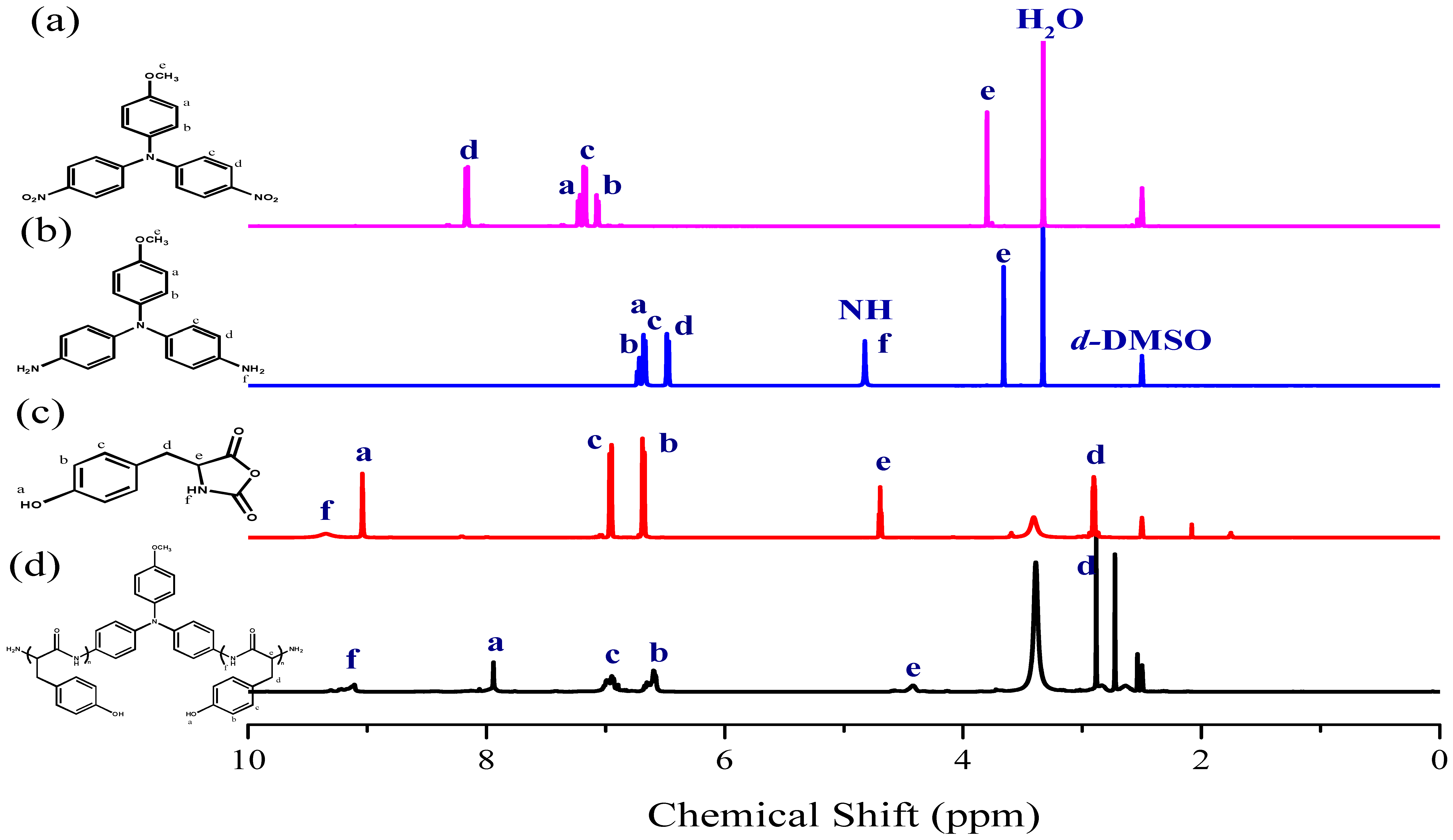
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental
2.1. Materials
2.1.1. 4,4′-Dinitro-4″-Methoxytriphenylamine
2.1.2. 4,4′-Diamino-4″-Methoxytriphenylamine
2.1.3. Tyr-NCA Monomer
2.1.4. PTyr-TPA Polypeptide
2.1.5. PTyr-TPA/P4VP Blends
2.2. Characterization
3. Results and Discussion
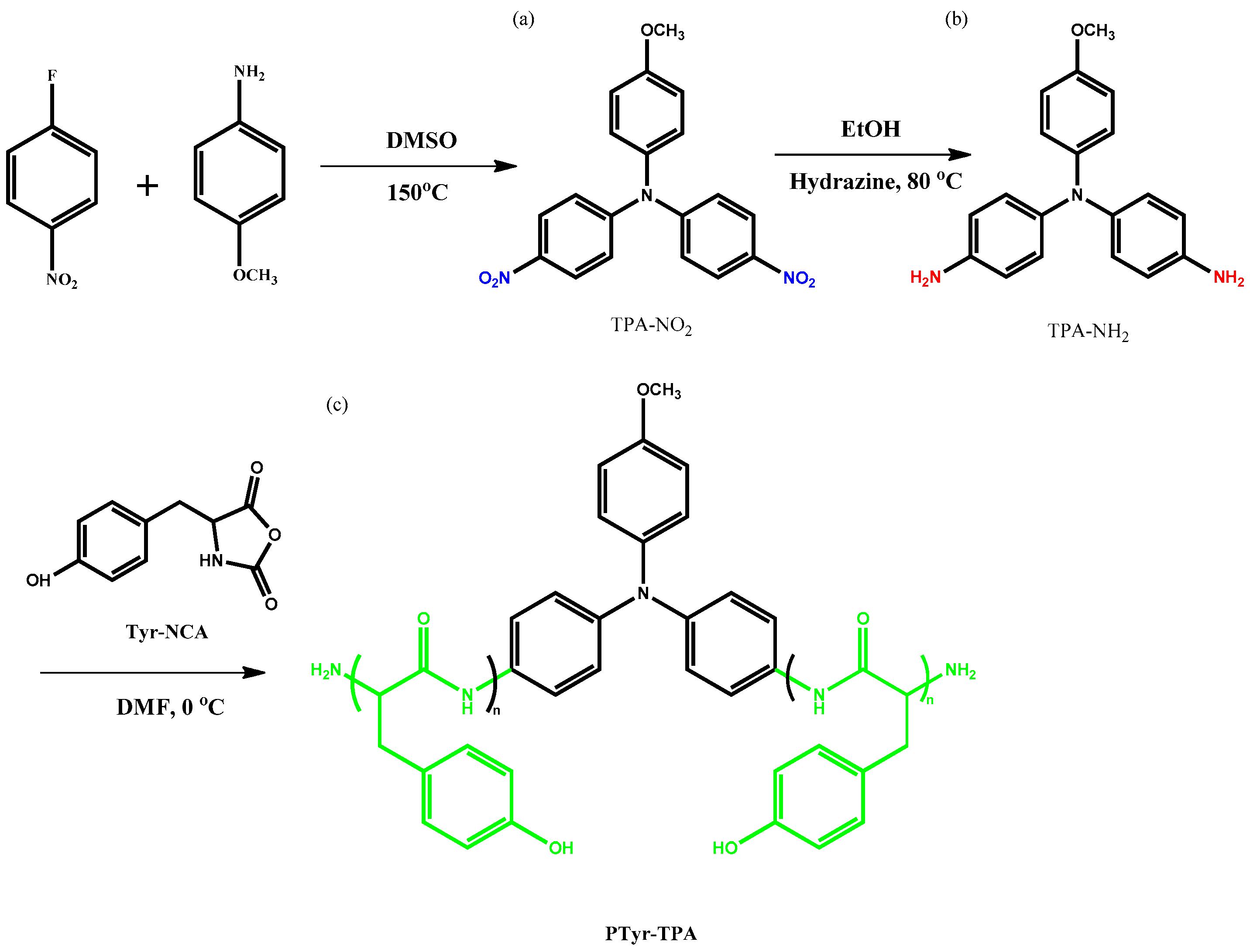
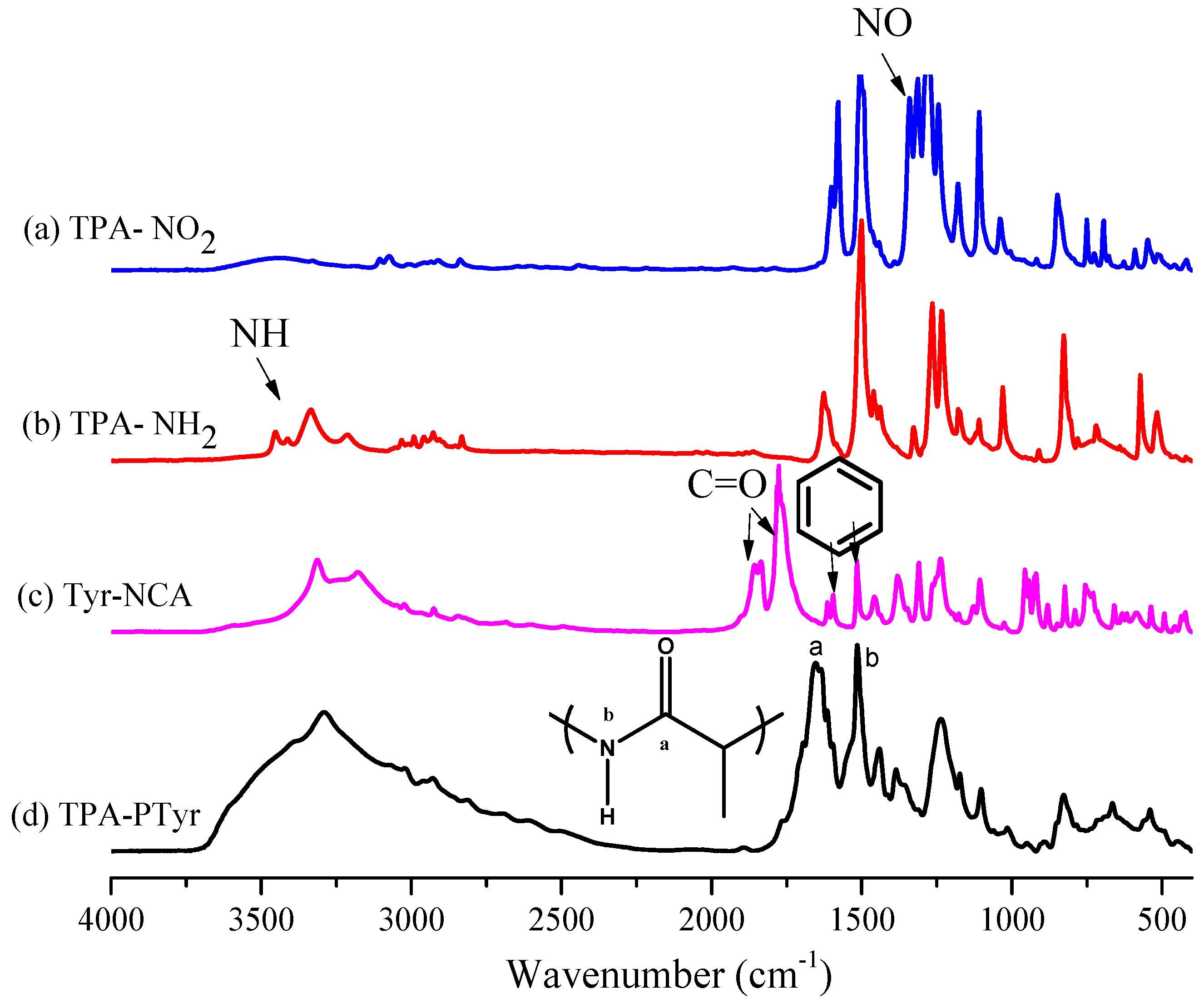
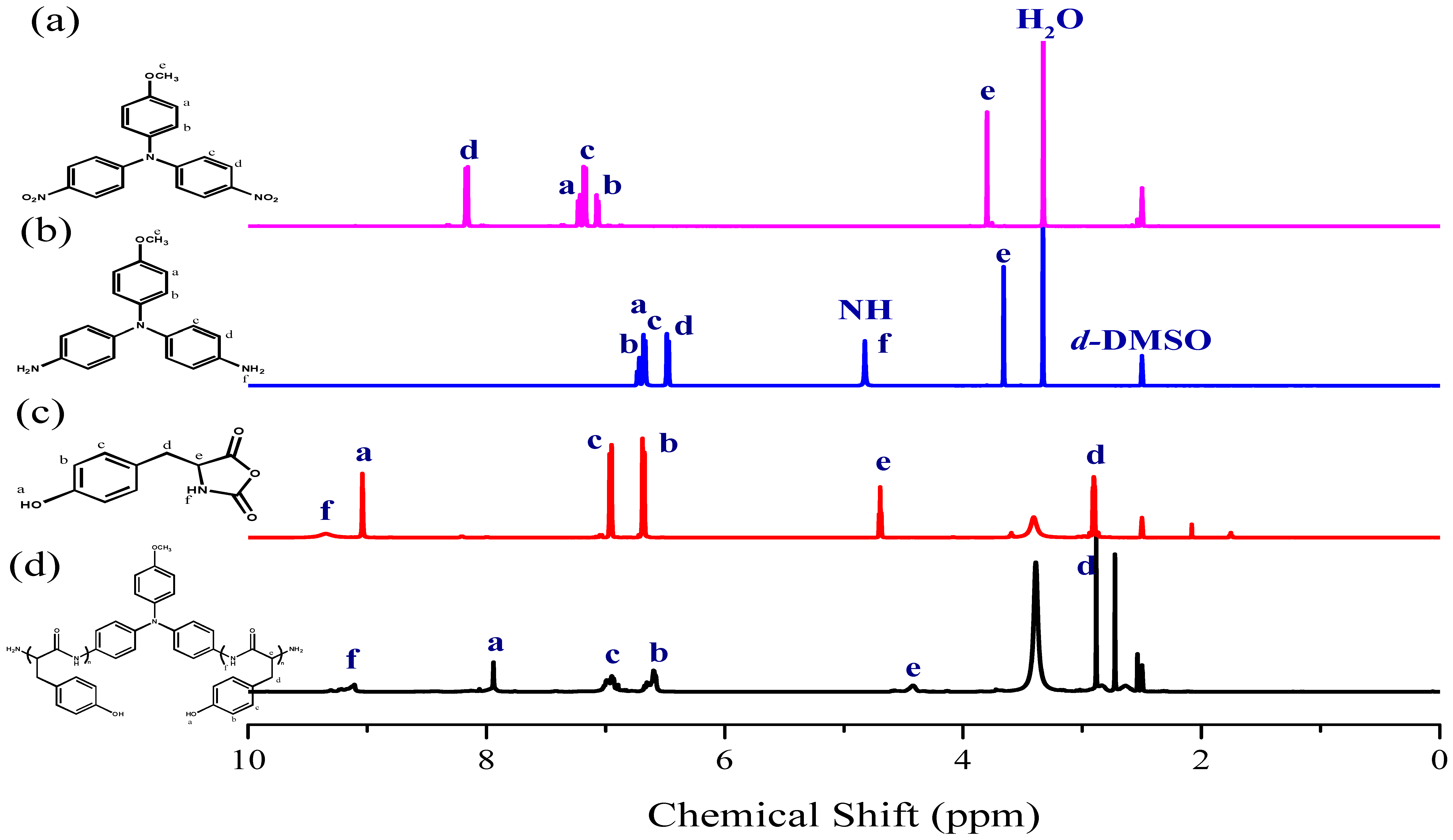
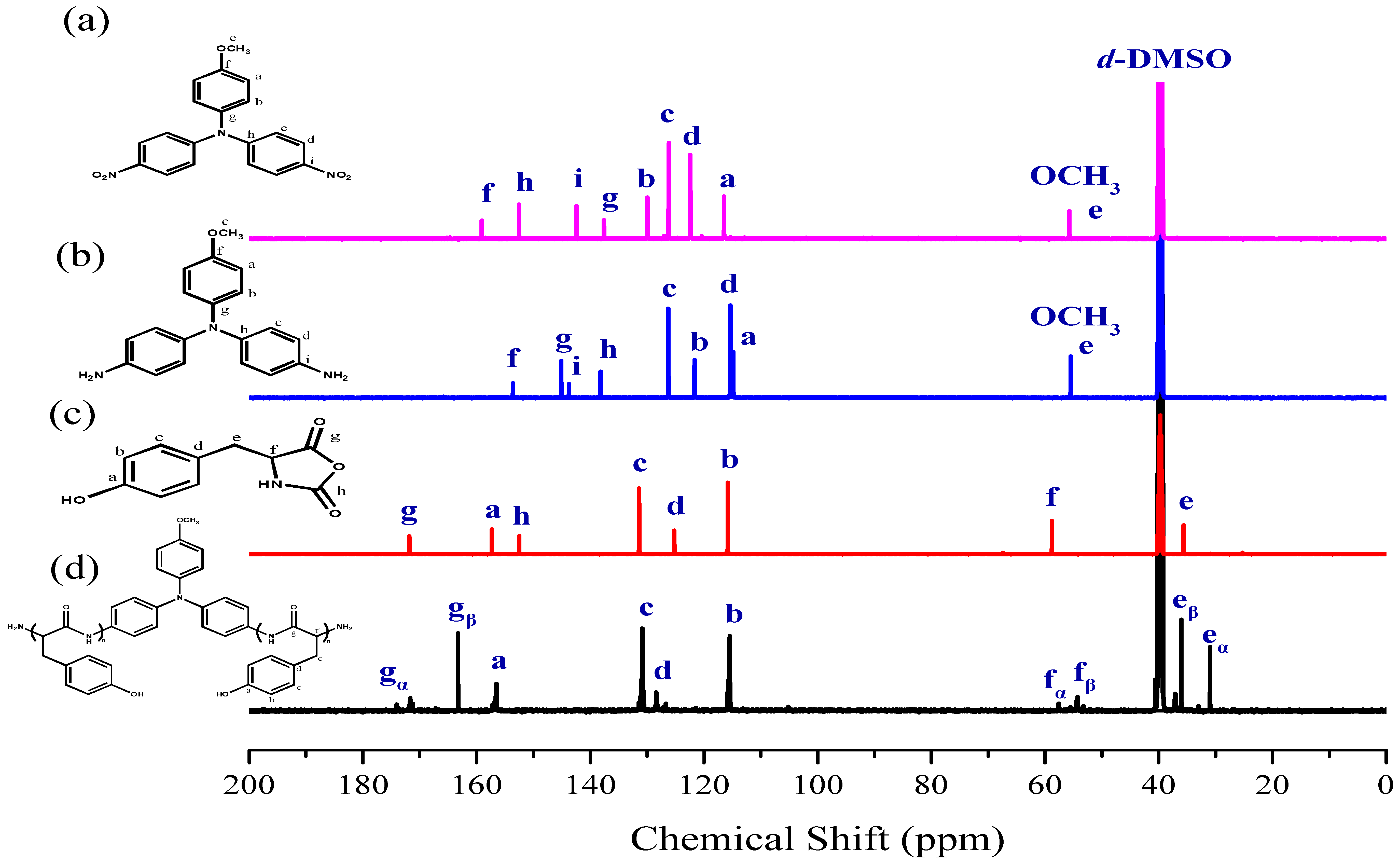
3.1. Synthesis of TPA-NH2
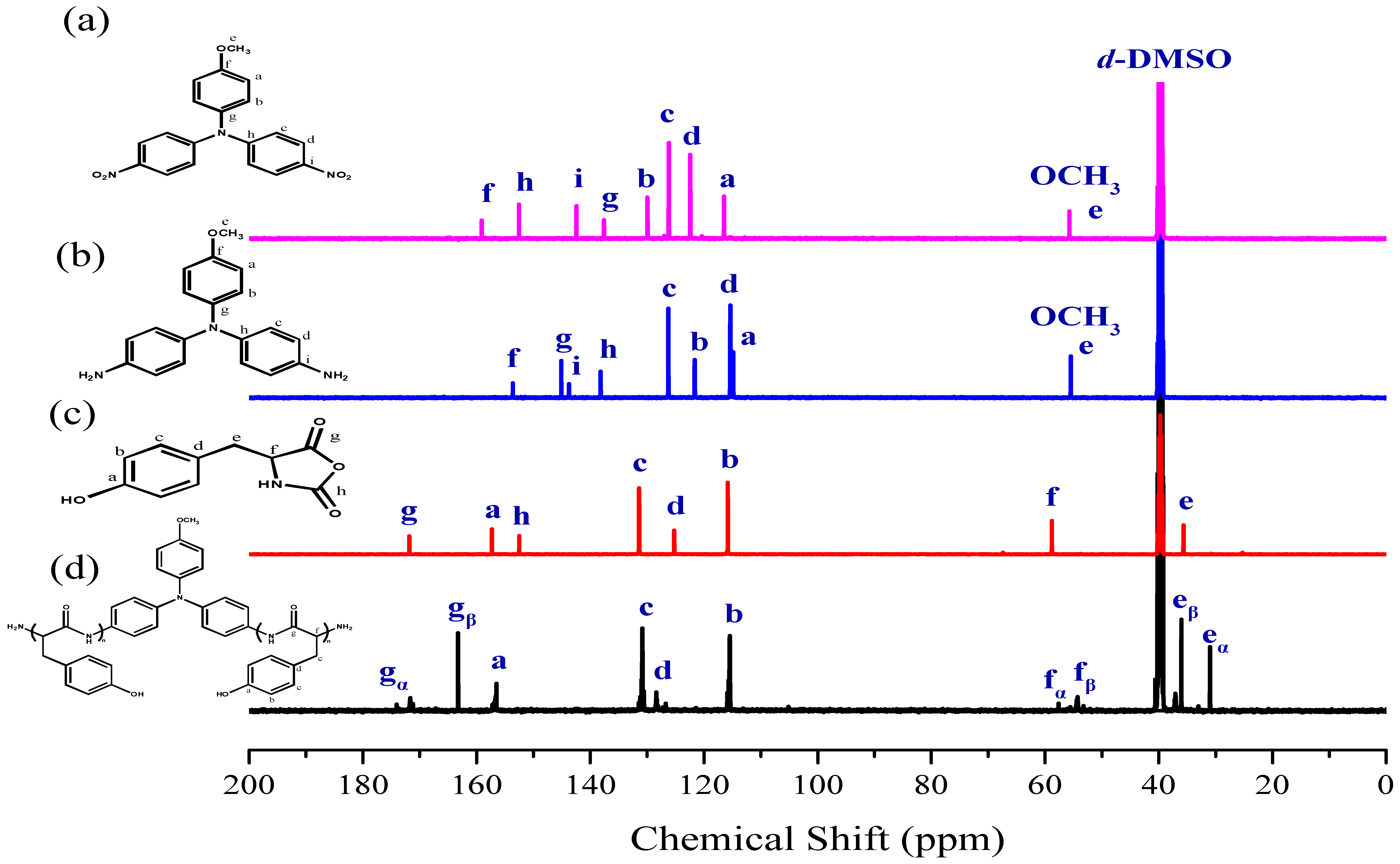
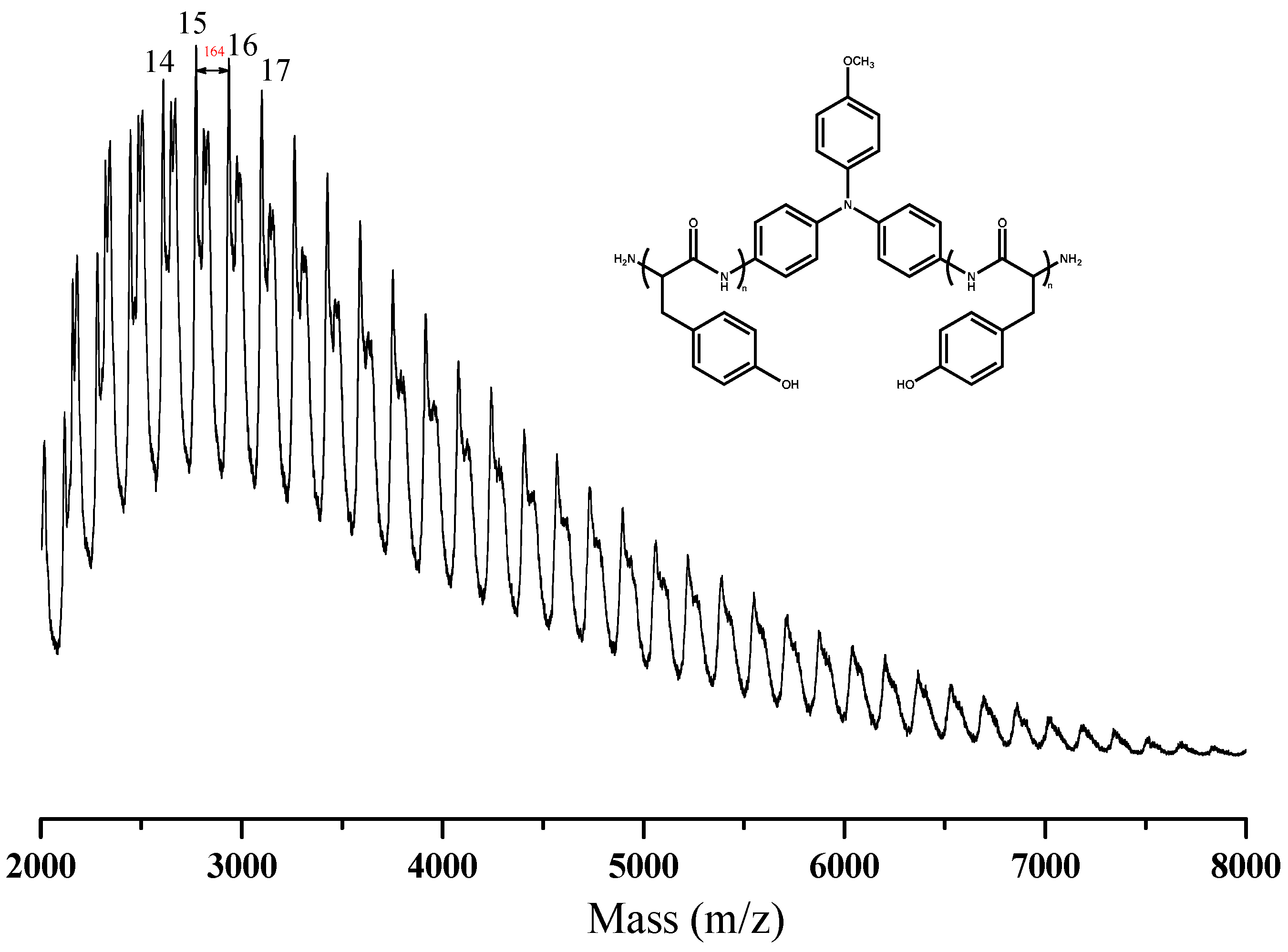
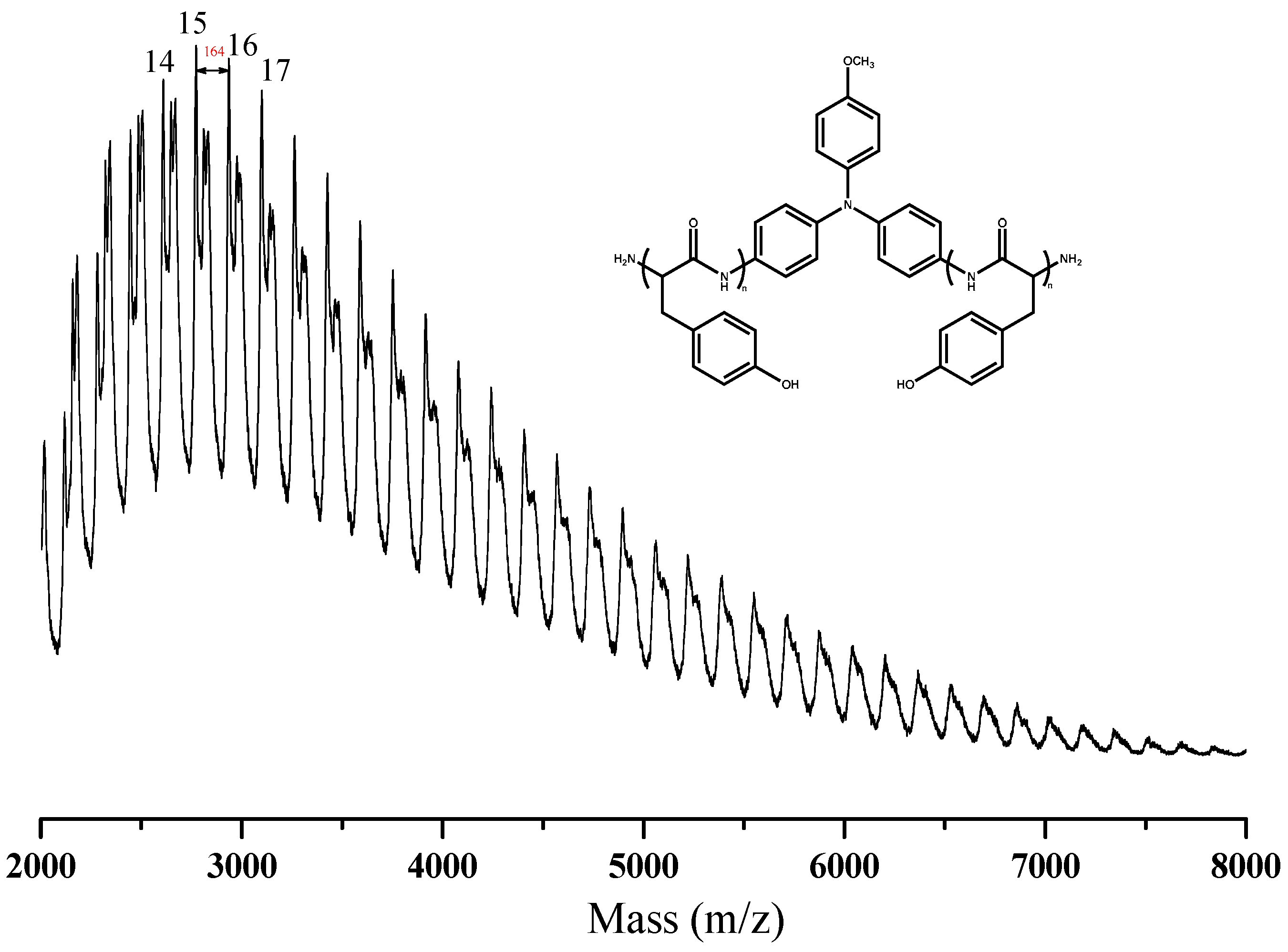
3.2. Synthesis of PTyr-TPA through NCA-ROP Polymerization
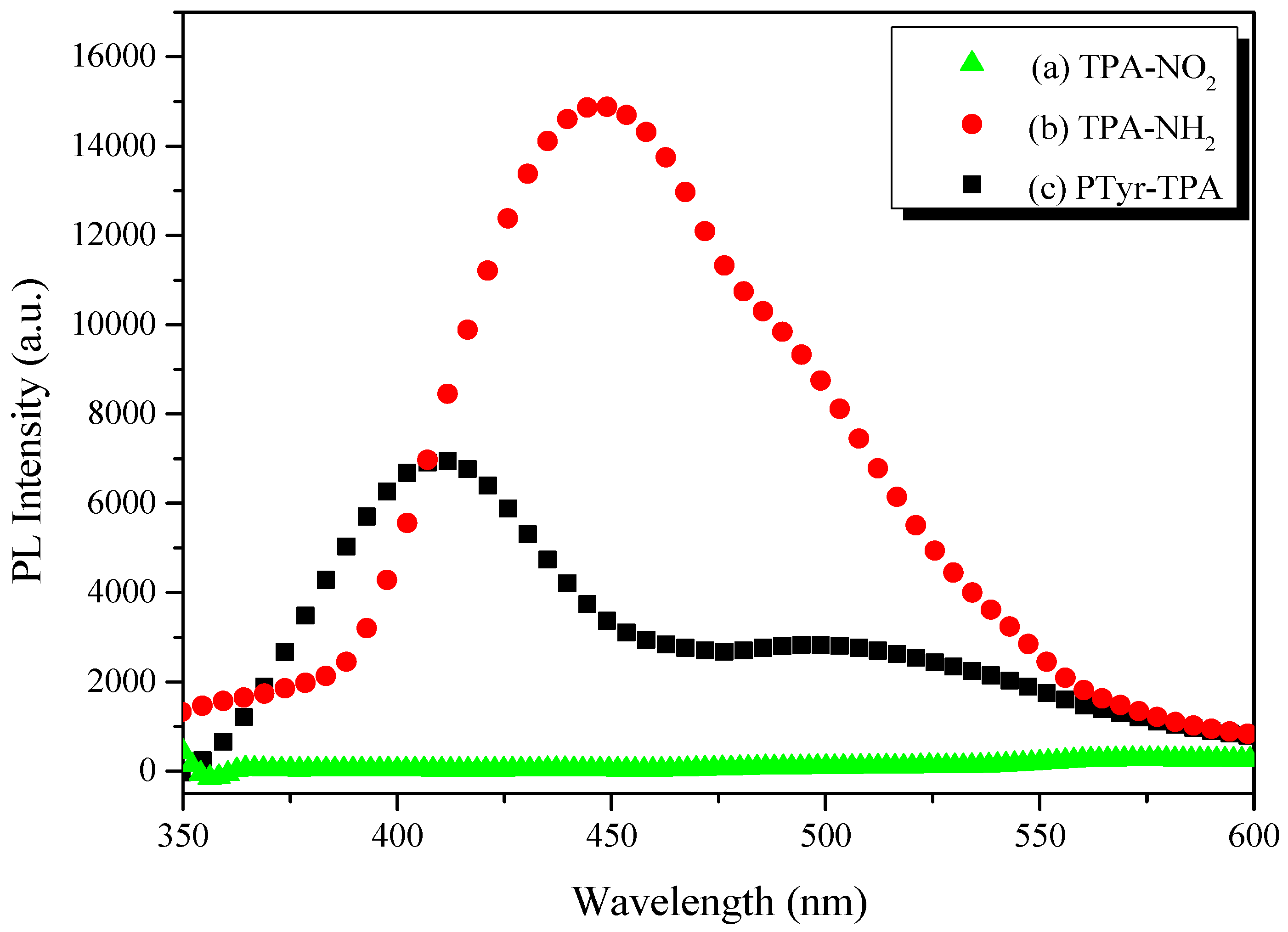
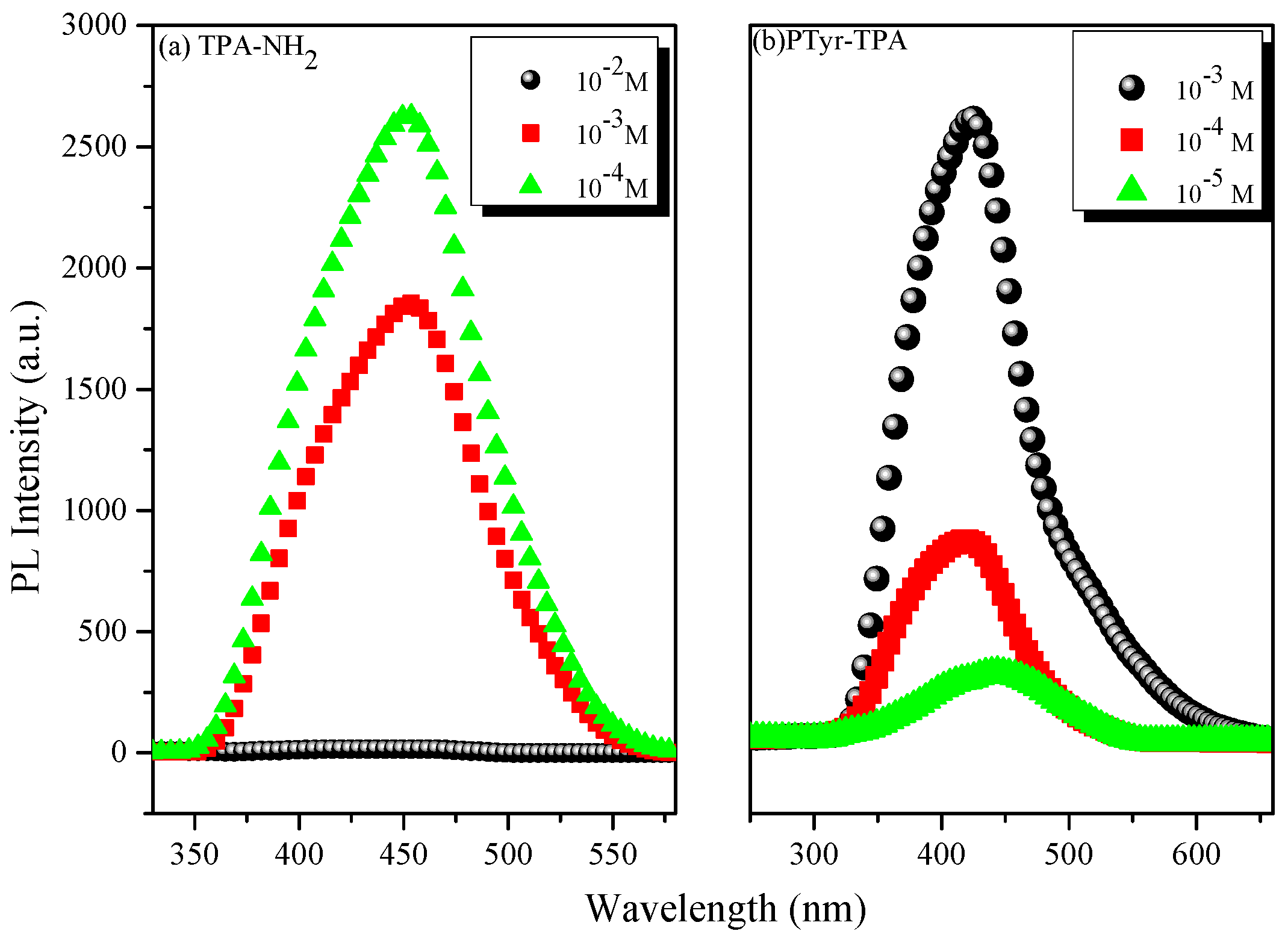
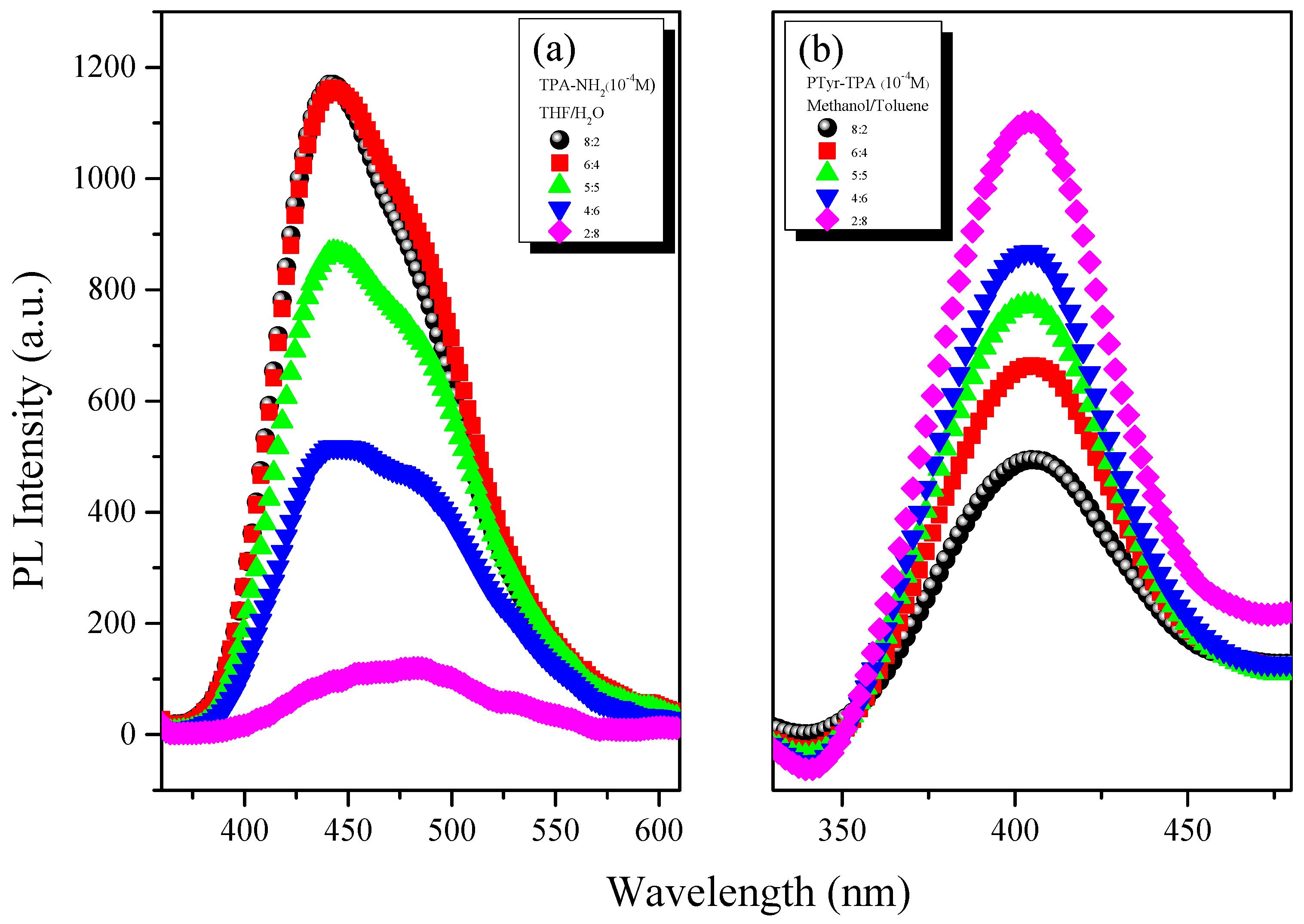
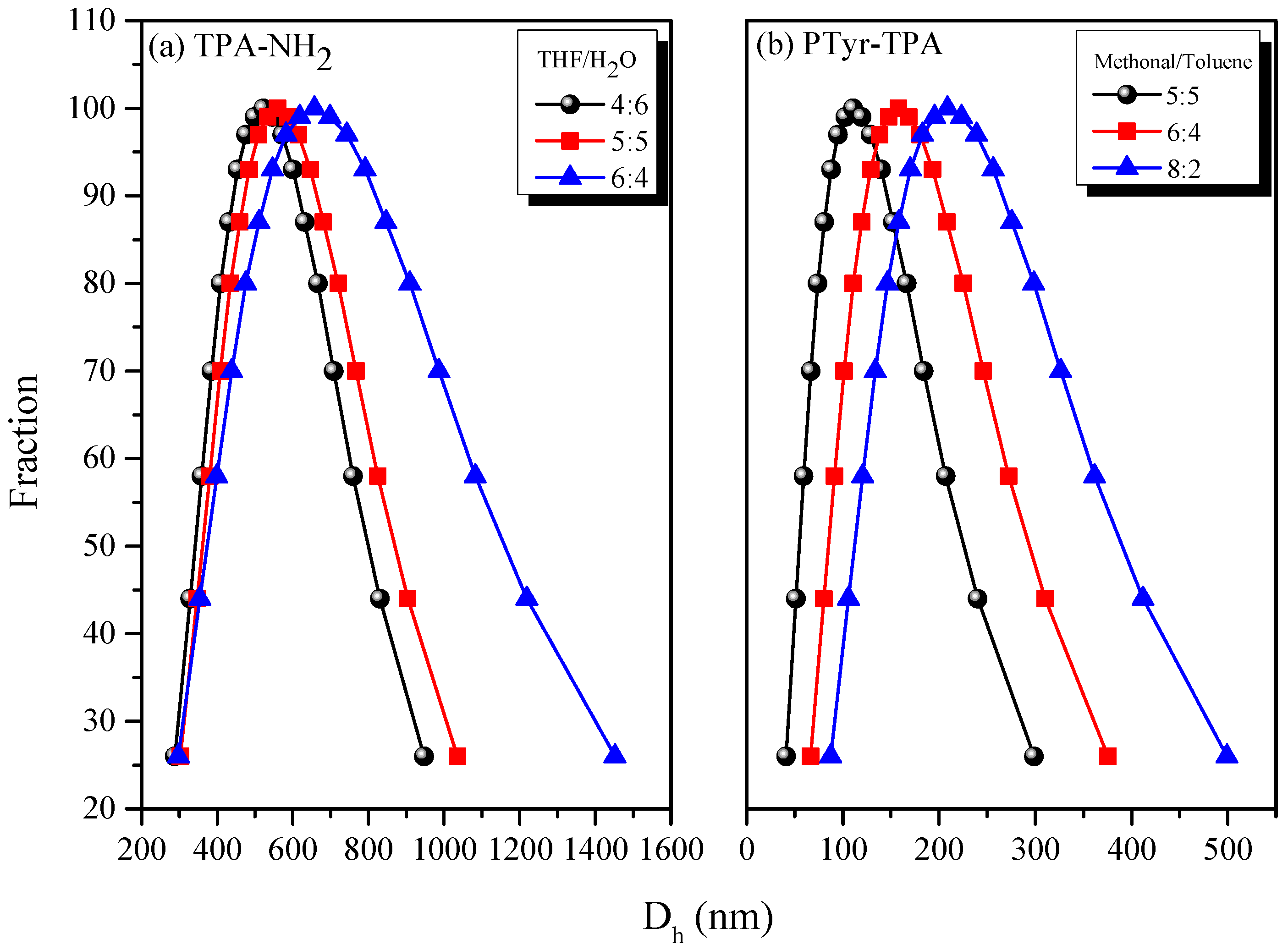
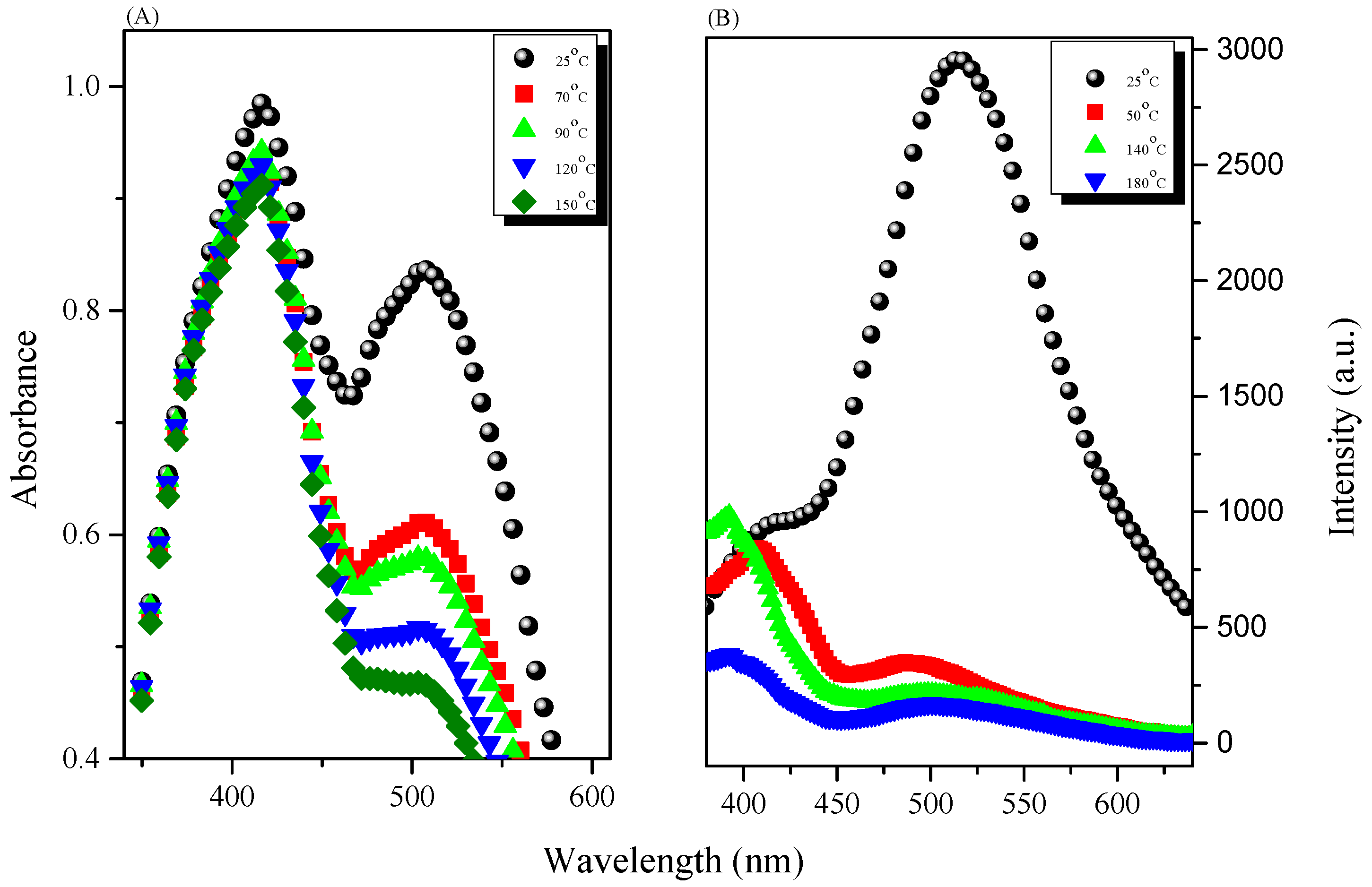
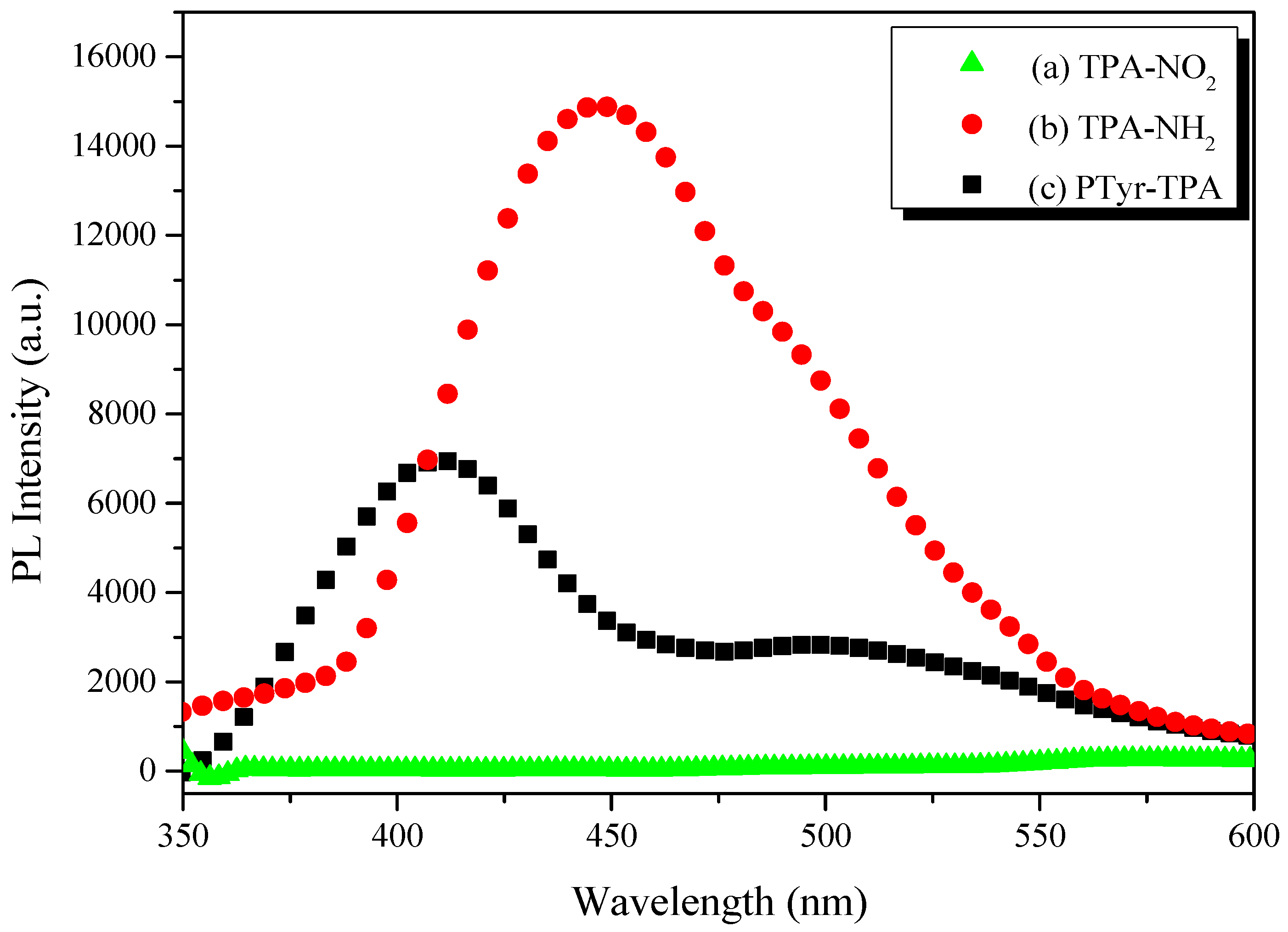
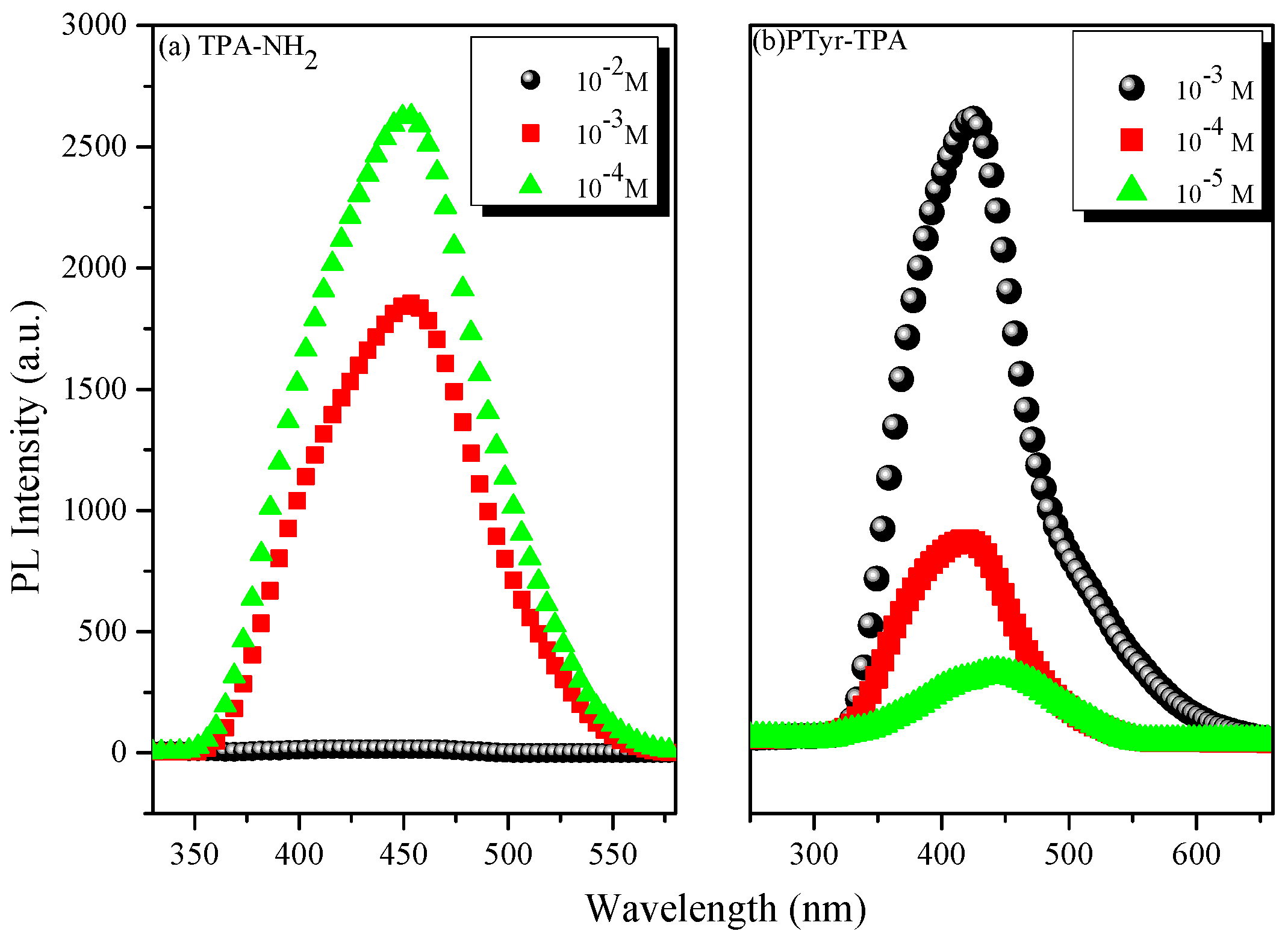
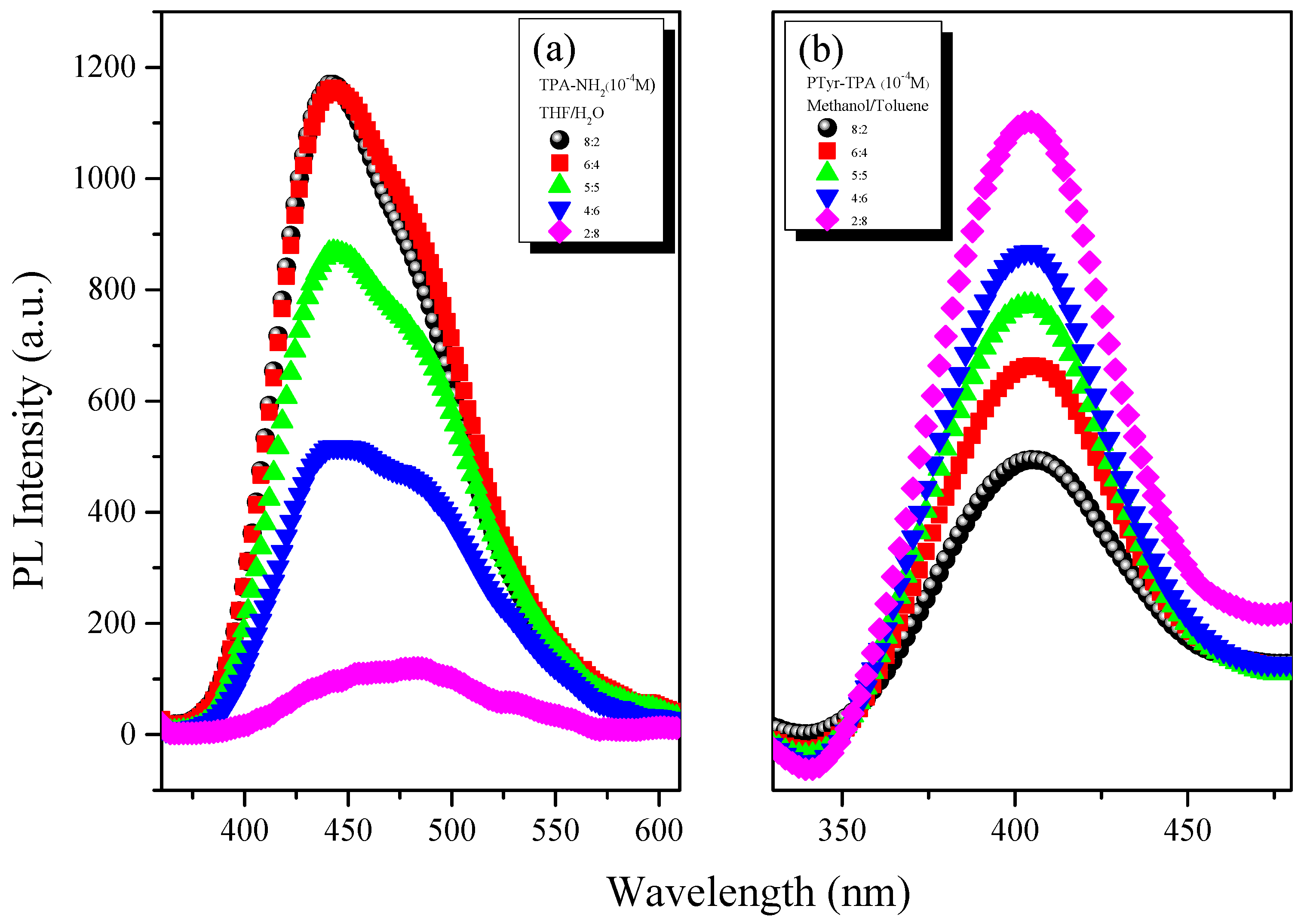
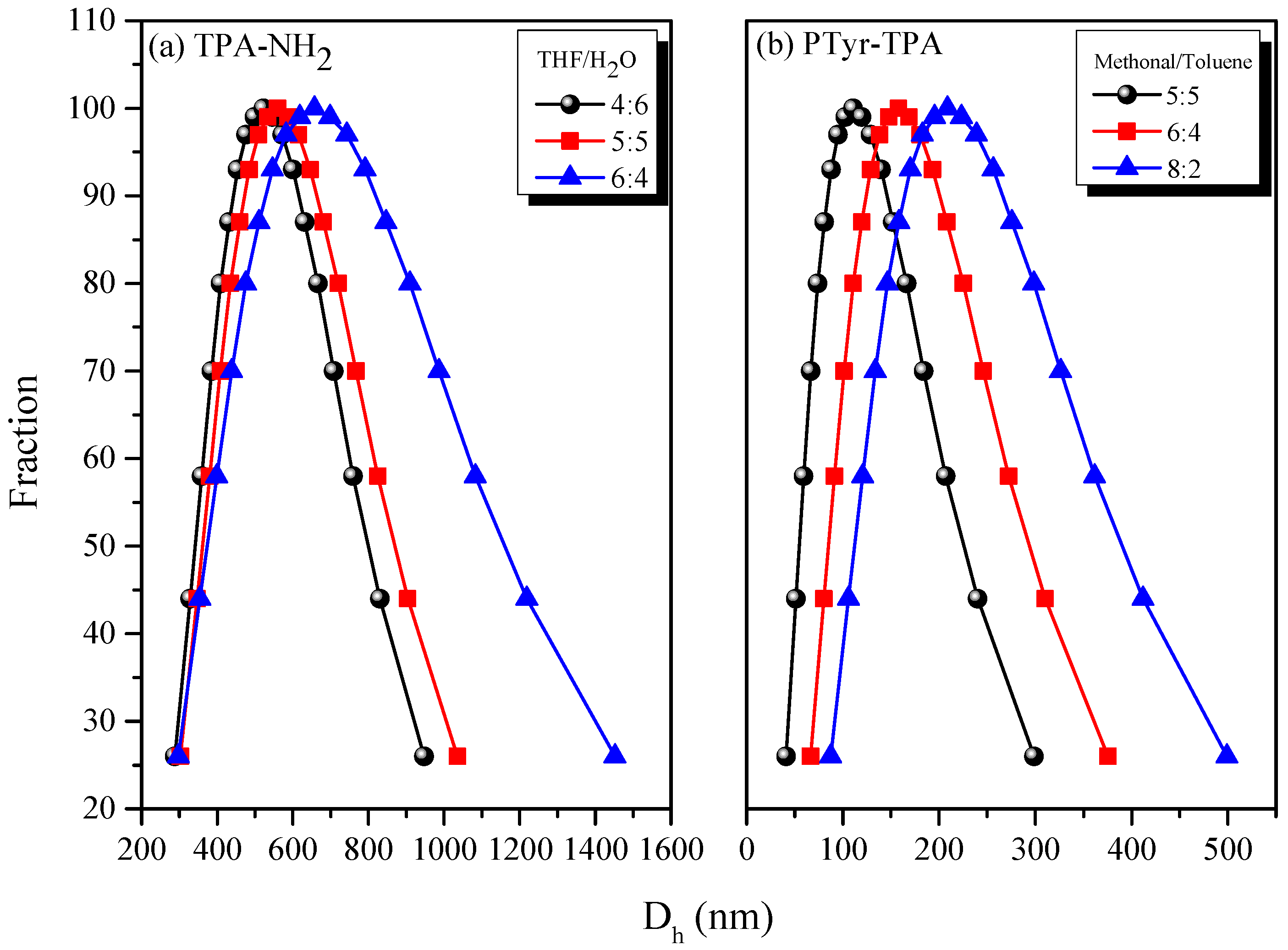
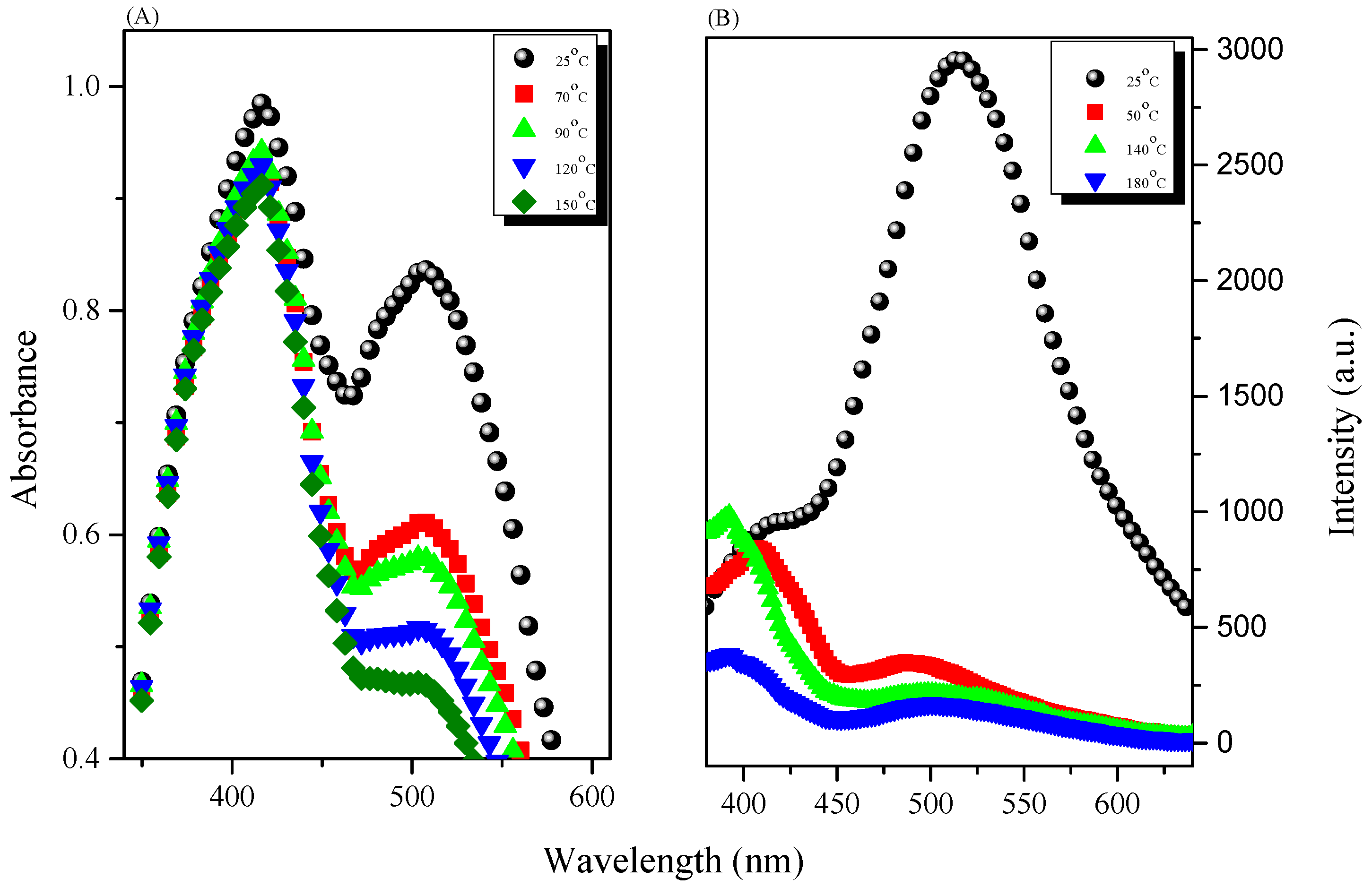
3.3. AIE Behavior
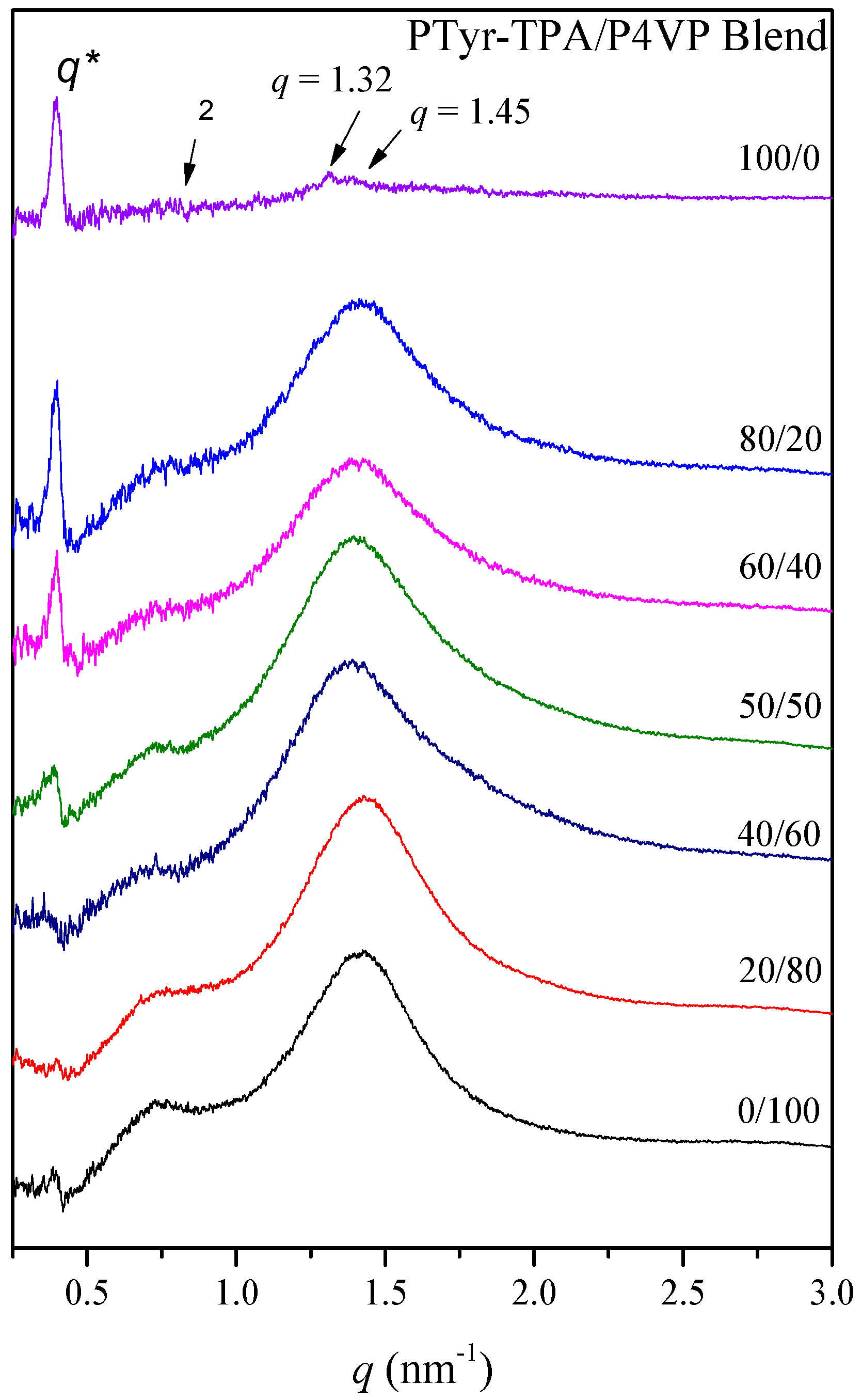
3.4. Physical Properties of PTyr-TPA/P4VP Blends
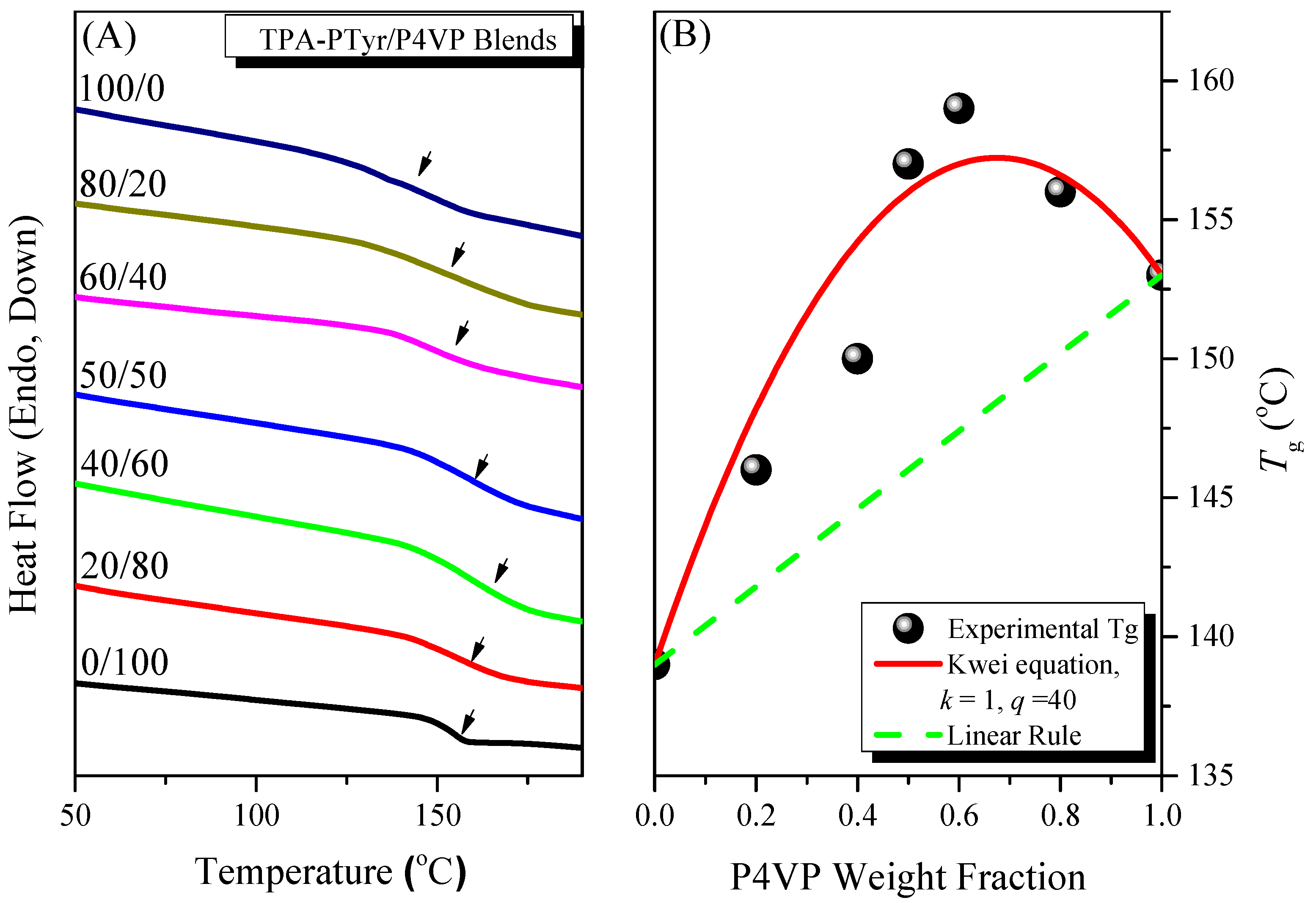
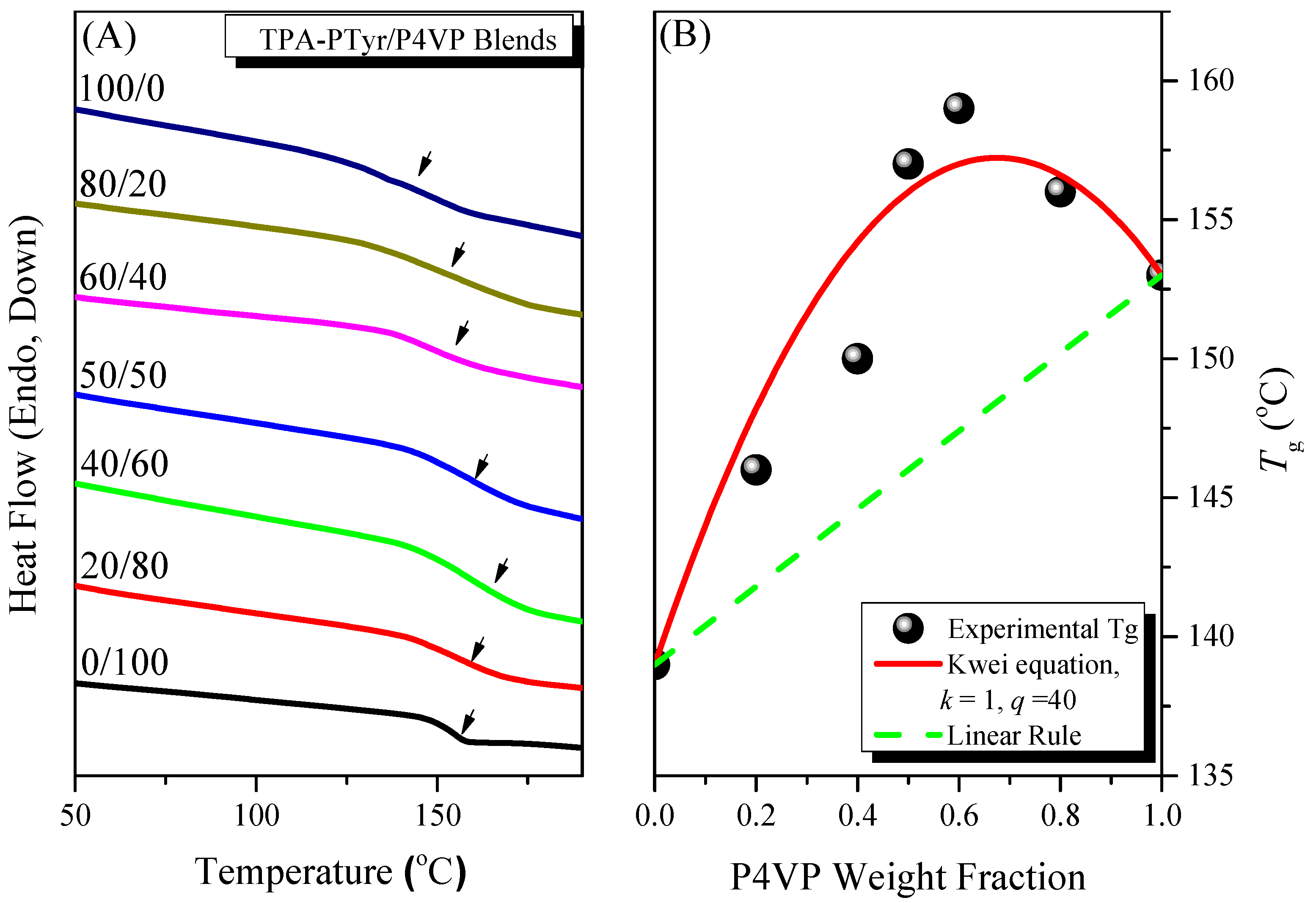
3.4.1. Thermal Properties of Binary PTyr-TPA/P4VP Blends
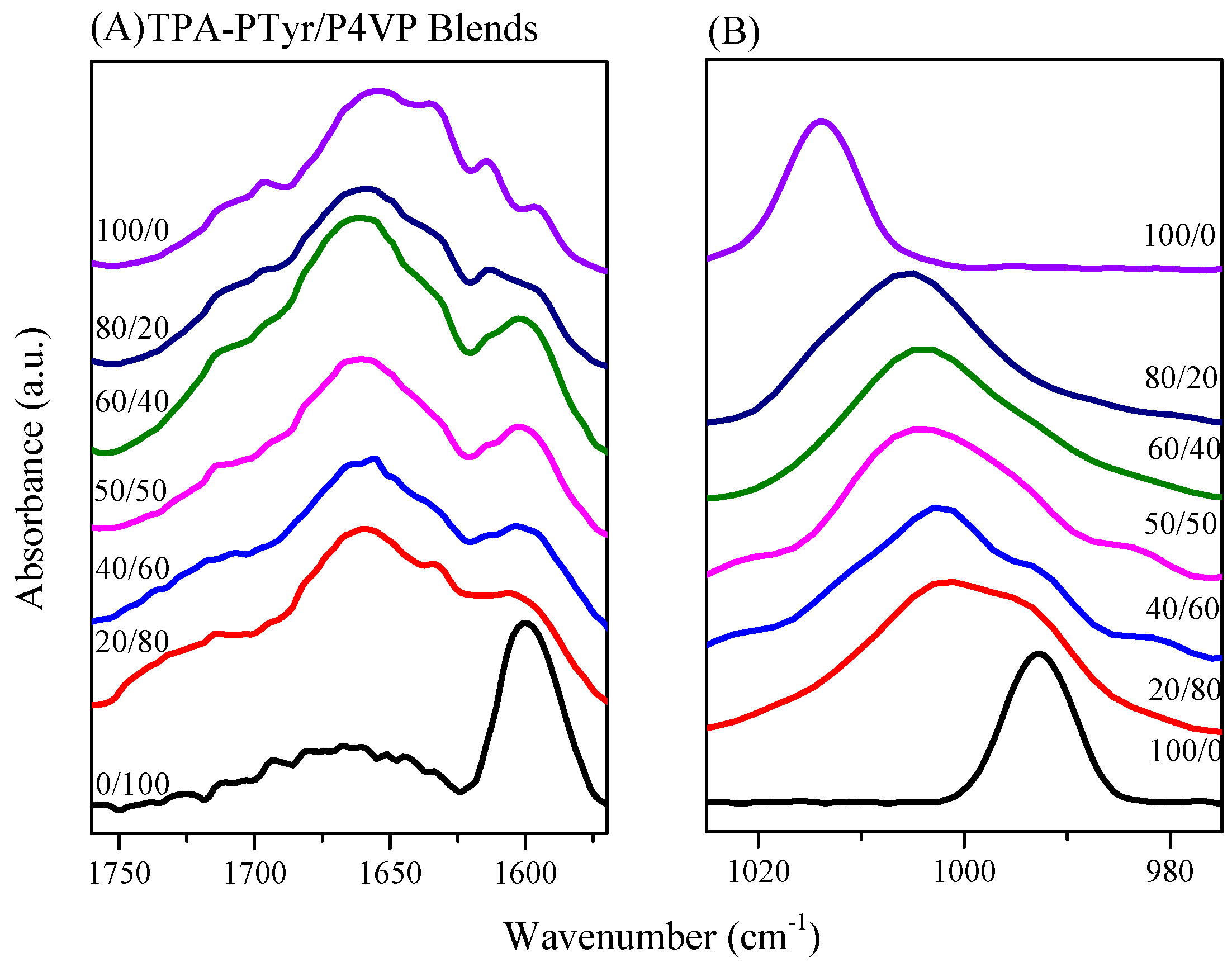
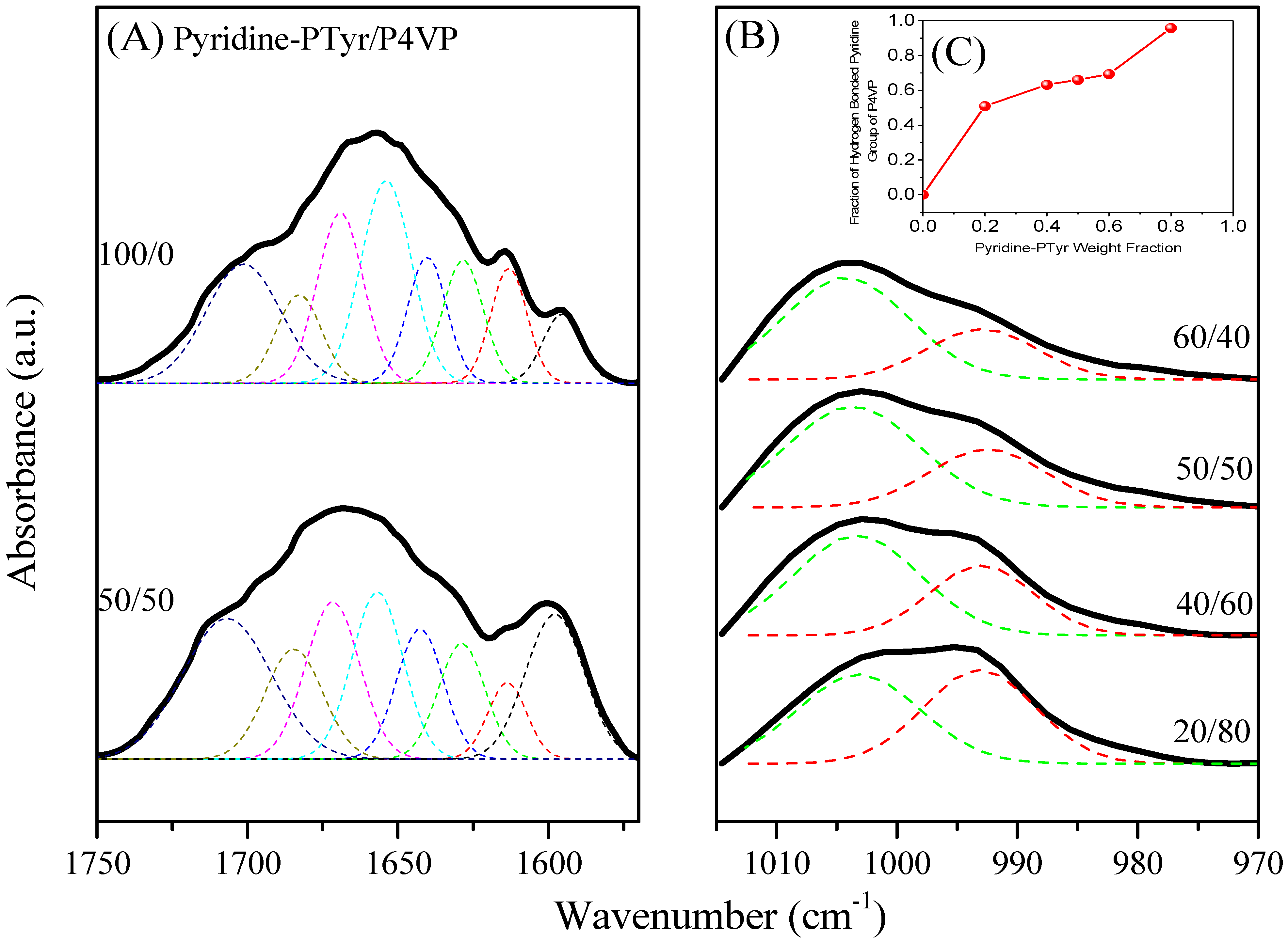
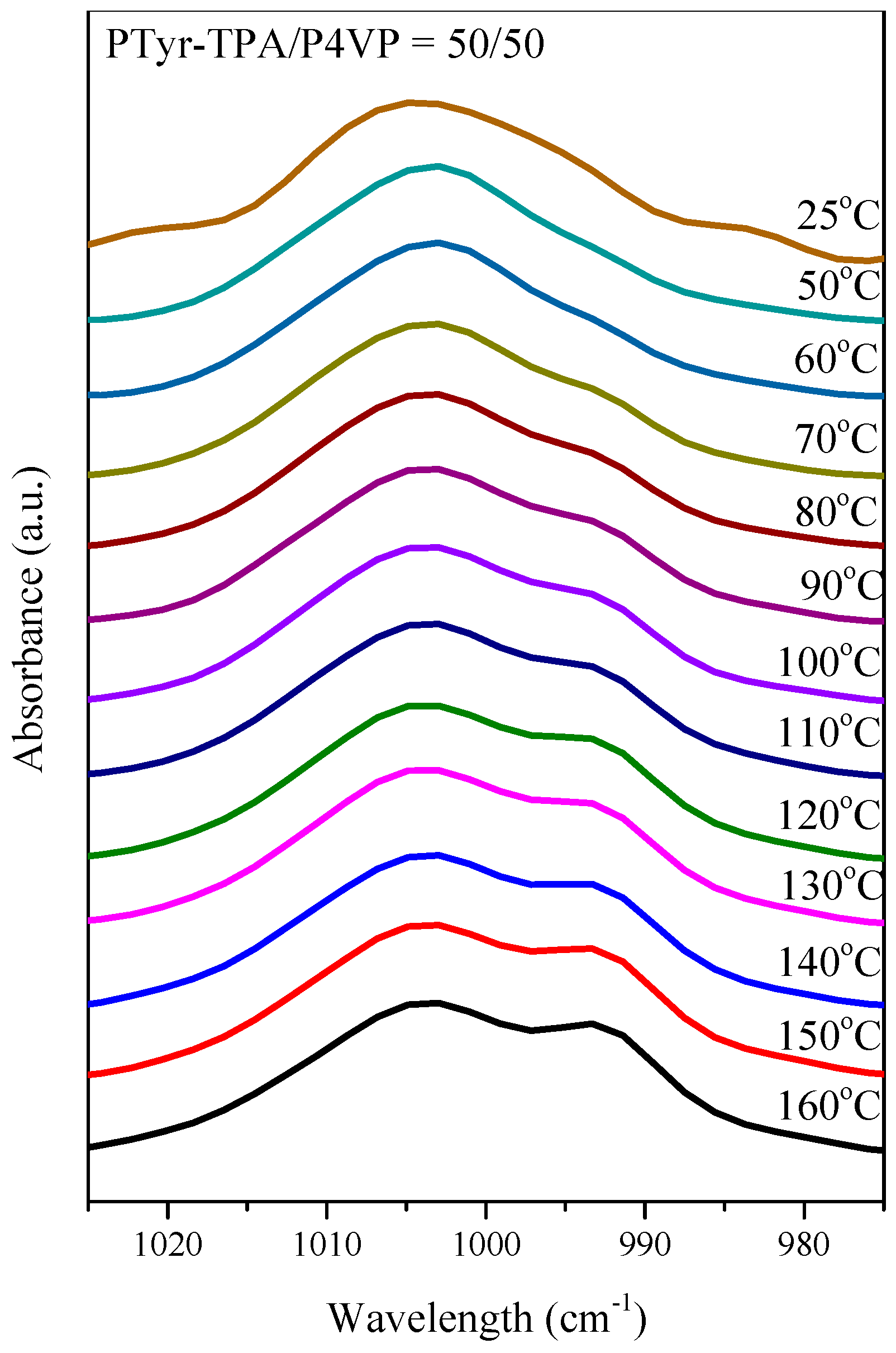
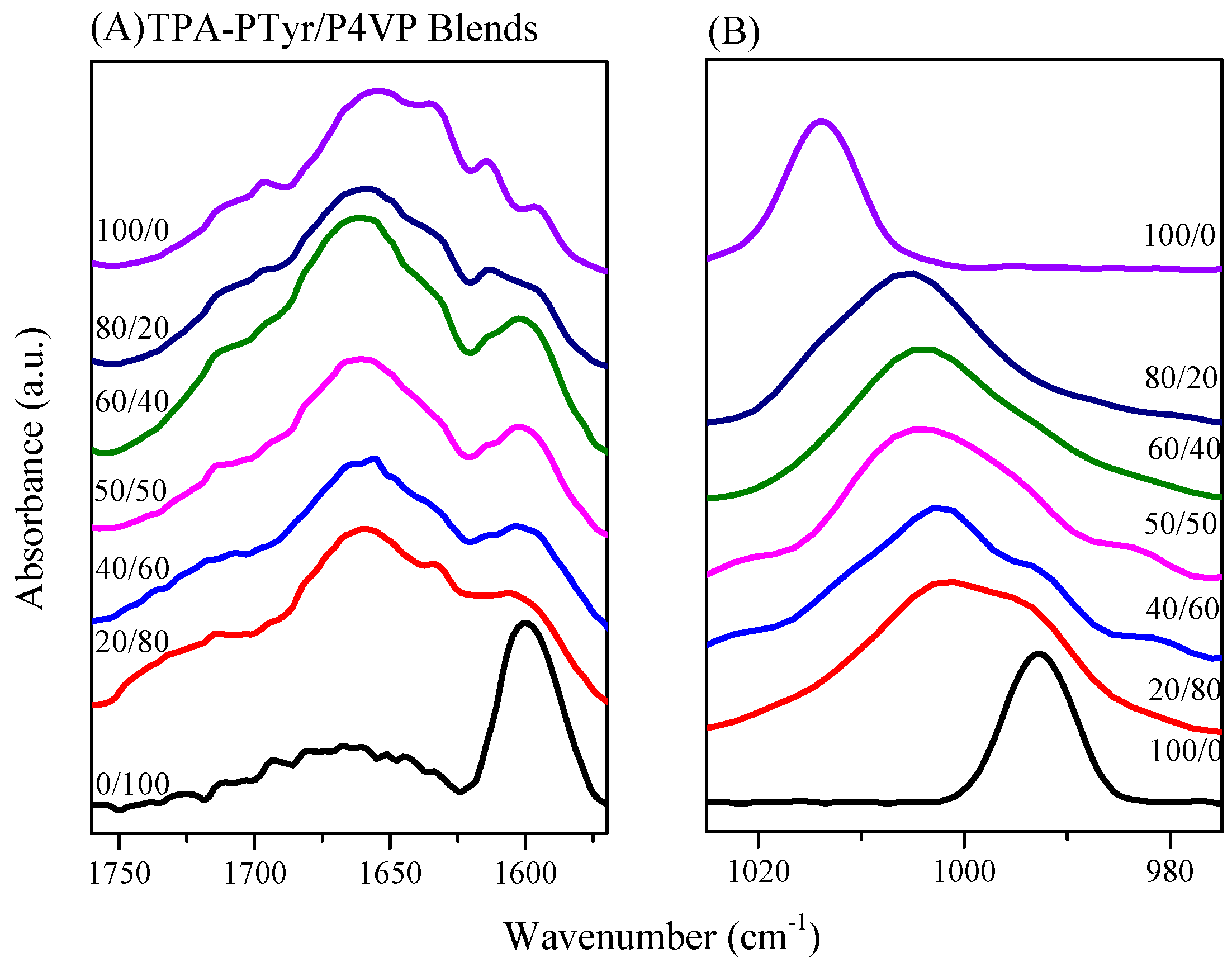
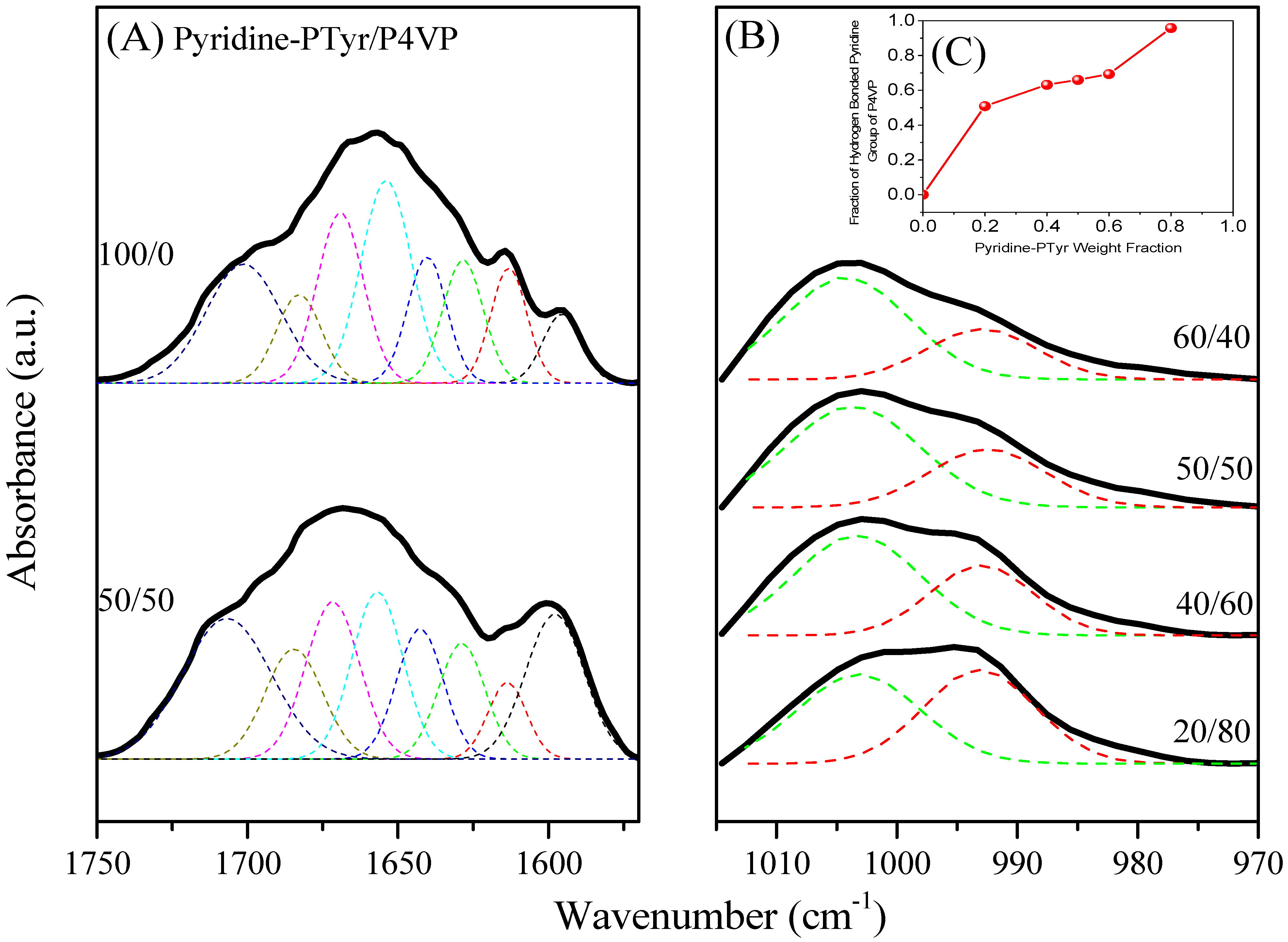
3.4.2. Secondary Structure and Hydrogen Bonding of PTyr-TPA/P4VP Blends
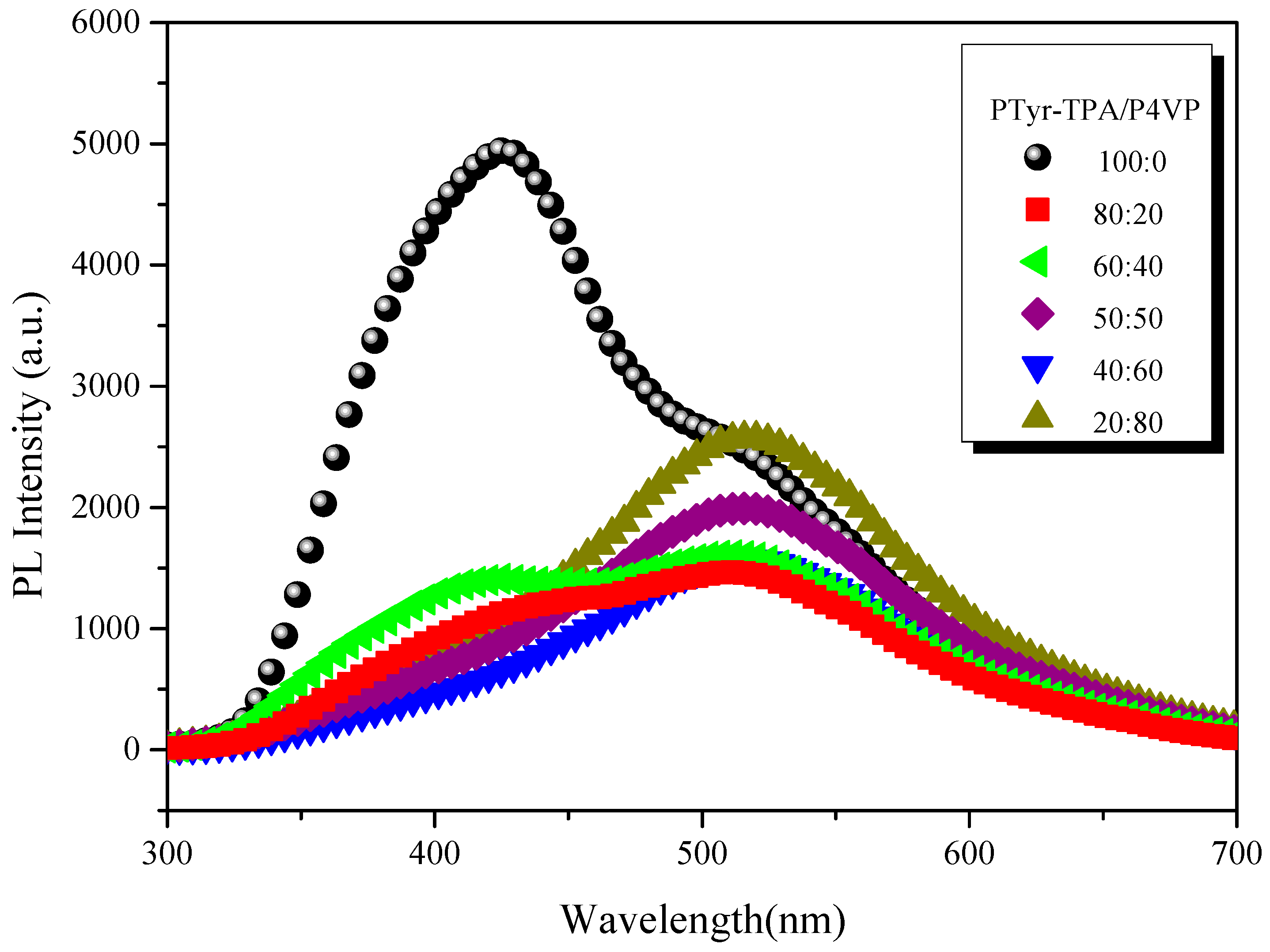
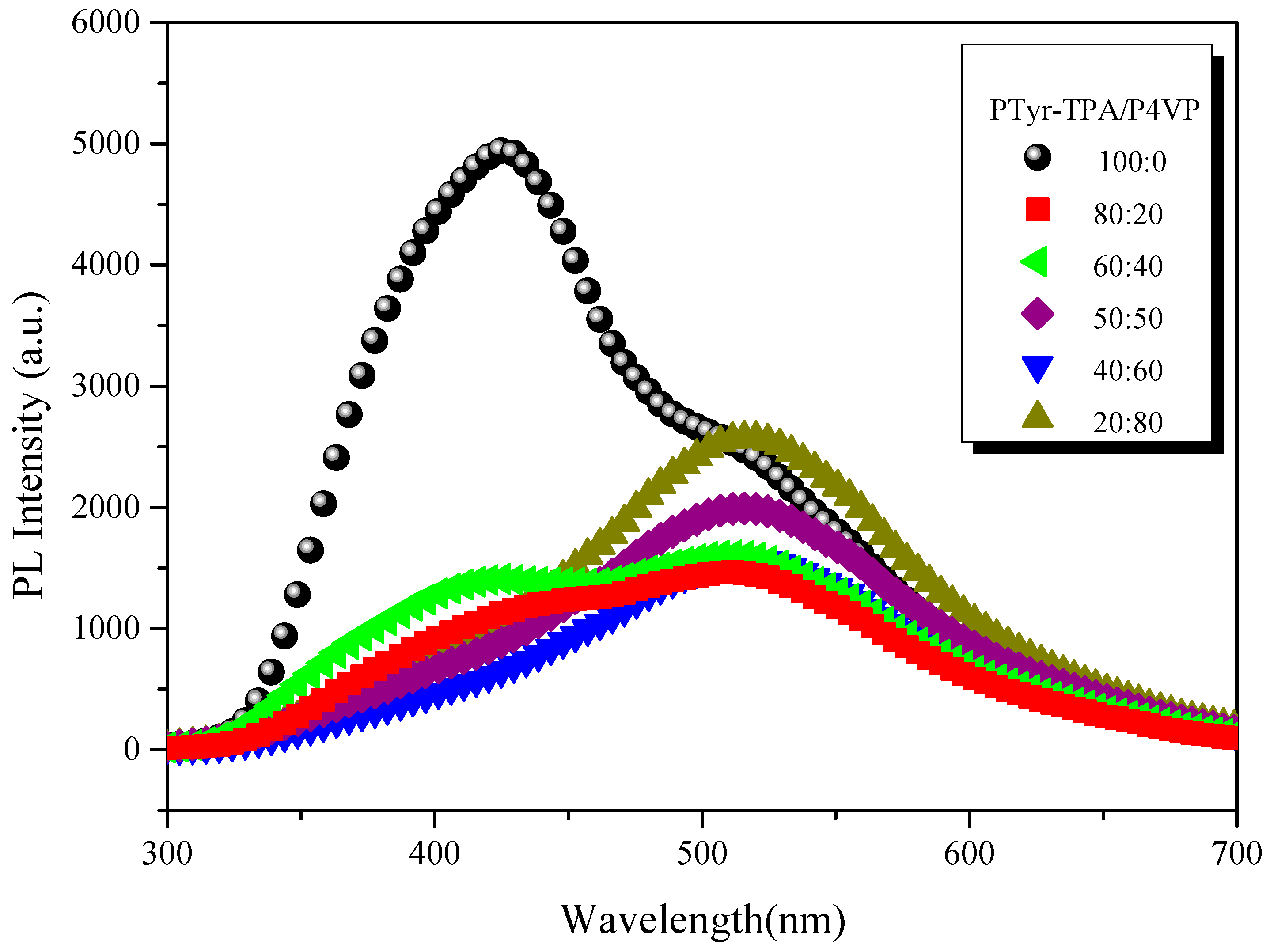
3.4.3. Emission Properties of PTyr-TPA/P4VP Blends
