
Self-Assembled Organic Materials for Photovoltaic Application


Abstract
:
1. Introduction
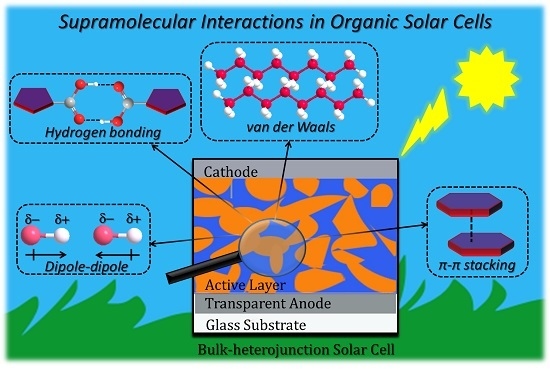
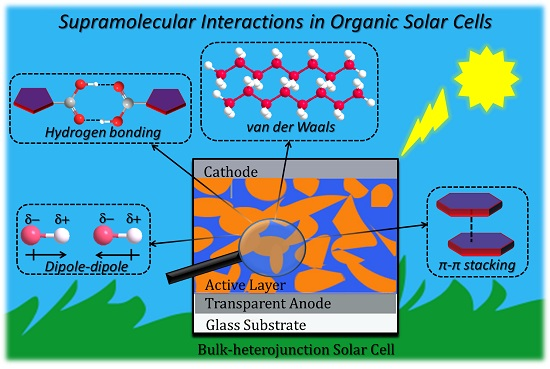
1.1. Supramolecular Interactions and Self-Assembly
1.1.1. Hydrogen Bonding
1.1.2. Ion-Ion Interactions
1.1.3. Ion-Dipole Interactions
1.1.4. Dipole-Dipole Interactions
1.1.5. Pi-Pi Stacking Interactions
1.1.6. Cation-Pi Interactions
1.1.7. Anion-Pi Interactions
1.1.8. Van Der Waals Forces
1.1.9. Hydrophobic Interactions
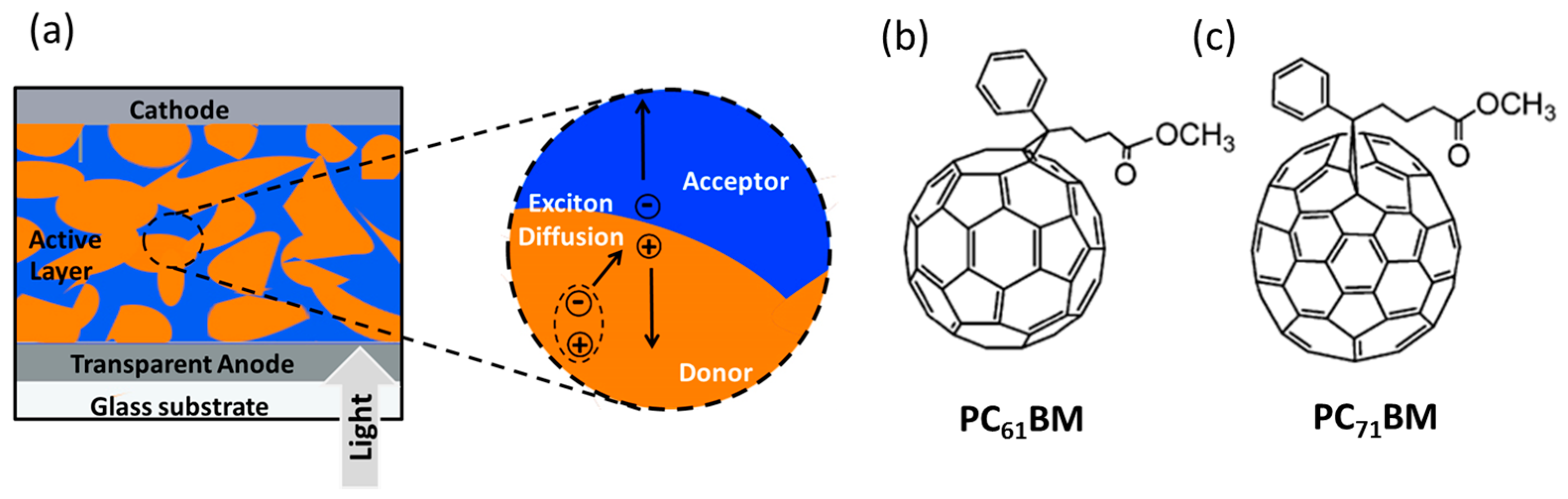
2. Small Molecules and Oligomers
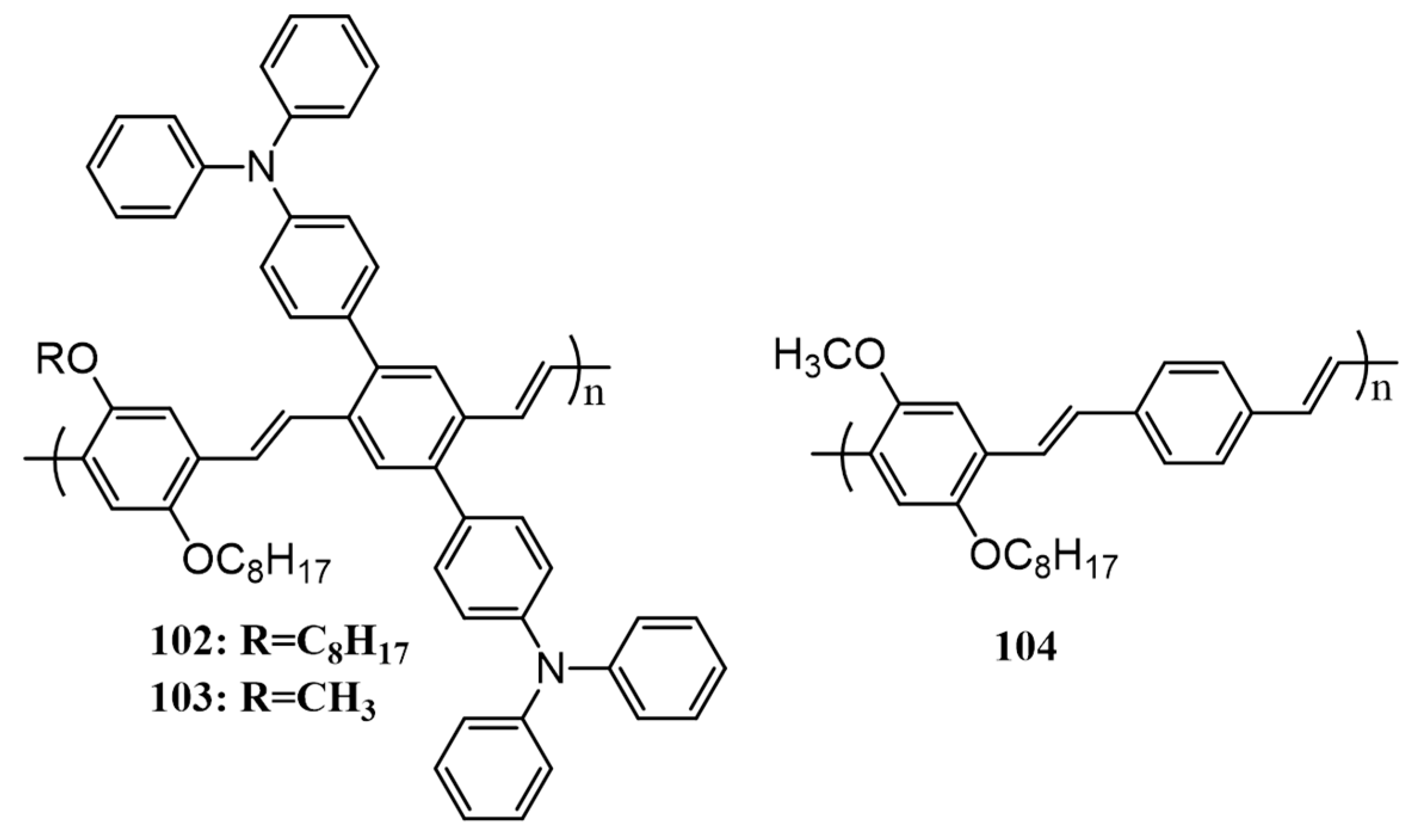
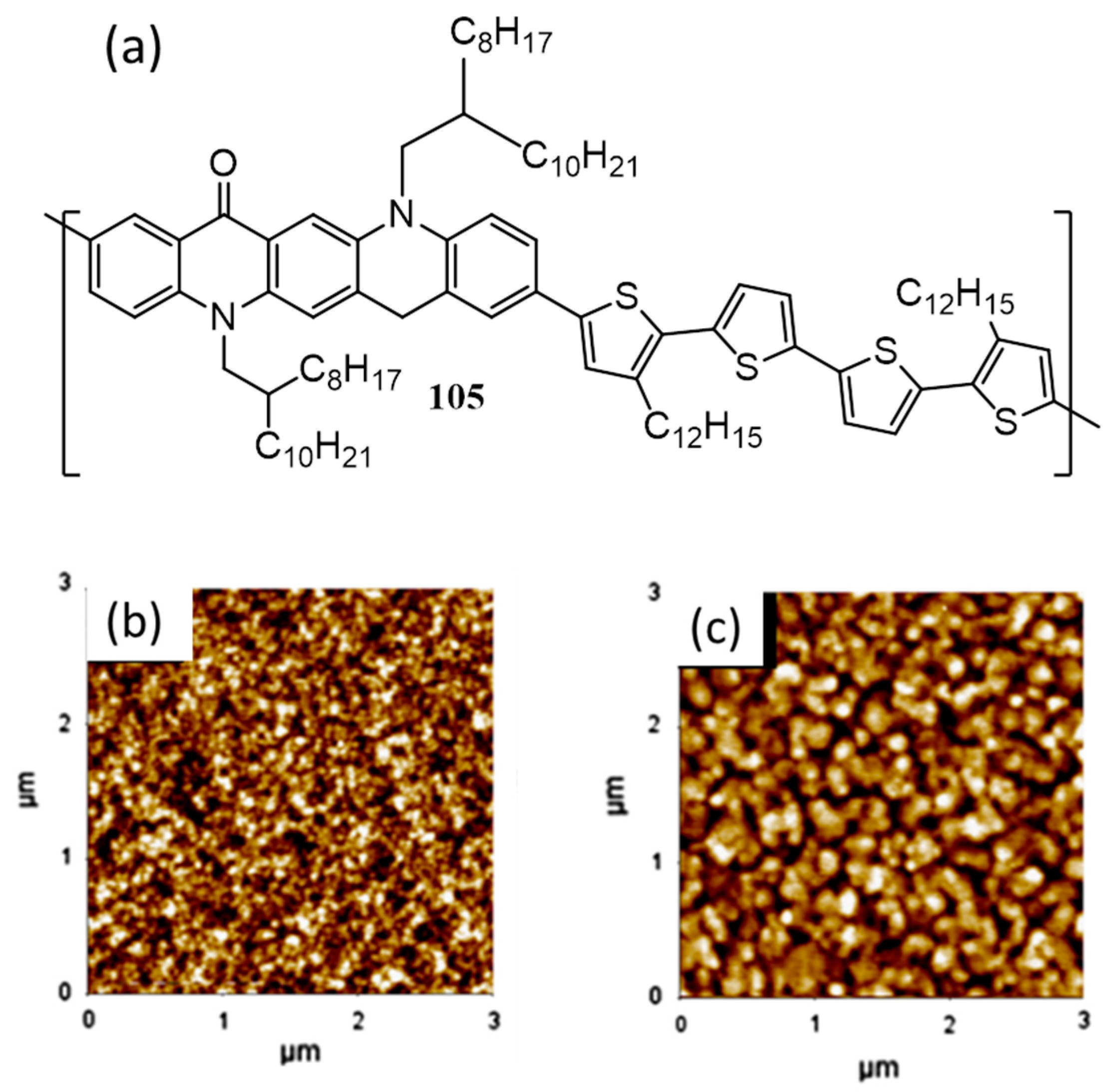
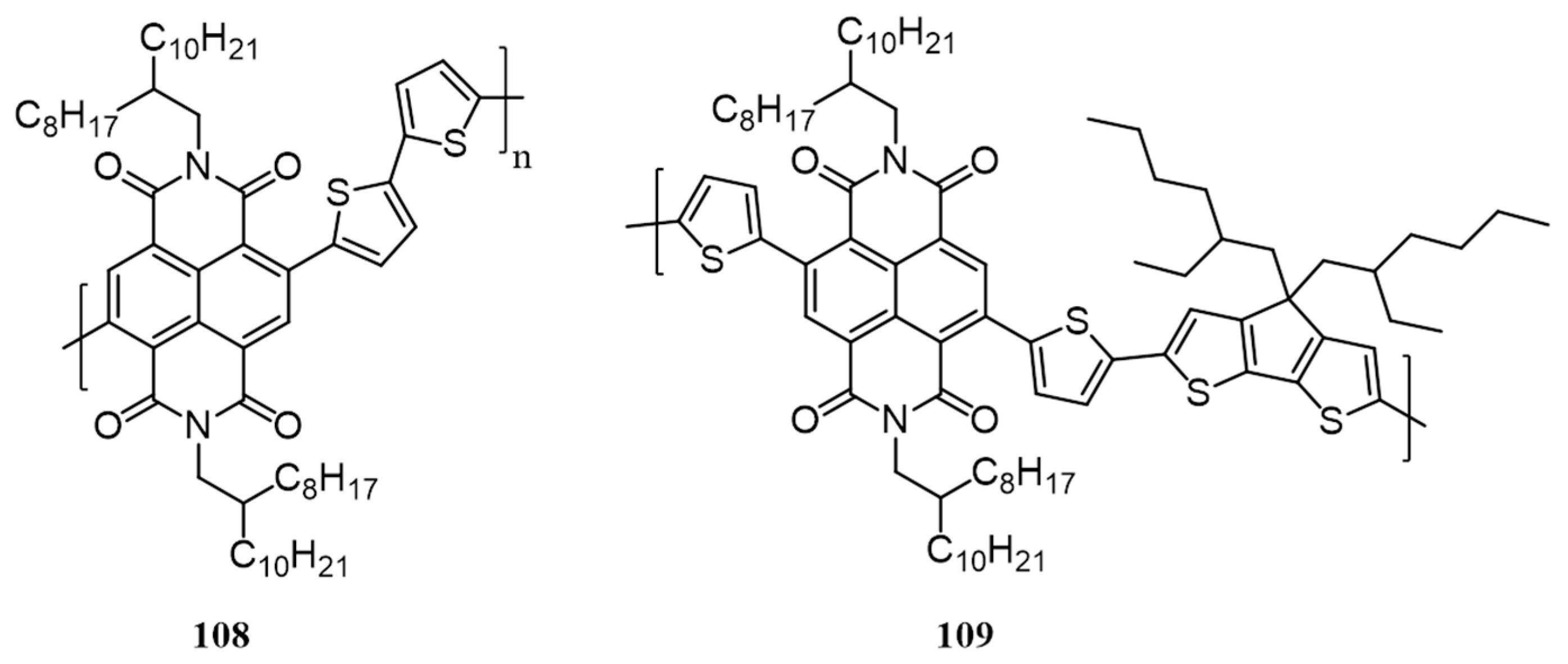
3. Polymers
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Cheng, Y.-B. Print Flexible Solar Cells. Nature 2016, 539, 488–489. [Google Scholar] [CrossRef] [PubMed]
- Nazeeruddin, M.K. Twenty-Five Years of Low-Cost Solar Cells. Nature 2016, 538, 463–464. [Google Scholar] [CrossRef] [PubMed]
- Banerji, N. Organic Photovoltaics: Pushing the Knowledge of Interfaces. Nat. Mater. 2017. [Google Scholar] [CrossRef] [PubMed]
- Major, J.D.; Treharne, R.E.; Phillips, L.J.; Durose, K. A Low-Cost Non-Toxic Post-Growth Activation Step for CdTe Solar Cells. Nature 2014, 511, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, C.; Cuevas, A.; De Wolf, S. High-Efficiency Crystalline Silicon Solar Cells: Status and Perspectives. Energy Environ. Sci. 2016, 9, 1552–1576. [Google Scholar] [CrossRef]
- Poplawsky, J.D.; Guo, W.; Paudel, N.; Ng, A.; More, K.; Leonard, D.; Yan, Y. Structural and Compositional Dependence of the CdTexSe1−x Alloy Layer Photoactivity in CdTe-Based Solar Cells. Nat. Commun. 2016, 7, 12537. [Google Scholar] [CrossRef] [PubMed]
- Darling, S.B.; You, F. The Case for Organic Photovoltaics. RSC Adv. 2013, 3, 17633–17648. [Google Scholar] [CrossRef]
- Service, R.F. Outlook Brightens for Plastic Solar Cells. Science 2011, 332, 293. [Google Scholar] [CrossRef] [PubMed]
- Kaltenbrunner, M.; White, M.S.; Głowacki, E.D.; Sekitani, T.; Someya, T.; Sariciftci, N.S.; Bauer, S. Ultrathin and Lightweight Organic Solar Cells with High Flexibility. Nat. Commun. 2012, 3, 770. [Google Scholar] [CrossRef] [PubMed]
- Angmo, D.; Larsen-olsen, T.T.; Krebs, F.C. Roll-to-Roll Fabrication of Polymer Solar Cells As the Performance in Terms of Power Conversion Efficiency and Operational. Mater. Today 2012, 15, 36–49. [Google Scholar]
- Wu, J.-S.; Cheng, S.-W.; Cheng, Y.-J.; Hsu, C.-S. Donor-Acceptor Conjugated Polymers Based on Multifused Ladder-Type Arenes for Organic Solar Cells. Chem. Soc. Rev. 2014, 44, 1113–1154. [Google Scholar] [CrossRef] [PubMed]
- Youn, H.; Park, H.J.; Guo, L.J. Organic Photovoltaic Cells: From Performance Improvement to Manufacturing Processes. Small 2015, 11, 2228–2246. [Google Scholar] [CrossRef] [PubMed]
- Brabec, C.; Scherf, U.; Dyakonov, V. Organic Photovoltaics: Materials, Device Physics, and Manufacturing Technologies, 2nd ed.; Willey: New York, NY, USA, 2014. [Google Scholar]
- Liu, D.; Zhu, Q.; Gu, C.; Wang, J.; Qiu, M.; Chen, W.; Bao, X.; Sun, M.; Yang, R. High-Performance Photovoltaic Polymers Employing Symmetry-Breaking Building Blocks. Adv. Mater. 2016, 28, 8490–8498. [Google Scholar] [CrossRef] [PubMed]
