Effect of the GO Reduction Method on the Dielectric Properties, Electrical Conductivity and Crystalline Behavior of PEO/rGO Nanocomposites

 , ,
, ,
Abstract
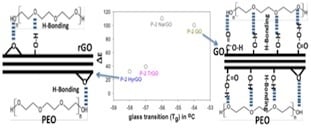
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Graphene Oxide (GO)
2.3. Synthesis of Reduced Graphene Oxide (rGO) by Different Reduction Methods
2.4. Fabrication of Polymer Nanocomposites by Solution Mixing
2.5. Characterization of the GO and rGO Fillers
2.6. Characterization of the Nanocomposites
3. Results and Discussion
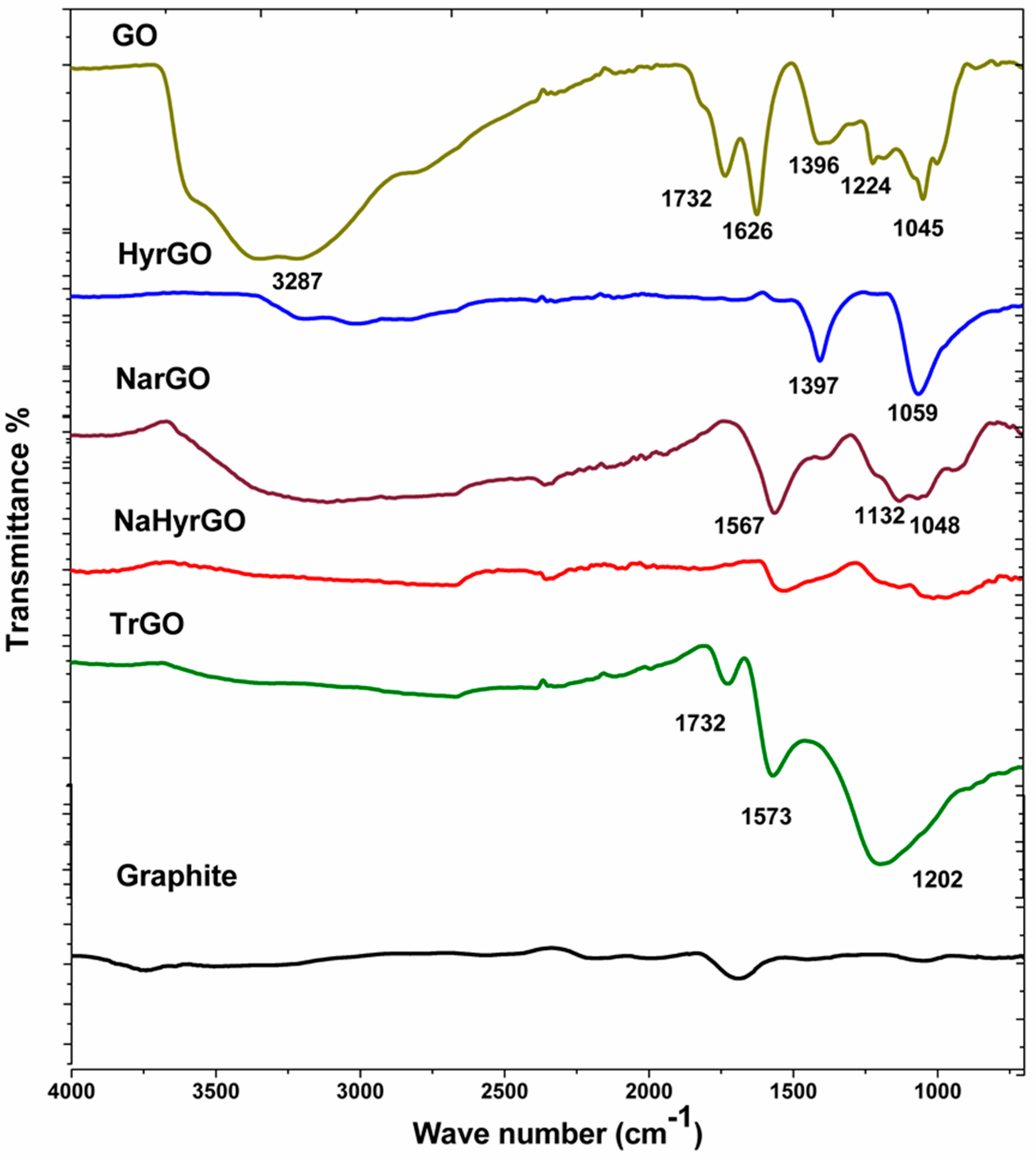
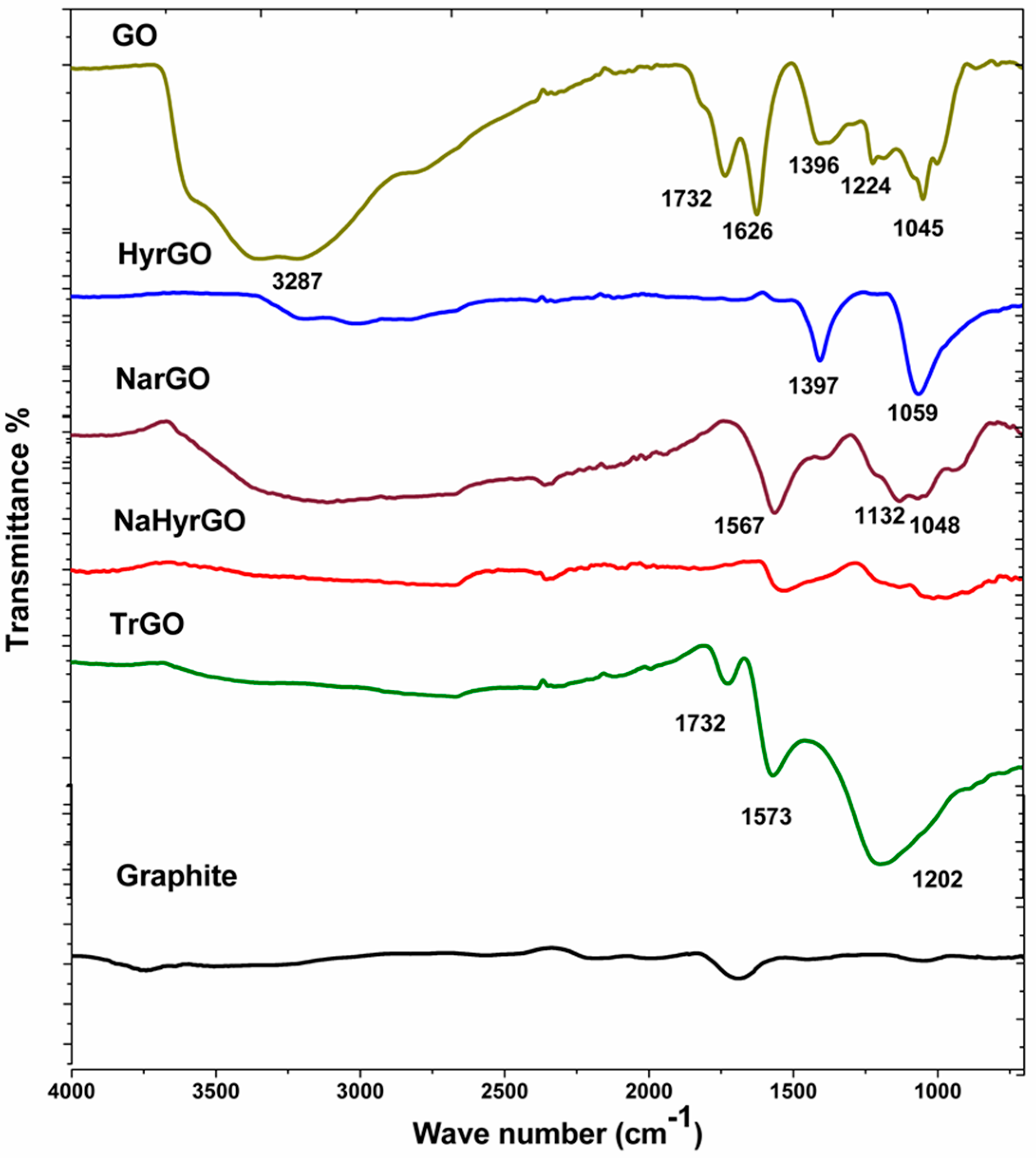
3.1. Analysis of FT-IR Spectra of Graphite, GO and Different rGOs
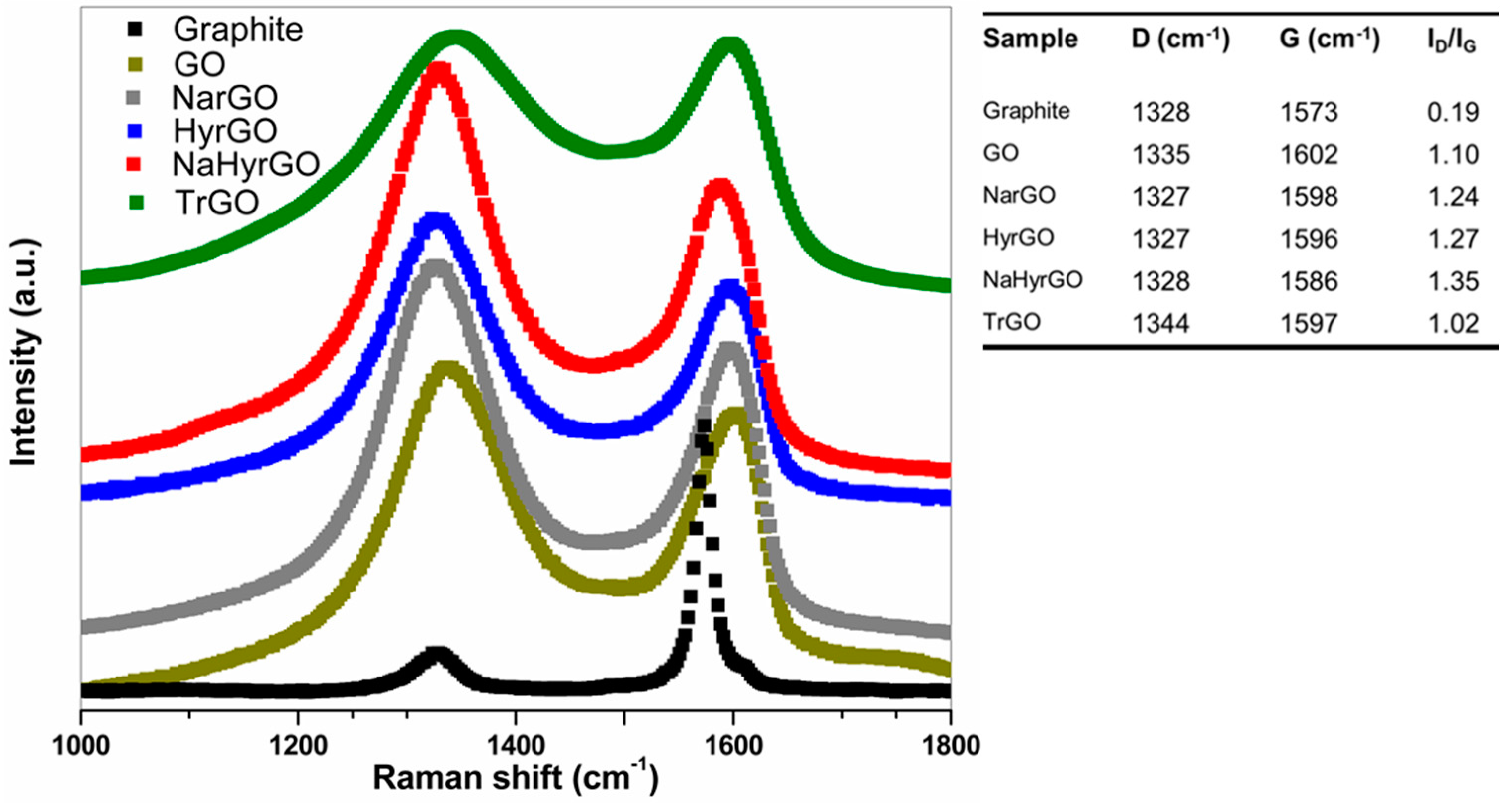
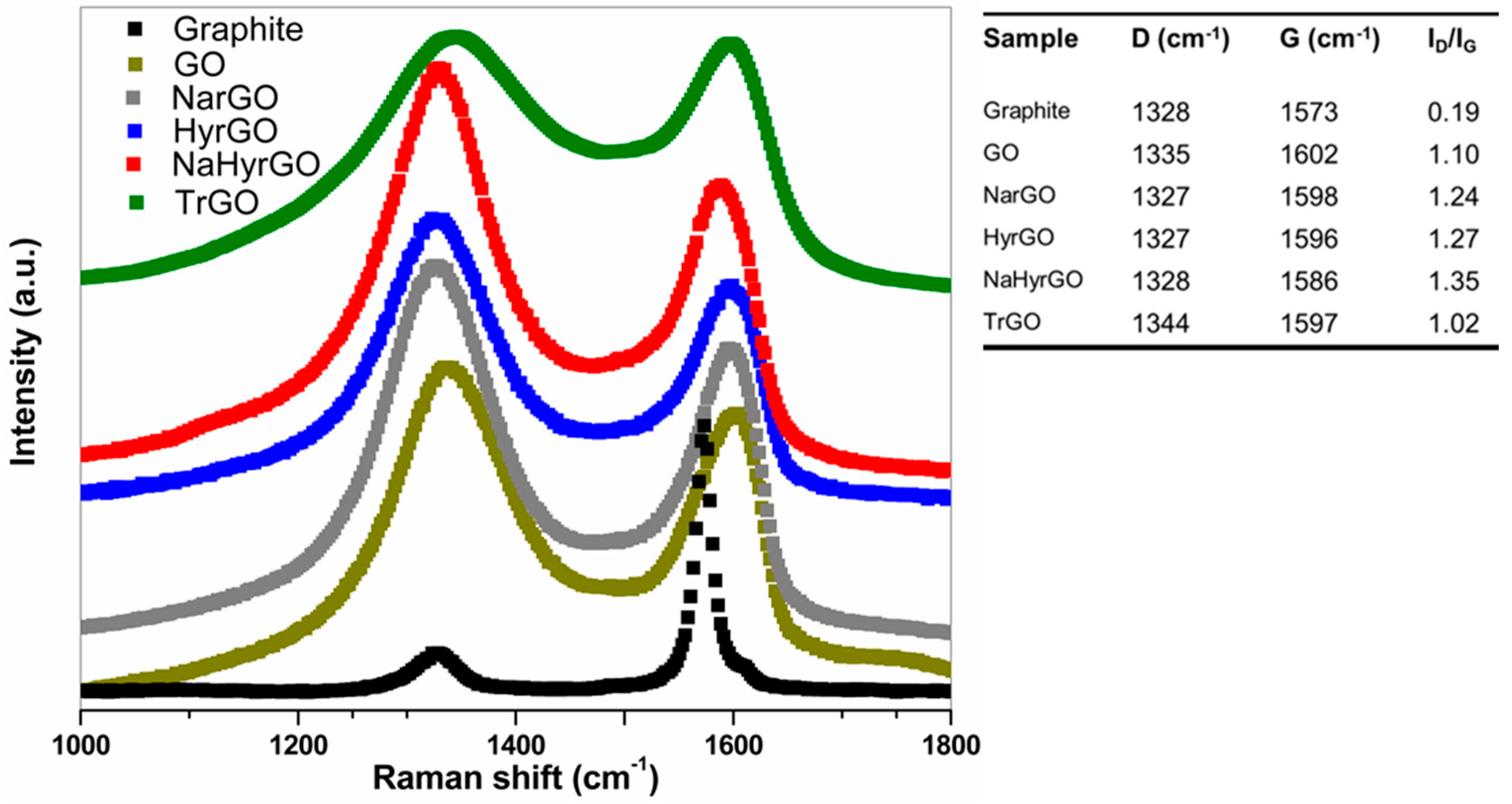
3.2. Analysis of Raman Spectra of Reduced GOs Synthesized by Different Methods
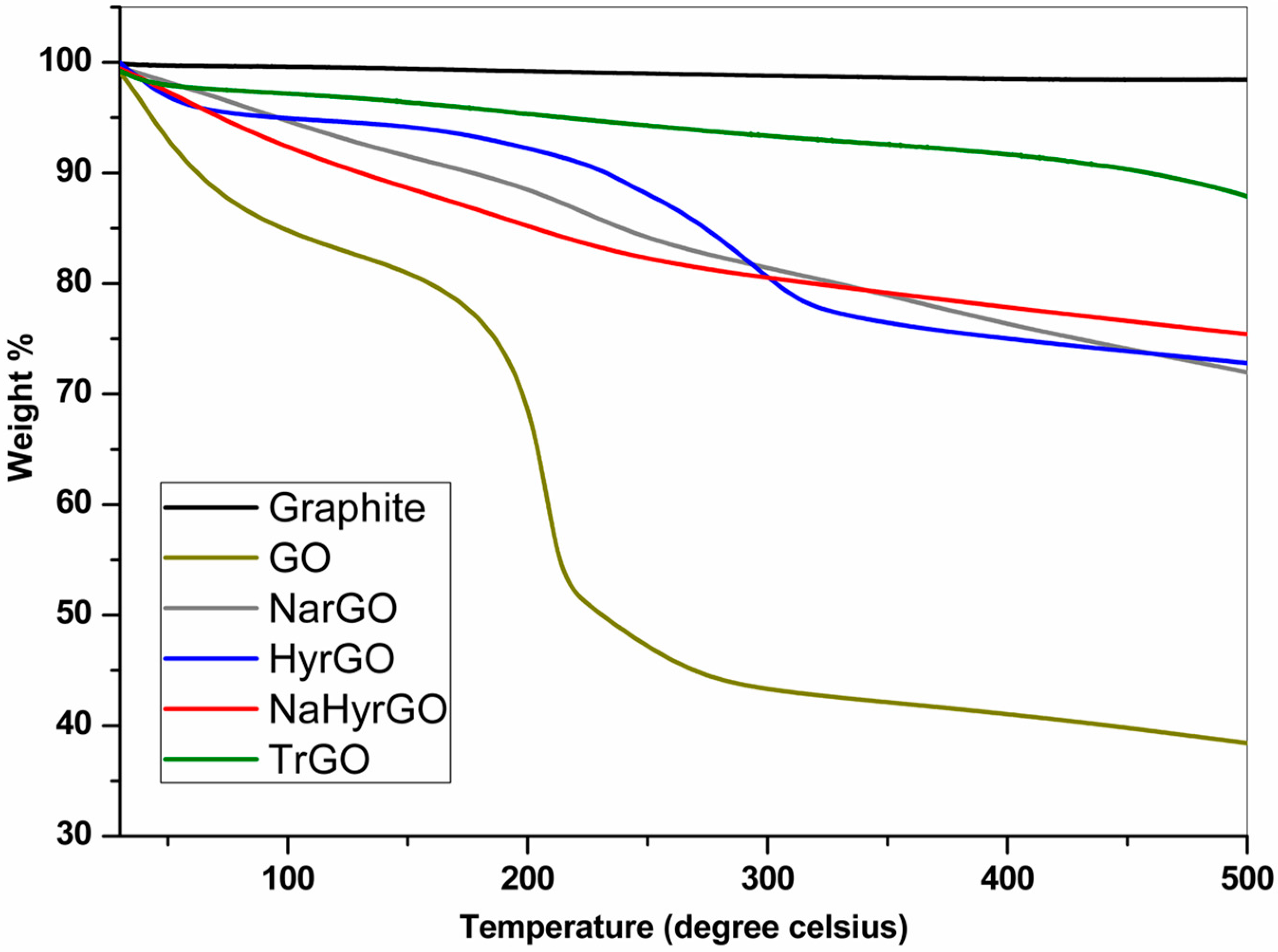
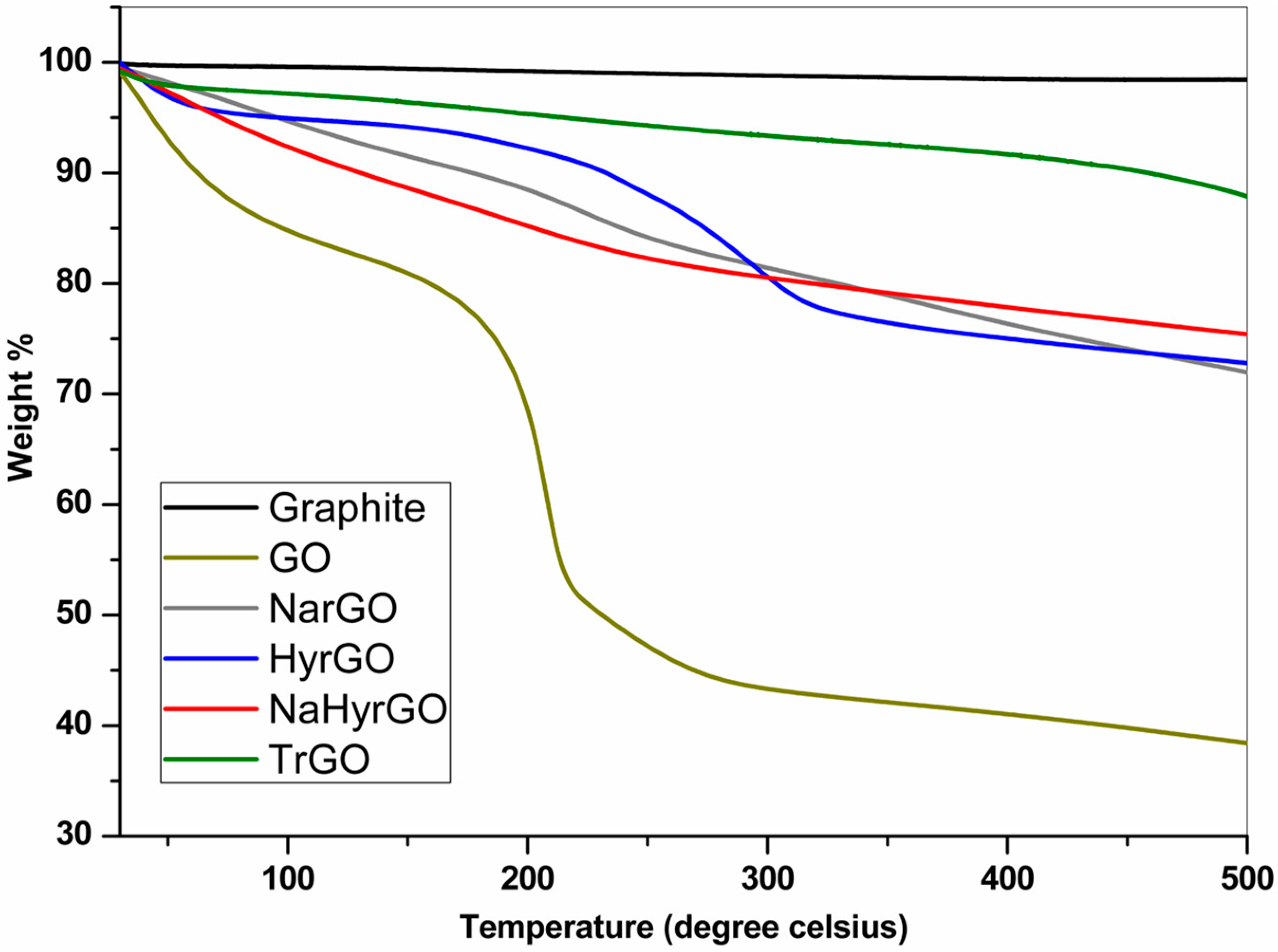
3.3. Thermal Degradation Behavior of Graphite, GO and rGOs
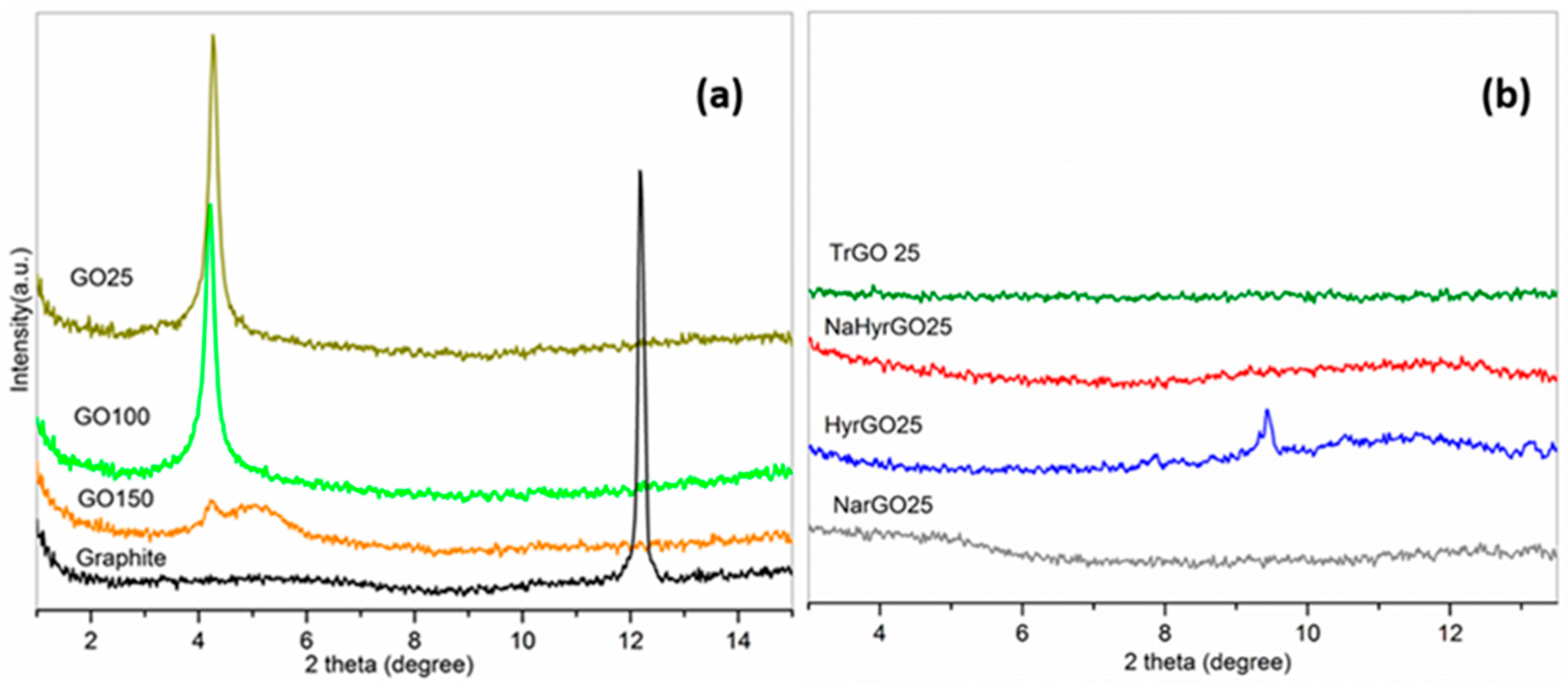
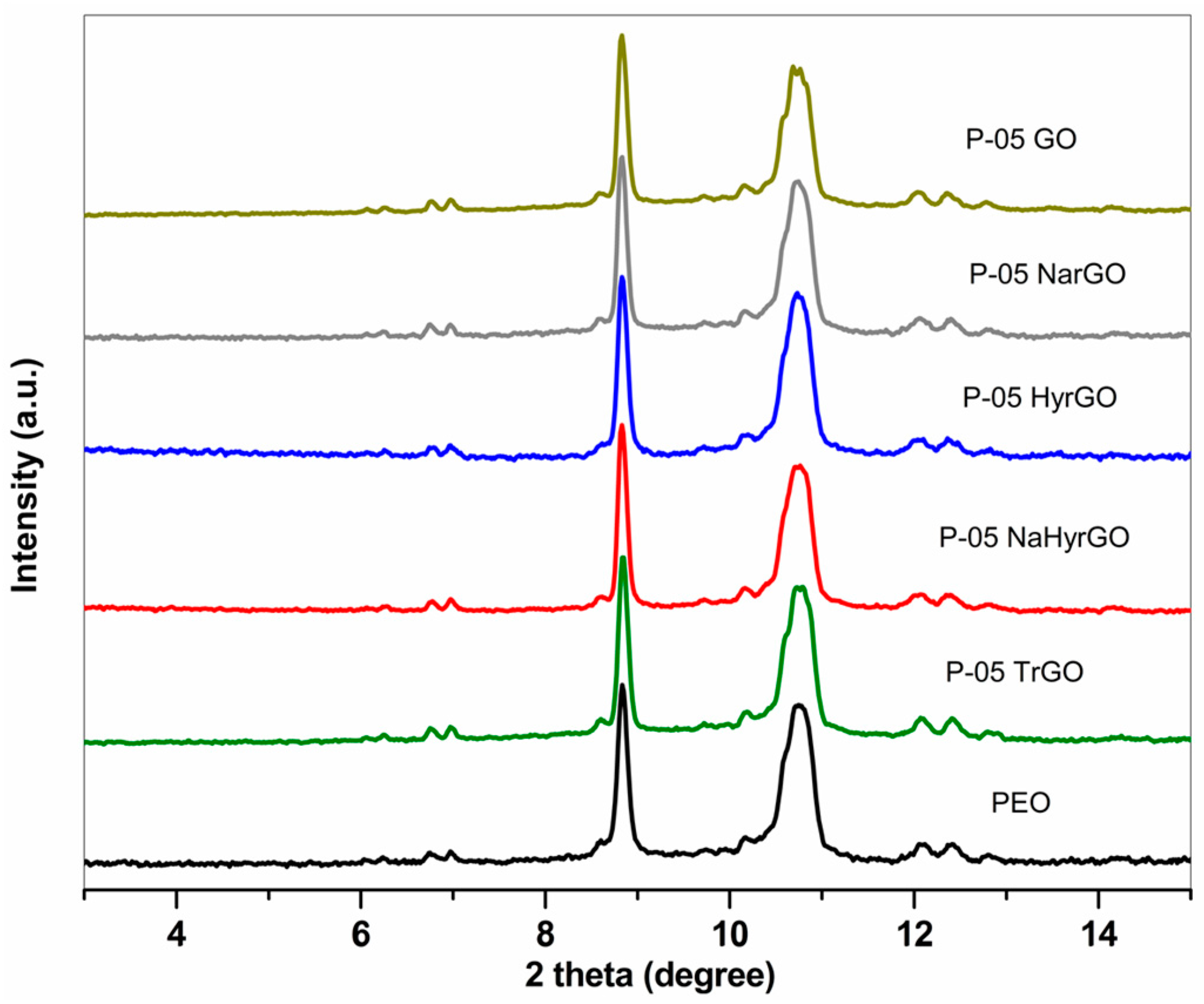
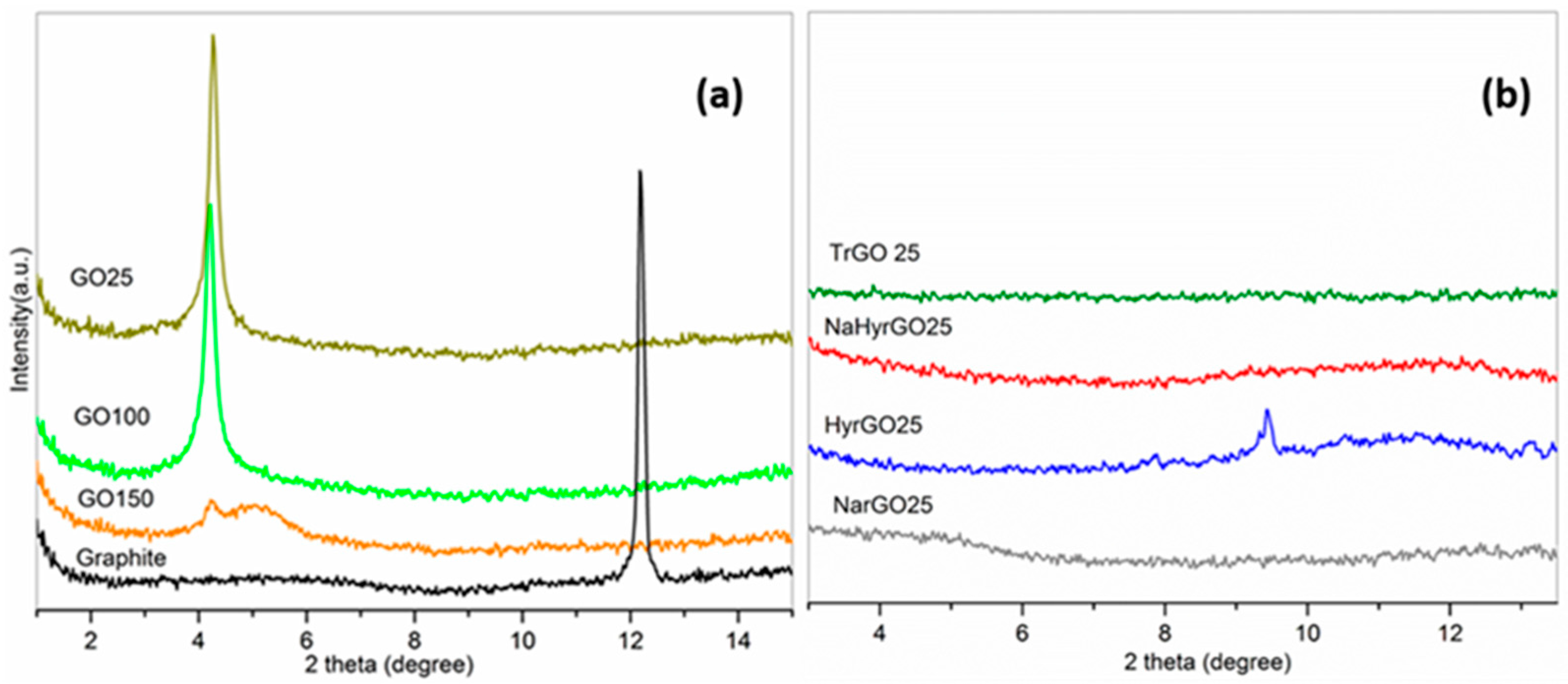
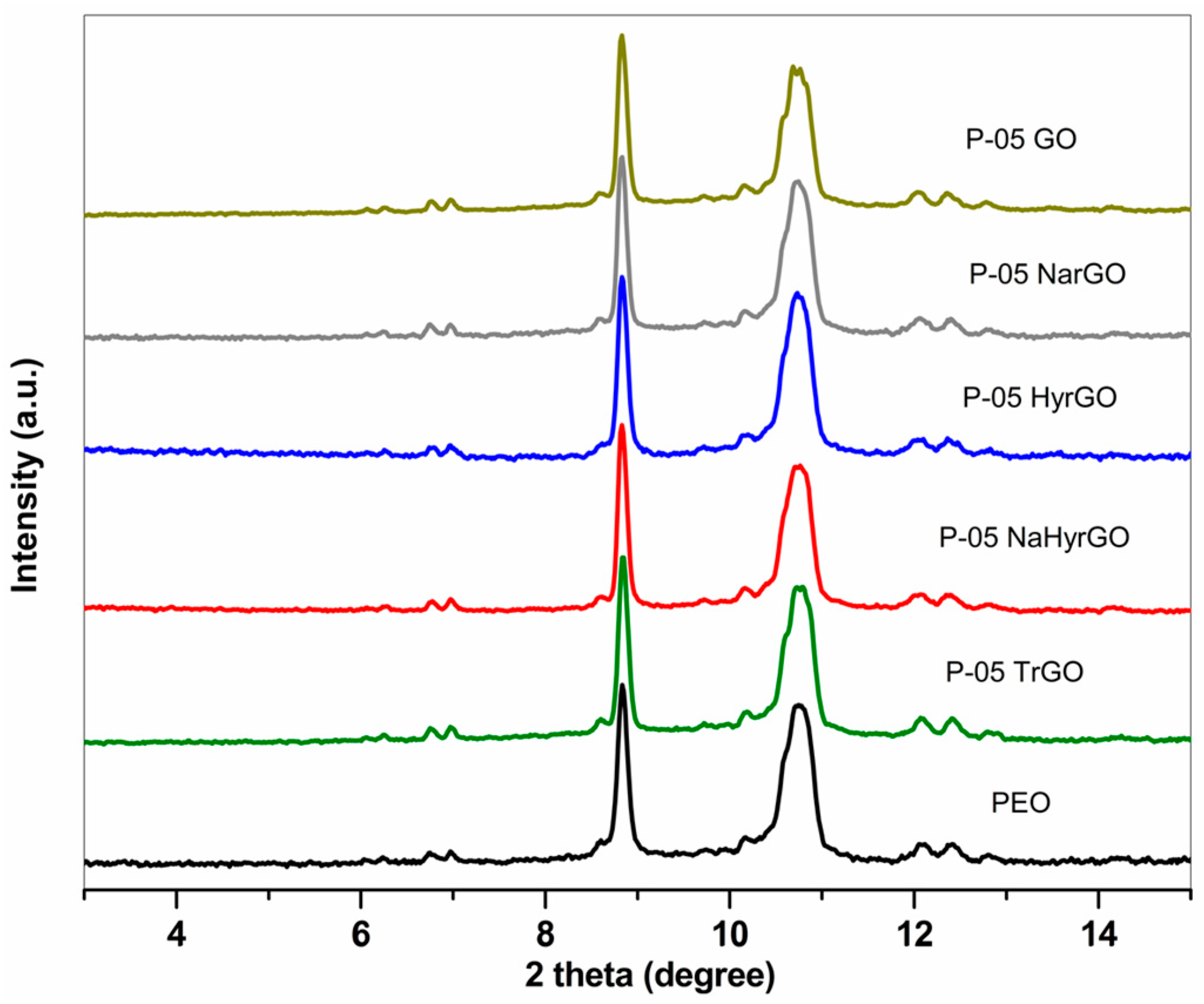
3.4. X-ray Diffraction Patterns of Graphite, GO and Various rGOs
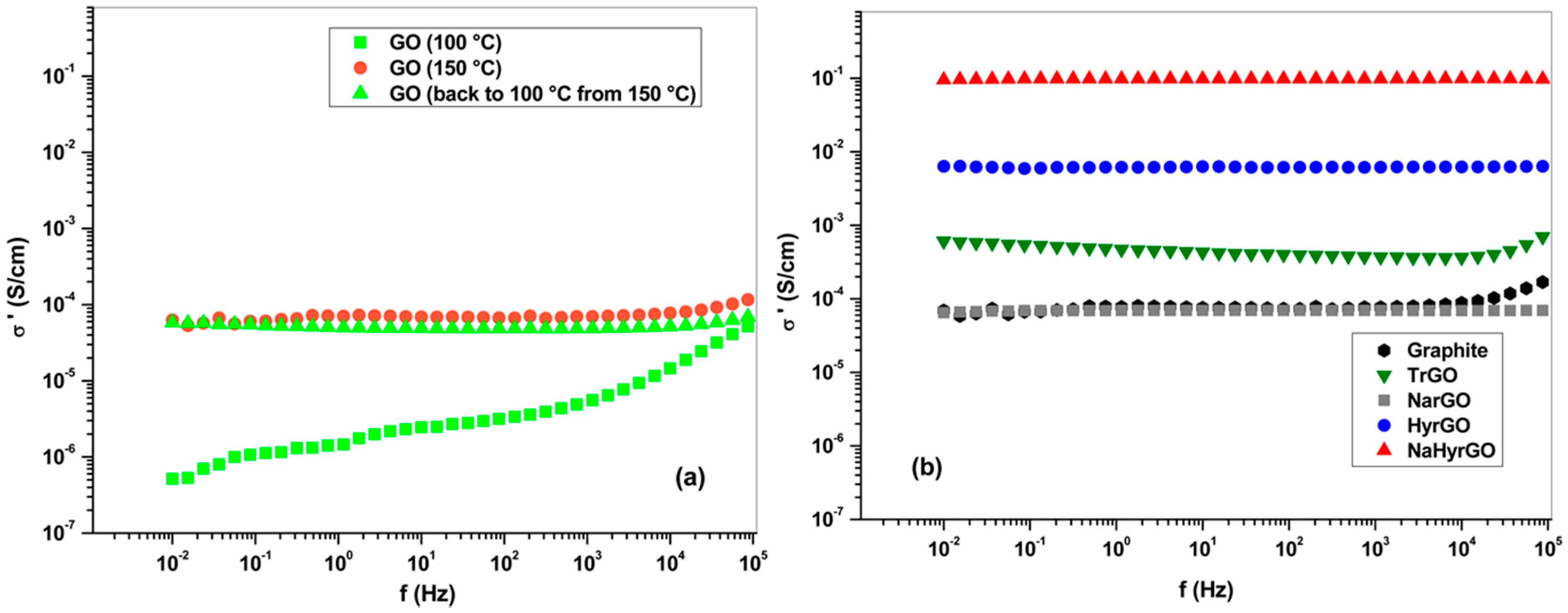
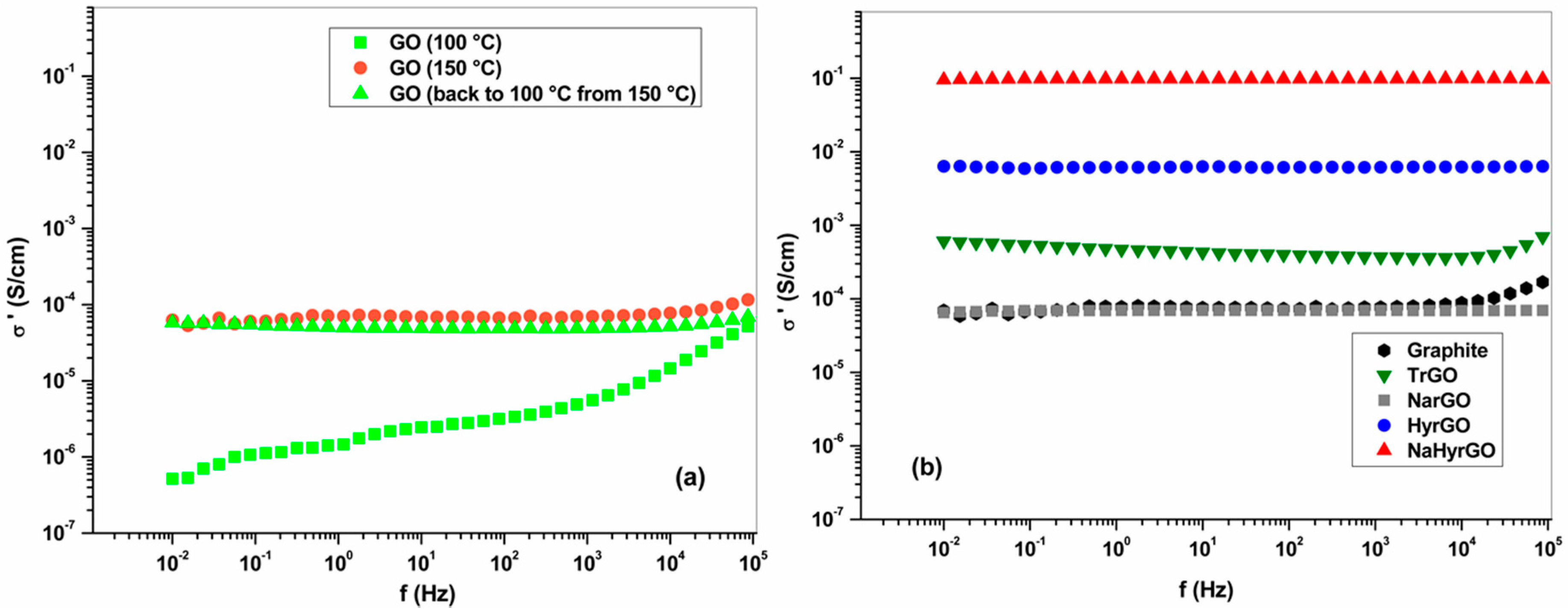
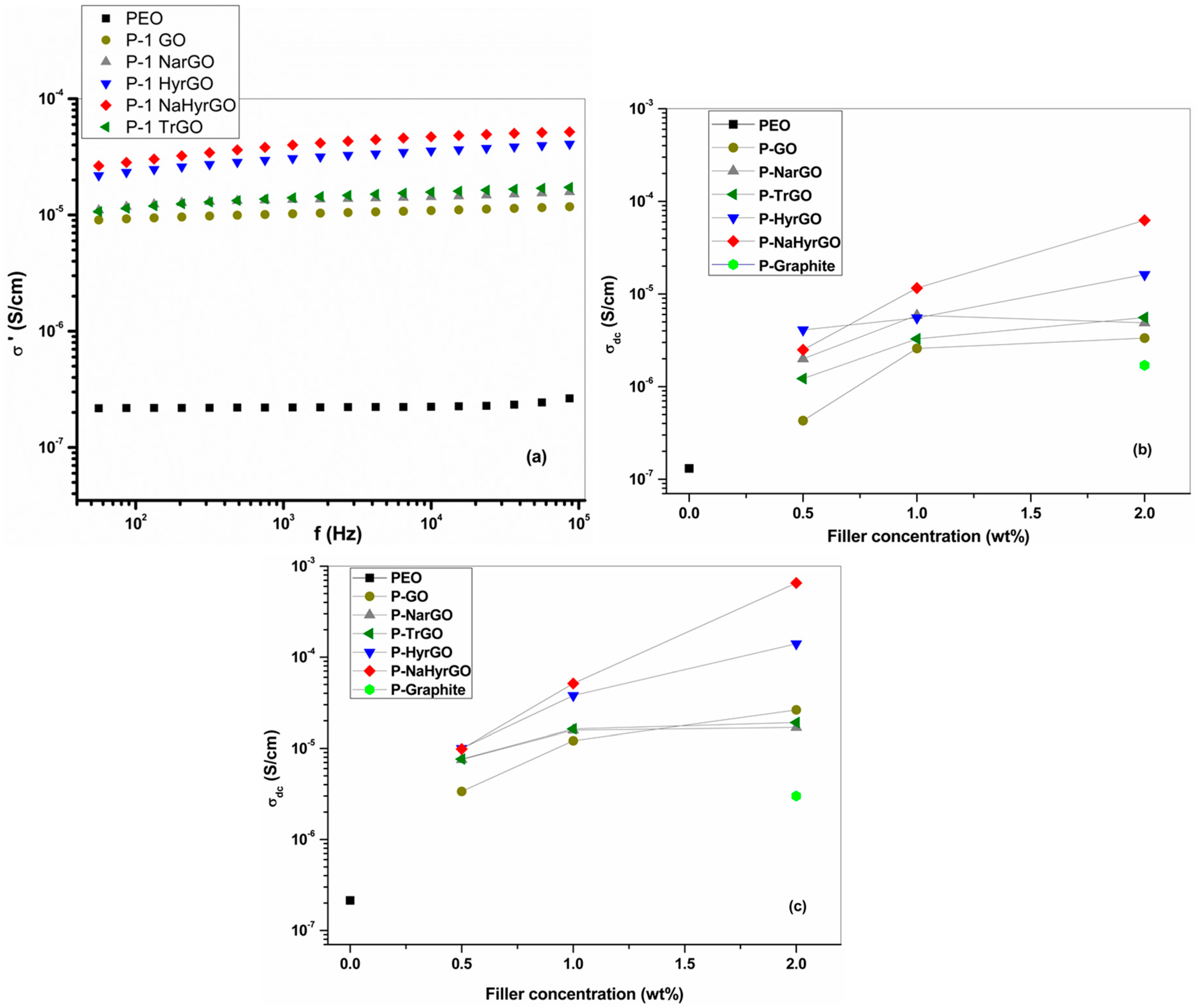
3.5. Bulk Electrical Conductivity of Graphite, GO and rGO Powders at Different Temperatures
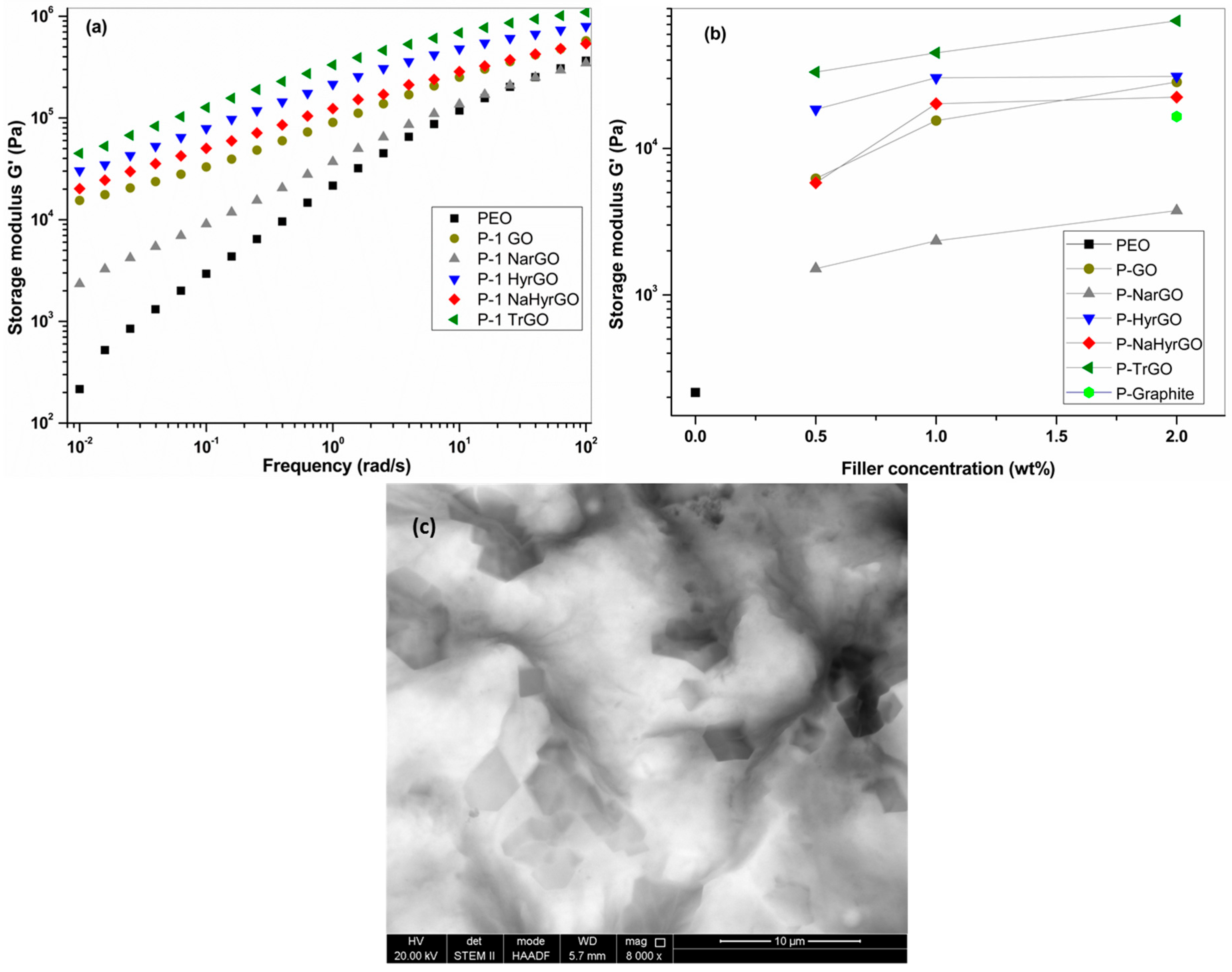
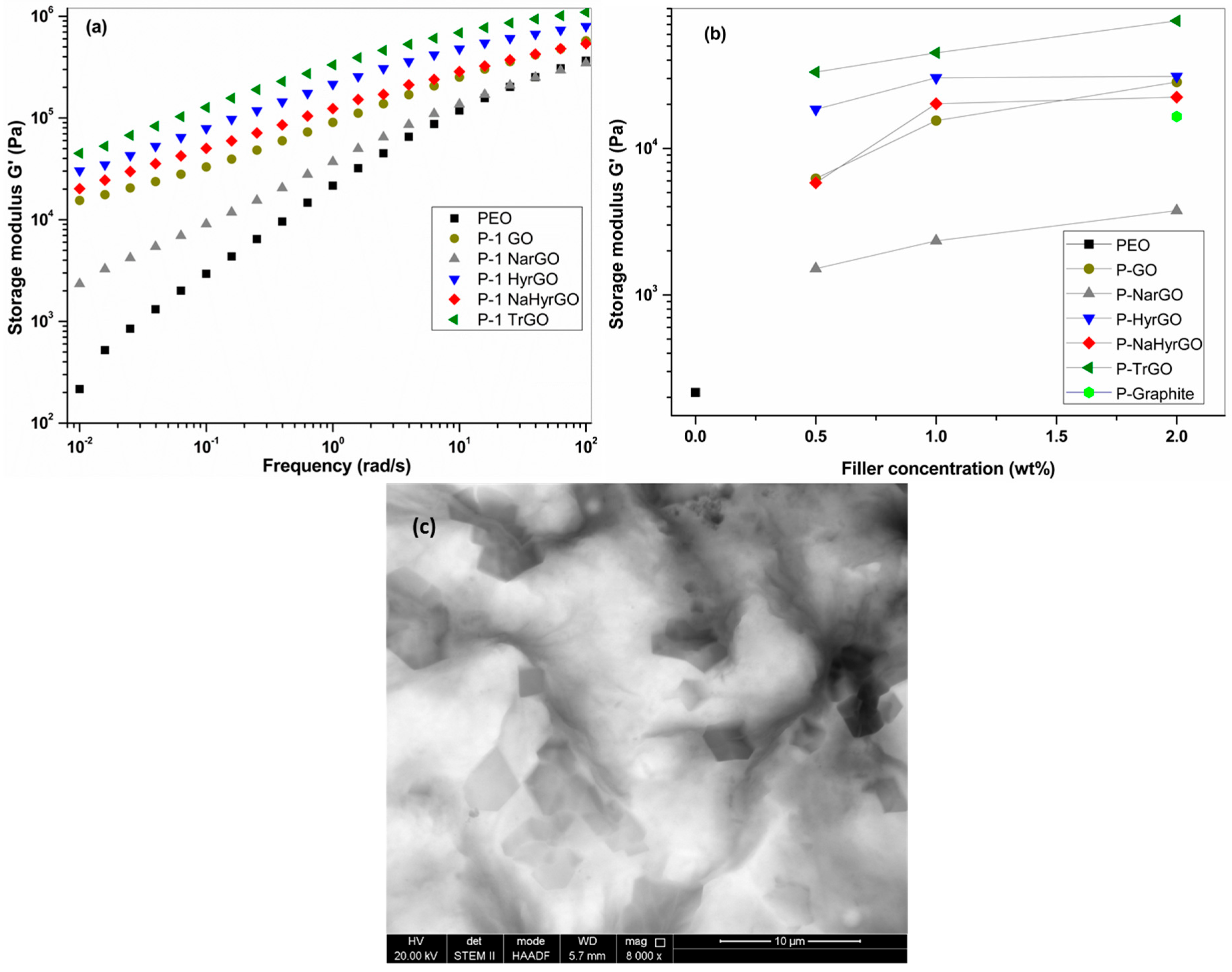
3.6. Effect of GO and Different Reduced GOs on the Linear Viscoelastic Behavior of PEO
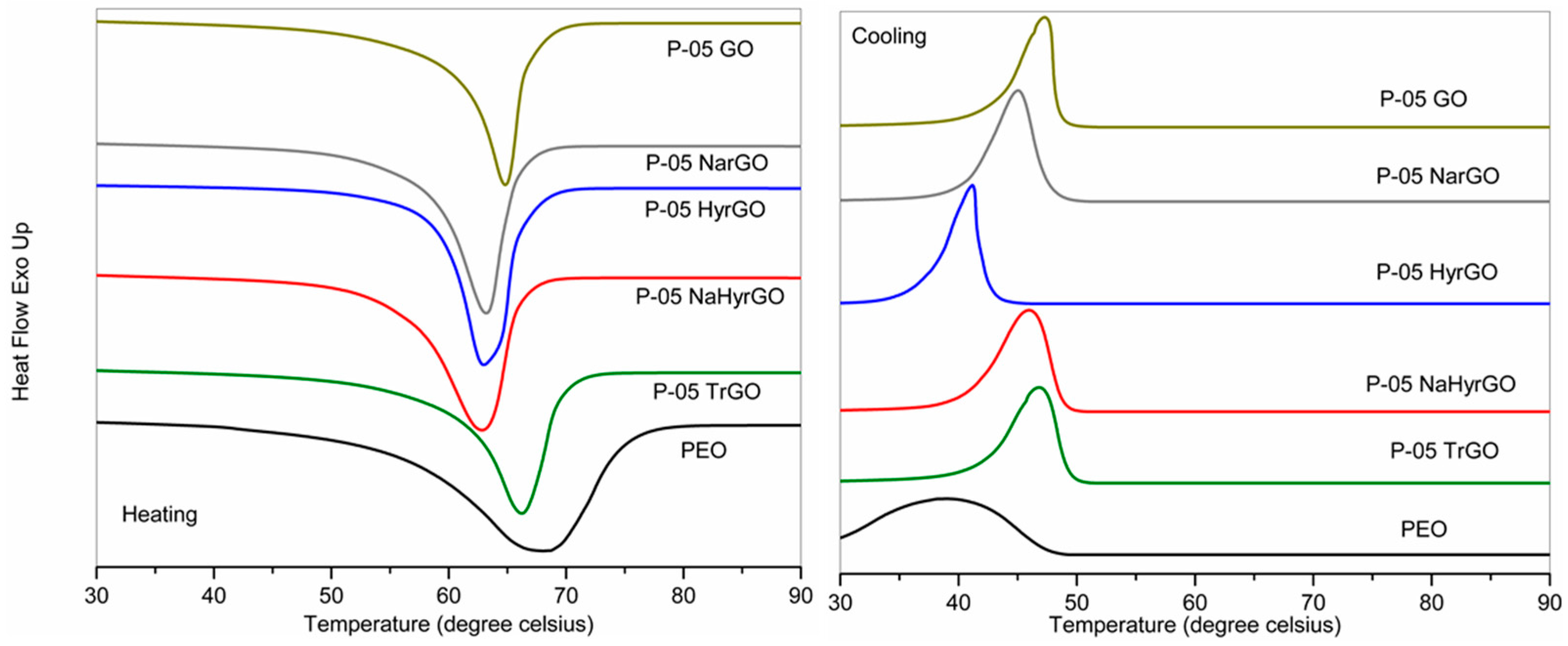
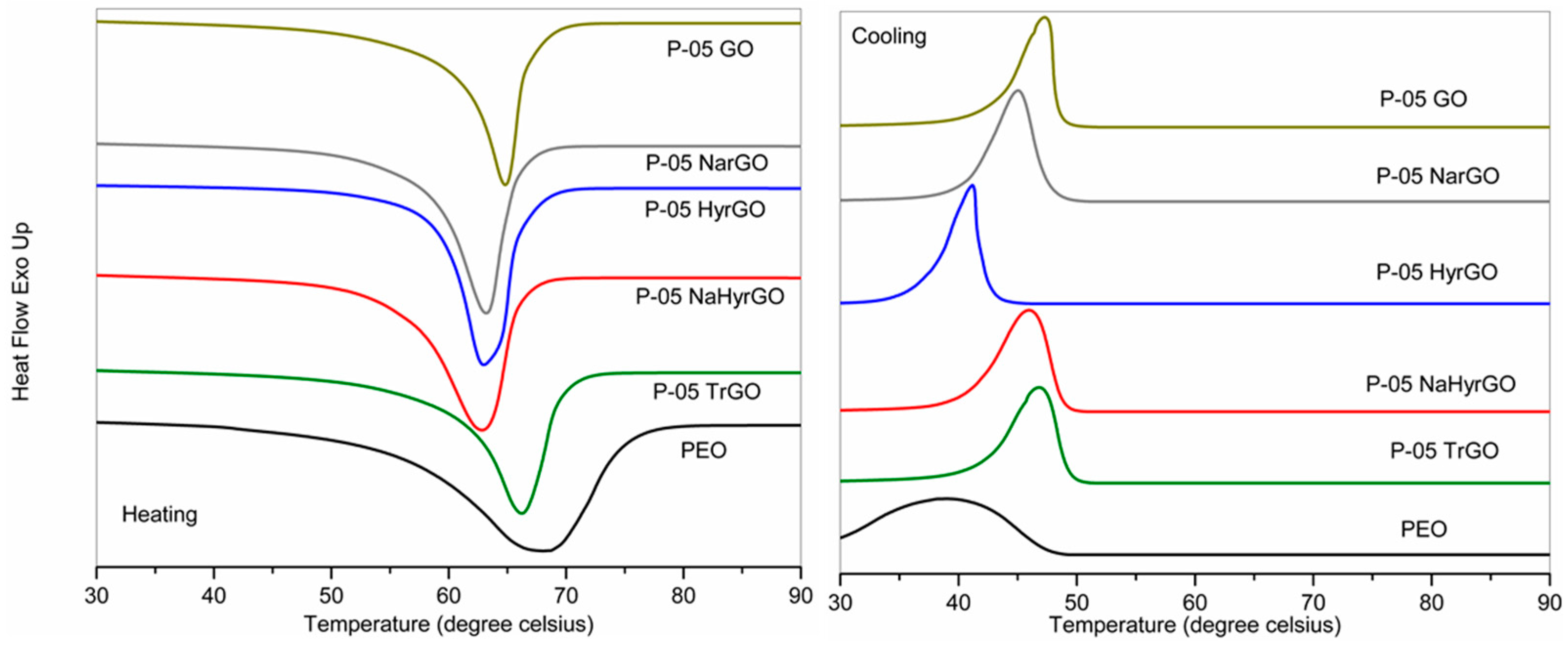
3.7. Effect of GO and Different rGOs on the Crystalline Behavior of PEO
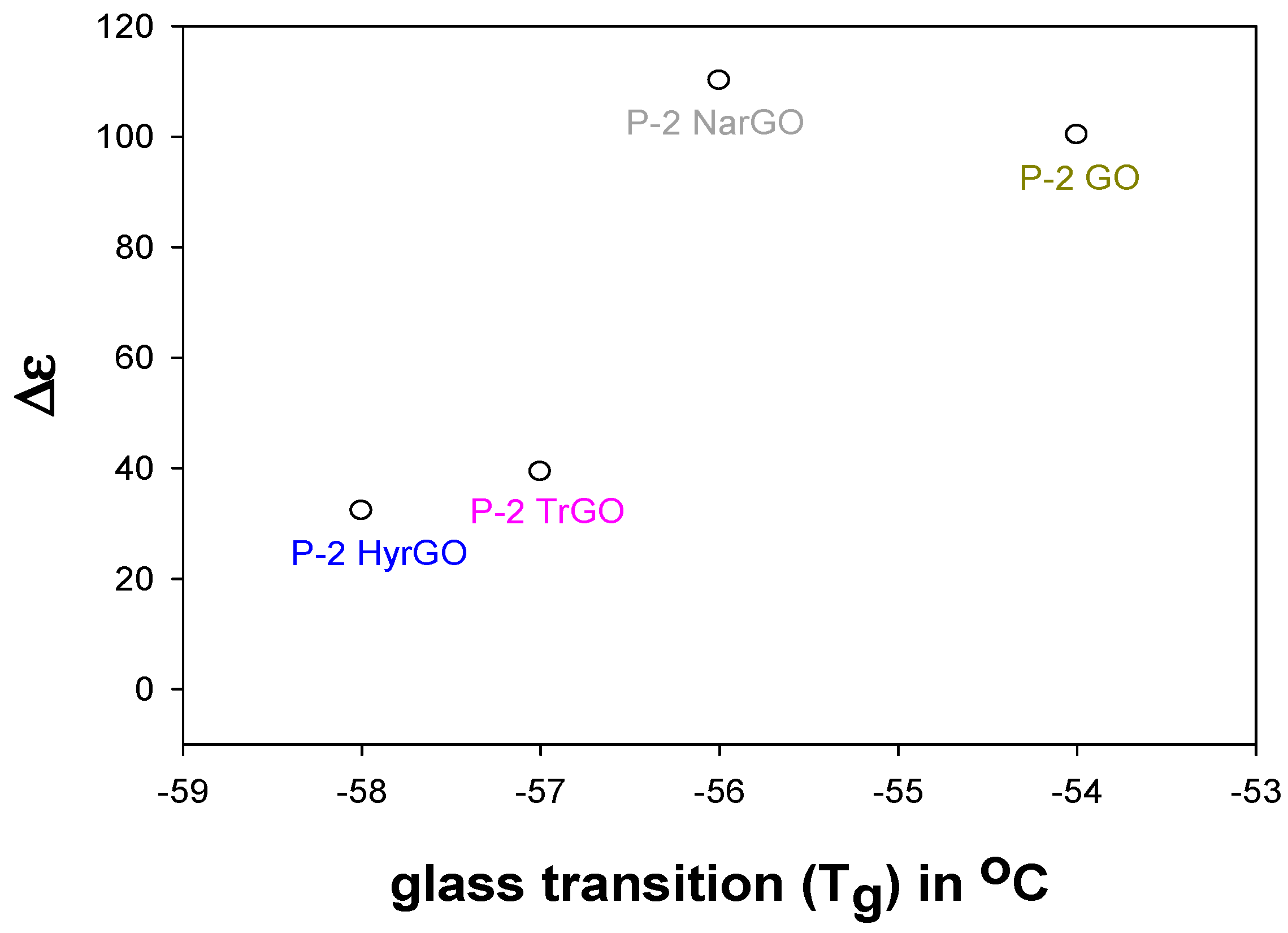
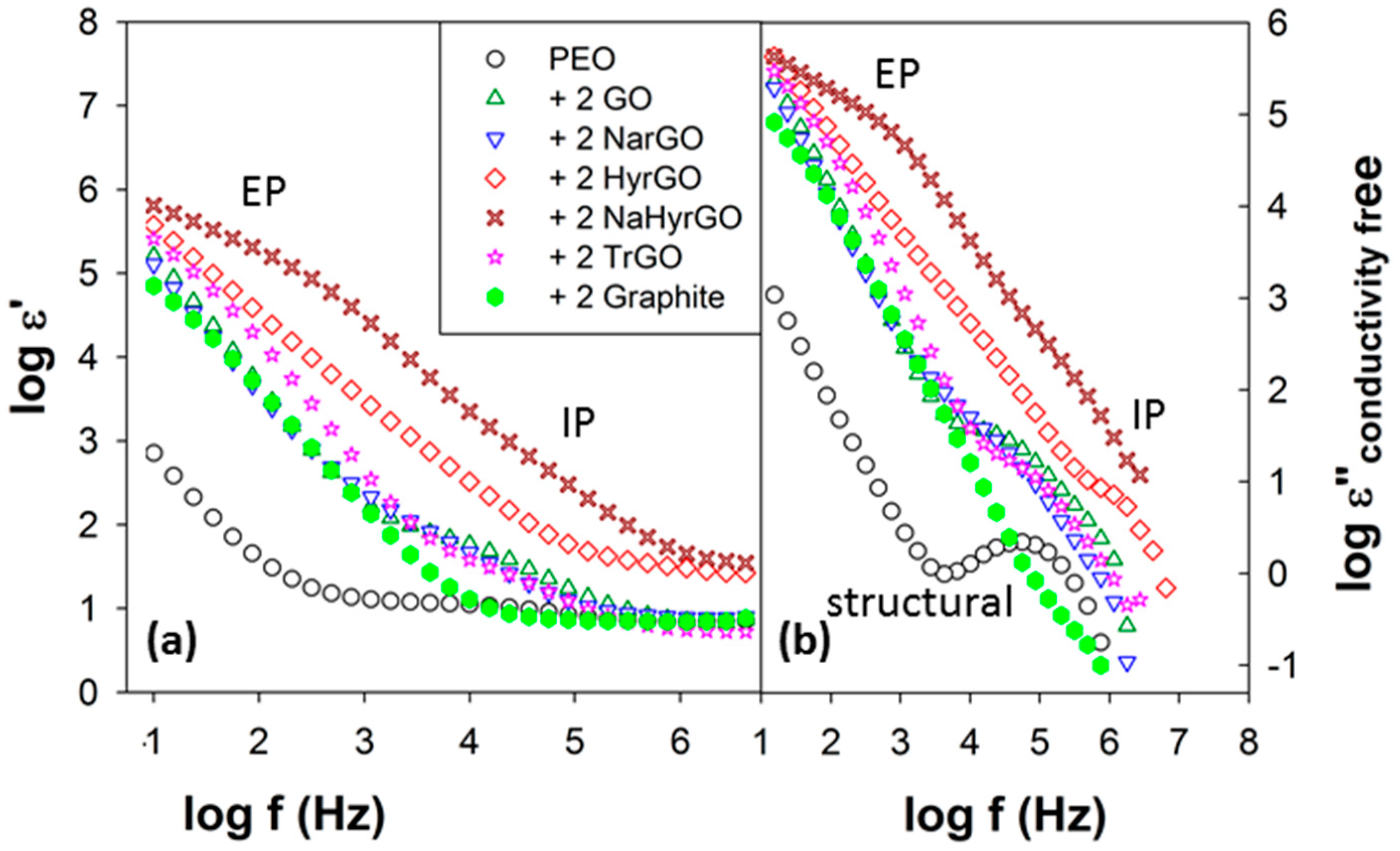
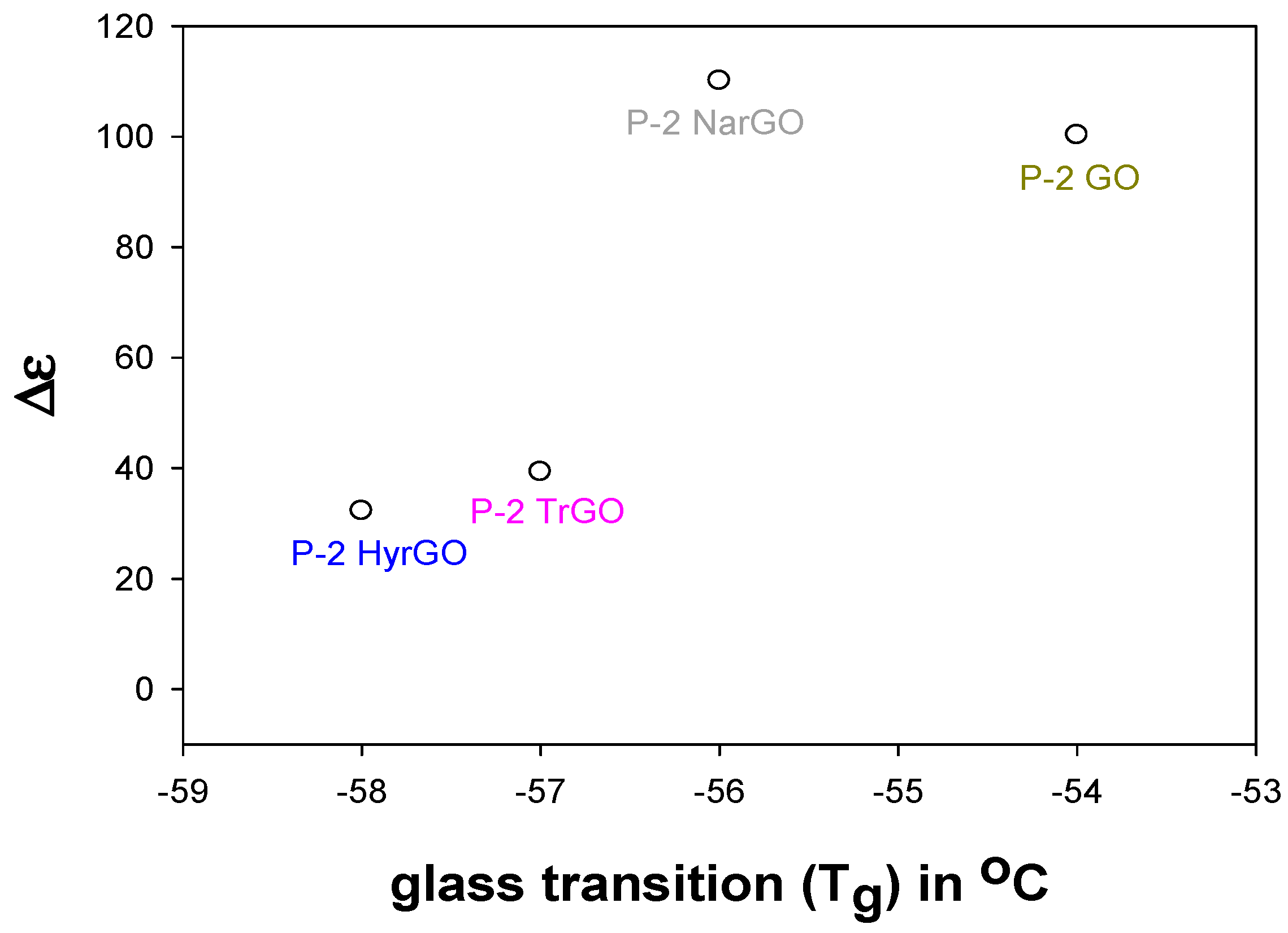
3.8. Effect of Polymer-Filler Interactions on the Glass Transition Temperature and the Dielectric Relaxation Strength of PEO-(r)GO Interfacial Polarization
3.9. Effect of GO and Different rGOs on the Electrical Conductivity of PEO
