Generation of Polymer Nanocomposites through Shear-Driven Aggregation of Binary Colloids

1
Department of Polymeric Materials and Engineering, School of Materials and Energy, Guangdong University of Technology, Guangzhou 510006, China
2
Institute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, 8093 Zurich, Switzerland
*
Author to whom correspondence should be addressed.
Polymers 2017, 9(11), 619; https://doi.org/10.3390/polym9110619
Submission received: 27 October 2017
/
Revised: 9 November 2017
/
Accepted: 12 November 2017
/
Published: 15 November 2017
(This article belongs to the Special Issue Polymer Nanocomposites)
Abstract
:Design of polymer nanocomposites has been an intense research topic in recent decades because hybrid nanomaterials are widely used in many fields. Throughout their development, there has often been a challenging issue how one can uniformly distribute nanoparticles (NPs) in a polymer matrix, avoiding their agglomeration. In this short review, we first introduce the theory of colloidal aggregation/gelation purely based on intense shear forces. Then, we illustrate a methodology for preparing polymer nanocomposites where the NPs (as fillers) are uniformly and randomly distributed inside a matrix of polymer NPs, based on intense shear-driven aggregation of binary colloids, without using any additives. Its feasibility has been demonstrated using two stable binary colloids composed of (1) poly-methyl methacrylate fillers and polystyrene NPs, and (2) graphene oxide sheets (fillers) and poly-vinylidene fluoride NPs. The mechanism leading to capturing and distribution of the fillers inside the polymer NP matrix has been illustrated, and the advantages of the proposed methodology compared with the other common methods are also discussed.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Nanocomposite materials have been widely applied to almost all fields of technology [1], particularly in biomedicine [2], dental and bone implants [3], therapeutics delivery, diagnostics, and treatment [4,5], membrane performance enhancement [6,7,8], coating industry [9,10,11], solid-state lighting, and photovoltaic devices [12,13]. Among them, imbedding nanoparticles (NPs) into polymer matrices to form polymer matrix nanocomposites can improve thermal, mechanical, electric, or optical properties of the polymers [1,14]. In those applications, it is especially important to uniformly and randomly distribute the NPs, as fillers, inside the polymer matrix, while avoiding aggregation among the NPs [15]. Different strategies have been developed in the literature, and among them, the following three are dominating in practical applications: solution mixing [16,17,18,19,20,21], melt compounding [16,19,22,23,24,25], and in situ polymerization [19,26,27,28,29]. Solution mixing is considered to be an effective technique for preparation of polymer nanocomposites, and it disperses fillers into a polymer matrix by simple mechanical stirring or high shear mixing [18,30]. This process often requires a large amount of organic solvent, except for water-based solutions [20,21], and the removal of the residual solvent often becomes crucial. In addition, fillers often re-aggregate during the process of slow solvent evaporation, and it is thus necessary to modify the NP surface to improve the compatibility between NPs and polymer matrix. In the case of melt blending, fillers are mixed with a polymer matrix at the molten state by using mixing equipment such as an extruder, internal mixer, and two-roll mill [22,31]. This process often requires high shear forces to fully mix the molten polymers and fillers, and no solvent is used during the process, making it an eco-friendly method for large scale production of polymer nanocomposites. However, at the molten state the high viscosity of the polymer may lead to poor dispersion of fillers [32,33]. In situ polymerization is another effective way to prepare uniformly distributed fillers inside a polymer matrix. In this process, the fillers are first uniformly dispersed in monomer solution in the presence of an initiator [26,29,34,35,36,37]. Initiated by radiation or thermal energy, in situ polymerization takes place, conferring strong interfacial interactions between the polymer chains and the fillers [29]. Compared with solution mixing and melt blending methods, in situ polymerization can obtain improved compatibility between fillers and polymer, thus better dispersion properties. Kim et al. [19] fabricated graphene-based polyurethane nanocomposites via melt blending, solution mixing, and in situ polymerization, the comparison of the results shows that melt blending would lead to the fillers to re-aggregate, while the other two processes could result in better dispersion of fillers throughout the polymer matrix.
Another important strategy for generation of nanocomposites is to use a colloidal route—Colloidal aggregation. In this way, the applied polymer is initially also in the form of NPs, thus, we have NPs, A and B, forming a binary colloidal system. Binary colloids have received great attention in recent years due to their potential applications in many industrial processes. Aggregation of binary colloids with different types, sizes, or properties has been found to be crucial in the fields such as waste water treatment [38,39], mineral flotation processes [40,41,42,43], cells and DNA analysis [44,45], and composite materials fabrication [46,47,48,49,50,51]. If A and B possess opposite charges, their electrostatic attraction can cause A-B aggregation, typically referred to as hetero-aggregation [52,53,54]. When the size of A and B is similar, the clusters formed from their aggregation are of irregular shape and low fractal dimension. Since the aggregation induced by electrostatic attraction is extremely fast, it is difficult to obtain uniform distribution of A and B within the clusters when the particle concentration is high. When B is much smaller than A, B can attach onto the surface of A, forming stable “core-shell” hetero-clusters [55,56]. When A and B possess charges of the same sign, their mixture is often colloidally stable, forming a new, binary colloid. In this case, one can effectively realize the A-B hetero-aggregation [47,48,50,51,53,57,58,59,60,61], by introducing electrolytes to screen the electrical double layer, adding bonding molecules or high molecular weight polymers to cause depletion aggregation, or varying pH to neutralize pH-sensitive charges. However, in most of these cases, it is rather difficult to control the hetero-aggregation processes to realize uniform and random distribution of the fillers inside the polymer matrix, while avoiding aggregation among the fillers.
Toward this aim, a new technique has been developed recently, which is based on intense shear-driven aggregation of binary colloids to uniformly and randomly distribute fillers into a polymer matrix and to avoid aggregation among the fillers [48,50]. Let us consider that the B NPs are the dominant polymeric component, being the elements eventually forming the matrix, and A NPs are the fillers to be distributed. The first key feature is that the binary colloids must be very stable at rest, thus warranting initial homogeneous mixing of A and B NPs at nanoscales. The second key feature is that the nanocomposite materials are generated through (purely) intense shear-driven aggregation of the binary colloids, without using any electrolytes. The feasibility of the above procedure has been demonstrated by experiments. In this work, we review the relevant intense shear-driven aggregation and its application to binary colloids to generate the polymer matrix nanocomposites.
2. Intense Shear-Driven Aggregation
The intensive shear-driven aggregation has been systematically investigated in recent years for unary (single component) colloids [62,63,64,65,66,67,68,69,70,71]. For a colloidal system that is rather stable under stagnant conditions, if we drive the system to pass through a microchannel system, where a sufficiently intense shear force is generated, the particles may overcome the repulsive interaction energy barrier, , leading to aggregation, which was first demonstrated in diluted dispersions [72,73]. When the particle volume fraction (φ) reaches a certain value, the clusters formed under the intense shear would connect to form a space-spanning network, resulting in a solid-like gel.
2.1. Main Phenomena Observed Experimentally
A schematic setup for conducting intense shear-driven aggregation, using a commercial device, HC-5000 homogenizer (Microfluidics, Westwood, MA, USA), equipped with a z-shaped microchannel (z-MC), is shown in Figure 1 a. When a liquid-like colloid is forced to pass through the z-MC at an extremely high shear rate (e.g., = 1 × 106 s−1), a solid-like (gel) transition occurs if φ is large enough (typically > 15%), as demonstrated in Figure 1b. Note that the cylindrical shape of the gel inside the bottles results from the tube connected to the outlet of the z-MC. There are important features for this shear-driven process. First, no additives are needed during the process, thus avoiding contamination of the final products. Second, the process can operate continuously, which is crucial for large scale industrial production.
The intense shear-driven aggregation kinetics and the time evolution of the cluster morphology have been studied using a polystyrene colloid, which has been forced to pass through the above mentioned z-MC many times, in the low range of φ, where, instead of gelation, only shear-driven aggregation occurs [71]. It was found that in this case a colloidal system after passing through the z-MC is composed of two distinct classes of clusters: Class 1, which is mainly composed of primary particles with small amount of small clusters made of two and three primary particles, referred to as doublets and triplets, respectively, and Class 2, which are big clusters with an average size at least two orders of magnitude larger than the primary particles, thus constituted of 103–104 primary particles [62,65,66]. The size and morphology of Class 2 clusters are controlled by breakage and restructuring induced by the intense shear, i.e., the size decreases as the shear rate increases. The size distribution is typically rather uniform.
