1. Introduction
The thermosetting phenol-formaldehyde (PF) resin is one of the most common adhesives in the wood industry. Although PF resin has been studied for over a century and acceptable products can be obtained, some challenges, such as a high curing temperature and low curing speed [
1,
2,
3], have not been addressed. To understand the chemistry of PF, the influences of the reaction conditions, including the catalysts, formaldehyde to phenol molar ratio (F/P), temperature, reactant-adding procedure etc., on the changes of the molecular composites of the resin have been investigated and discussed [
3,
4,
5,
6,
7,
8,
9], but the experimental results have not been well rationalized due to the complexity of the PF chemistry and difficulties in having insight into the involved reactions, especially the microscopic mechanisms. Therefore, the theory of PF resin synthesis has been developed slowly during the past decades.
The kinetics and mechanisms of the individual reactions in the PF system are apparently the key issues and also the foundation for understanding the chemistry of PF. However, by surveying the literature from the middle of the last century to recent years, discrepant results for the kinetics and mechanisms of the base-catalyzed condensation reactions between hydroxymethylphenols (HMPs) have been reported [
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19]. In some earlier studies, the condensations of HMPs were reported to be first-order [
9,
10,
11], pseudo-first-order or second-order [
12,
13]. These reports implied or clearly pointed out that the condensations occurred in an S
N2 (bimolecular nucleophilic substitution)-type mechanism. Namely, the reactions occurred between undissociated and dissociated (ionized) HMPs, dissociated and dissociated HMPs, and undissociated and undissociated HMPs. Later in 1983, Jones observed the self-condensation of trihydroxymethylphenol (THMP) to be first-order and proposed a quinone methide (QM) intermediate hypothesis to explain the kinetics [
14]. In 2001, Higuchi et al. confirmed that the self-condensation of 2-hydroxymethylphenol (2-HMP) is kinetically of genuine first-order and the QM hypothesis seems to hold in such a reaction [
15]. However, in their continuous studies on 4-hydroxymethylphenol (4-HMP) [
16], 2,4-dihydroxymethylphenol (2,4-DHMP) [
17] and 2,4,6-trihydroxymethylphenol (2,4,6-THMP) [
18], the condensations appeared to be of fractional order, ranging from 1.1~2.0. Further, the reaction order varied with the change of the NaOH/HMP molar ratio and with the change of the HMP concentration. Based on these results, Higuchi et al. implied that, beside the unimolecular mechanism which is represented by the QM hypothesis, bimolecular reactions such as S
N2 may also contribute to the reaction [
16,
17,
18]. A very recent study [
19] also suggested that both the QM and S
N2 mechanisms work in the condensations. Obviously, the very fundamental issue in PF resin chemistry has not been addressed.
The discrepant conclusions drawn from the kinetics experiments are mainly due to the complex nature of the PF reaction system. On the other hand, the reactive intermediates are generally short lived and they cannot be directly observed or monitored by experimental methods. Fortunately, theoretical calculations on the computer based on developed quantum chemistry theory allow us to investigate each individual reaction and can provide us with a clear picture of how the reactants evolve to intermediates and finally to products.
In this study, detailed theoretical calculations were performed on the model base-catalyzed reaction between phenol and hydroxymethylphenol to clarify the key issues that have not been addressed. First, how is the quinine methide intermediate formed? How is it reactive in condensation reactions? Second, is the SN2 mechanism that was proposed previously also possible? If yes, which one is more favorable? Finally, are there any other new mechanisms?
The reaction pattern and mechanisms identified for phenol in this study may be extended to other phenolic compounds, such as resorsinol and tannin [
20], because they have a similar phenolic functional group, although the reactivity and molecular weight of these compounds are different from phenol.
3. Results and Discussion
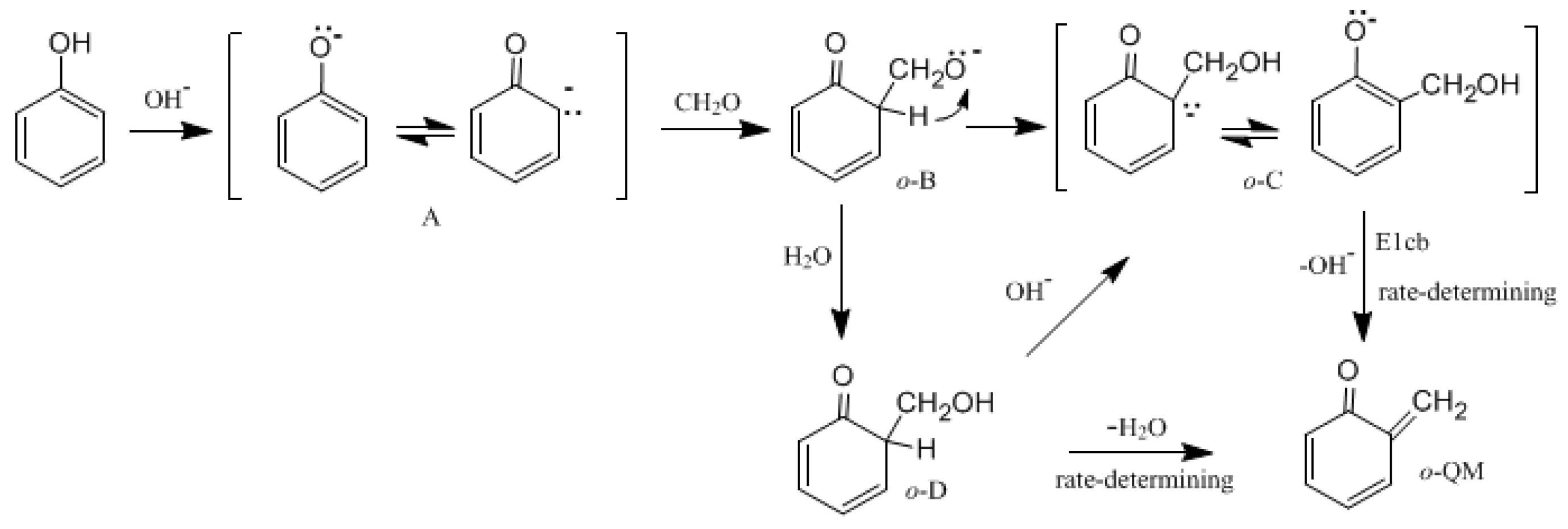
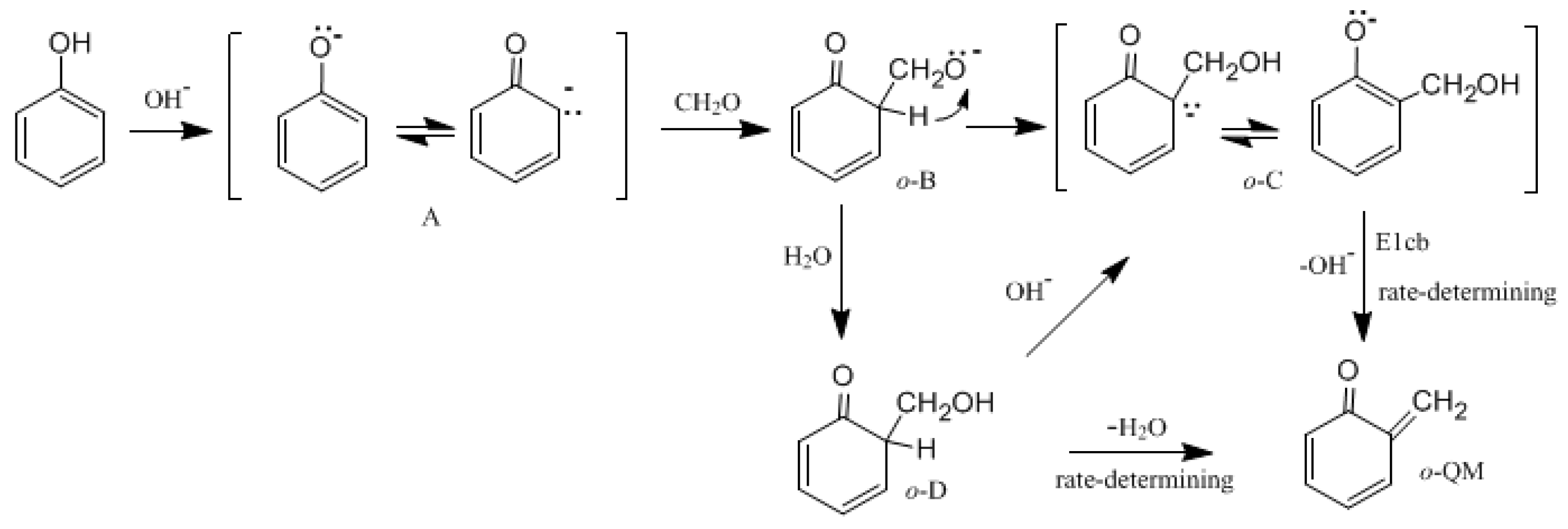
Figure 1 shows the possible mechanisms for the formation of the
ortho-quinone methide (
o-QM) intermediate.