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Du, J.; Cheng, H.M. The fabrication, properties, and uses of graphene/polymer composites. Macro Mol. Chem. Phys. 2012, 213, 1060–1077. [Google Scholar] [CrossRef]
- Huang, X.; Yin, Z.; Wu, S.; Qi, X.; He, Q.; Zhang, Q.; Yan, Q.; Boey, F.; Zhang, H. Graphene-based materials: Synthesis, characterization, properties, and applications. Small 2011, 7, 1876–1902. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Abdala, A.A.; Macosko, C.W. Graphene/polymer nanocomposites. Macromolecules 2010, 43, 6515–6530. [Google Scholar] [CrossRef]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Potts, J.R.; Dreyer, D.R.; Bielawski, C.W.; Ruoff, R.S. Graphene-based polymer nanocomposites. Polymer 2011, 52, 5–25. [Google Scholar] [CrossRef]
- Zhu, Y.; Murali, S.; Cai, W.; Li, X.; Suk, J.W.; Potts, J.R.; Ruoff, R.S. Graphene and graphene oxide: Synthesis, properties, and applications. Adv. Mater. 2010, 22, 3906–3924. [Google Scholar] [CrossRef] [PubMed]
- Bagri, A.; Mattevi, C.; Acik, M.; Chabal, Y.J.; Chhowalla, M.; Shenoy, V.B. Structural evolution during the reduction of chemically derived graphene oxide. Nat. Chem. 2010, 2, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Tang, G.; Zhang, H.-B.; Yu, Z.-Z.; Yavari, F.; Koratkar, N.; Lim, S.-H.; Lee, M.-W. In Situ thermal reduction of graphene oxide for high electrical conductivity and low percolation threshold in polyamide 6 nanocomposites. Compos. Sci. Technol. 2012, 72, 284–289. [Google Scholar] [CrossRef]
- Li, H.; Pang, S.; Wu, S.; Feng, X.; Mullen, K.; Bubeck, C. Layer-by-layer assembly and UV photoreduction of graphene–polyoxometalate composite films for electronics. J. Am. Chem. Soc. 2011, 133, 9423–9429. [Google Scholar] [CrossRef] [PubMed]
- Mural, P.K.S.; Sharma, M.; Madras, G.; Bose, S. A critical review on in situ reduction of graphene oxide during preparation of conducting polymeric nanocomposites. RSC Adv. 2015, 5, 32078–32087. [Google Scholar] [CrossRef]
- Thomassin, J.-M.; Trifkovic, M.; Alkarmo, W.; Detrembleur, C.; Jérôme, C.; Macosko, C. Poly (methyl methacrylate)/graphene oxide nanocomposites by a precipitation polymerization process and their dielectric and rheological characterization. Macromolecules 2014, 47, 2149–2155. [Google Scholar] [CrossRef]