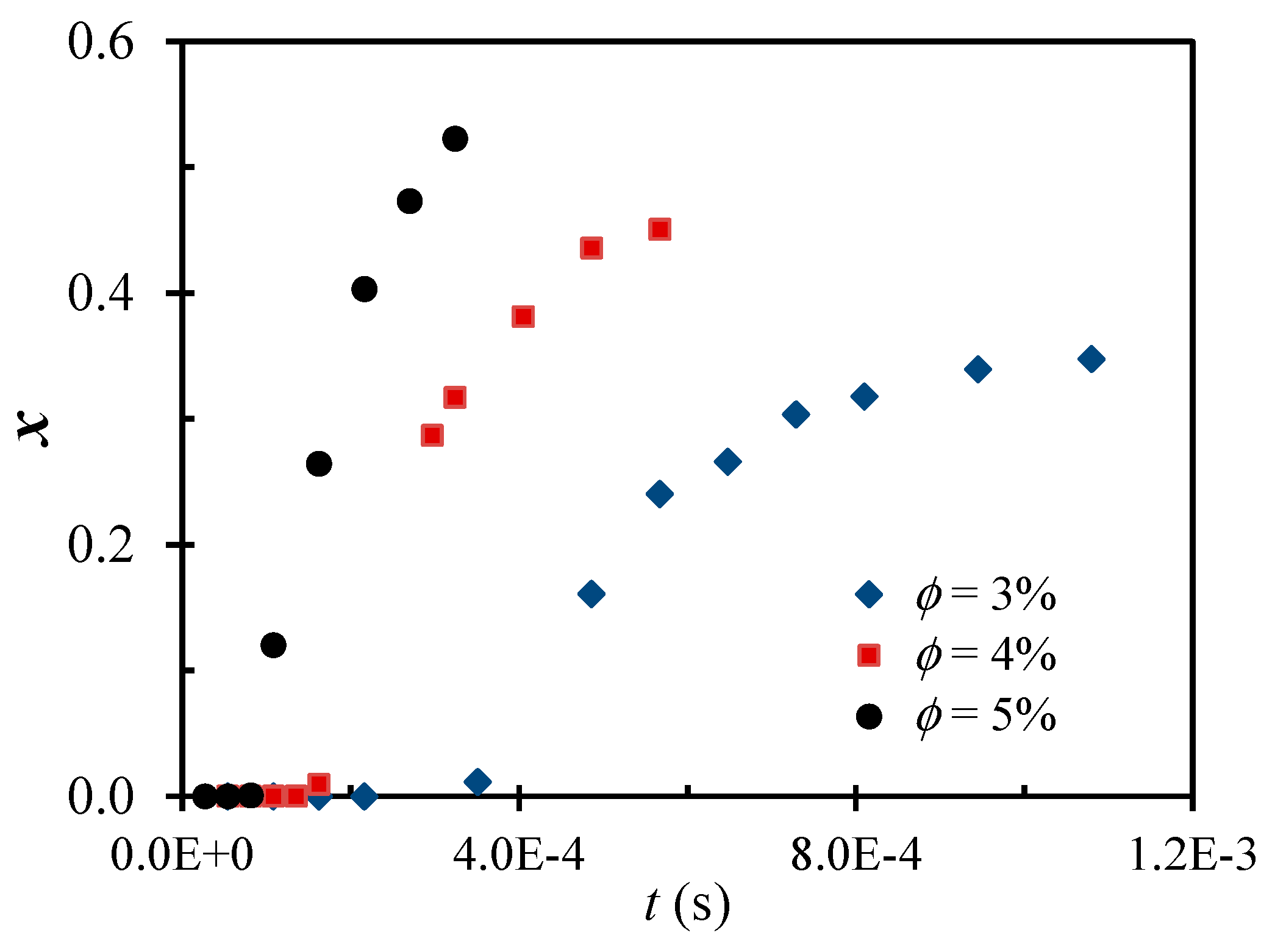
Figure 2 shows the shear-driven aggregation of the polystyrene colloid in the z-MC, particularly, the evolution of the primary particle conversion (x) to big (Class 2) clusters as a function of time. Note that the time here is a cumulative (residence) time from forcing the same colloid to repeatedly pass through the z-MC many times. It is seen that the x evolution is typically composed of three stages: induction, sharp increase and slow increase to reach a plateau. In the induction stage, the x value is negligible. In the stage where x increases sharply with the pass number (i.e., the shearing time), the average size of Class 2 clusters increases also sharply, often characterized by an overshooting. In the last stage where the x value increases slowly and eventually reaches a plateau, the average size of Class 2 clusters decreases to also reach a plateau. The fractal dimension of Class 2 clusters increases with the shearing time from the initial value of to reach . Thus, the final clusters generated by the intense shear are typically rather compact.
It should be mentioned that the plateau value of x reached at large shearing times at each particle volume fraction cannot be explained at the present stage, which happened also for the intense shear-driven aggregation of the other colloidal systems [50]. In principle, as a second-order kinetics, the intense shear-driven aggregation of the primary particles to big clusters should continue until reaching 100% conversion. To understand this behavior, new experiments have been designed and the investigation is continuing in our lab.
2.2. Theoretical Background of the Intense Shear-Driven Aggregation
A theory has been developed to better understand the shear-driven event [63,67,69]. We start with the simplest case, the doublet formation. For interactive particles embedded in a linear velocity field, the stationary particle concentration field c(x) can be written following the Smoluchowski equation
where D is the mutual diffusion coefficient of the particles (D = 2D0G(x), where D0 is the diffusion coefficient of an isolated particle, and G(x) is the hydrodynamic correction for viscous retardation), kB is Boltzmann’s constant, T is the absolute temperature, UT(x) is the colloidal interaction energy, η is the viscosity of the solvent, a is the particle radius, and v(x) is the flow velocity. To solve Equation (1), the boundary conditions for the irreversible shear-driven aggregation are given by
where the δ value can be determined from the boundary layer approximation [63]
where κ is the Debye length, and Peclet number, Pe, is defined as,
with the shear rate. From Equation (1), the derived kinetic constant or kernel, k1,1, for the aggregation between two particles forming a doublet can be expressed as,
where is the effective velocity for aggregation. The k1,1 values predicated from Equation (5) have been verified by comparing them with those obtained from numerical simulations of the full convective diffusion equation, Equation (1) [63].
After proper simplification and approximation in the frame of the DLVO (Derjaguin-Landau-Verwey-Overbeek) interactions, Equation (5) can reduce to the following Arrhenius form
where , and α is a geometrical parameter. From Equation (6), it can be clearly seen that at small Pe values, the exponent can be negative so that the aggregation rate is rather small. As the Pe value increases, the collision rate increases, and once Pe increases to a certain critical value, Pecr, which can be defined by setting the exponent in Equation (6) equal to zero as
the shear force (Pe) plays a prominent role. In particular, when Pe << Pecr, the colloidal interaction barrier (UT,max) plays the dominant role, and the aggregation rate (k1,1) increases as UT,max decreases, corresponding to the Brownian motion controlled aggregation. When Pe >> Pecr, the shear force takes over the dominant role, and the effect of UT,max becomes negligible. The k1,1 value increases exponentially with Pe, corresponding to the shear controlled regime.
It should be particularly noted that from Equation (4), Pe is proportional to the radius of the particles, a, to a power of 3, indicating that the particle or cluster size has substantial influence on the aggregation rate. During the shear-driven event, the formed doublets progressively grow to larger clusters. Then, even though initially one has Pe << Pecr, as the cluster size increases progressively with time to reach a critical radius, acr, the situation, Pe >> Pecr, occurs, leading to an exponential increase in k1,1, thus, self-acceleration. The critical cluster radius, acr is given by
The above self-accelerating aggregation predicted by the theory is in excellent agreement with the experimental results in Figure 2. At each particle volume fraction, the induction stage corresponds to the time needed to reach the critical value, acr and then after the radius of the clusters has reached acr, the conversion to big clusters increases sharply, i.e., the aggregation accelerates. The acr value has been identified experimentally and confirmed to increase as the shear rate decreases [71].