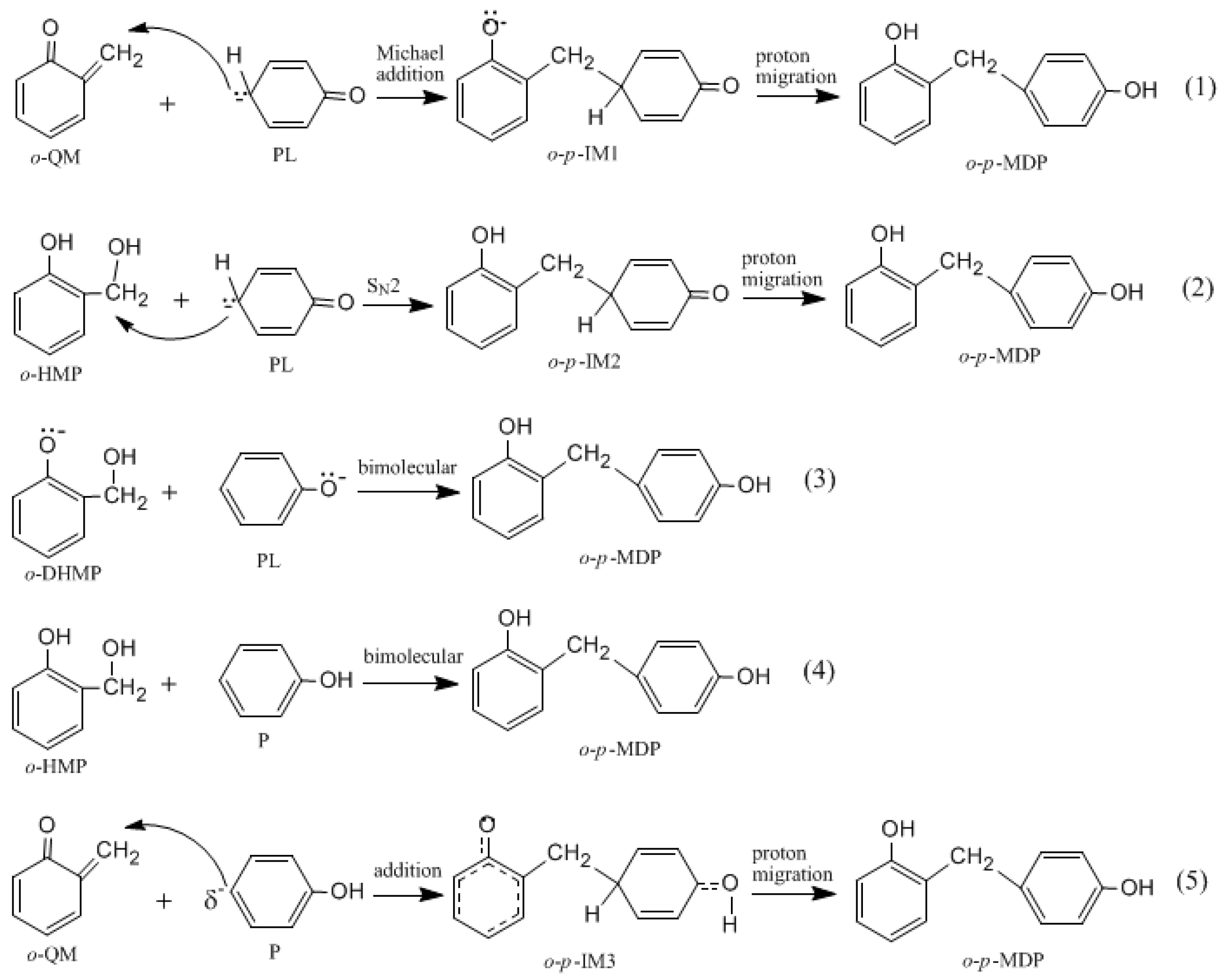
Figure 2 shows the possible mechanisms for the condensation reactions. The first pathway leading to
o-QM is A →
o-B →
o-C →
o-QM. Another pathway is A →
o-B →
o-D →
o-C →
o-QM. Both the pathways must pass the intermediate
o-C. Via the E1cb loss of OH
−, the
o-QM can be formed and this is the rate-determining step. Such a mechanism is proposed in some literature [
15,
16,
17,
18,
19]. However, we considered another mechanism where intra-molecular water elimination from the intermediate
o-D also leads to
o-QM. The para-quinone methide (
p-QM) can also be formed via similar mechanisms. Note that a water-elimination mechanism was also proposed by Higuchi et al. [
15]. However, the initial species was thought to be a neutral HMP. We performed calculations on this mechanism, but it was not identified.
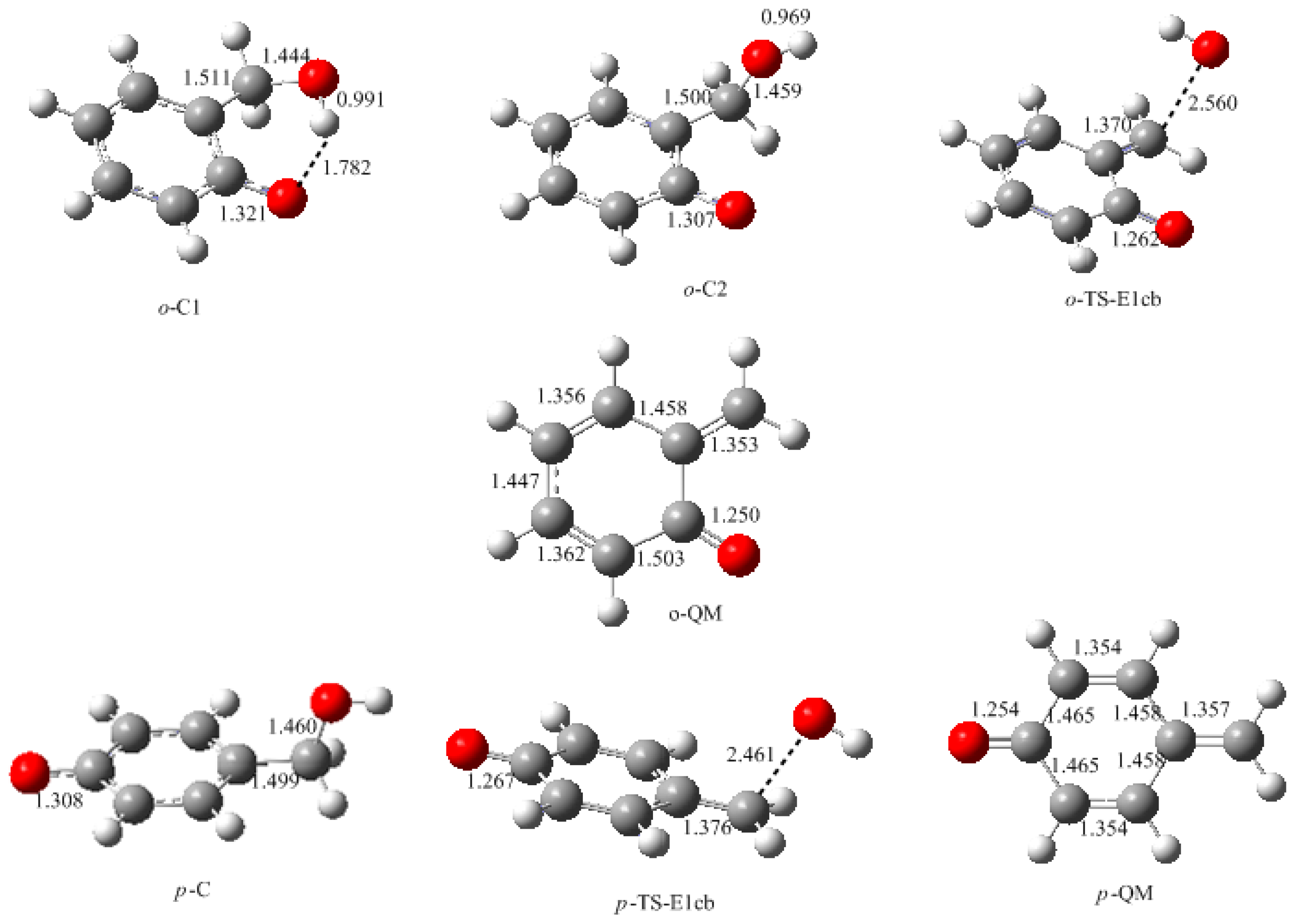
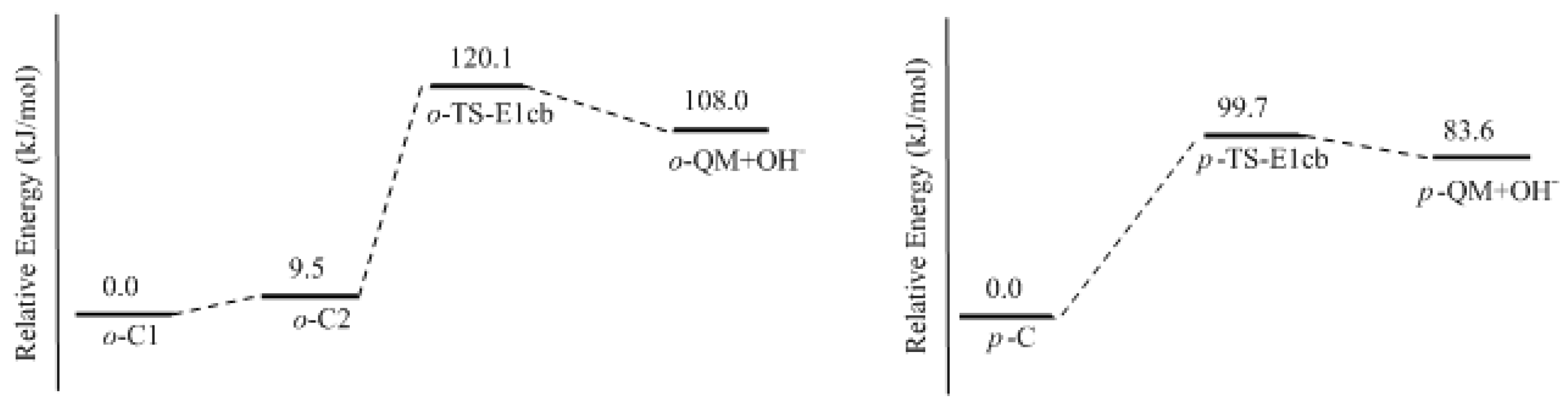
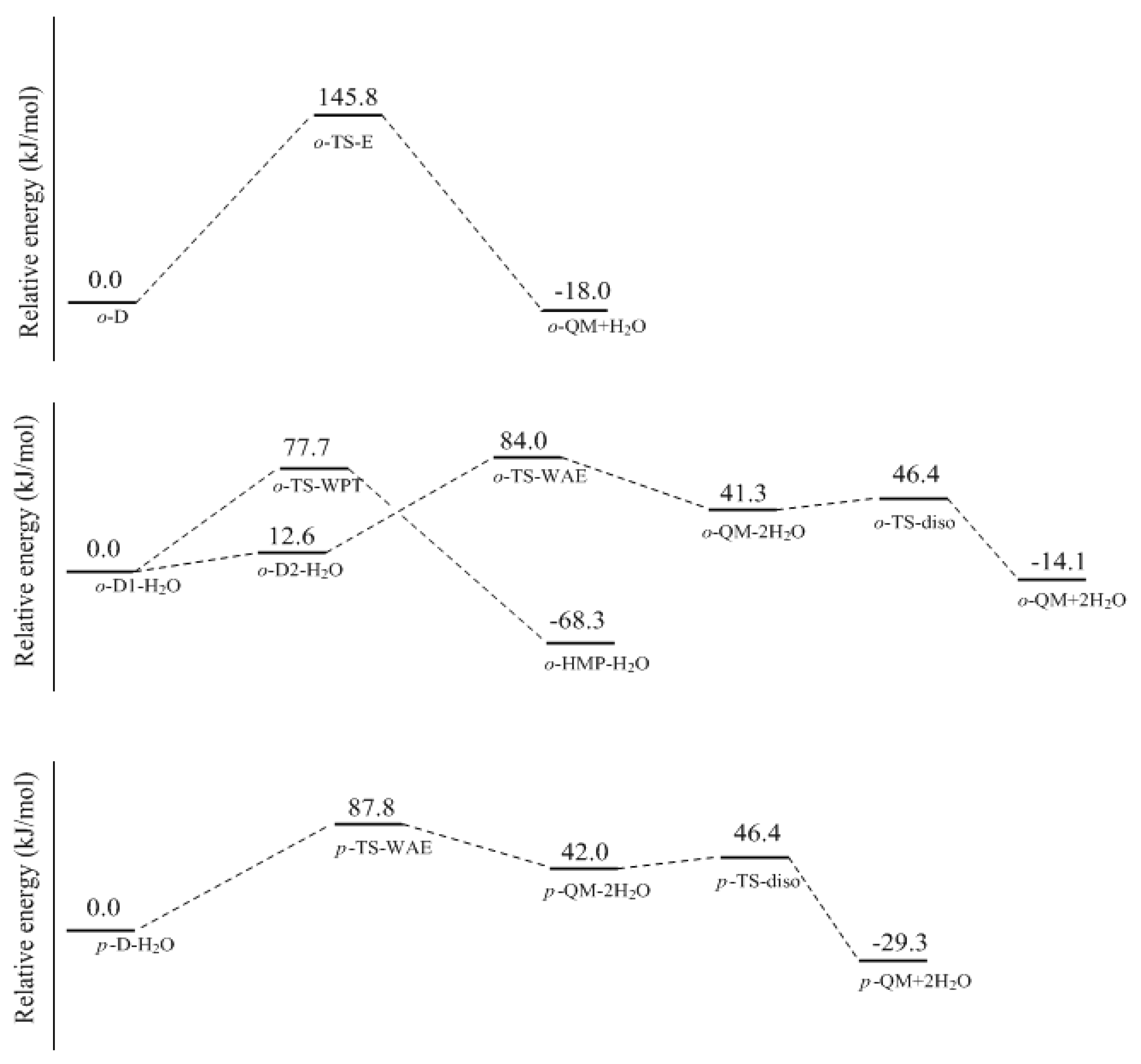
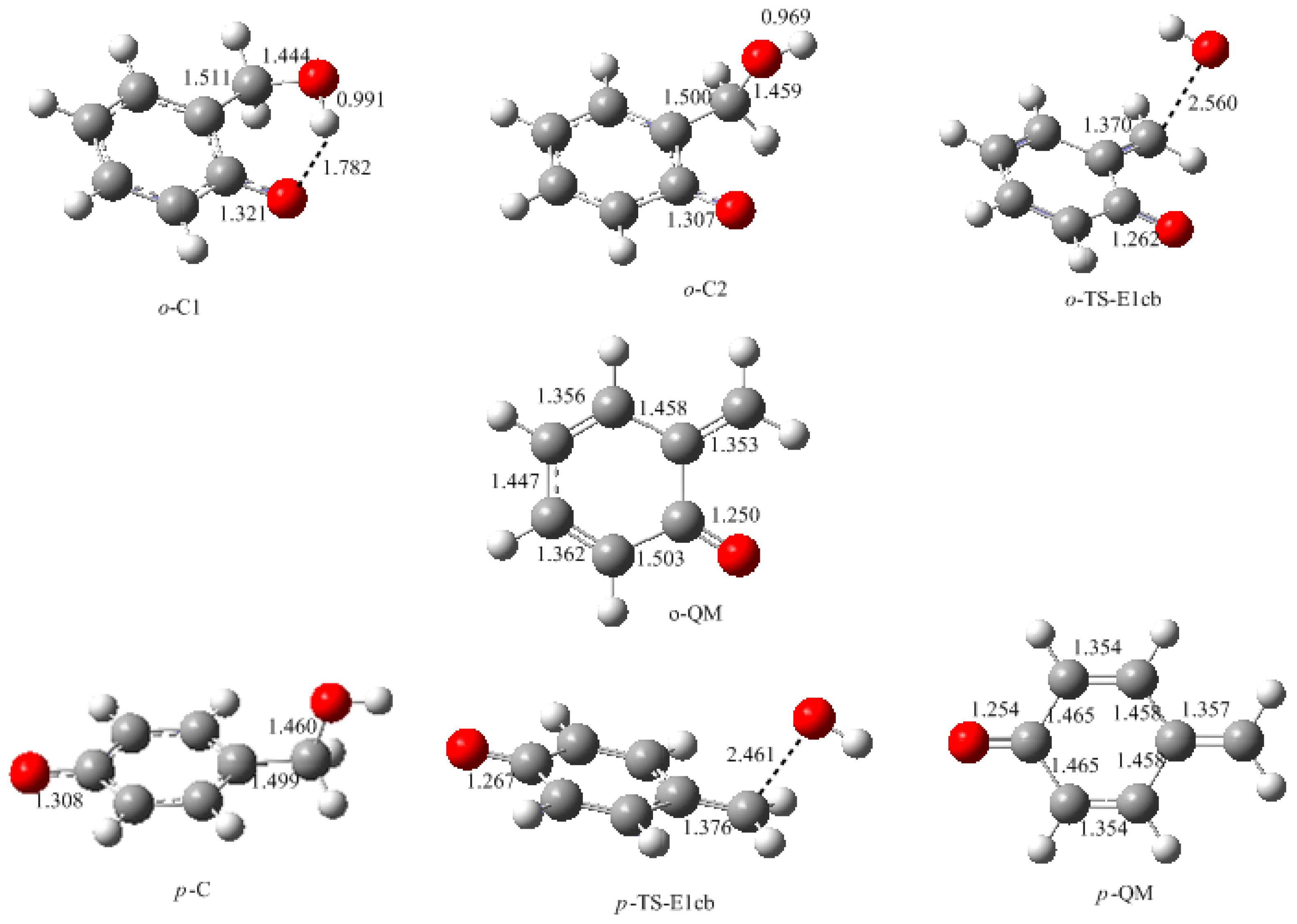
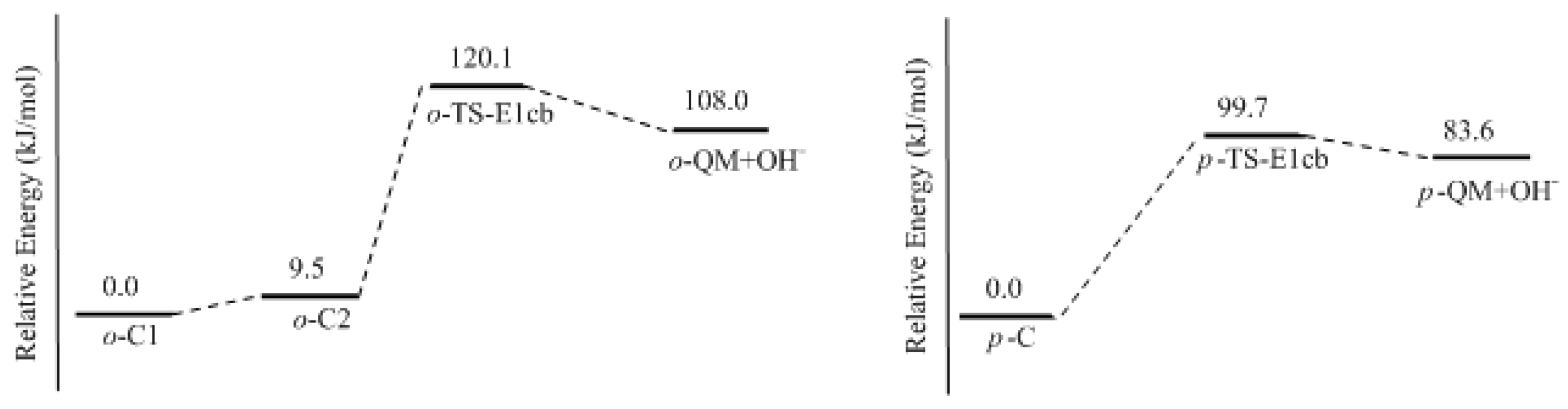
In this work, the rate-determining step of E1cb and water elimination was calculated and the obtained structures are shown in
Figure 3. The calculated potential energy profiles are given in
Figure 4. The
o-C1 and
o-C2 are two structures of intermediate C, and the former is 9.5 kJ/mol more stable than the latter because a hydrogen bond (1.782 Å) can be formed between the
o-hydroxymethyl group and the carbonyl oxygen. Via the transition state
o-TS-E1cb, the OH
− can be eliminated and produce
o-QM. The potential energy barrier of this step was calculated be 120.1 kJ/mol. For the formation of
p-QM, this barrier was lowered to 99.7 kJ/mol. Note that
o-QM and
p-QM are isomers and our calculations suggest that the latter is more stable by 24.4 kJ/mol.