- Bin, H.; Gao, L.; Zhang, Z.; Yang, Y.; Zhang, Y.; Zhang, C.; Chen, S.; Xue, L.; Yang, C.; Xiao, M.; et al. 11.4% Efficiency Non-Fullerene Polymer Solar Cells with Trialkylsilyl Substituted 2D-Conjugated Polymer as Donor. Nat. Commun. 2016, 7, 13651. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Zhang, Y.; Zhang, J.; Wang, Z.; Zhu, L.; Fang, J.; Xia, B.; Wang, Z.; Lu, K.; Ma, W.; et al. Fluorination-Enabled Optimal Morphology Leads to over 11% Efficiency for Inverted Small-Molecule Organic Solar Cells. Nat. Commun. 2016, 7, 13740. [Google Scholar] [CrossRef] [PubMed]
- Holliday, S.; Ashraf, R.S.; Wadsworth, A.; Baran, D.; Yousaf, S.A.; Nielsen, C.B.; Tan, C.; Dimitrov, S.D.; Shang, Z.; Gasparini, N.; et al. High-Efficiency and Air-Stable P3HT-Based Polymer Solar Cells with a New Non-Fullerene Acceptor. Nat. Commun. 2016, 7, 11585. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Li, C.; Yang, F.; Zhang, J.; Wang, Z.; Wei, Z.; Li, W. An Electron Acceptor with Porphyrin and Perylene Bisimides for Efficient Non-Fullerene Solar Cells. Angew. Chem. Int. Ed. 2017, 56, 2694–2698. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.D.; Cruickshank, C.; Foged, S.; Thorsen, J.; Krebs, F.C. Solar Energy Materials & Solar Cells Business, Market and Intellectual Property Analysis of Polymer Solar Cells. Sol. Energy Mater. Sol. Cells 2010, 94, 1553–1571. [Google Scholar]
- The future is light: Organic solar film by Heliatek. Available online: http://www.heliatek.com (accessed on 10 January 2017).
- Konark.com. Available online: http://www.konarka.com (accessed on 10 January 2017).
- Tang, C.W. Two-Layer Organic Photovoltaic Cell. Appl. Phys. Lett. 1986, 48, 183–185. [Google Scholar] [CrossRef]
- Kaur, N.; Singh, M.; Pathak, D.; Wagner, T.; Nunzi, J.M. Organic Materials for Photovoltaic Applications: Review and Mechanism. Synth. Met. 2014, 190, 20–26. [Google Scholar] [CrossRef]
- Wohrle, B.D.; Meissner, D. Organic Solar Cells. Adv. Mater. 1991, 3, 129–138. [Google Scholar] [CrossRef]
- Halls, J.J.M.; Pichler, K.; Friend, R.H.; Moratti, S.C.; Holmes, A.B. Exciton Diffusion and Dissociation in a Poly(p-phenylenevinylene)/C60 Heterojunction Photovoltaic Cell. Appl. Phys. Lett. 2012, 68, 3120–3122. [Google Scholar] [CrossRef]
- Peumans, P.; Forrest, S.R. Very-High-Efficiency Double-Heterostructure Copper Phthalocyanine/C60 Photovoltaic Cells. Appl. Phys. Lett. 2001, 79, 126–128. [Google Scholar] [CrossRef]
- Xue, J.; Uchida, S.; Rand, B.P.; Forrest, S.R. 4.2% Efficient Organic Photovoltaic Cells with Low Series Resistances. Appl. Phys. Lett. 2004, 84, 3013–3015. [Google Scholar] [CrossRef]
- Xue, J.; Rand, B.P.; Uchida, S.; Forrest, S.R. Mixed Donor-Acceptor Molecular Heterojunctions for Photovoltaic Applications. II. Device Performance. J. Appl. Phys. 2005, 12, 1249031–1249039. [Google Scholar] [CrossRef]
- Fitzner, R.; Reinold, E.; Mishra, A.; Mena-osteritz, E.; Ziehlke, H.; Körner, C.; Leo, K.; Riede, M.; Weil, M.; Tsaryova, O.; et al. Dicyanovinyl-Substituted Oligothiophenes: Structure—Property Relationships and Application in Vacuum—Processed Small-Molecule Organic Solar Cells. Adv. Energy Mater. 2011, 21, 897–910. [Google Scholar] [CrossRef]