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alconel, S.N.; Baas, A.S.; Maynard, H.D. FDA-approved poly(ethylene glycol)–protein conjugate drugs. Polym. Chem. 2011, 2, 1442–1448. [Google Scholar] [CrossRef]
- Gauthier, M.A.; Klok, H.A. Peptide/protein–polymer conjugates: Synthetic strategies and design concepts. Chem. Commun. 2008, 23, 2591–2611. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Pang, X.H.; Dong, C.M. Dual Stimuli-Responsive Supramolecular Polypeptide-Based Hydrogel and Reverse Micellar Hydrogel Mediated by Host–Guest Chemistry. Adv. Funct. Mater. 2010, 20, 579–586. [Google Scholar] [CrossRef]
- Peng, S.M.; Chen, Y.; Hua, C.; Dong, C.M. Dendron-like Polypeptide/Linear Poly(ethylene oxide) Biohybrids with Both Asymmetrical and Symmetrical Topologies Synthesized via the Combination of Click Chemistry and Ring-Opening Polymerization. Macromolecules 2009, 42, 104–113. [Google Scholar] [CrossRef]
- Deming, T.J. Polypeptide Materials: New synthetic methods and applications. Adv. Mater. 1997, 9, 299–311. [Google Scholar] [CrossRef]
- Zhang, X.; Li, J.; Li, W.; Zhang, A. Synthesis and Characterization of Thermo- and pH-Responsive Double-Hydrophilic Diblock Copolypeptides. Biomacromolecules 2007, 8, 3557–3567. [Google Scholar] [CrossRef] [PubMed]
- Kricheldorf, H.R. Polypeptides and 100 Years of Chemistry of α-Amino Acid N-Carboxyanhydrides. Angew. Chem. Int. Ed. 2006, 45, 5752–5784. [Google Scholar] [CrossRef] [PubMed]
- Deming, T.J. Synthetic polypeptides for biomedical applications. Prog. Polym. Sci. 2007, 32, 858–875. [Google Scholar] [CrossRef]