However, the formation of QM will not definitely experience intermediate
o-C or
p-C if the initial reactants are phenol and formaldehyde. An alternative mechanism that works via water elimination from
o-D or
p-D is also possible. The calculated results for this mechanism are shown in
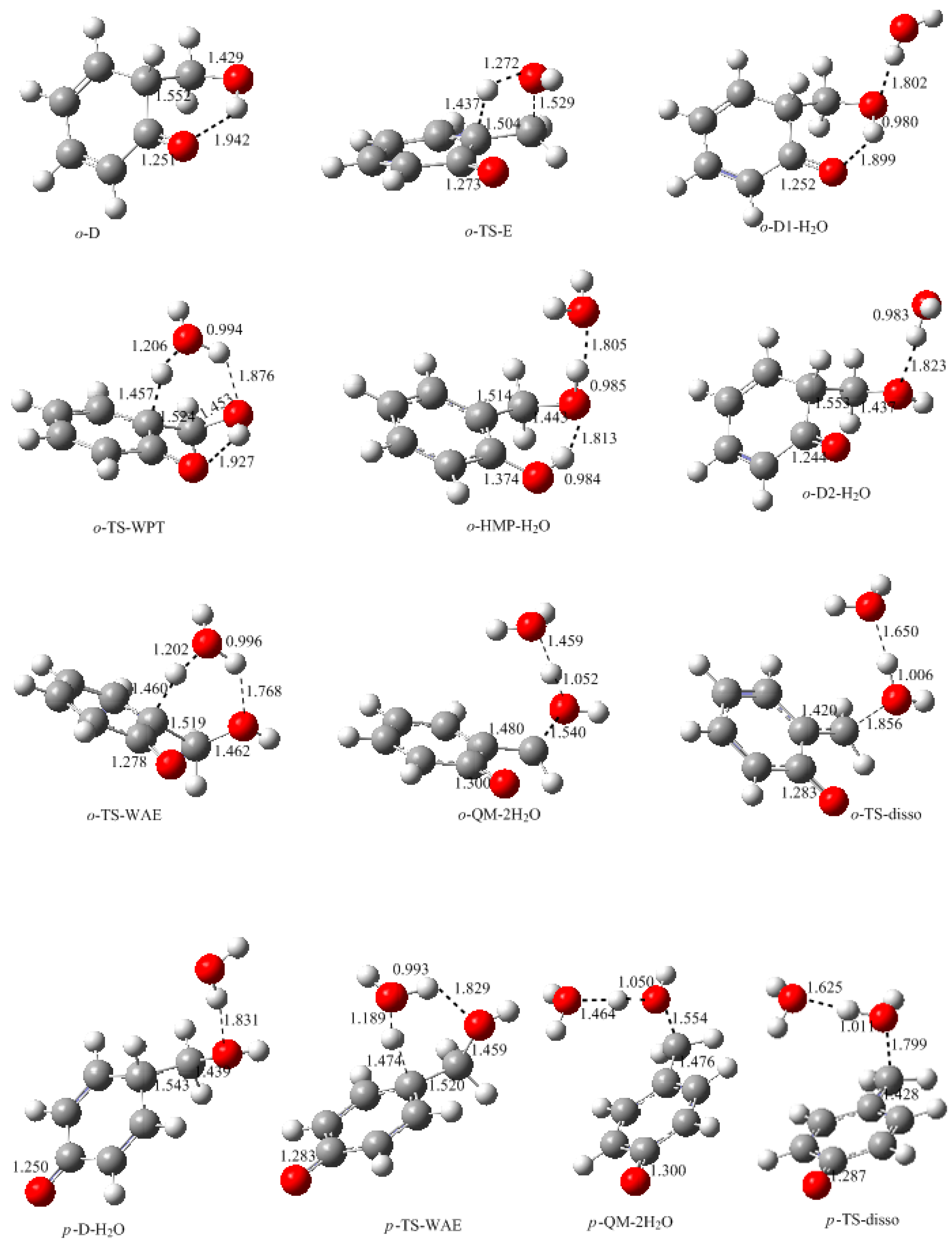
Figure 5 and
Figure 6. From the
ortho-position intermediate
o-D in which an intra-molecular bond is also formed, a water molecule can be eliminated via the four-member transition state TS-E, directly leading to
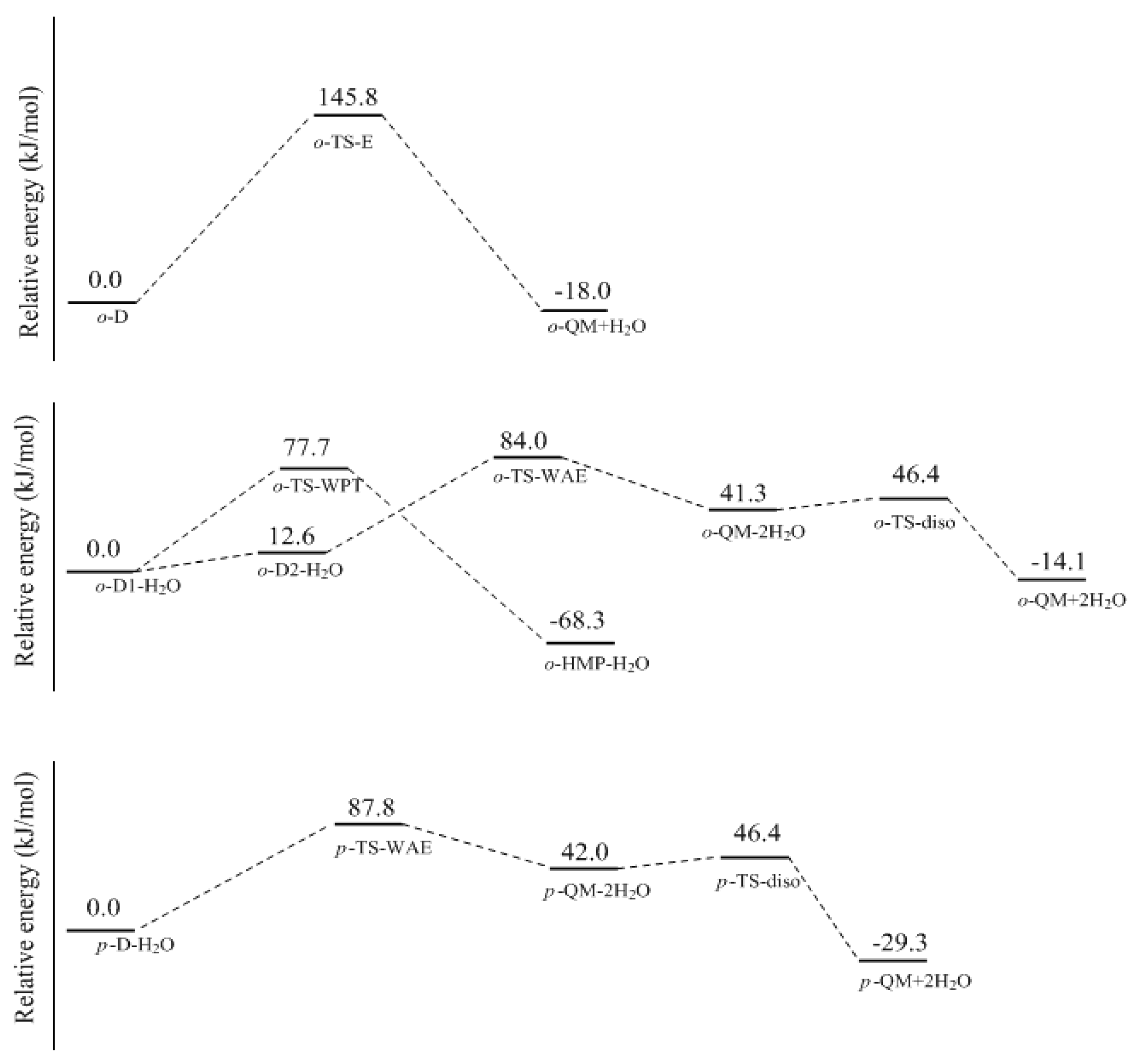
o-QM. The energy barrier of this reaction was calculated to be 145.8 kJ/mol. Obviously, such a high barrier is not consistent with the kinetics measurement. However, in aqueous solution, the water molecule may play the role of catalyst. Therefore, we put a water molecule in the theoretical model to simulate the catalytic effect. The
o-D1-H
2O is the obtained intermediate complex formed through the intermolecular hydrogen bonding interaction between
o-D1 and H
2O. Based on this complex, the six-member water-mediated proton transfer transition state
o-TS-WPT was identified. The vibration mode of the imaginary frequency clearly showed the proton transfer from the carbon to the water oxygen, and from the water to the hydroxyl oxygen. It is much like the expected transition state. However, IRC calculation demonstrated that once a proton shifted to the hydroxyl oxygen, another proton on the hydroxyl group simultaneously moved to the carbonyl oxygen due to the strong intra-molecular hydrogen bonding effect, leading to
o-HMP-H
2O which can be viewed as a complex of
o-HMP and H
2O. Therefore, the
o-TS-WPT does not correspond to the water elimination and formation of