- Liu, H.; Yang, W.; He, F.; Li, Y.; Yang, X.; Zhang, K. Graphene-based composite with microwave absorption property prepared by in situ reduction. Polym. Compos. 2014, 35, 461–467. [Google Scholar] [CrossRef]
- Bai, X.; Zhai, Y.; Zhang, Y. Green approach to prepare graphene-based composites with high microwave absorption capacity. J. Phys. Chem. C 2011, 115, 11673–11677. [Google Scholar] [CrossRef]
- Su, Y.J.; Xie, G.Z.; Chen, J.; Du, H.F.; Zhang, H.L.; Yuan, Z.; Ye, Z.B.; Du, X.S.; Taia, H.L.; Jiang, Y.D. Reduced graphene oxide-polyethylene oxide hybrid films for toluene sensing at room temperature. RSC Adv. 2016, 6, 97840–97847. [Google Scholar] [CrossRef]
- Cheng, S.; Smith, D.M.; Li, C.Y. Anisotropic ion transport in a poly(ethylene oxide)-liclo4 solid state electrolyte templated by graphene oxide. Macromolecules 2015, 48, 4503–4510. [Google Scholar] [CrossRef]
- Kammoun, M.; Berg, S.; Ardebili, H. Flexible thin-film battery based on graphene-oxide embedded in solid polymer electrolyte. Nanoscale 2015, 7, 17516–17522. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.W.; Lee, K.S.; Lee, Y.W.; Bang, J.H. Poly(ethylene oxide)/graphene oxide nanocomposites: Structure, properties and shape memory behavior. Polym. Bull. 2015, 72, 1937–1948. [Google Scholar] [CrossRef]
- Jagtap, S.B.; Kushwaha, R.K.; Ratna, D. Novel green method of preparation of a poly (ethylene oxide)/graphene nanocomposite using organic salt assisted dispersion. RSC Adv. 2015, 5, 30555–30563. [Google Scholar] [CrossRef]
- Tong, Y.; Lin, Y.; Wang, S.D.; Song, M. A study of crystallisation of poly (ethylene oxide) and polypropylene on graphene surface. Polymer 2015, 73, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Alazmi, A.; Rasul, S.; Patole, S.P.; Costa, P. Comparative study of synthesis and reduction methods for graphene oxide. Polyhedron 2016, 116, 153–161. [Google Scholar] [CrossRef]
- Rasul, S.; Alazmi, A.; Jaouen, K.; Hedhili, M.N.; Costa, P. Rational design of reduced graphene oxide for superior performance of supercapacitor electrodes. Carbon 2017, 111, 774–781. [Google Scholar] [CrossRef]