- Li, P.C.; Lin, Y.C.; Chen, M.; Kuo, S.W. Self-assembled structures from Pegylated polypeptide block copolymers synthesized using a combination of ATRP, ROP, and click chemistry. Soft Matter 2013, 9, 11257–11269. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Z.; Li, Z. Thermoresponsive Polypeptides from Pegylated Poly-l-glutamates. Biomacromolecules 2011, 12, 2859–2863. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Shen, Y.; Fu, W.; Li, Z. Thermoresponsive Oligo(ethylene glycol) Functionalized Poly-l-cysteine. Macromolecules 2013, 46, 3753–3760. [Google Scholar] [CrossRef]
- Hua, C.; Dong, C.M.; Wei, Y. Versatile Strategy for the Synthesis of Dendronlike Polypeptide/Linear Poly(ε-caprolactone) Block Copolymers via Click Chemistry. Biomacromolecules 2009, 10, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.W.; Lee, H.F.; Huang, C.F.; Huang, C.J.; Chang, F.C. Synthesis and Self-Assembly of Helical Polypeptide-Random Coil Amphiphilic Diblock Copolymer. J. Polym. Sci. 2008, 46, 3108–3119. [Google Scholar] [CrossRef]
- Lin, Y.C.; Kuo, S.W. Polypeptide/Multi-Walled Carbon Nanotubes Hybrid Complex through Noncovalent Bonding Interactions. J. Polym. Sci. Part A 2014, 52, 321–329. [Google Scholar] [CrossRef]
- Huang, C.W.; Mohamed, M.G.; Zhu, C.Y.; Kuo, S.W. Functional Supramolecular Polypeptides Involving π–π Stacking and Multiple Hydrogen Bonding Interactions: A Conformation Study toward Carbon Nanotubes (CNTs) Dispersion. Macromolecules 2016, 49, 5374–5385. [Google Scholar] [CrossRef]
- Lin, Y.C.; Kuo, S.W. Hierarchical self-assembly and secondary structures of linear polypeptides graft onto POSS in the side chain through click chemistry. Polym. Chem. 2012, 3, 162–171. [Google Scholar] [CrossRef]
- Lin, Y.C.; Kuo, S.W. Hierarchical self-assembly structures of POSS-containing polypeptide block copolymers synthesized using a combination of ATRP, ROP and click chemistry. Polym. Chem. 2012, 3, 882–891. [Google Scholar] [CrossRef]