- Lin, C.; Liu, S.; Lee, C.; Hunag, J.; Su, W.; Chiu, T.; Chen, C.; Lee, J. Open-Circuit Voltage and Efficiency Improvement of Subphthalocyanine-Based Organic Photovoltaic Device through Deposition Rate Control. Sol. Energy Mater. Sol. Cells 2012, 103, 69–75. [Google Scholar] [CrossRef]
- Sariciftci, N.S.; Smilowitz, L.; Heeger, A.J.; Wudi, F. Photoinduced Electron Transfer from a Conducting Polymer to Buckminsterfullerene. Science 1988, 258, 1474–1476. [Google Scholar] [CrossRef] [PubMed]
- Lehn, J.M. Cryptates: Inclusion Complexes of Macropolycyclic Receptor Molecules. Pure Appl. Chem. 1978, 50, 871–892. [Google Scholar] [CrossRef]
- Steed, J.W.; Atwood, J.L. Supramolecular Chemistry; John Wiley and Sons Ltd.: London, UK, 2009. [Google Scholar]
- Nernst, W. Verteilung eines Stoffes zwischen zwei Lösungsmitteln und zwischen Lösungsmittel und Dampfraum. Z. Phys. Chem. 1891, 8, 110–139. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Pranata, J. Importance of Secondary Interactions in Triply Hydrogen Bonded Complexes: Guanine-Cytosine vs. Uracil-2,6-Diaminopyridine. J. Am. Chem. Soc. 2010, 112, 2008–2010. [Google Scholar] [CrossRef]
- Pranata, J.; Wierschke, S.G.; Jorgensen, W.L. OPLS Potential Functions for Nucleotide Bases. Relative Association Constants of Hydrogen-Bonded Base Pairs in Chloroform. J. Am. Chem. Soc. 1991, 113, 2810–2819. [Google Scholar] [CrossRef]
- Hunter, C.A.; Sanders, J.K.M. The Nature of Pi-Pi Interactions. J. Am. Chem. Soc. 1990, 112, 5525–5534. [Google Scholar] [CrossRef]
- Demeshko, S.; Dechert, S.; Meyer, F. Anion-π Interactions in a Carousel Copper(II)-Triazine Complex. J. Am. Chem. Soc. 2004, 126, 4508–4509. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Nikiforov, M.P.; Darling, S.B. Morphology Characterization in Organic and Hybrid Solar Cells. Energy Environ. Sci. 2012, 5, 8045–8074. [Google Scholar] [CrossRef]
- Huang, Y.; Kramer, E.J.; Heeger, A.J.; Bazan, G.C. Bulk Heterojunction Solar Cells: Morphology and Performance Relationships. Chem. Rev. 2014, 114, 7006–7043. [Google Scholar] [CrossRef] [PubMed]
- Bassani, D.M.; Jonusauskaite, L.; Lavie-cambot, A.; Mcclenaghan, N.D.; Pozzo, J.; Ray, D.; Vives, G. Harnessing Supramolecular Interactions in Organic Solid-State Devices: Current Status and Future Potential. Coord. Chem. Rev. 2010, 254, 2429–2445. [Google Scholar] [CrossRef]
- Troshin, P.A.; Sariciftci, N.S. Supramolecular Chemistry for Organic Photovoltaics; John Wiley & Sons, Ltd.: London, UK, 2012. [Google Scholar]
- Broggi, A.; Tomasi, I.; Bianchi, L.; Marrocchi, A.; Vaccaro, L. Small Molecular Aryl Acetylenes: Chemically Tailoring High-Efficiency Organic Semiconductors for Solar Cells and Field-Effect Transistors. Chempluschem 2014, 79, 486–507. [Google Scholar] [CrossRef]
- Haruk, A.M.; Mativetsky, J.M. Supramolecular Approaches to Nanoscale Morphological Control in Organic Solar Cells. Int. J. Mol. Sci. 2015, 16, 13381–13406. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Li, Y.; Zhan, X. Small Molecule Semiconductors for High-Efficiency Organic Photovoltaics. Chem. Soc. Rev. 2012, 41, 4245–4272. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Bäuerle, P. Small Molecule Organic Semiconductors on the Move: Promises for Future Solar Energy Technology. Angew. Chem. Int. Ed. Engl. 2012, 51, 2020–2067. [Google Scholar] [CrossRef] [PubMed]