- Marcano, D.C.; Kosynkin, D.V.; Berlin, J.M.; Sinitskii, A.; Sun, Z.; Slesarev, A.; Alemany, L.B.; Lu, W.; Tour, J.M. Improved synthesis of graphene oxide. ACS Nano 2010, 4, 4806–4814. [Google Scholar] [CrossRef] [PubMed]
- WooáLee, J.; BináKim, S. Enhanced Cr(VI) removal using iron nanoparticle decorated graphene. Nanoscale 2011, 3, 3583–3585. [Google Scholar]
- Stankovich, S.; Dikin, D.A.; Piner, R.D.; Kohlhaas, K.A.; Kleinhammes, A.; Jia, Y.; Wu, Y.; Nguyen, S.T.; Ruoff, R.S. Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon 2007, 45, 1558–1565. [Google Scholar] [CrossRef]
- Song, N.-J.; Chen, C.-M.; Lu, C.; Liu, Z.; Kong, Q.-Q.; Cai, R. Thermally reduced graphene oxide films as flexible lateral heat spreaders. J. Mater. Chem. A 2014, 2, 16563–16568. [Google Scholar] [CrossRef]
- Kremer, F.; Schönhals, A. Broadband Dielectric Spectroscopy; Springer: Berlin, Germany, 2003. [Google Scholar]
- Wübbenhorst, M.; van Turnhout, J. Analysis of complex dielectric spectra. I. One-dimensional derivative techniques and three-dimensional modelling. J. Non-Cryst. Solids 2002, 305, 40–49. [Google Scholar] [CrossRef]
- Samir, M.A.S.A.; Alloin, F.; Sanchez, J.-Y.; Dufresne, A. Cellulose nanocrystals reinforced poly (oxyethylene). Polymer 2004, 45, 4149–4157. [Google Scholar] [CrossRef]
- Vidotto, G.; Levy, D.; Kovacs, A. Cristallisation et fusion des polymères autoensemencés. Kolloid Z. Z. Polym. 1969, 230, 289–305. [Google Scholar] [CrossRef]
- Szabó, T.; Berkesi, O.; Forgó, P.; Josepovits, K.; Sanakis, Y.; Petridis, D.; Dékány, I. Evolution of surface functional groups in a series of progressively oxidized graphite oxides. Chem. Mater. 2006, 18, 2740–2749. [Google Scholar] [CrossRef]
- Gao, C.; Yu, X.-Y.; Xu, R.-X.; Liu, J.-H.; Huang, X.-J. Alooh-reduced graphene oxide nanocomposites: One-pot hydrothermal synthesis and their enhanced electrochemical activity for heavy metal ions. ACS Appl. Mater. Interfaces 2012, 4, 4672–4682. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Wang, X.; Chang, R.; Zhao, X. Polyaniline decorated graphene sheet suspension with enhanced electrorheology. Soft Matter 2012, 8, 294–297. [Google Scholar] [CrossRef]