3. Applications of the Shear-Driven Aggregation to Binary Colloids
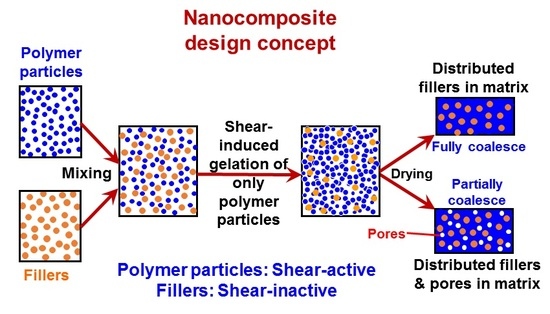
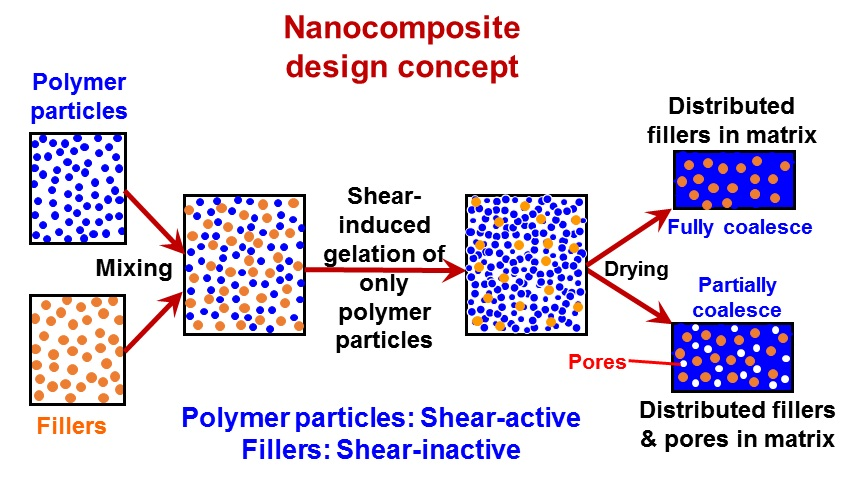
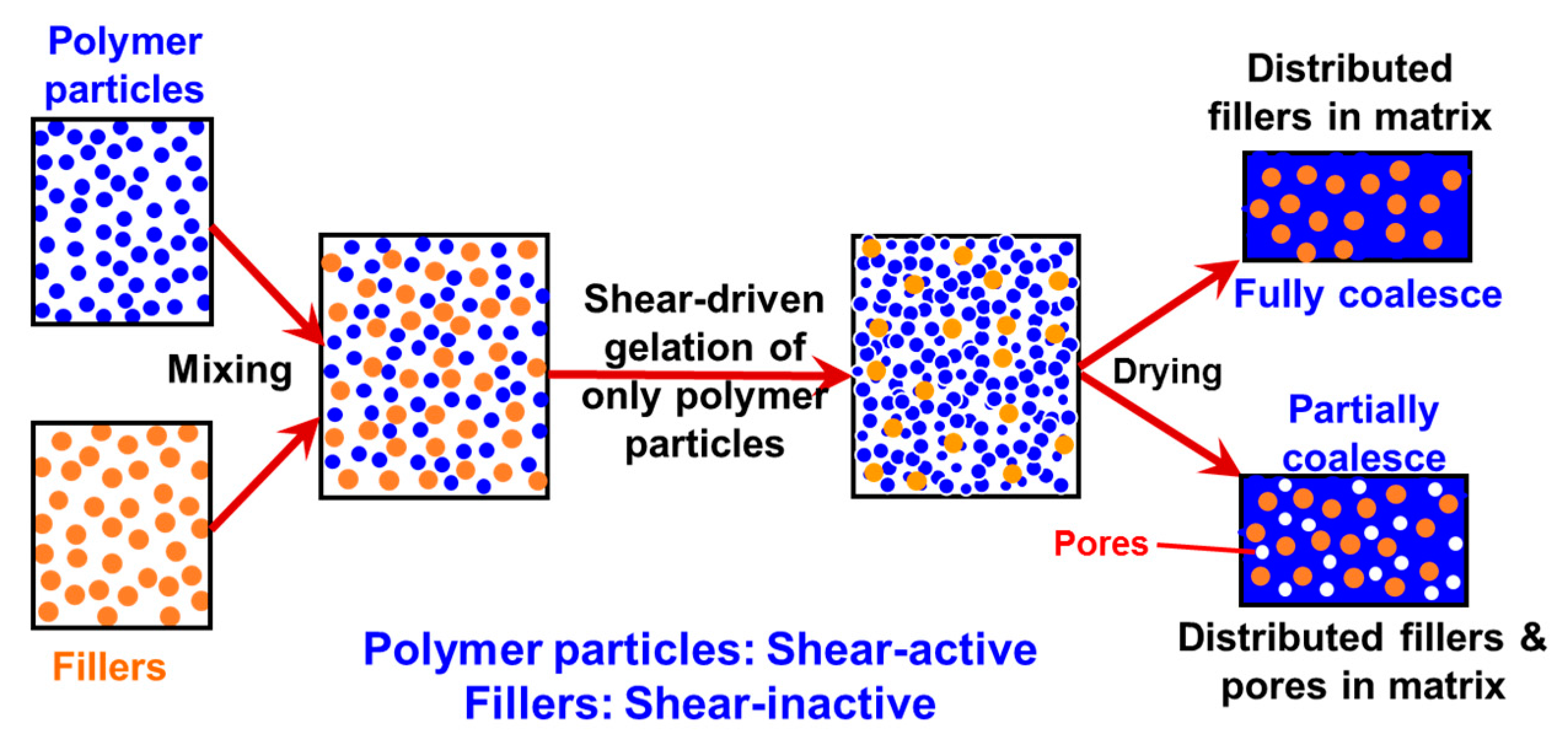
Recently, in the frame of preparation of nanocomposites where the fillers have to be uniformly and randomly distributed inside a polymer matrix, a general methodology has been developed [48,50], which is purely based on the intense shear-driven aggregation discussed above but starting with binary colloids, without using any additives. The setup used for the shear-driven aggregation of the binary colloids is the same as that sketched in Figure 1a. The design concept is shown in Figure 3 and described as follows:
- (1)
- It starts with two colloids: fillers (A) NP dispersion and polymer (B) NP dispersion, both of which have the same sign of charges;
- (2)
- When the two colloids mix, a stable binary colloid is formed, where A and B NPs are homogeneously distributed at nanoscales;
- (3)
- The polymer (B) is the dominant colloid, and the volume fraction of the fillers (A) is smaller;
- (4)
- The colloidal interactions are controlled such that the B (polymer) NPs are unstable under the used shear rate (referred to as shear active) and can aggregate to form clusters or gels. Instead, the A (fillers) NPs are very stable under the same shear rate and the shear-driven aggregation does not occur, referred to as shear-inactive.
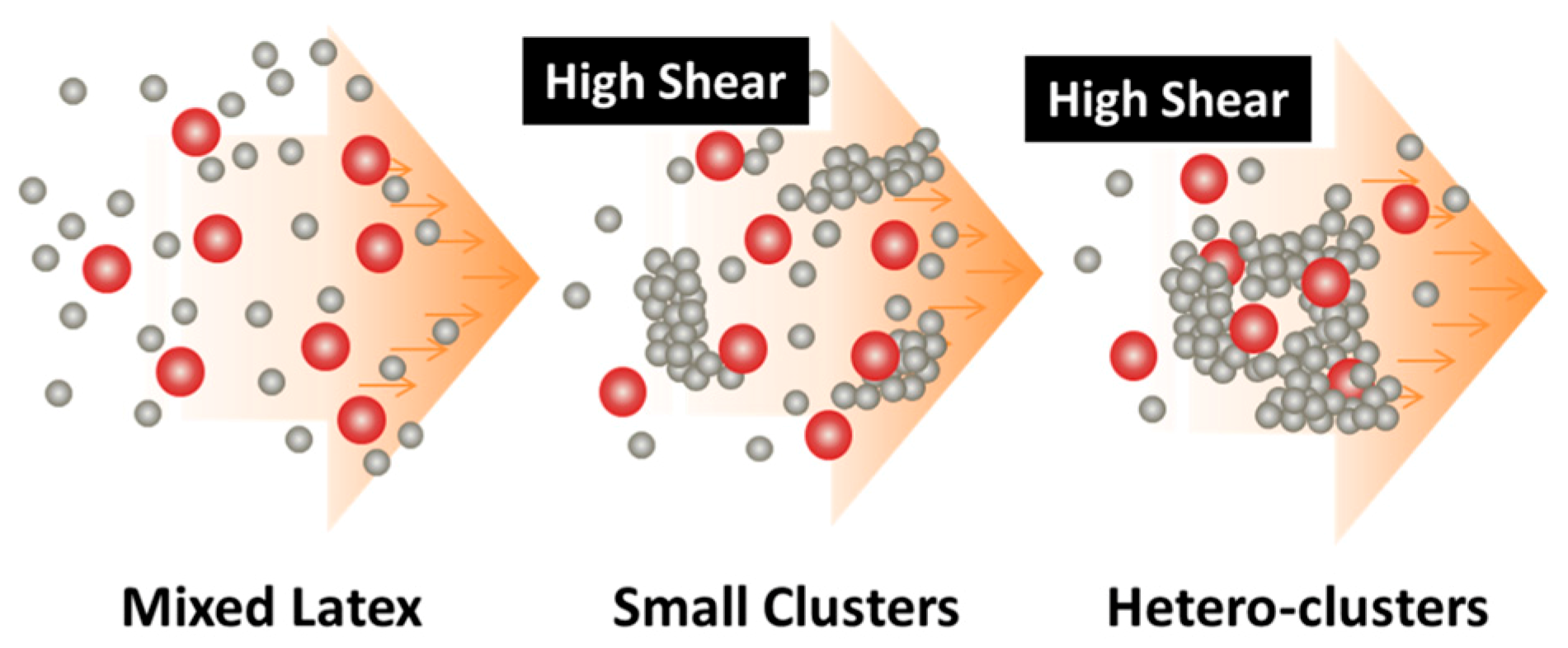
Thus, when the binary colloid passes through the z-MC as shown in Figure 1a at an extremely high shear rate and at a significantly high particle volume fraction (typically φB > 15%), the binary colloid will be converted to a solid-like gel after passing through the z-MC. It should be particularly noticed that since the residence time of the colloid in the z-MC is very short, in μs scales, the extremely fast gelation of the B NPs can basically freeze the initially distributed A NPs (fillers) inside the formed gel. It is this frozen mechanism that avoids the possibility of aggregation among the A NPs, being the key novelty of the proposed methodology.
After the solid-like gel is formed and dried at a temperature larger than the melting point of the polymer (B), the B NPs can fully melt to form a matrix where the fillers (A) NPs are uniformly and randomly distributed with negligible aggregation among the A NPs. If one properly controls the drying temperature such that the polymer particles are only partially coalesced, there is the possibility of having desired pores inside the nanocomposites. In the following subsections, we show two examples, where we demonstrate that, based on the design concept in Figure 3, the intense shear-driven aggregation of binary colloids can indeed successfully distribute fillers homogeneously inside the polymeric NP gels.
3.1. Distribution of PMMA Particles into PS NP Matrix
The first example is a model system where poly-methyl methacrylate (PMMA) NPs are distributed in the matrix of polystyrene (PS) NPs [48]. Two types of colloids, the PMMA and PS NP aqueous dispersions, have been first synthesized separately. The PS NPs are charged with the fixed negative charges, –OSO3– and can undergo the shear-driven aggregation (shear-active), at a shear rate of = 1.5 × 106 s−1. The PMMA NPs are also charged with –OSO3– In addition, the PMMA particles possess certain hydrophilicity, because of the surface ester groups from the MMA monomers, which favor the tendency of forming ordered water layers [75]. Thus, the PMMA particle surface possesses substantial short-range, repulsive hydration forces. These non-DLVO forces together with the DLVO interactions from the charges provide an extremely high energy barrier and very shallow primary minimum [74], such that aggregation among the PMMA particles under the same shear rate does not occur. For example, the prepared PMMA colloid with the radius of the primary particles, aPMMA = 75 nm, does not undergo any aggregation after passing through the z-MC at = 1.5 × 106 s−1, even for more than 20 times at a particle volume fraction larger than 20%.