o-QM, but corresponds to the simultaneous transfer of three protons and the formation of
o-HMP. Despite that, in contrast to TS-E, the water transfer energy barrier was significantly lowered to 77.7 kJ/mol.
Obviously, the proton transfer from the hydroxyl group to the carbonyl group is due to the strong intra-molecular hydrogen bonding effect. If this effect is destroyed, water elimination may occur. By rotating the hydroxyl group and canceling the hydrogen bonding effect, the complex
o-D2-H
2O, which is 12.6 kJ/mol less stable than
o-D1-H
2O, was identified. After this complex, the water-aided elimination transition state
o-TS-WAE was located. The IRC calculation showed that the proton on the carbon transferred to the hydroxyl group with the mediation of water, tending to eliminate two water molecules. Thus,
o-QM-2H
2O was the resulting intermediate. Further calculations confirmed that two water molecules can be eliminated via the dissociation transition state
o-TS-disso, resulting in the formation of
o-QM. As shown in
Figure 6, the transition state
o-TS-WAE represents the rate-determining step, and the energy barrier relative to
o-D1-H
2O was predicted to be 84.0 kJ/mol, which is significantly lower than the barrier of TS-E, clearly indicating the catalytic effect of water. Compared with the E1cb mechanism, this mechanism is also much more favorable. However, due to the intra-molecular hydrogen bonding effect, a large portion of the complex formed between
o-D1 and H
2O may exist mainly in the form of
o-D1-H
2O, instead of
o-D2-H
2O. As revealed above, the former evolved to
o-HMP, not
o-QM. Therefore, the inhibitory effect of hydrogen bonding on the formation of
o-QM works here. The
para-position QM (
p-QM) is not kinetically more favorable than