- Hsiao, M.-C.; Liao, S.-H.; Lin, Y.-F.; Wang, C.-A.; Pu, N.-W.; Tsai, H.-M.; Ma, C.-C.M. Preparation and characterization of polypropylene-graft-thermally reduced graphite oxide with an improved compatibility with polypropylene-based nanocomposite. Nanoscale 2011, 3, 1516–1522. [Google Scholar] [CrossRef] [PubMed]
- Dresselhaus, M.S.; Jorio, A.; Hofmann, M.; Dresselhaus, G.; Saito, R. Perspectives on carbon nanotubes and graphene raman spectroscopy. Nano Lett. 2010, 10, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Kudin, K.N.; Ozbas, B.; Schniepp, H.C.; Prud’Homme, R.K.; Aksay, I.A.; Car, R. Raman spectra of graphite oxide and functionalized graphene sheets. Nano Lett. 2008, 8, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, Y.; Tian, Y.; Yang, Z.; Xiao, Q.; Guo, X.; Jing, L.; Zhao, Y.; Yan, Y.; Feng, J. Insight into the capacitive properties of reduced graphene oxide. ACS Appl. Mater. Interfaces 2014, 6, 2248–2254. [Google Scholar] [CrossRef] [PubMed]
- Marciano, O.; Gonen, S.; Levy, N.; Teblum, E.; Yemini, R.; Nessim, G.D.; Ruthstein, S.; Elbaz, L. Modulation of oxygen content in graphene surfaces using temperature-programmed reductive annealing: Electron paramagnetic resonance and electrochemical study. Langmuir 2016, 32, 11672–11680. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, D.R.; Park, S.; Bielawski, C.W.; Ruoff, R.S. The chemistry of graphene oxide. Chem. Soc. Rev. 2010, 39, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bao, Q.; Tang, L.A.L.; Zhong, Y.; Loh, K.P. Hydrothermal dehydration for the “green” reduction of exfoliated graphene oxide to graphene and demonstration of tunable optical limiting properties. Chem. Mater. 2009, 21, 2950–2956. [Google Scholar] [CrossRef]
- Shen, J.; Hu, Y.; Shi, M.; Lu, X.; Qin, C.; Li, C.; Ye, M. Fast and facile preparation of graphene oxide and reduced graphene oxide nanoplatelets. Chem. Mater. 2009, 21, 3514–3520. [Google Scholar] [CrossRef]