Then, the PS and PMMA NP colloids are mixed to form a new binary colloid, which is stable at rest. When this binary colloid is forced to pass through the z-MC, the shear-driven aggregation/gelation can also occur, and the formed gels are composed of both the ‘shear-active’ PS and ‘shear-inactive’ PMMA particles. Moreover, in the case of low particle volume fractions where no gelation occurs, the aggregated binary system, similar to the unary system, is also composed of two distinct classes of clusters: Class 1, which is mainly composed of the primary PS and PMMA NPs; and Class 2, which are big clusters (mainly made of the PS NPs) with an average size at least two orders of magnitude larger than the primary NPs. Figure 4 compares typical morphology of the clusters formed after shear-driven aggregation of the pure PS colloid and of the PMMA/PS (aPMMA = 75 nm, aPS = 21 nm) binary colloid. It is seen that the general morphology is rather similar in Figure 4a,b, because in both cases it results from aggregation of PS NPs. The only difference is that in Figure 4b the PMMA particles are uniformly and randomly distributed (captured) within the clusters of the PS particles.
In addition, it was found that the presence of the PMMA NPs does not affect significantly the shear-driven aggregation kinetics of the PS NPs nor the average cluster size at low volume fractions, and the PMMA NPs behave as inert fillers. Therefore, the above results confirm the proposed shear-driven aggregation mechanism of the binary colloids. As schematically shown in Figure 5, since the shear-driven aggregation of the shear-active PS NPs takes place in micron seconds in the z-MC, the initially distributed PMMA NPs in the binary colloid, though shear-inactive, do not have time to escape from the PS aggregation process and are captured inside the clusters. The PMMA NP capture efficiency may reach 100% at low PMMA volume fractions, but it can be significantly lower at high PMMA volume fractions, particularly when the PMMA/PS size ratio is very large. On the other hand, if the volume fraction of the shear-active PS NPs in the binary system is high enough, such that the shear-driven gelation occurs after the binary colloid passing through the MC just one time, the PMMA NP capture efficiency can naturally reach 100%.
3.2. Distribution of GO Sheets into PVDF NP Matrix
The same methodology has been further verified by Sheng et al. [50], for a binary colloidal dispersion composed of graphene oxide (GO) sheets and poly-vinylidene fluoride (PVDF) NPs. In this case, the GO sheets are fillers. They are shear-inactive, because there are many oxygen-containing groups (e.g., –OH and –COOH) on their edges and epoxide groups on their basal planes. These groups are rather hydrophilic, leading to the GO sheets being extremely stable under the intense shear rate. Instead, the (dominant component) PVDF NPs are shear-active, because of extreme hydrophobicity of the polymer. After passing the binary dispersion through the z-MC device at an extremely high shear rate (~106 s−1), aggregation/gelation of the shear-active PVDF NPs occurs, leading to uniform capture and distribution of the shear-inactive GO sheets inside the PVDF NP matrix, as demonstrated by the SEM pictures in Figure 6. Therefore, this example demonstrates again that through the intense shear-driven aggregation of binary colloids, one can effectively distribute uniformly and randomly the shear inactive fillers inside a polymer matrix, leading to well-defined nanocomposites.
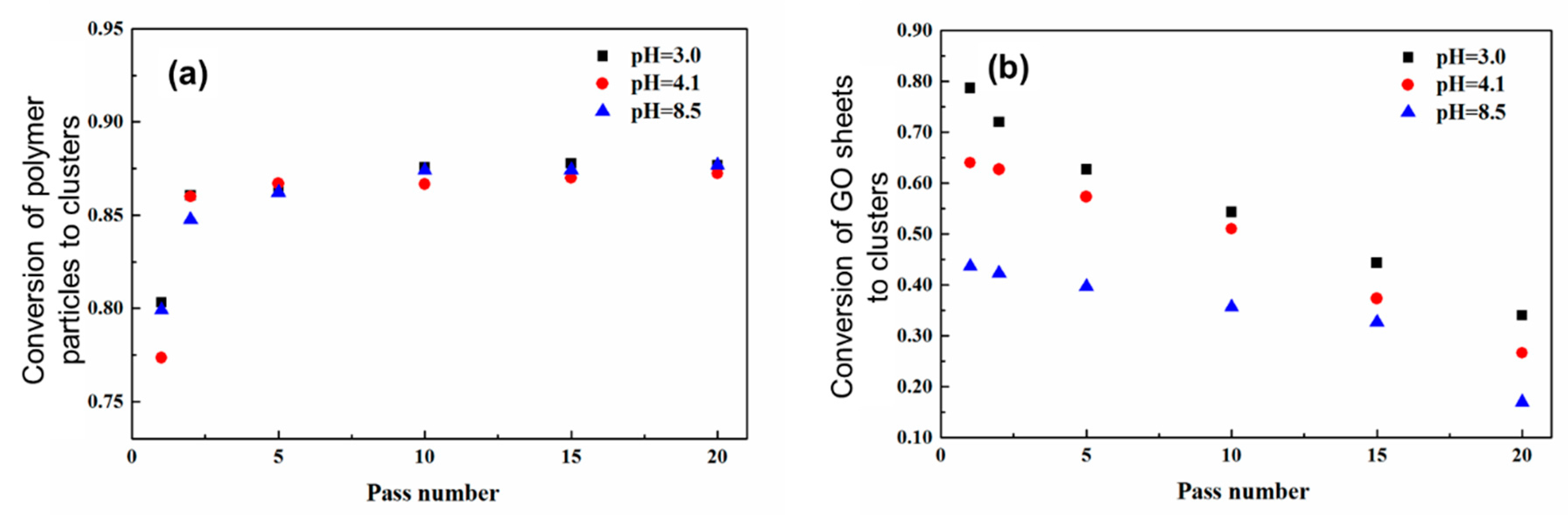
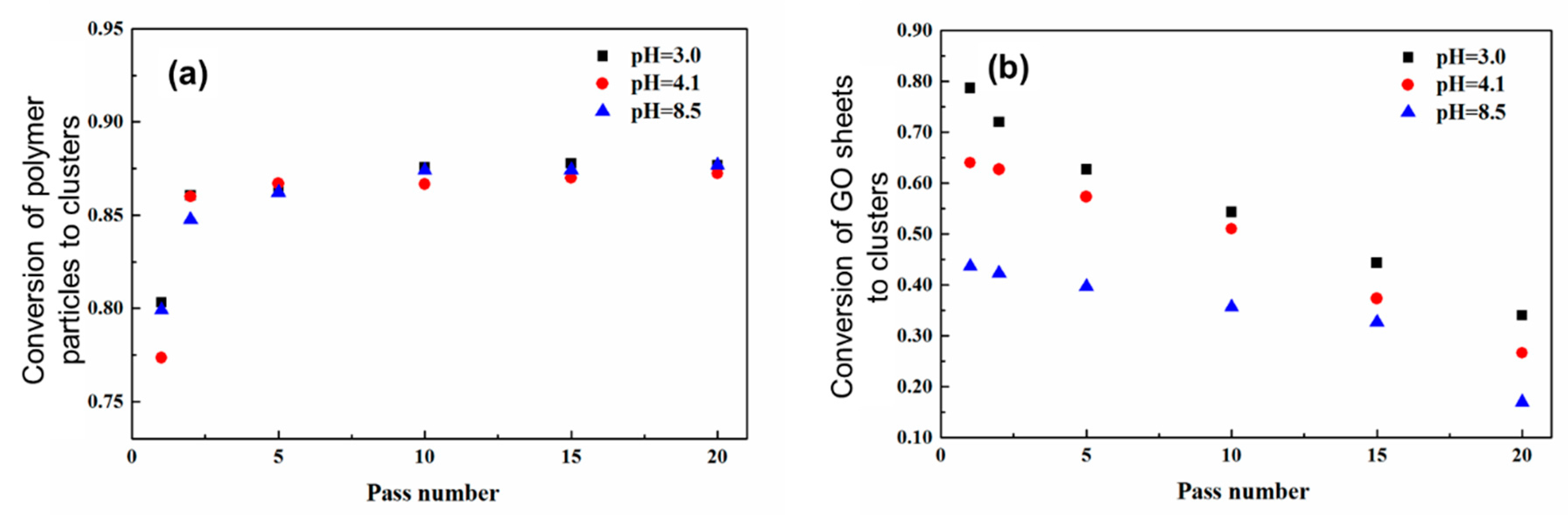
In the case of low initial particle volume fractions where only partial aggregation-instead of complete gelation-occurs after the binary colloid passes through the z-MC one time, it is observed that the GO/PVDF NP ratio inside the formed Class 2 clusters is lower than that in the initial binary colloid. Moreover, if one allows the aggregated system to repeatedly pass through the z-MC, although the conversion of the PVDF NPs to Class 2 clusters increases with the pass number, as shown in Figure 7a, the amount of the captured GO sheets inside the clusters decreases monotonically with the pass number, as reported in Figure 7b. This arises because of two main reasons. First, the GO sheets are shear inactive and only captured (instead of aggregated) inside the PVDF NP clusters during the extremely fast shear-driven aggregation. Second, along the shear-driven aggregation in the z-MC, there are also cluster breakage and restructuring, and these processes provide the opportunity to the captured GO sheets to escape from the Class 2 clusters. In addition, it can be observed from Figure 7b that the fraction of the GO sheets inside the clusters decreases as pH increases. This is considered to be related to some specific interactions induced by the H⋅⋅⋅F halogen bonding between the –CF2– groups of the PVDF NPs and the –COOH groups on the GO sheet [76,77]. In particular, the –COOH groups on the GO sheets would be progressively deprotonated as pH increases, and this would reduce the H⋅⋅⋅F halogen bonding interactions, thus promoting the escape of the GO sheets from Class 2 clusters during the shear-driven aggregation/breakage. This phenomenon indicates that during the preparation of the nanocomposites through the shear-driven aggregation of binary colloids, one should also consider the roles played by the various interactions among the functional groups present on the surface of the two colloidal identities.