- Kan, B.; Li, M.; Zhang, Q.; Liu, F.; Wan, X.; Wang, Y.; Ni, W.; Long, G.; Yang, X.; Feng, H.; et al. A Series of Simple Oligomer-like Small Molecules Based on Oligothiophenes for Solution-Processed Solar Cells with High Efficiency. J. Am. Chem. Soc. 2015, 137, 3886–3893. [Google Scholar] [CrossRef] [PubMed]
- El-ghayoury, A.; Schenning, A.P.H.J.; van Hal, P.A.; van Duren, J.K.J.; Janssen, Â.A.J.; Meijer, E.W. Supramolecular Hydrogen-Bonded Oligo(p-Phenylene Vinylene) Polymers. Angew. Chem. Int. Ed. 2001, 40, 3660–3663. [Google Scholar] [CrossRef]
- Huang, C.; Mcclenaghan, N.D.; Kuhn, A.; Hofstraat, J.W.; Bassani, D.M. Enhanced Photovoltaic Response in Hydrogen-Bonded All-Organic Devices. Org. Lett. 2005, 7, 3409–3412. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Ali, A.; Bilic, A.; Gao, M.; Hegedus, K.; Singh, B.; Watkins, S.E.; Wilson, G.J.; Bach, U.; Evans, R.A. Absorption Enhancement of Oligothiophene Dyes through the Use of a Cyanopyridone Acceptor Group in Solution-Processed Organic Solar Cells. Chem. Commun. 2012, 48, 1889–1891. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.J.; Subbiah, J.; Holmes, A.B. Enhancement of Efficiency in Organic Photovoltaic Devices Containing Self-Complementary Hydrogen-Bonding Domains. Beilstein J. Org. Chem. 2013, 9, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Aytun, T.; Barreda, L.; Ruiz-carretero, A.; Lehrman, J.A.; Stupp, S.I. Improving Solar Cell Efficiency through Hydrogen Bonding: Method for Tuning Active Layer Morphology. Chem. Mater. 2015, 27, 1201–1209. [Google Scholar] [CrossRef]
- Yagai, S.; Suzuki, M.; Lin, X.; Gushiken, M.; Noguchi, T.; Karatsu, T.; Kitamura, A.; Saeki, A.; Seki, S.; Kikkawa, Y.; et al. Supramolecular Engineering of Oligothiophene Nanorods without Insulators: Hierarchical Association of Rosettes and Photovoltaic Properties. Chem. Eur. J. 2014, 20, 16128–16137. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, H.; Lin, X.; Kizaki, T.; Prabhu, D.D.; Silly, F.; Kajitani, T.; Fukushima, T.; Nakayama, K.; Yagai, S. Hydrogen-Bonded Oligothiophene Rosettes with a Benzodithiophene Terminal Unit: Self-Assembly and Application to Bulk Heterojunction Solar Cells. Chem. Commun. 2016, 52, 7874–7877. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, Y.; Li, C.; Li, W.; Zhou, C.; Liu, H.; Bo, Z.; Li, Y. New Methanofullerenes Containing Amide as Electron Acceptor for Construction Photovoltaic Devices. J. Phys. Chem. C 2009, 113, 21970–21975. [Google Scholar] [CrossRef]
- Siram, R.B.K.; Tandy, K.; Horecha, M.; Formanek, P.; Stamm, M.; Gevorgyan, S.; Krebs, F.C.; Kiriy, A.; Meredith, P.; Burn, P.L.; et al. Synthesis and Self-Assembly of Donor-Acceptor-Donor Based Oligothiophenes and Their Optoelectronic Properties. J. Phys. Chem. C 2011, 115, 14369–14376. [Google Scholar] [CrossRef]
- Siram, R.B.K.; Stephen, M.; Ali, F.; Patil, S. Investigation of Phase Separation in Bulk Heterojunction Solar Cells via Supramolecular Chemistry. J. Phys. Chem. C 2013, 117, 9129–9136. [Google Scholar] [CrossRef]