- Panteny, S.; Stevens, R.; Bowen, C.R. The frequency dependent permittivity and ac conductivity of random electrical networks. Ferroelectrics 2005, 319, 199–208. [Google Scholar] [CrossRef]
- Marinho, B.; Ghislandi, M.; Tkalya, E.; Koning, C.E.; de With, G. Electrical conductivity of compacts of graphene, multi-wall carbon nanotubes, carbon black, and graphite powder. Powder Technol. 2012, 221, 351–358. [Google Scholar] [CrossRef]
- Vermant, J.; Ceccia, S.; Dolgovskij, M.K.; Maffettone, P.L.; Macosko, C.W. Quantifying dispersion of layered nanocomposites via melt rheology. J. Rheol. 2007, 51, 429–450. [Google Scholar] [CrossRef]
- Macosko, C. Rheology: Principles, Measurements, and Applications; VCH: New York, NY, USA, 1994. [Google Scholar]
- Chrissopoulou, K.; Andrikopoulos, K.S.; Fotiadou, S.; Bollas, S.; Karageorgaki, C.; Christofilos, D.; Voyiatzis, G.A.; Anastasiadis, S.H. Crystallinity and chain conformation in PEO/layered silicate nanocomposites. Macromolecules 2011, 44, 9710–9722. [Google Scholar] [CrossRef]
- Homminga, D.; Goderis, B.; Dolbnya, I.; Reynaers, H.; Groeninckx, G. Crystallization behavior of polymer/montmorillonite nanocomposites. Part I. Intercalated poly(ethylene oxide)/montmorillonite nanocomposites. Polymer 2005, 46, 11359–11365. [Google Scholar] [CrossRef]
- Xu, C.; Wu, D.; Lv, Q.; Yan, L. Crystallisation temperature as the probe to detect polymer-filler compatibility in the poly(ε-caprolactone) composites with acetylated cellulose nanocrystal. J. Phys. Chem. C 2017, 121, 18615–18624. [Google Scholar] [CrossRef]
- Perez, R.A.; Lopez, J.V.; Hoskins, J.N.; Zhang, B.; Grayson, S.M.; Casas, M.T.; Puiggali, J.; Müller, A.J. Nucleation and antinucleation effects of functionalized carbon nanotubes on cyclic and linear poly(ε-caprolactones). Macromolecules 2014, 47, 3553–3566. [Google Scholar] [CrossRef]
- Konishi, T.; Sakatsuji, W.; Fukao, K.; Miyamoto, Y. Temperature dependence of lamellar thickness in isothermally crystallized poly(butylene terephthalate). Macromolecules 2016, 49, 2272–2280. [Google Scholar] [CrossRef]