- Kuo, S.W.; Lee, H.F.; Huang, W.J.; Jeong, K.U.; Chang, F.C. Solid state and solution self-assembly of helical polypeptides tethered to polyhedral oligomeric silsesquioxanes. Macromolecules 2009, 42, 1619–1626. [Google Scholar] [CrossRef]
- Kuo, S.W.; Tsai, H.T. Control of peptide secondary structure on star shape polypeptides tethered to polyhedral oligomeric silsesquioxane nanoparticle through click chemistry. Polymer 2010, 51, 5695–5704. [Google Scholar] [CrossRef]
- Lin, Y.C.; Kuo, S.W. Self-assembly and secondary structures of linear polypeptides tethered to polyhedral oligomeric silsesquioxane nanoparticles through click chemistry. J. Polym. Sci. Part A 2011, 49, 2127–2137. [Google Scholar] [CrossRef]
- Painter, P.C.; Tang, W.L.; Graf, J.F.; Thomson, B.; Colema, M.M. Formation of Molecular Composites through Hydrogen-Bonding Interactions. Macromolecules 1991, 24, 3929–3936. [Google Scholar] [CrossRef]
- Deng, X.; Hao, J.; Yuan, M.; Xiong, C.; Zhao, S. Miscibility, thermal behaviour, morphology and mechanical properties of binary blends of poly [(R)-3-hydroxybutyrate] with poly (γ-benzyl-l-glutamate). Polym. Int. 2001, 50, 37–44. [Google Scholar] [CrossRef]
- Aoi, K.; Nakamura, R.; Okada, M. Polypeptide-synthetic polymer hybrids, 2 Miscibility of poly(vinyl alcohol) with polysarcosine. Macromol. Chem. Phys. 2000, 201, 1059–1066. [Google Scholar] [CrossRef]
- Kuo, S.W.; Chen, C.J. Using Hydrogen-Bonding Interactions To Control the Peptide Secondary Structures and Miscibility Behavior of Poly(l-glutamate) s with Phenolic Resin. Macromolecules 2011, 44, 7315–7326. [Google Scholar] [CrossRef]
- Kuo, S.W.; Chen, C.J. Functional Polystyrene Derivatives Influence the Miscibility and Helical Peptide Secondary Structures of Poly(γ-benzyl l-glutamate). Macromolecules 2012, 45, 2442–2452. [Google Scholar] [CrossRef]