On the other hand, in Figure 7a, the conversion of the PVDF NPs to Class 2 clusters, xp, does not change significantly with pH. This arises because the PVDF NPs are shear-active, and their aggregation is purely shear-controlled [63,65]. In the shear-controlled regime, the evolution of the aggregation is independent of the surface charge or potential and governed by the value of the Peclet number, Pe, defined by Equation (4).
The above results indicate that to effectively capture the GO sheets inside the PVDF NP clusters, one should let the binary colloid pass through the z-MC only one time, instead of many times. As mentioned above, if a solid-like gel of the binary colloid can be formed after passing through the z-MC just one time, all the GO sheets can be captured inside the gel, leading to 100% capture efficiency. Such an observation is particularly important, as an essential guideline, in practical applications of the developed methodology to form nanocomposites.
4. Concluding Remarks and Perspectives
In this short review, we have presented recent advances in our knowledge about the aggregation of unary and binary colloids. We have introduced the aggregation and gelation of colloids purely based on intense shear force and the relevant theory and experimental validation. Then, we have described how to apply the intense shear-driven aggregation technique to binary colloids to generate nanocomposite materials, where A NPs as fillers are uniformly and randomly distributed inside a matrix of B polymer NPs. We have also illustrated the advantages of the developed methodology compared with the other common methods.
The feasibility of the proposed methodology has been demonstrated using two stable binary colloids: one is composed of PMMA NPs as the fillers and PS NPs as the matrix, and another of GO sheets (fillers) and PVDF NPs. In the first case, the PS NPs alone undergo the shear-driven aggregation (shear-active), while the PMMA fillers alone do not (shear-inactive). It was observed that the shear-driven aggregation of the PMMA/PS binary colloids can occur, and the formed clusters or gels are composed of both components. More importantly, the PMMA fillers are uniformly and randomly distributed inside PS NP matrix. Similar results were also obtained in the case of GO/PVDF binary systems, and the gelation of the shear-active PVDF NPs is able to uniformly capture and distribute the shear-inactive GO sheets inside the PVDF NP gel matrix. All the results confirm the feasibility of the proposed methodology.
The shear-driven aggregation mechanism of the binary colloids for the filler capturing and distribution has been illustrated, which would help broaden the means of fabricating nanocomposites through the binary colloidal route. Typical applications include incorporation of fillers (e.g., graphene nanosheets, clays, or silica or oxide NPs) into the polymer NPs matrix to generate highly electronically conductive materials, high-performance gas barrier composite membranes, composite separators for lithium-ion batteries, etc. Studies regarding these applications have been started in our groups, and the preliminary results for the properties of the generated nanocomposites are very encouraging.
Acknowledgments
This work is financially supported from the Swiss National Science Foundation (Grant No. 200020_165917).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schadler, L.S. Polymer-based and polymer-filled nanocomposites. In Nanocomposite Science and Technology; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2003; pp. 77–153. [Google Scholar]
- Chan, W.C.W.; Nie, S. Quantum dot bioconjugates for ultrasensitive nonisotopic detection. Science 1998, 281, 2016–2018. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.C.; Ko, C.-C.; Douglas, W.H. Preparation of hydroxyapatite-gelatin nanocomposite. Biomaterials 2003, 24, 2853–2862. [Google Scholar] [CrossRef]
- Sershen, S.R.; Westcott, S.L.; Halas, N.J.; West, J.L. Temperature-sensitive polymer-nanoshell composites for photothermally modulated drug delivery. J. Biomed. Mater. Res. 2000, 51, 293–298. [Google Scholar] [CrossRef]
- Misra, S.K.; Valappil, S.P.; Roy, I.; Boccaccini, A.R. Polyhydroxyalkanoate (pha)/inorganic phase composites for tissue engineering applications. Biomacromolecules 2006, 7, 2249–2258. [Google Scholar] [CrossRef] [PubMed]
- Elimelech, M.; Phillip, W.A. The future of seawater desalination: Energy, technology, and the environment. Science 2011, 333, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Mahendra, S.; Lyon, D.Y.; Brunet, L.; Liga, M.V.; Li, D.; Alvarez, P.J.J. Antimicrobial nanomaterials for water disinfection and microbial control: Potential applications and implications. Water Res. 2008, 42, 4591–4602. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.; Joy, N.; Aparna, E.P.; Vijayan, R. Polymer grafted inorganic nanoparticles, preparation, properties, and applications: A review. Polym. Rev. 2014, 54, 268–347. [Google Scholar] [CrossRef]
- Chattopadhyay, D.K.; Raju, K.V.S.N. Structural engineering of polyurethane coatings for high performance applications. Prog. Polym. Sci. 2007, 32, 352–418. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Z.; Xu, X.; Zhu, X.; Men, X.; Zhou, X. Superhydrophilic-superoleophobic coatings. J. Mater. Chem. 2012, 22, 2834–2837. [Google Scholar] [CrossRef]
- Jhaveri, J.H.; Murthy, Z.V.P. A comprehensive review on anti-fouling nanocomposite membranes for pressure driven membrane separation processes. Desalination 2016, 379, 137–154. [Google Scholar] [CrossRef]
- McDonald, S.A.; Konstantatos, G.; Zhang, S.; Cyr, P.W.; Klem, E.J.D.; Levina, L.; Sargent, E.H. Solution-processed pbs quantum dot infrared photodetectors and photovoltaics. Nat. Mater. 2005, 4, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Schaefer, J.L.; Yang, Z.; Tu, Z.; Archer, L.A. 25th anniversary article: Polymer-particle composites: Phase stability and applications in electrochemical energy storage. Adv. Mater. 2014, 26, 201–234. [Google Scholar] [CrossRef] [PubMed]
- Althues, H.; Henle, J.; Kaskel, S. Functional inorganic nanofillers for transparent polymers. Chem. Soc. Rev. 2007, 36, 1454–1465. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.J.M.; Wilmer, C.E.; Soh, S.; Grzybowski, B.A. Nanoscale forces and their uses in self-assembly. Small 2009, 5, 1600–1630. [Google Scholar] [CrossRef] [PubMed]
- Ke, K.; Wang, Y.; Liu, X.-Q.; Cao, J.; Luo, Y.; Yang, W.; Xie, B.-H.; Yang, M.-B. A comparison of melt and solution mixing on the dispersion of carbon nanotubes in a poly(vinylidene fluoride) matrix. Compos. Part B 2012, 43, 1425–1432. [Google Scholar] [CrossRef]
- Yousefi, N.; Gudarzi, M.M.; Zheng, Q.; Aboutalebi, S.H.; Sharif, F.; Kim, J.-K. Self-alignment and high electrical conductivity of ultralarge graphene oxide-polyurethane nanocomposites. J. Mater. Chem. 2012, 22, 12709–12717. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Q.; Chen, D.; Lu, P. Enhanced mechanical properties of graphene-based poly(vinyl alcohol) composites. Macromolecules 2010, 43, 2357–2363. [Google Scholar] [CrossRef]
- Kim, H.; Miura, Y.; Macosko, C.W. Graphene/polyurethane nanocomposites for improved gas barrier and electrical conductivity. Chem. Mater. 2010, 22, 3441–3450. [Google Scholar] [CrossRef]