o-QM, although its formation is not influenced by the hydrogen bonding effect. As shown in
Figure 5 and
Figure 6, the
p-TS-WAE was the identified transition state for the
para-position water-aided elimination mechanism. The energy barrier represented by this transition state was 87.8 kJ/mol, slightly higher than that of
o-TS-WAE. However, the addition between phenol and formaldehyde at the
para-position is generally faster than the addition at the
ortho-position. Therefore, the
p-QM may have a higher concentration in the solution and the following condensation occurs mainly between the free
ortho-postion of the aromatic ring and
p-QM, leading to the
o-
p condensation structure. This can rationalize the experimentally observed result that the
o-
p condensation structure was predominant.
In the kinetics study of Higuchi et al. [
15,
16], the starting reactant was 2-hydroxymethylphenol (2-HMP) or 4-hydroxymethylphenol (4-HMP) and they estimated the activation energy barriers for the formation of
o-QM and
p-QM as 103 ± 5 and 78 ± 5 kJ/mol. This seems to agree with our calculations for the E1cb mechanism in which the
o-QM is energetically less favorable. However,
o-QM or
p-QM may not be directly produced via the E1cb mechanism, because the
o-C or
p-C intermediate, which can be viewed as dissociated HMP, can evolve to intermediate
o-D or
p-D. As discussed above,
o-TS-WAE and
p-TS-WAE have a close potential energy barrier of 84.0 and 87.0 kJ/mol, respectively, but due to the intra-molecular hydrogen bonding effect, a portion of
o-D-H
2O will evolve to
o-HMP instead of
o-QM. As a result, a lower reaction rate and higher activation energy will be measured. This is in agreement with the conjecture of Higuchi et al. that the hydrogen bonding in 2-HMP has an inhibitory effect on
o-QM formation.