- Lu, Y.S.; Lin, Y.C.; Kuo, S.W. Separated Coil and Chain Aggregation Behaviors on the Miscibility and Helical Peptide Secondary Structure of Poly(tyrosine) with Poly(4-vinylpyridine). Macromolecules 2012, 45, 6547–6556. [Google Scholar] [CrossRef]
- Mohamed, M.G.; Lu, F.H.; Hong, J.L.; Kuo, S.W. Strong Emission of 2,4,6-Triphenylpyridine-Functinalized Polytyrosine and Hydrogen-Bonding Interactions with Poly(4-vinylpyridine). Polym. Chem. 2015, 6, 6340–6350. [Google Scholar] [CrossRef]
- Lu, Y.S.; Bastakoti, B.P.; Pramanik, M.; Yamauchi, Y.; Kuo, S.W. Self-Assembly Structures of Poly(styrene-b-4-vinyl pyridine) Diblock Copolymer Blending with Poly(tyrosine) in Mercury Adsorption Application. RSC Adv. 2016, 6, 106866–106872. [Google Scholar] [CrossRef]
- Huang, C.W.; Chang, F.C.; Chu, Y.L.; Lai, C.C.; Lin, T.E.; Zhu, C.Y.; Kuo, S.W. A Solvent-Resistant Azide-based Hole Injection/Transporting Conjugated Polymer for Fluorescence and Phosphorescence Light-Emitting Diodes. J. Mater. Chem. C 2015, 3, 8142–8151. [Google Scholar] [CrossRef]
- Shih, H.K.; Chen, Y.H.; Chu, Y.L.; Cheng, C.C.; Chang, F.C.; Zhu, C.Y.; Kuo, S.W. Photo-Crosslinking of Pendent Uracil Units Provides Supramolecular Hole Injection/Transport Conducting Polymers for Highly Efficient Light-Emitting Diodes. Polymers 2015, 7, 804–818. [Google Scholar] [CrossRef]
- Wu, W.C.; Lai, H.J. Preparation of thermo-responsive electrospun nanofibers containing rhodamine-based fluorescent sensor for Cu2+ detection. J. Polym. Res. 2016, 23, 223. [Google Scholar] [CrossRef]
- Xavier, M.; Fabien, P.; Thilo, L.D.; Maxime, D.; Marcel, B.P.; Paul, A.A.; Shimon, W. Properties of Fluorescent Semiconductor Nanocrystals and their Application to Biological Labeling. Single Mol. 2001, 2, 261–276. [Google Scholar]
- Chenais, S.; Forget, S. Recent advances in solid-state organic lasers. Polym. Int. 2012, 61, 390–406. [Google Scholar] [CrossRef] [Green Version]