- Cavallaro, G.; Lazzara, G.; Milioto, S. Dispersions of nanoclays of different shapes into aqueous and solid biopolymeric matrices. Extended physicochemical study. Langmuir 2011, 27, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Makaremi, M.; Pasbakhsh, P.; Cavallaro, G.; Lazzara, G.; Aw, Y.K.; Lee, S.M.; Milioto, S. Effect of morphology and size of halloysite nanotubes on functional pectin bionanocomposites for food packaging applications. ACS Appl. Mater. Interfaces 2017, 9, 17476–17488. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Huang, J.; Yang, L.; Zhang, N.; Jin, G.; Qu, J. In-situ thermal reduction and effective reinforcement of graphene nanosheet/poly (ethylene glycol)/poly(lactic acid) nanocomposites. Polym. Adv. Technol. 2014, 25, 1515–1522. [Google Scholar] [CrossRef]
- Wang, C.; Liu, Z.; Wang, S.; Zhang, Y. Preparation and properties of octadecylamine modified graphene oxide/styrene-butadiene rubber composites through an improved melt compounding method. J. Appl. Polym. Sci. 2016, 133, 42907. [Google Scholar] [CrossRef]
- Mofokeng, J.P.; Luyt, A.S. Morphology and thermal degradation studies of melt-mixed pla/phbv biodegradable polymer blend nanocomposites with tio2 as filler. J. Appl. Polym. Sci. 2015, 132, 42138. [Google Scholar] [CrossRef]
- El Achaby, M.; Arrakhiz, F.-E.; Vaudreuil, S.; el Kacem Qaiss, A.; Bousmina, M.; Fassi-Fehri, O. Mechanical, thermal, and rheological properties of graphene-based polypropylene nanocomposites prepared by melt mixing. Polym. Compos. 2012, 33, 733–744. [Google Scholar] [CrossRef]
- Tripathi, S.; Saini, P.; Gupta, D.; Choudhary, V. Electrical and mechanical properties of pmma/reduced graphene oxide nanocomposites prepared via in situ polymerization. J. Mater. Sci. 2013, 48, 6223–6232. [Google Scholar] [CrossRef]
- Wang, J.; Xu, C.; Hu, H.; Wan, L.; Chen, R.; Zheng, H.; Liu, F.; Zhang, M.; Shang, X.; Wang, X. Synthesis, mechanical, and barrier properties of ldpe/graphene nanocomposites using vinyl triethoxysilane as a coupling agent. J. Nanopart. Res. 2011, 13, 869–878. [Google Scholar] [CrossRef]
- Wang, J.; Hu, H.; Wang, X.; Xu, C.; Zhang, M.; Shang, X. Preparation and mechanical and electrical properties of graphene nanosheets–poly(methyl methacrylate) nanocomposites via in situ suspension polymerization. J. Appl. Polym. Sci. 2011, 122, 1866–1871. [Google Scholar] [CrossRef]
- Jin, L.; Wu, H.; Morbidelli, M. Synthesis of water-based dispersions of polymer/TiO2 hybrid nanospheres. Nanomaterials 2015, 5, 1454. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bai, L.; Feng, G.; Yang, X.Y.; Lv, M.J.; Zhang, C.A.; Hu, H.; Wang, X.B. Thermal reduced graphene based poly(ethylene vinyl alcohol) nanocomposites: Enhanced mechanical properties, gas barrier, water resistance, and thermal stability. Ind. Eng. Chem. Res. 2013, 52, 16745–16754. [Google Scholar] [CrossRef]
- Mittal, G.; Dhand, V.; Rhee, K.Y.; Park, S.-J.; Lee, W.R. A review on carbon nanotubes and graphene as fillers in reinforced polymer nanocomposites. J. Ind. Eng. Chem. 2015, 21, 11–25. [Google Scholar] [CrossRef]
- Taguet, A.; Cassagnau, P.; Lopez-Cuesta, J.M. Structuration, selective dispersion and compatibilizing effect of (nano)fillers in polymer blends. Prog. Polym. Sci. 2014, 39, 1526–1563. [Google Scholar] [CrossRef]
- Istrate, O.M.; Paton, K.R.; Khan, U.; O’Neill, A.; Bell, A.P.; Coleman, J.N. Reinforcement in melt-processed polymer-graphene composites at extremely low graphene loading level. Carbon 2014, 78, 243–249. [Google Scholar] [CrossRef]
- Sheng, X.; Xie, D.; Wang, C.; Zhang, X.; Zhong, L. Synthesis and characterization of core/shell titanium dioxide nanoparticle/polyacrylate nanocomposite colloidal microspheres. Colloid Polym. Sci. 2015, 294, 463–469. [Google Scholar] [CrossRef]
- Sheng, X.; Xie, D.; Cai, W.; Zhang, X.; Zhong, L.; Zhang, H. In situ thermal reduction of graphene nanosheets based poly(methyl methacrylate) nanocomposites with effective reinforcements. Ind. Eng. Chem. Res. 2015, 54, 649–658. [Google Scholar] [CrossRef]
- Liu, H.T.; Zeng, X.F.; Zhao, H.; Chen, J.F. Highly transparent and multifunctional polymer nanohybrid film with superhigh zno content synthesized by a bulk polymerization method. Ind. Eng. Chem. Res. 2012, 51, 6753–6759. [Google Scholar] [CrossRef]
- Meng, X.; Wu, H.; Storti, G.; Morbidelli, M. Effect of dispersed polymeric nanoparticles on the bulk polymerization of methyl methacrylate. Macromolecules 2016, 49, 7758–7766. [Google Scholar] [CrossRef]
- Wang, H.; Dong, Y.-N.; Zhu, M.; Li, X.; Keller, A.A.; Wang, T.; Li, F. Heteroaggregation of engineered nanoparticles and kaolin clays in aqueous environments. Water Res. 2015, 80, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Labille, J.; Harns, C.; Bottero, J.-Y.; Brant, J. Heteroaggregation of titanium dioxide nanoparticles with natural clay colloids. Environ. Sci. Technol. 2015, 49, 6608–6616. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, F.; Wang, Z.; Cao, X.; Xing, B. Heteroaggregation of graphene oxide with minerals in aqueous phase. Environ. Sci. Technol. 2015, 49, 2849–2857. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Phenrat, T.; Tilton, R.D.; Lowry, G.V. Effect of kaolinite, silica fines and ph on transport of polymer-modified zero valent iron nano-particles in heterogeneous porous media. J. Colloid. Interface Sci. 2012, 370, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.A.; McCaffery, J.M.; Chen, K.L. Heteroaggregation of multiwalled carbon nanotubes and hematite nanoparticles: Rates and mechanisms. Environ. Sci. Technol. 2012, 46, 5912–5920. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Silva, M.; Uribe-Salas, A.; Waters, K.E.; Finch, J.A. Zeta potential study of pentlandite in the presence of serpentine and dissolved mineral species. Miner. Eng. 2016, 85, 66–71. [Google Scholar] [CrossRef]
- Armstrong, R.; Friedrich, V.L.; Holmes, K.V.; Dubois-Dalcq, M. In vitro analysis of the oligodendrocyte lineage in mice during demyelination and remyelination. J. Cell Biol. 1990, 111, 1183–1195. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.-S.; Davis, L.M.; Shows, T.B. Mapping small DNA sequences by fluorescence in situ hybridization directly on banded metaphase chromosomes. Proc. Natl. Acad. Sci. USA 1990, 87, 6223–6227. [Google Scholar] [CrossRef] [PubMed]
- Di Michele, L.; Zaccone, A.; Eiser, E. Analytical theory of polymer-network-mediated interaction between colloidal particles. Proc. Natl. Acad. Sci. USA 2012, 109, 10187–10192. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Pedrero, F.; Alousque, F.; de Gaudemaris, B.; Berriot, J.; Gaboriaud, F.; Bremond, N.; Bibette, J. Making an elastomeric composite material via the heteroaggregation of a binary colloidal dispersion. Soft Matter 2012, 8, 8752–8757. [Google Scholar] [CrossRef]
- Meng, X.; Wu, H.; Morbidelli, M. Shear-driven aggregation of binary colloids for randomly distributing nanoparticles in a matrix. Soft Matter 2016, 12, 3696–3702. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Conrad, J.C. Dynamics of confined depletion mixtures of polymers and bidispersed colloids. Soft Matter 2013, 9, 10617–10626. [Google Scholar] [CrossRef]
- Sheng, X.; Xie, D.; Zhang, X.; Zhong, L.; Wu, H.; Morbidelli, M. Uniform distribution of graphene oxide sheets into a poly-vinylidene fluoride nanoparticle matrix through shear-driven aggregation. Soft Matter 2016, 12, 5876–5882. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Zuo, K.; Wang, Z.; Zhang, L.; Liu, L.; Guo, B. Using a green method to develop graphene oxide/elastomers nanocomposites with combination of high barrier and mechanical performance. Compos. Sci. Technol. 2014, 92, 1–8. [Google Scholar] [CrossRef]