- Kim, K.; Yu, H.; Kang, H.; Kang, D.J.; Cho, C.; Cho, H.; Oh, J.H.; Kim, B.J. Influence of Intermolecular Interactions of Electron Donating Small Molecules on Their Molecular Packing and Performance in Organic Electronic Devices. J. Mater. Chem. A 2013, 1, 14538–14547. [Google Scholar] [CrossRef]
- Yassin, A.; Leriche, P.; Allain, M.; Roncali, J. Donor–acceptor–donor (D–A–D) Molecules Based on Isoindigo as Active Material for Organic Solar Cells. New J. Chem. 2013, 37, 502–507. [Google Scholar] [CrossRef]
- Xiao, Z.; Sun, K.; Subbiah, J.; Ji, S.; Jones, D.J.; Wong, W.W.H. Hydrogen Bonding in Bulk Heterojunction Solar Cells: A Case Study. Sci. Rep. 2014, 4, 5701. [Google Scholar] [CrossRef] [PubMed]
- Fechtenkötter, A.; Saalwächter, K.; Harbison, M.A.; Müllen, K.; Spiess, H.W. Highly Ordered Columnar Structures from Hexa-Peri-hexabenzocoronenes—Synthesis, X-ray Diffraction, and Solid-State Heteronuclear Multiple-Quantum NMR Investigations. Angew. Chem. Int. Ed. 1999, 38, 3039–3042. [Google Scholar] [CrossRef]
- Schön, J.H.; Kloc, C.; Batlogg, B. Perylene: A Promising Organic Field-Effect Transistor Material. Appl. Phys. Lett. 2000, 77, 3776–3778. [Google Scholar] [CrossRef]
- Schmidt-mende, L.; Fechtenkotter, A.; Mullen, K.; Moons, E.; Friend, R.H.; MacKenzie, J.D. Self-Organized Discotic Liquid Crystals for High-Efficiency Organic Photovoltaics. Science 2001, 293, 1119–1122. [Google Scholar] [CrossRef] [PubMed]
- Bagnis, D.; Beverina, L.; Huang, H.; Silvestri, F.; Yao, Y.; Yan, H.; Pagani, G.A.; Marks, T.J.; Facchetti, A. Marked Alkyl- vs. Alkenyl-Substitutent Effects on Squaraine Dye Solid-State Structure, Carrier Mobility, and Bulk-Heterojunction Solar Cell Efficiency. J. Am. Chem. Soc. 2010, 132, 4074–4075. [Google Scholar] [CrossRef] [PubMed]
- Lee, O.P.; Yiu, A.T.; Beaujuge, P.M.; Woo, C.H.; Holcombe, T.W.; Millstone, J.E.; Douglas, J.D.; Chen, M.S.; Fréchet, J.M.J. Efficient Small Molecule Bulk Heterojunction Solar Cells with High Fill Factors via Pyrene-Directed Molecular Self-Assembly. Adv. Mater. 2011, 23, 5359–5363. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, R.S.; Kronemeijer, J.; James, I.; Sirringhaus, H.; McCulloch, I. A New Thiophene Substituted Isoindigo Based Copolymer for High Performance Ambipolar Transistors. Chem. Commun. 2012, 48, 3939–3941. [Google Scholar] [CrossRef] [PubMed]
- Odajima, T.; Ashizawa, M.; Konosu, Y.; Matsumoto, H.; Mori, T. Impact of Molecular Planarity on Electronic Devices in Thienoisoindigo-Based Organic Semiconductors. J. Mater. Chem. C 2014, 2, 10455–10467. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, Y.; Zhang, Q.; Gao, X. Non-Fullerene Small Molecule Acceptors Based on Perylene Diimides. J. Mater. Chem. A 2016, 4, 17604–17622. [Google Scholar] [CrossRef]
- Fernández-lázaro, F.; Zink-Lorre, N.; Sastre-Santos, A. Perylenediimides as Non-Fullerene Acceptors in Bulk-Heterojunction Solar Cells (BHJSCs). J. Mater. Chem. A 2016, 4, 9336–9346. [Google Scholar] [CrossRef]