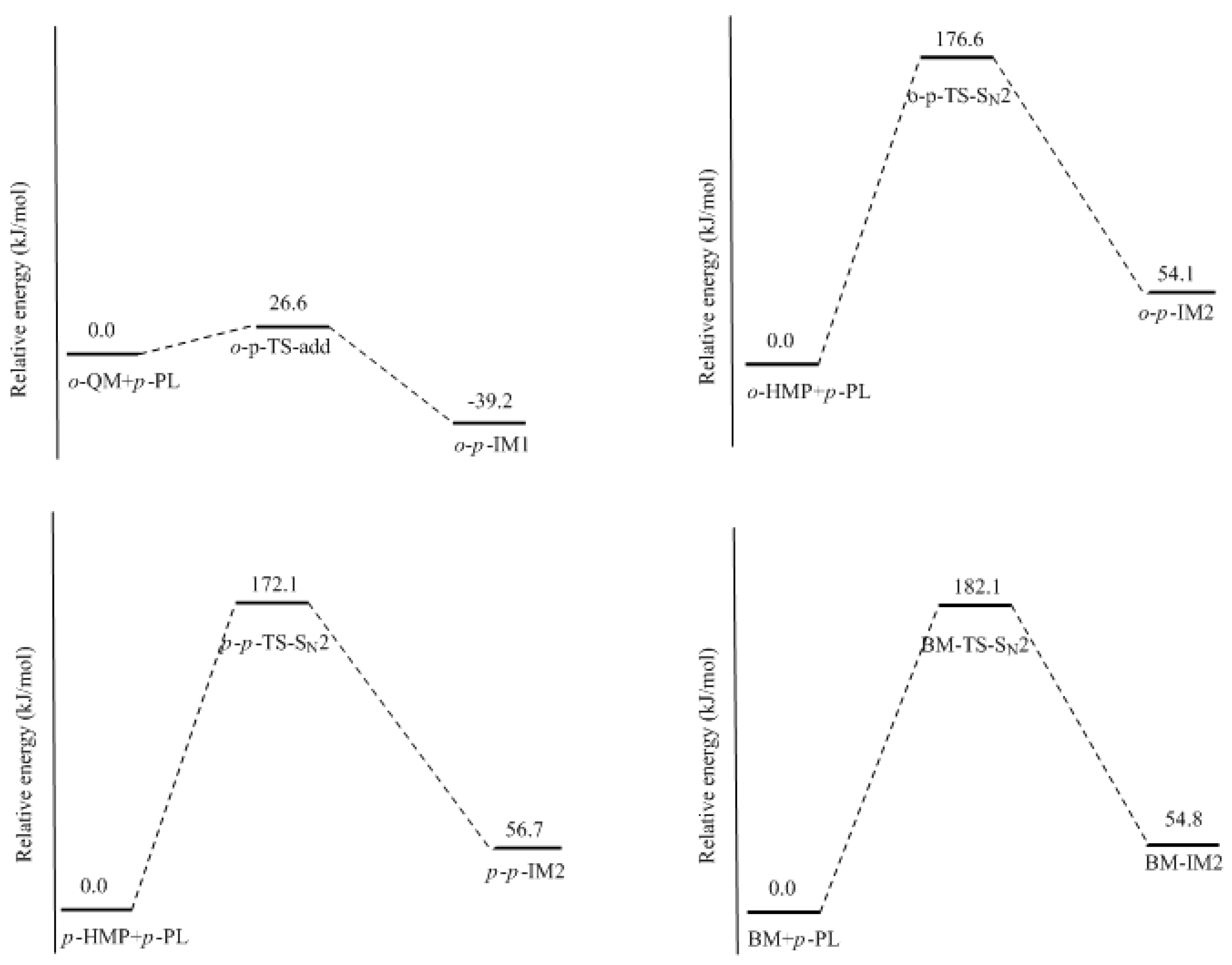
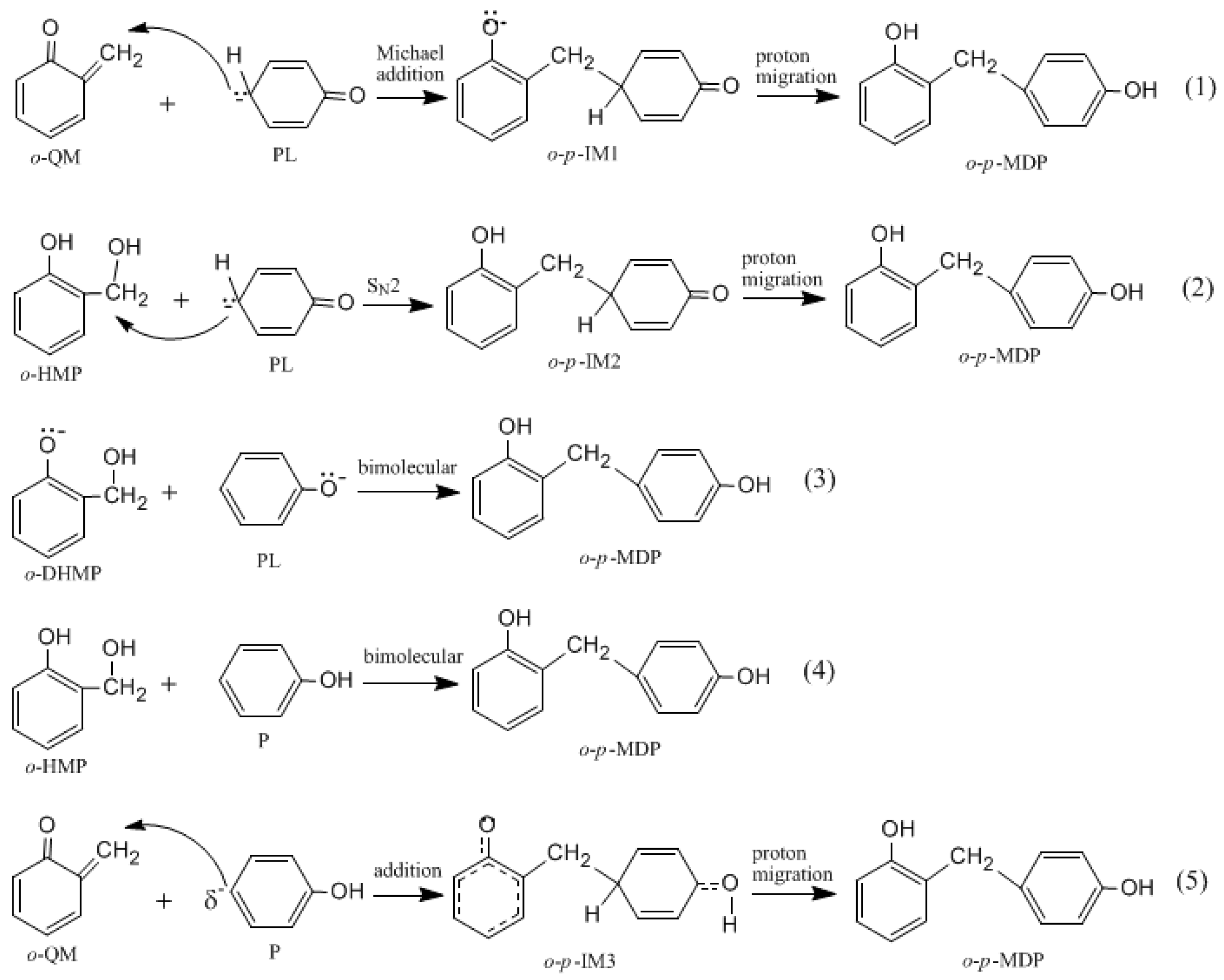
It was assumed in the literature that the condensation reaction of a phenolate ion (PL) or dissociated HMP with QM was much faster than the formation of QM [
14,
15,
16,
17,
18,
19]. To confirm this, we also performed calculations on the reaction (1) in
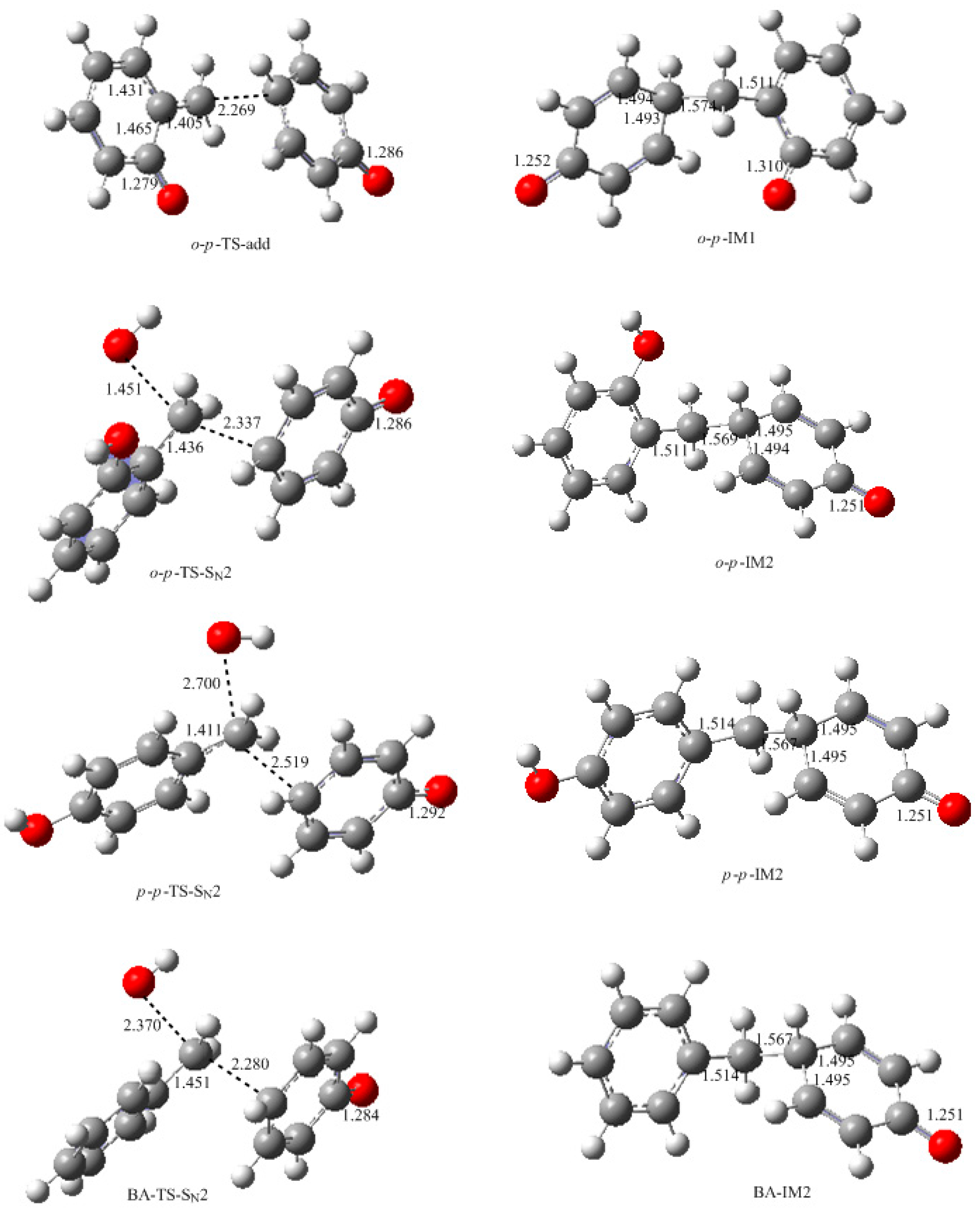
Figure 2. As shown in
Figure 7, an addition-like transition state,
o-
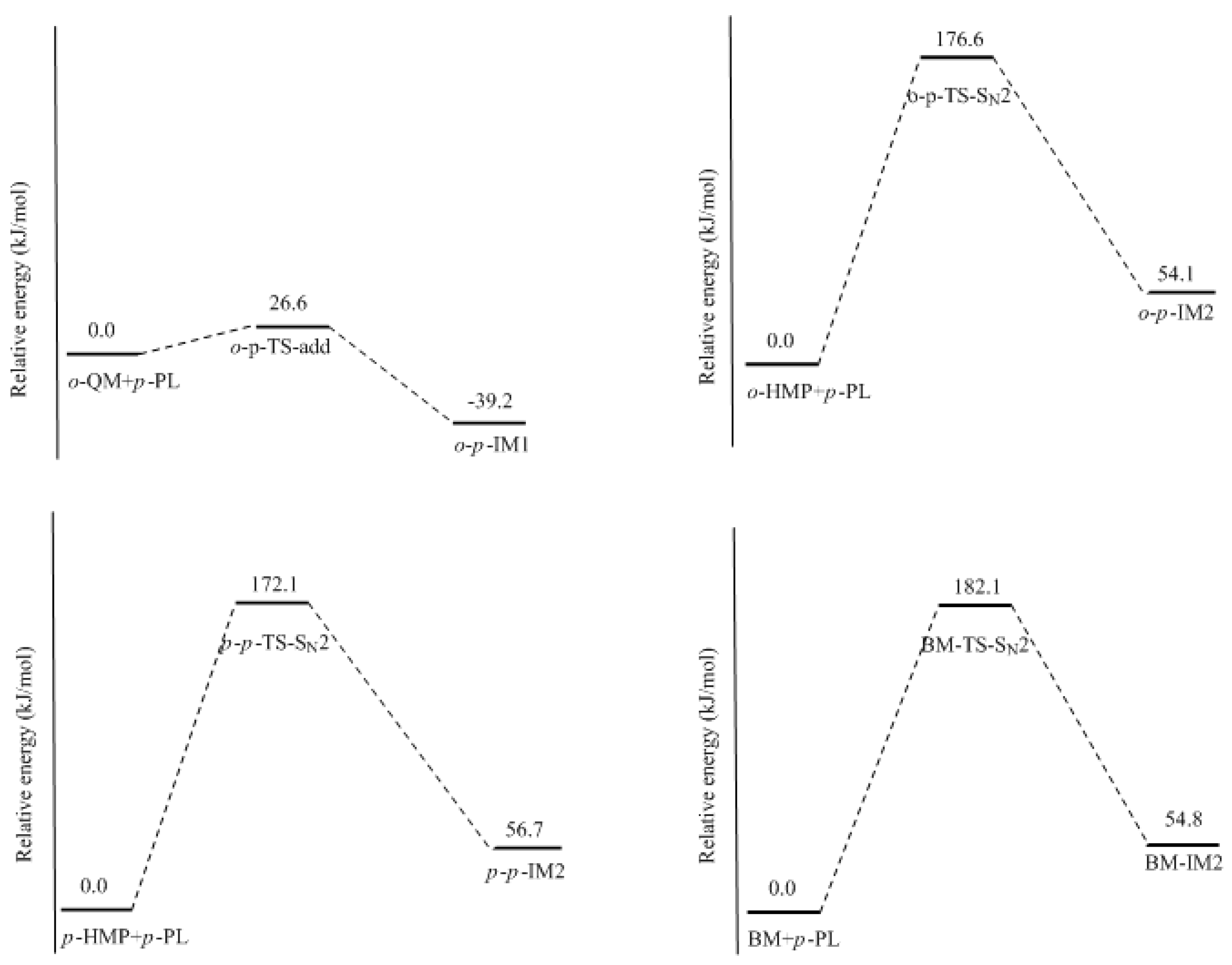
p-TS-add, was located. In fact, this is the typical Michael addition. The potential energy profile in
Figure 8 shows this step has a small barrier of 26.6 kJ/mol, indicating this reaction is indeed much faster than QM formation. Proton migrations after the intermediate
o-
p-IM1, leading to
o-
p-methylenediphenol (
o-
p-MDP), were also assumed to be fast reactions. Therefore, further theoretical calculations were not carried out.
The S
N2 mechanism shown in
Figure 2 as reaction (2) was also suggested by some early reports [
9,
10,
11,
12,
13]. However, in the study of Higuchi et al. on the self-condensation of 2-HMP (
o-HMP) [
15], the authors concluded that the reaction is of genuine first-order and any bimolecular mechanism should be ruled out. To clarify the competitive relationship of the QM and S
N2 mechanisms, we performed calculations on the S
N2 reaction. In
Figure 7, the S
N2 transition state
o-
p-TS-S
N2 demonstrates that the collision between the
o-HMP and the
para-position of dissociated phenol (phenolate ion) can lead to the removal of OH
−, forming the intermediate
o-
p-IM2. The energy barrier of this reaction was predicted to be 172.1 kJ/mol, which is about 70 and 90 kJ/mol higher than that of the two QM mechanisms, respectively. Obviously, such a mechanism is not competitive over the QM mechanism. This results rationalized the kinetics results of Higuchi et al. on the self-condensation of 2-HMP. However, in their study on the self-condensation of 4-HMP (
p-HMP) [
16], the reaction order was found to be 1.3 and they thought a bimolecular reaction may be involved. Subsequently, the S
N2 mechanism was proposed again. To confirm whether 4-HMP is more reactive than 2-HMP in the S