- Kim, A.Y.; Berg, J.C. Fractal heteroaggregation of oppositely charged colloids. J. Colloid Interface Sci. 2000, 229, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Islam, A.M.; Chowdhry, B.Z.; Snowden, M.J. Heteroaggregation in colloidal dispersions. Adv. Colloid Interface Sci. 1995, 62, 109–136. [Google Scholar] [CrossRef]
- Lopez-Lopez, J.M.; Schmitt, A.; Moncho-Jorda, A.; Hidalgo-Alvarez, R. Stability of binary colloids: Kinetic and structural aspects of heteroaggregation processes. Soft Matter 2006, 2, 1025–1042. [Google Scholar] [CrossRef]
- Furusawa, K.; Anzai, C. Heterocoagulation behaviour of polymer latices with spherical silica. Colloids Surf. 1992, 63, 103–111. [Google Scholar] [CrossRef]
- Ottewill, R.H.; Schofield, A.B.; Waters, J.A.; Williams, N.S.J. Preparation of core-shell polymer colloid particles by encapsulation. Colloid Polym. Sci. 1997, 275, 274–283. [Google Scholar] [CrossRef]
- Grossiord, N.; Loos, J.; Regev, O.; Koning, C.E. Toolbox for dispersing carbon nanotubes into polymers to get conductive nanocomposites. Chem. Mater. 2006, 18, 1089–1099. [Google Scholar] [CrossRef]
- Thomassin, J.-M.; Trifkovic, M.; Alkarmo, W.; Detrembleur, C.; Jérôme, C.; Macosko, C. Poly(methyl methacrylate)/graphene oxide nanocomposites by a precipitation polymerization process and their dielectric and rheological characterization. Macromolecules 2014, 47, 2149–2155. [Google Scholar] [CrossRef]
- Hui, D.; Nawaz, M.; Morris, D.P.; Edwards, M.R.; Saunders, B.R. Study of ph-triggered heteroaggregation and gel formation within mixed dispersions. J. Colloid Interface Sci. 2008, 324, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Kobayashi, M.; Skarba, M.; Mu, C.; Galletto, P.; Borkovec, M. Heteroaggregation in binary mixtures of oppositely charged colloidal particles. Langmuir 2006, 22, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.J.; Steenkeste, K.; Canette, A.; Eloy, M.; Brosson, D.; Gaboriaud, F.; Fontaine-Aupart, M.-P. Natural rubber–filler interactions: What are the parameters? Langmuir 2015, 31, 12437–12446. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zaccone, A.; Tsoutsoura, A.; Lattuada, M.; Morbidelli, M. High shear-induced gelation of charge-stabilized colloids in a microchannel without adding electrolytes. Langmuir 2009, 25, 4715–4723. [Google Scholar] [CrossRef] [PubMed]
- Zaccone, A.; Wu, H.; Gentili, D.; Morbidelli, M. Theory of activated-rate processes under shear with application to shear-induced aggregation of colloids. Phys. Rev. E 2009, 80, 051404. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Xie, D.L.; Wu, H.; Braun, L.; Morbidelli, M. Effect of primary particle morphology on the structure of gels formed in intense turbulent shear. Langmuir 2010, 26, 6643–6649. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Tsoutsoura, A.; Lattuada, M.; Zaccone, A.; Morbidelli, M. Effect of temperature on high shear-induced gelation of charge-stabilized colloids without adding electrolytes. Langmuir 2010, 26, 2761–2768. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.L.; Wu, H.; Zaccone, A.; Braun, L.; Chen, H.Q.; Morbidelli, M. Criticality for shear-induced gelation of charge-stabilized colloids. Soft Matter 2010, 6, 2692–2698. [Google Scholar] [CrossRef]
- Zaccone, A.; Gentili, D.; Wu, H.; Morbidelli, M. Shear-induced reaction-limited aggregation kinetics of brownian particles at arbitrary concentrations. J. Chem. Phys. 2010, 132, 134903. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.L.; Arosio, P.; Wu, H.; Morbidelli, M. Effect of surfactants on shear-induced gelation and gel morphology of soft strawberry-like particles. Langmuir 2011, 27, 7168–7175. [Google Scholar] [CrossRef] [PubMed]
- Zaccone, A.; Gentili, D.; Wu, H.; Morbidelli, M.; Del Gado, E. Shear-driven solidification of dilute colloidal suspensions. Phys. Rev. Lett. 2011, 106, 138301. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Wu, H.; Morbidelli, M. Snapshotted glass and gel transitions of stable colloidal dispersions after shear-driven aggregation in a microchannel. Soft Matter 2015, 11, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Wu, H.; Morbidelli, M. Kinetics and cluster morphology evolution of shear-driven aggregation of well-stabilized colloids. Langmuir 2015, 31, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Zaccone, A.; Wu, H.; Lattuada, M.; Morbidelli, M. Charged molecular films on brownian particles: Structure, interactions, and relation to stability. J. Phys. Chem. B 2008, 112, 6793–6802. [Google Scholar] [CrossRef] [PubMed]
- Zaccone, A.; Wu, H.; Lattuada, M.; Morbidelli, M. Correlation between colloidal stability and surfactant adsorption/association phenomena studied by light scattering. J. Phys. Chem. B 2008, 112, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Morbidelli, M. Gelation of polymeric nanoparticles. Particuology 2014, 14, 1–11. [Google Scholar] [CrossRef]
- Parsegian, V.A.; Zemb, T. Hydration forces: Observations, explanations, expectations, questions. Curr. Opin. Colloid Interface Sci. 2011, 16, 618–624. [Google Scholar] [CrossRef]
- Li, Y.; Hu, J.; He, J.; Gao, L. The graphene oxide polymer composites with high breakdown field strength and energy storage ability. In Proceedings of the International Congress on Energy Efficiency and Energy Related Materials (ENEFM 2013), Antalya, Turkey, 9–12 October 2013; Oral, A.Y., Bahsi, Z.B., Ozer, M., Eds.; Springer International Publishing: New York, NY, USA, 2014; Volume 155, pp. 431–438. [Google Scholar]
- El Achaby, M.; Arrakhiz, F.Z.; Vaudreuil, S.; Essassi, E.M.; Qaiss, A. Piezoelectric β-polymorph formation and properties enhancement in graphene oxide—Pvdf nanocomposite films. Appl. Surf. Sci. 2012, 258, 7668–7677. [Google Scholar] [CrossRef]
Figure 1.
A z-shaped microchannel (z-MC) setup (a) for conducting intense shear-driven aggregation/gelation of colloidal dispersion, and (b) intense shear-driven transition from liquid-like colloids to solid-like gels at the shear rate, = 1 × 106 s–1, at different particle volume fractions (φ) and different operation temperatures. Reproduced with permission from [65,74]. Copyright (2010) American Chemical Society and (2014) Elsevier.
Figure 1.
A z-shaped microchannel (z-MC) setup (a) for conducting intense shear-driven aggregation/gelation of colloidal dispersion, and (b) intense shear-driven transition from liquid-like colloids to solid-like gels at the shear rate, = 1 × 106 s–1, at different particle volume fractions (φ) and different operation temperatures. Reproduced with permission from [65,74]. Copyright (2010) American Chemical Society and (2014) Elsevier.

Figure 2.
Time evolution of the conversion of polystyrene NPs to big clusters through the intense shear-driven aggregation at a fixed shear rate, = 1.5 × 106 s−1, and various particle volume fractions. Reproduced with permission from [71]. Copyright (2014) American Chemical Society.
Figure 2.
Time evolution of the conversion of polystyrene NPs to big clusters through the intense shear-driven aggregation at a fixed shear rate, = 1.5 × 106 s−1, and various particle volume fractions. Reproduced with permission from [71]. Copyright (2014) American Chemical Society.
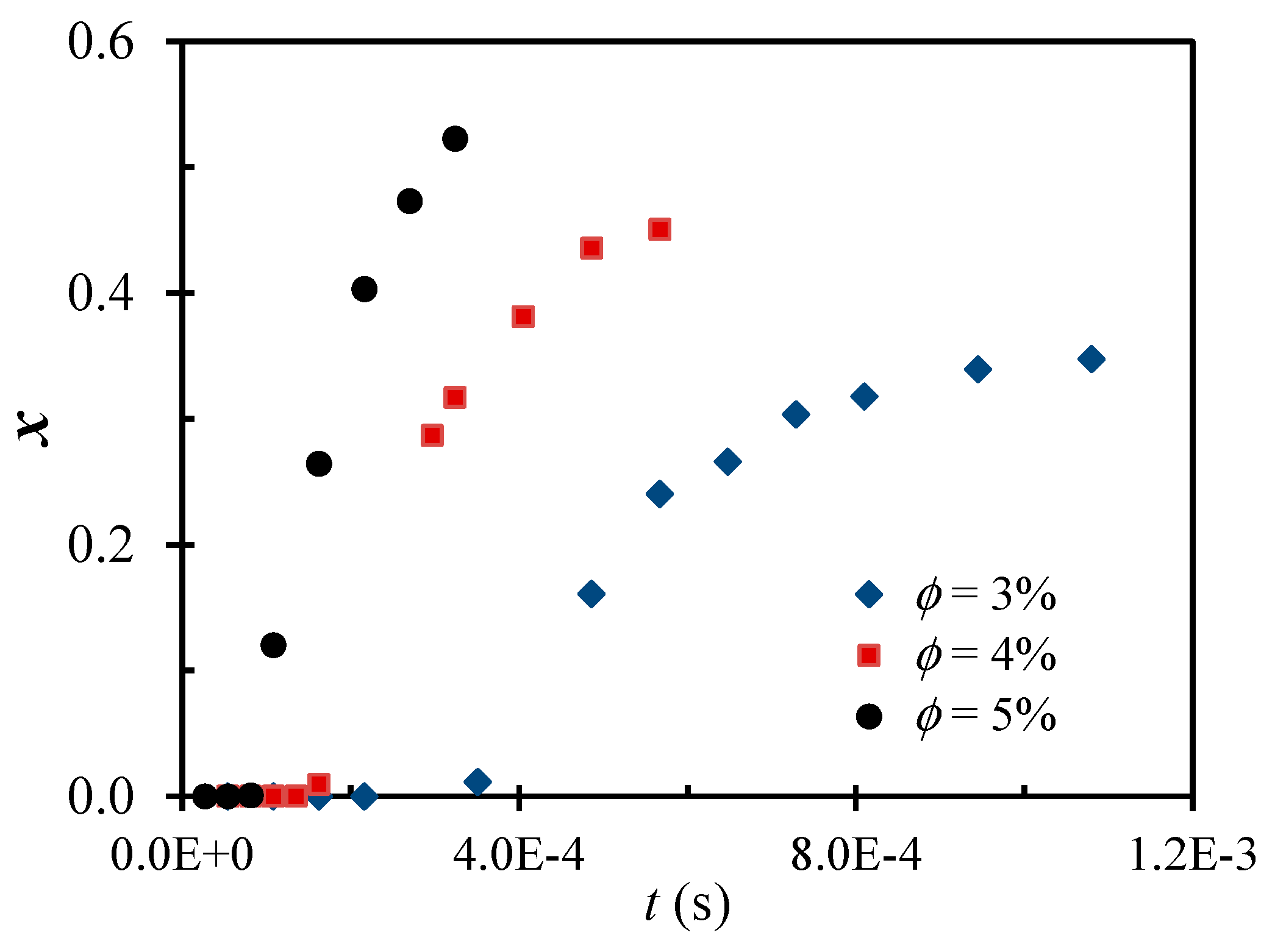
Figure 3.
Design concept for fabrication of nanocomposites via purely shear-driven aggregation/gelation of binary colloids.
Figure 3.
Design concept for fabrication of nanocomposites via purely shear-driven aggregation/gelation of binary colloids.