- Jenekhe, S.A.; Osaheni, J.A. Excimers and exciplexes of conjugated polymers. Science 1994, 265, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Friend, R.H.; Gymer, R.W.; Holmes, A.B.; Burroughes, J.H.; Marks, R.N.; Taliani, C.; Bradley, D.D.C.; Dos Santos, D.A.; Brédas, J.L.; Lögdlund, M.; et al. Electroluminescence in conjugated polymers. Nature 1999, 397, 121–128. [Google Scholar] [CrossRef]
- Pu, K.Y.; Liu, B. Conjugated Polyelectrolytes as Light-Up Macromolecular Probes for Heparin Sensing. Adv. Funct. Mater. 2009, 19, 277–284. [Google Scholar] [CrossRef]
- Zhang, X.; Cui, M.; Zhou, R.; Chen, C.; Zhang, G. Facile Synthesis of β-Diketone Alcohols for Combined Functionality: Initiation, Catalysis, and Luminescence. Macromol. Rapid Commun. 2014, 35, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Palmer, G.M.; Dewhirst, M.W.; Fraser, C.L. A dual-emissive-materials design concept enables tumour hypoxia imaging. Nat. Mater. 2009, 8, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Xie, Z.; Lam, J.W.Y.; Cheng, L.; Chen, H.; Qiu, C.; Kwok, H.S.; Zhan, X.; Liu, Y.; Zhu, D.; et al. Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole. Chem. Commun. 2001, 18, 1740–1741. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, S.; Lam, J.W.Y.; Lu, P.; Zhong, Y.; Wong, K.S.; Kwok, H.S.; Tang, B.Z. Creation of highly efficient solid emitter by decorating pyrene core with AIE-active tetraphenylethene peripheries. Chem. Commun. 2010, 46, 2221–2223. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhong, Y.; Lam, J.W.Y.; Lu, P.; Hong, Y.; Yu, Y.; Yue, Y.; Faisal, M.; Sung, H.H.Y.; Williams, I.D.; et al. Hyperbranched Conjugated Polysiloles: Synthesis, Structure, Aggregation-Enhanced Emission, Multicolor Fluorescent Photopatterning, and Superamplified Detection of Explosives. Macromolecules 2010, 43, 4921–4936. [Google Scholar] [CrossRef]