- Rajaram, S.; Shivanna, R.; Kandappa, S.K.; Narayan, K.S. Nonplanar Perylene Diimides as Potential Alternatives to Fullerenes in Organic Solar Cells. J. Phys. Chem. Lett. 2012, 3, 2405–2408. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Ye, L.; Li, X.; Xiao, C.; Tan, F.; Zhao, W.; Hou, J.; Wang, Z. Bay-Linked Perylene Bisimides as Promising Non-Fullerene Acceptors for Organic Solar Cells. Chem. Commun. 2014, 50, 1024–1026. [Google Scholar] [CrossRef] [PubMed]
- Hartnett, P.E.; Matte, R.H.S.S.; Eastham, N.D.; Jackson, N.E.; Wu, Y.; Chen, L.X.; Ratner, M.A.; Chang, R.P.H.; Hersam, M.C.; Wasielewski, M.R.; et al. Ring-Fusion as a Perylenediimide Dimer Design Concept for High-Performance Non-Fullerene Organic Photovoltaic Acceptors. Chem. Sci. 2016, 7, 3543–3555. [Google Scholar] [CrossRef]
- Eaton, S.W.; Shoer, L.E.; Karlen, S.D.; Dyar, S.M.; Margulies, E.A.; Veldkamp, B.S.; Ramanan, C.; Hartzler, D.A.; Savikhin, S.; Marks, T.J.; et al. Singlet Exciton Fission in Polycrystalline Thin Films of a Slip-Stacked Perylenediimide. J. Am. Chem. Soc. 2013, 135, 14701–14712. [Google Scholar] [CrossRef] [PubMed]
- Hartnett, P.E.; Timalsina, A.; Matte, H.S.S.R.; Zhou, N.; Guo, X.; Zhao, W.; Facchetti, A.; Chang, R.P.H.; Hersam, M.C.; Wasielewski, M.R.; et al. Slip-Stacked Perylenediimides as an Alternative Strategy for High Efficiency Nonfullerene Acceptors in Organic Photovoltaics. J. Am. Chem. Soc. 2014, 136, 16345–16356. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, T.; Gopal, A.; Saeki, A.; Seki, S.; Nair, V.C. p/n-Polarity of Thiophene Oligomers in Photovoltaic Cells: Role of Molecular vs. Supramolecular Properties. Phys. Chem. Chem. Phys. 2015, 17, 10630–10639. [Google Scholar] [CrossRef] [PubMed]
- De Bettignies, R.; Nicolas, Y.; Blanchard, P.; Levillain, E.; Nunzi, J.-M.; Roncali, J. Planarized Star-Shaped Oligothiophenes as a New Class of Organic Semiconductors for Heterojunctions Solar Cell. Adv. Mater. 2003, 15, 1939–1943. [Google Scholar] [CrossRef]
- Ripaud, E.; Rousseau, T.; Leriche, P.; Roncali, J. Unsymmetrical Triphenylamine-Oligothiophene Hybrid Conjugated Systems as Donor Materials for High-Voltage Solution-Processed Organic Solar Cells. Adv. Energy Mater. 2011, 1, 540–545. [Google Scholar] [CrossRef] [Green Version]
- Aytun, T.; Santos, P.J.; Bruns, C.J.; Huang, D.; Koltonow, A.R.; Cruz, M.O.; Stupp, S.I. Self-Assembling Tripodal Small-Molecule Donors for Bulk Heterojunction Solar Cells. J. Phys. Chem. C 2016, 120, 3602–3611. [Google Scholar] [CrossRef]
- Wurthner, F.; Bauer, C.; Stepanenko, V.; Yagai, S. A Black Perylene Bisimide Super Gelator with an Unexpected J-Type Absorption Band. Adv. Mater. 2008, 20, 1695–1698. [Google Scholar] [CrossRef]
- Lindner, S.M.; Thelakkat, M. Nanostructures of N-Type Organic Semiconductor in a P-Type Matrix via Self-Assembly of Block Copolymers. Macromolecules 2004, 37, 8832–8835. [Google Scholar] [CrossRef]
- Wicklein, A.; Ghosh, S.; Sommer, M.; Wurthner, F.; Thelakkat, M. Self-Assembly of Semiconductor Organogelator Nanowires for Photoinduced Charge Separation. ACS Nano 2009, 3, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.; Tevis, I.D.; Tayi, A.S.; Cui, H.; Stupp, S.I. Semiconducting Nanowires from Hairpin-Shaped Self-Assembling Sexithiophenes. J. Phys. Chem. B 2010, 114, 14778–14786. [Google Scholar] [CrossRef] [PubMed]