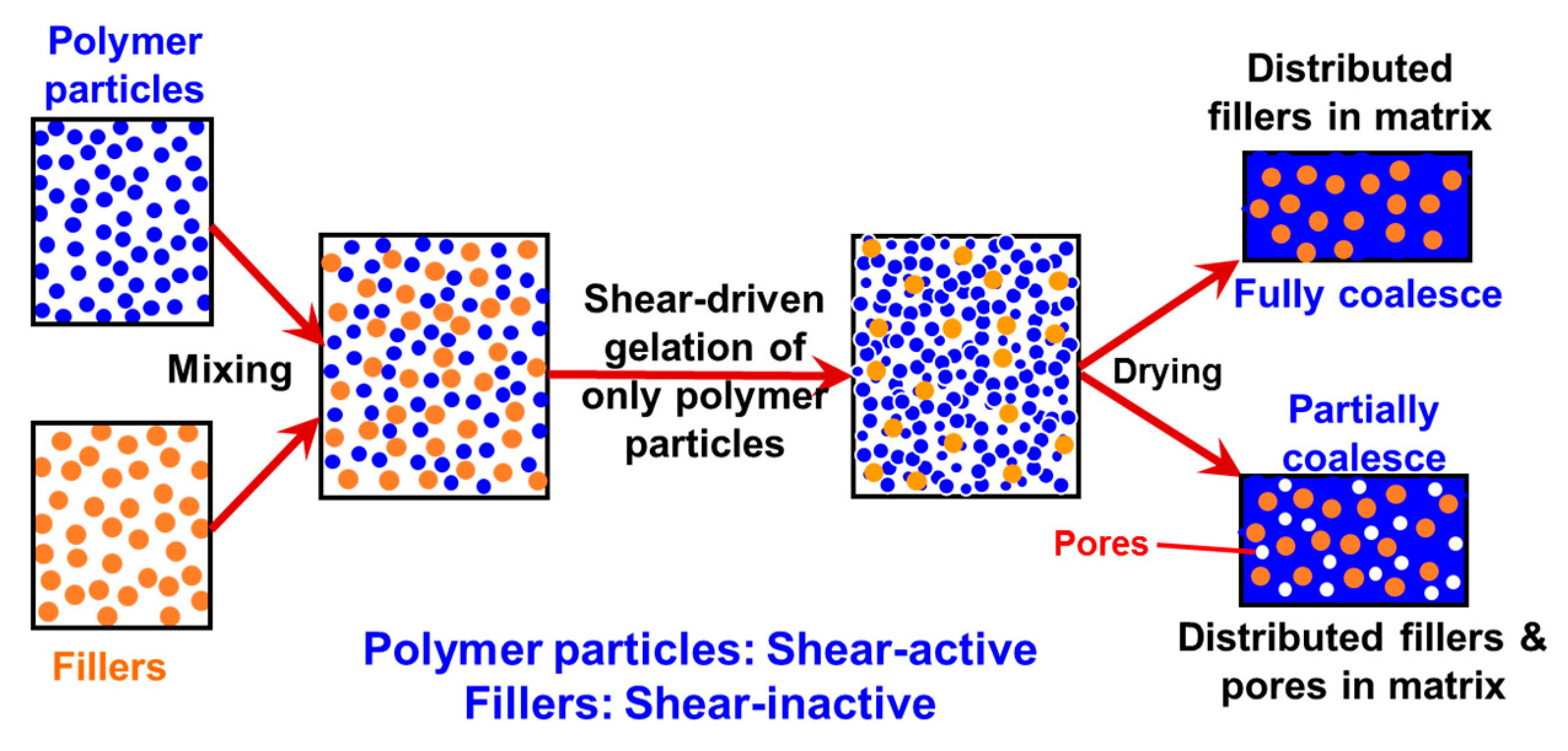
Figure 4.
SEM pictures of the clusters formed after shear-driven aggregation of (a) pure PS dispersion and (b) the PMMA + PS binary dispersion. Reproduced with permission from [48]. Copyright (2016) The Royal Society of Chemistry.
Figure 4.
SEM pictures of the clusters formed after shear-driven aggregation of (a) pure PS dispersion and (b) the PMMA + PS binary dispersion. Reproduced with permission from [48]. Copyright (2016) The Royal Society of Chemistry.

Figure 5.
A schematic representation of the shear-driven aggregation mechanism of the PS/PMMA binary colloid, where the small PS NPs are shear active, while the PMMA NPs are shear inactive. Reproduced with permission from [48]. Copyright (2016) The Royal Society of Chemistry.
Figure 5.
A schematic representation of the shear-driven aggregation mechanism of the PS/PMMA binary colloid, where the small PS NPs are shear active, while the PMMA NPs are shear inactive. Reproduced with permission from [48]. Copyright (2016) The Royal Society of Chemistry.
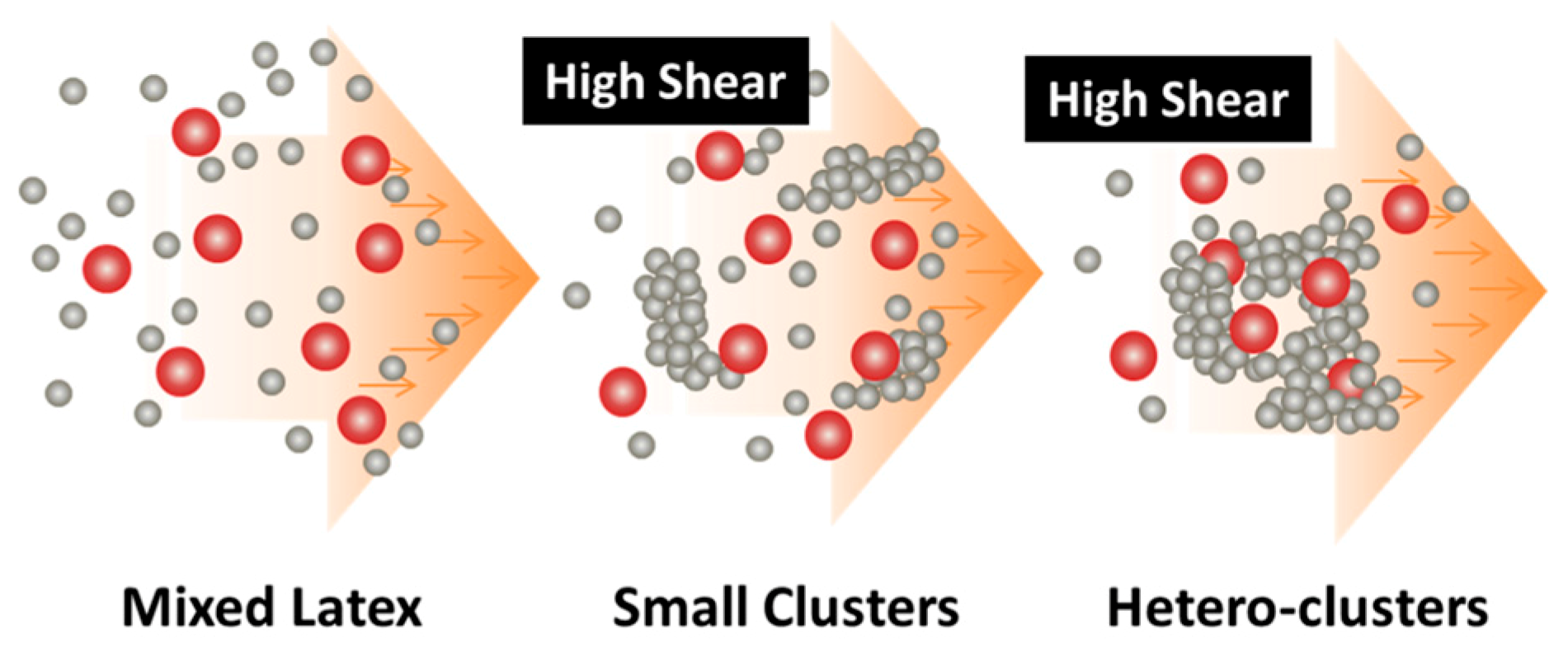
Figure 6.
SEM images of the clusters formed by the GO/PVDF binary colloid after passing through the z-MC one time: (a) typical shape of the clusters and (b) the dispersed GO sheets inside the clusters, as indicated by the red arrows. Reproduced with permission from [50]. Copyright (2016) The Royal Society of Chemistry.
Figure 6.
SEM images of the clusters formed by the GO/PVDF binary colloid after passing through the z-MC one time: (a) typical shape of the clusters and (b) the dispersed GO sheets inside the clusters, as indicated by the red arrows. Reproduced with permission from [50]. Copyright (2016) The Royal Society of Chemistry.

Figure 7.
Conversion of the polymer (PVDF) particles (a) and the GO sheets (b) to big clusters, as a function of the number of passes. The volume fraction of PVDF NPs = 2.9%, and the volume fraction of the GO sheets with respect to the polymer = 3.0%. Reproduced with permission from [50]. Copyright (2016) The Royal Society of Chemistry.
Figure 7.
Conversion of the polymer (PVDF) particles (a) and the GO sheets (b) to big clusters, as a function of the number of passes. The volume fraction of PVDF NPs = 2.9%, and the volume fraction of the GO sheets with respect to the polymer = 3.0%. Reproduced with permission from [50]. Copyright (2016) The Royal Society of Chemistry.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sheng, X.; Zhang, L.; Wu, H. Generation of Polymer Nanocomposites through Shear-Driven Aggregation of Binary Colloids. Polymers 2017, 9, 619. https://doi.org/10.3390/polym9110619
AMA Style
Sheng X, Zhang L, Wu H. Generation of Polymer Nanocomposites through Shear-Driven Aggregation of Binary Colloids. Polymers. 2017; 9(11):619. https://doi.org/10.3390/polym9110619
Chicago/Turabian StyleSheng, Xinxin, Li Zhang, and Hua Wu. 2017. "Generation of Polymer Nanocomposites through Shear-Driven Aggregation of Binary Colloids" Polymers 9, no. 11: 619. https://doi.org/10.3390/polym9110619
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.