N2 reaction, the
para-
para reaction was also calculated and the transition state is shown in
Figure 7 as
p-
p-TS-S
N2. The barrier of it was calculated to be 172.1 kJ/mol. Therefore, the S
N2 mechanism for 4-HMP is also not competitive over the QM mechanism. To rationalize their results, they thought the bimolecular reactions between two dissociated 4-HMPs or between two undissociated 4-HMPs, which are shown in
Figure 2 as reaction (3) and (4), may be possible. However, they also doubted the rationality of the S
N2 reaction mechanism in these two types of reactions, and finally they concluded that some unknown mechanisms may be at work. We also performed calculations on reaction (3) and (4), but they were not identified.
To further confirm the role of the S
N2 mechanism, we have did a simple experiment on a model reaction where the phenol was reacted with benzyl alcohol under the conditions of pH = 10, 90 °C. The
13C NMR determination was performed for analyzing the reaction products. Benzyl alcohol (BA) is structurally similar to HMP in the aspect of the aromatic hydroxymethyl group but the QM intermediate cannot be formed from it. The S
N2 mechanism works if the condensed structure can be formed. However, the
13C NMR spectrum did not indicate any condensation structure or other products within 180 min. This is why we did not show the experimental results here. To theoretically compare the reactivity of BA with HMP, we calculated the S
N2 reaction between BA and the phenolate ion. The structure of the transition state and the reaction potential energy profile are also given in
Figure 7 and
Figure 8, respectively. The energy barrier for this reaction was calculated to be 182.1 kJ/mol, close to that of the reaction between the HMP and phenolate ion. This result suggests that the S
N2 reaction is very unlikely to occur. Note that the nulceophile in this study is always an unsubstituted phenolate ion, not dissociated (ionized) HMP. Theoretically, dissociated HMP should be a weaker nucleophile due to the electron-withdrawing effect of the hydroxymethyl group. Therefore, the S
N2 reaction between dissociated HMP and undissociated HMP is more impossible.