- Li, S.T.; Lin, Y.C.; Kuo, S.W.; Chuang, W.T.; Hong, J.L. Aggregation induced emission enhancement in relation to the secondary structures of poly (γ-benzyl-l-glutamate) containing a fluorescent tetraphenylthiophene moiety. Polym. Chem. 2012, 3, 2393–2402. [Google Scholar] [CrossRef]
- Shih, K.Y.; Lin, Y.C.; Hsiao, T.S.; Deng, S.L.; Kuo, S.W.; Hong, J.L. Amorphous and crystalline blends from polytyrosine and pyridine-functionalized anthracene: Hydrogen-bond interactions, conformations, intramolecular charge transfer and aggregation-induced emission. Polym. Chem. 2014, 5, 5765–5774. [Google Scholar] [CrossRef]
- Lin, R.C.; Mohamed, M.G.; Kuo, S.W. Benzoxazine/Triphenylamine-Based Dendrimers Prepared through Facile One-Pot Mannich Condensations. Macromol. Rapid Commun. 2017, 38, 1700251. [Google Scholar] [CrossRef] [PubMed]
- Lodge, T.P.; McLeish, T.C.B. Self-concentrations and effective glass transition temperatures in polymer blends. Macromolecules 2000, 33, 5278–5284. [Google Scholar] [CrossRef]
- Kwei, T.K. The effect of hydrogen bonding on the glass transition temperatures of polymer mixtures. J. Polym. Sci. Part C Polym. Lett. 1984, 22, 307–313. [Google Scholar] [CrossRef]
- Kuo, S.W.; Tung, P.H.; Chang, F.C. Syntheses and the study of strongly hydrogen-bonded poly (vinylphenol-b-vinylpyridine) diblock copolymer through anionic polymerization. Macromolecules 2006, 39, 9388–9395. [Google Scholar] [CrossRef]
- Lee, J.Y.; Painter, P.C.; Coleman, M.M. Hydrogen bonding in polymer blends. 4. Blends involving polymers containing methacrylic acid and vinylpyridine groups. Macromolecules 1988, 21, 954–960. [Google Scholar] [CrossRef]