- Tevis, I.D.; Tsai, W.; Palmer, L.C.; Aytun, T.; Stupp, S.I. Grooved Nanowires from Self-Assembling Hairpin Molecules for Solar Cells. ACS Nano 2012, 6, 2032–2040. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-carretero, A.; Aytun, T.; Bruns, C.J.; Newcomb, C.J.; Tsai, W.; Stupp, S.I. Stepwise Self-Assembly to Improve Solar Cell Morphology. J. Mater. Chem. A 2013, 1, 11674–11681. [Google Scholar] [CrossRef]
- Lam, K.H.; Foong, T.; Ooi, Z.E.; Zhang, J.; Grimsdale, A.C.; Lam, Y.M. Enhancing the Performance of Solution-Processed Bulk- Heterojunction Solar Cells Using Hydrogen-Bonding-Induced Self-Organization of Small Molecules. ACS Appl. Mater. Interfaces 2013, 5, 13265–13274. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, N.M.; Deppisch, M.; Würthner, F.; Lademann, H.W.A.; Deing, K.; Meerholz, K. Bulk Heterojunction Organic Solar Cells Based on Merocyanine Colorants. Chem. Commun. 2008, 48, 6489–6491. [Google Scholar] [CrossRef] [PubMed]
- Bürckstümmer, H.; Kronenberg, N.M.; Gsanger, M.; Stolte, M.; Meerholz, K.; Würthner, F. Tailored Merocyanine Dyes for Solution-Processed BHJ Solar Cells. J. Mater. Chem. 2010, 20, 240–243. [Google Scholar] [CrossRef]
- Würthner, F.; Yao, S. Dipolar Dye Aggregates: A Problem for Nonlinear Optics, but a Chance for Supramolecular Chemistry. Angew. Chem. Int. Ed. 2000, 39, 1978–1981. [Google Scholar] [CrossRef]
- Bürckstümmer, H.; Tulyakova, E.V.; Deppisch, M.; Lenze, M.R.; Kronenberg, N.M.; Gsänger, M.; Stolte, M.; Meerholz, K.; Würthner, F. Efficient Solution-Processed Bulk Heterojunction Solar Cells by Antiparallel Supramolecular Arrangement of Dipolar Donor-Acceptor Dyes. Angew. Chem. Int. Ed. 2011, 50, 11628–11632. [Google Scholar] [CrossRef] [PubMed]
- Takacs, C.J.; Sun, Y.; Welch, G.C.; Perez, L.A.; Liu, X.; Wen, W.; Bazan, G.C.; Heeger, A.J. Solar Cell Efficiency, Self-Assembly, and Dipole–Dipole Interactions of Isomorphic Narrow-Bandgap Molecules. J. Am. Chem. Soc. 2012, 134, 16597–16606. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Welch, G.C.; Leong, W.L.; Takacs, C.J.; Bazan, G.C.; Heeger, A.J. Solution-Processed Small-Molecule Solar Cells with 6.7% Efficiency. Nat. Mater. 2012, 11, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Henson, Z.B.; Welch, G.C.; Poll, T.; Bazan, G.C. Pyridalthiadiazole-Based Narrow Band Gap Chromophores. J. Am. Chem. Soc. 2012, 134, 3766–3779. [Google Scholar] [CrossRef] [PubMed]
- Welch, G.C.; Perez, L.A.; Hoven, C.V.; Zhang, Y.; Dang, X.; Sharenko, A.; Toney, M.F.; Kramer, E.J.; Nguyen, D.T.; Bazan, G.C. A Modular Molecular Framework for Utility in Small-Molecule Solution-Processed Organic Photovoltaic Devices. J. Mater. Chem. 2011, 21, 12700–12709. [Google Scholar] [CrossRef]
- Shirakawa, H.; Louis, E.J.; Macdiarmid, A.G.; Chiang, C.K.; Heeger, A.J. Synthesis of Electrically Conducting Organic Polymers: Halogen Derivtives of Polyacetylene, (CH)x. J. Chem. Soc. Chem. Commun. 1977, 16, 578–580. [Google Scholar] [CrossRef]
- Liang, T.; Chiang, I.; Yang, P.; Kekuda, D.; Chu, C.; Lin, H. Supramolecular Assembly of H-Bonded Side-Chain Polymers Containing Conjugated Pyridyl H-Acceptor Pendants and Various Low-Band-Gap H-Donor Dyes Bearing Cyanoacrylic Acid Groups for Organic Solar Cell Applications. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 5998–6013. [Google Scholar] [CrossRef]