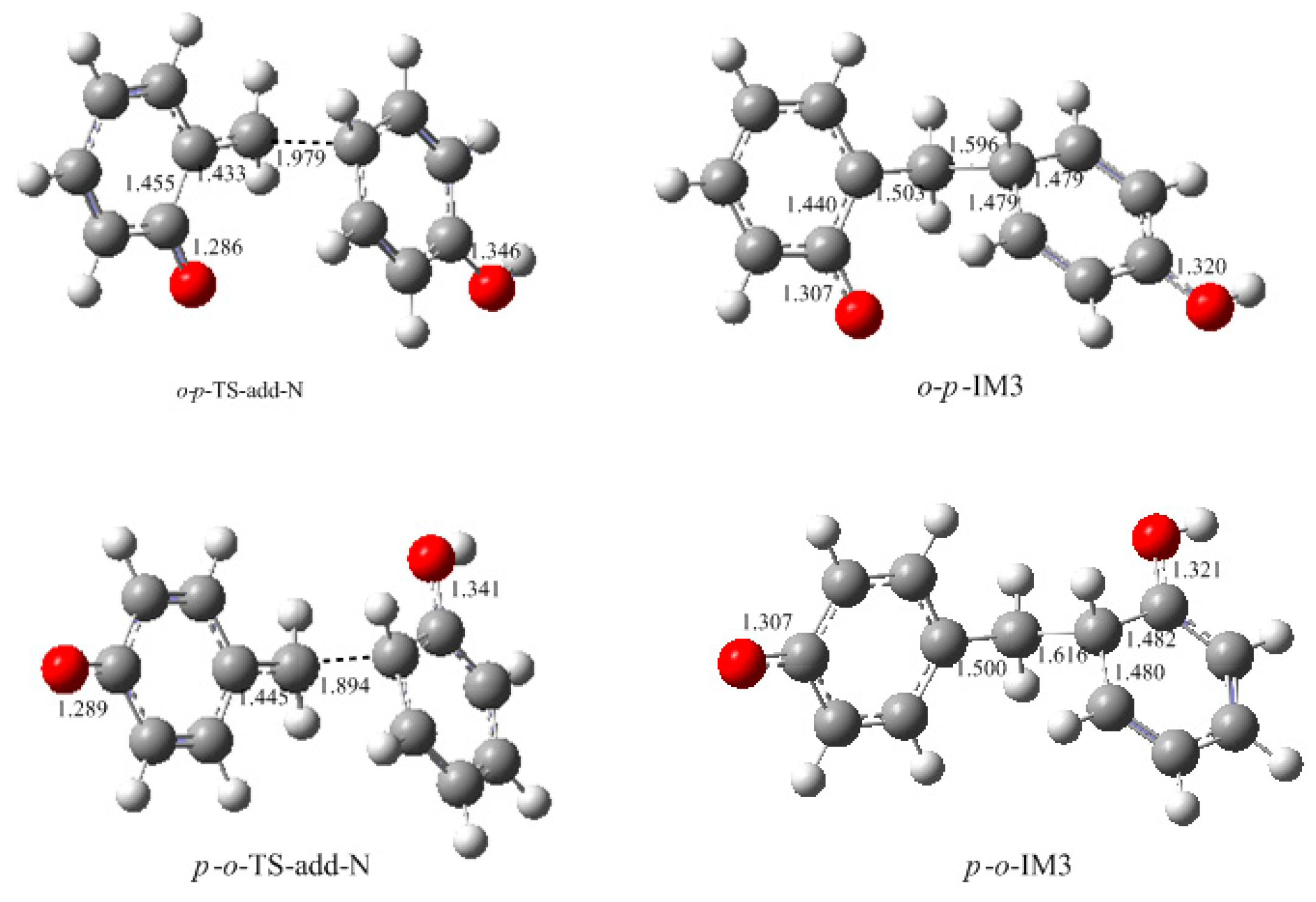
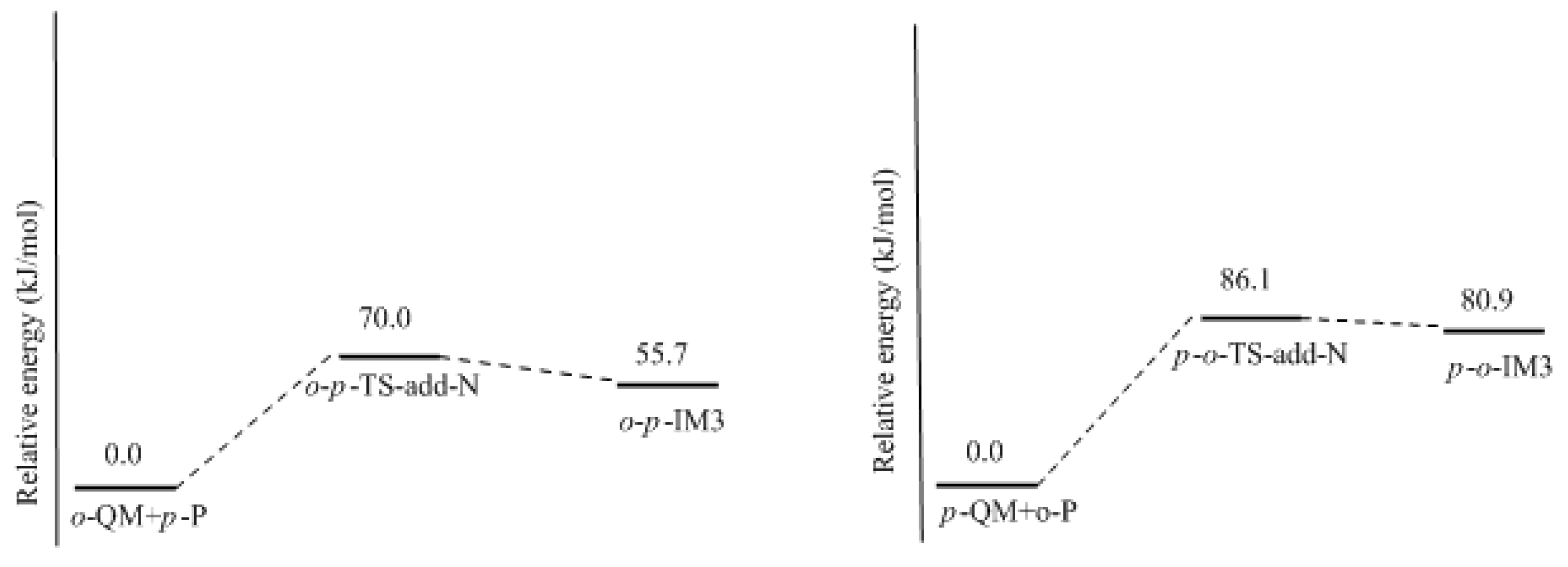
How to explain the experimentally observed bimolecular feature? We considered another possible mechanism. That is the reaction between QM and a neutral phenol or HMP. In such a reaction, the neutral phenol or HMP is a weaker nucleophile than the dissociated species. Moreover, the phenol is a weak acid, and even with the presence of a base such as NaOH, a portion of the phenol or HMP is still in neutral form. Higuchi et al. found that the bimolecular reaction rate varied with the change of the NaOH/4-HMP molar ratio [
16]. Especially when the molar ratio was higher than 0.5, the reaction rate began to decrease. This implies that some kind of reaction that involves the neutral species may occur. Based on this context, we proposed a new mechanism where the QM intermediate reacts with the neutral phenol. The results are given in
Figure 9 and
Figure 10. The energy barrier for the reaction between
o-QM and the
p-position of the neutral phenol was 70.6 kJ/mol, which is still lower than the barrier of
o-TS-WAE. The rate-determining step was still the formation of
o-QM. The energy barrier for the reaction between
p-QM and the
ortho-position of neutral phenol was calculated to be 86.1 kJ/mol, which is closer to the barrier of 87.8 kJ/mol for
p-QM formation. When we alternated the neutral phenol to 4-HMP, this barrier was calculated to be 87.6 kJ/mol (not shown in
Figure 10 due to the similarity), suggesting the substituent effect on this reaction is very weak. It is possible that the theoretical calculation underestimated the barrier of this reaction to some extent due to the difficulties in simulating the real solution for the theoretical solvent model. If this reaction becomes the rate-determining step, the overall reaction will appear as a bimolecular feature. To say the least, even the barrier of the addition step is really slightly lower, they are very close indeed. Therefore, it is not safe to experimentally judge that the reaction is unimolecular.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}