- Kuo, S.W.; Lin, C.L.; Chang, F.C. The study of hydrogen bonding and miscibility in poly (vinylpyridines) with phenolic resin. Polymer 2002, 43, 3943–3949. [Google Scholar] [CrossRef]
- Bercea, M.; Morariu, S.; Teodorescu, M. Rheological investigation of poly(vinyl alcohol)/poly(N-vinyl pyrrolidone) mixtures in aqueous solution and hydrogel state. J. Polym. Res. 2016, 23, 142. [Google Scholar] [CrossRef]
- Papadopoulous, P.; Floudas, G.; Klok, H.A.; Schnell, I.; Pakula, T. Self-Assembly and Dynamics of Poly(γ-benzyl-l-glutamate) Peptides. Biomacromolecules 2004, 5, 81–91. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, Y.; Hielscher, R.; Voicescu, M.; Gross, J.; Hellwig, P. On the specificity of the amide VI band for the secondary structure of proteins. Vib. Spectrosc. 2011, 55, 258–266. [Google Scholar] [CrossRef]
- Auer, H.E.; McKnight, R.P. Two classes of beta pleated-sheet conformation in poly(l-tyrosine): A model for tertiary structure in native proteins. Biochemistry 1978, 14, 2798–2805. [Google Scholar] [CrossRef]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jheng, Y.-R.; Mohamed, M.G.; Kuo, S.-W. Supramolecular Interactions Induce Unexpectedly Strong Emissions from Triphenylamine-Functionalized Polytyrosine Blended with Poly(4-vinylpyridine). Polymers 2017, 9, 503. https://doi.org/10.3390/polym9100503
Jheng Y-R, Mohamed MG, Kuo S-W. Supramolecular Interactions Induce Unexpectedly Strong Emissions from Triphenylamine-Functionalized Polytyrosine Blended with Poly(4-vinylpyridine). Polymers. 2017; 9(10):503. https://doi.org/10.3390/polym9100503
Chicago/Turabian StyleJheng, Yu-Ru, Mohamed Gamal Mohamed, and Shiao-Wei Kuo. 2017. "Supramolecular Interactions Induce Unexpectedly Strong Emissions from Triphenylamine-Functionalized Polytyrosine Blended with Poly(4-vinylpyridine)" Polymers 9, no. 10: 503. https://doi.org/10.3390/polym9100503