- Karan, N.; Pradhan, D.; Thomas, R.; Natesan, B.; Katiyar, R. Solid polymer electrolytes based on polyethylene oxide and lithium trifluoro-methane sulfonate (PEO–LiCF3SO3): Ionic conductivity and dielectric relaxation. Solid State Ion. 2008, 179, 689–696. [Google Scholar] [CrossRef]
- Leys, J.; Wübbenhorst, M.; Menon, C.P.; Rajesh, R.; Thoen, J.; Glorieux, C.; Nockemann, P.; Thijs, B.; Binnemans, K.; Longuemart, S. Temperature dependence of the electrical conductivity of imidazolium ionic liquids. J. Chem. Phys. 2008, 128, 064509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharati, A.; Wubbenhorst, M.; Moldenaers, P.; Cardinaels, R. Dielectric properties of phase-separated blends containing a microcapacitor network of carbon nanotubes: Compatibilization by a random or block copolymer. Macromolecules 2017, 50, 3856–3868. [Google Scholar] [CrossRef]
- Zhang, J.; Mine, M.; Zhu, D.; Matsuo, M. Electrical and dielectric behaviors and their origins in the three-dimensional polyvinyl alcohol/MWCNT composites with low percolation threshold. Carbon 2009, 47, 1311–1320. [Google Scholar] [CrossRef]
- Havriliak, S.; Havriliak, S.J. Dielectric and Mechanical Relaxation in Materials: Analysis, Interpretation, and Application to Polymers; Hanser Publishers: Munich, Germany; Vienna, Austria; New York, NY, USA, 1997. [Google Scholar]
- Putson, C.; Jaaoh, D.; Muensit, N. Interface polarization effect on dielectric and electrical properties of polyurethane (PU)/polyaniline (PANI) polymer composites. Adv. Mater. Res. 2013, 770, 275–278. [Google Scholar] [CrossRef]
- Kilbride, B.E.; Coleman, J.N.; Fraysse, J.; Fournet, P.; Cadek, M.; Drury, A.; Hutzler, S.; Roth, S.; Blau, W.J. Experimental observation of scaling laws for alternating current and direct current conductivity in polymer-carbon nanotube composite thin films. J. Appl. Phys. 2002, 92, 4024–4030. [Google Scholar] [CrossRef]
- Skipa, T.; Lellinger, D.; Bohm, W.; Saphiannikova, M.; Alig, I. Influence of shear deformation on carbon nanotube networks in polycarbonate melts: Interplay between build-up and destruction of agglomerates. Polymer 2010, 51, 201–210. [Google Scholar] [CrossRef]
- Li, Q.; Xue, Q.; Gao, X.; Zheng, Q. Temperature dependence of the electrical properties of the carbon nanotube/polymer composites. Express Polym. Lett. 2009, 3, 769–777. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Tc (°C) | Tonset cryst. (°C) | Tm (°C) | ΔHm (J/g) | χc (%) (DSC) | χc (%) (XRD) |
|---|---|---|---|---|---|---|
| PEO | 39.2 | 48.5 | 68.2 | 132.6 | 64.7 | 66.9 |
| P-05 GO | 47.3 | 49.9 | 64.8 | 106.6 | 52.0 | 55.0 |
| P-05 NarGO | 45.0 | 49.5 | 63.2 | 122.0 | 59.8 | 63.6 |
| P-05 TrGO | 46.8 | 50.4 | 66.2 | 106.6 | 52.2 | 58.7 |
| P-05 HyrGO | 41.2 | 45.5 | 63.0 | 132.3 | 64.8 | 71.0 |
| P-05 NaHyrGO | 46.0 | 50.0 | 62.8 | 119.8 | 58.7 | 65.0 |
| P-2 GO | 46.9 | 51.5 | 65.3 | 102.1 | 50.8 | 60.7 |
| P-2NarGO | 43.7 | 51.9 | 65.9 | 105.6 | 52.5 | 60.8 |
| P-2 TrGO | 48.0 | 52.2 | 66.7 | 103.5 | 51.5 | 60.7 |
| P-2 HyrGO | 44.6 | 49.4 | 62.4 | 127.5 | 63.5 | 65.8 |
| P-2 NaHyrGO | 44.3 | 50.2 | 61.3 | 111.6 | 55.5 | 63.1 |
| P-2 Graphite | 48.2 | 52.2 | 67.5 | 138.0 | 68.7 | / |
| Samples | σ’150/σ’100 (Fillers) | σ’150/σ’100 (PEO and Filled Composites) |
|---|---|---|
| PEO | - | 1.6 |
| P-2 GO | 42.8 | 7.9 |
| P-2 NarGO | 1.2 | 3.4 |
| P-2 HyrGO | 1.5 | 8.6 |
| P-2 NaHyrGO | 1.4 | 10.5 |
| P-2 TrGO | 1.4 | 3.4 |
| P-2 Graphite | 15.7 | 1.7 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malas, A.; Bharati, A.; Verkinderen, O.; Goderis, B.; Moldenaers, P.; Cardinaels, R. Effect of the GO Reduction Method on the Dielectric Properties, Electrical Conductivity and Crystalline Behavior of PEO/rGO Nanocomposites. Polymers 2017, 9, 613. https://doi.org/10.3390/polym9110613
Malas A, Bharati A, Verkinderen O, Goderis B, Moldenaers P, Cardinaels R. Effect of the GO Reduction Method on the Dielectric Properties, Electrical Conductivity and Crystalline Behavior of PEO/rGO Nanocomposites. Polymers. 2017; 9(11):613. https://doi.org/10.3390/polym9110613
Chicago/Turabian StyleMalas, Asish, Avanish Bharati, Olivier Verkinderen, Bart Goderis, Paula Moldenaers, and Ruth Cardinaels. 2017. "Effect of the GO Reduction Method on the Dielectric Properties, Electrical Conductivity and Crystalline Behavior of PEO/rGO Nanocomposites" Polymers 9, no. 11: 613. https://doi.org/10.3390/polym9110613