- Sahu, D.; Padhy, H.; Patra, D.; Kekuda, D.; Chu, C.; Chiang, I.; Lin, H. Synthesis and Application of H-Bonded Cross-Linking Polymers Containing a Conjugated Pyridyl H-Acceptor Side-Chain Polymer and Various Carbazole-Based H-Donor Dyes Bearing Symmetrical Cyanoacrylic Acids for Organic Solar Cells. Polymer 2010, 51, 6182–6192. [Google Scholar] [CrossRef]
- Yao, K.; Chen, L.; Li, F.; Wang, P.; Chen, Y. Cooperative Assembly Donor À Acceptor System Induced by Intermolecular Hydrogen Bonds Leading to Oriented Nanomorphology for Optimized Photovoltaic Performance. J. Phys. Chem. C 2012, 116, 714–721. [Google Scholar] [CrossRef]
- Nakabayashi, K.; Mori, H. Donor-Acceptor Block Copolymers: Synthesis and Solar Cell Applications. Materials 2014, 7, 3274–3290. [Google Scholar] [CrossRef]
- Lee, Y.; Gomez, E.D. Challenges and Opportunities in the Development of Conjugated Block Copolymers for Photovoltaics. Macromolecules 2015, 48, 7385–7395. [Google Scholar] [CrossRef]
- Topham, P.D.; Parnell, A.J.; Hiorns, R.C. Block Copolymer Strategies for Solar Cell Technology. J. Polym. Sci. Part B Polym. Phys. 2011, 49, 1131–1156. [Google Scholar] [CrossRef]
- Darling, S.B. Block Copolymers for Photovoltaics. Energy Environ. Sci. 2009, 2, 1266–1273. [Google Scholar] [CrossRef]
- Lin, Y.; Verduzco, R. Synthesis and Process-Dependent Film Structure of All-Conjugated Copolymers for Organic Photovoltaics; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2014. [Google Scholar]
- Kipp, D.; Verduzco, R.; Ganesan, V. Block Copolymer Compatibilizers for Ternary Blend Polymer Bulk Heterojunction Solar Cells—An Opportunity for Computation Aided Molecular Design. Mol. Syst. Des. Eng. 2016, 1, 353–369. [Google Scholar] [CrossRef]
- Janssen, G.; Aguirre, A.; Goovaerts, E.; Vanlaeke, P.; Poortmans, J.; Manca, J. Optimization of Morphology of P3HT/PCBM Films for Organic Solar Cells: Effects of Thermal Treatments and Spin Coating Solvents. Eur. Phys. J. Appl. Phys. 2007, 37, 287–290. [Google Scholar] [CrossRef]
- Lin, Y.; Lim, J.A.; Wei, Q.; Mannsfeld, S.C.B.; Briseno, A.L.; Watkins, J.J. Cooperative Assembly of Hydrogen-Bonded Diblock Copolythiophene/Fullerene Blends for Photovoltaic Devices with Well-Defined Morphologies and Enhanced Stability. Chem. Mater. 2012, 24, 622–632. [Google Scholar] [CrossRef]
- Patra, D.; Ramesh, M.; Sahu, D.; Padhy, H.; Chu, C.; Wei, K.; Lin, H. Enhancement of Photovoltaic Properties in Supramolecular Polymer Networks Featuring a Solar Cell Main-Chain Polymer H-Bonded with Conjugated Cross-Linkers. Polymer 2012, 53, 1219–1228. [Google Scholar] [CrossRef]
- Li, F.; Yager, K.G.; Dawson, N.M.; Yang, J.; Malloy, K.J.; Qin, Y. Complementary Hydrogen Bonding and Block Copolymer Self-Assembly in Cooperation toward Stable Solar Cells with Tunable Morphologies. Macromolecules 2013, 46, 9021–9031. [Google Scholar] [CrossRef]
- Guo, C.; Lin, Y.; Witman, M.D.; Smith, K.A.; Wang, C.; Hexemer, A.; Strzalka, J.; Gomez, E.D.; Verduzco, R. Conjugated Block Copolymer Photovoltaics with near 3% Efficiency through Microphase Separation. Nano Lett. 2013, 13, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Kipp, D.; Mok, J.; Strzalka, J.; Darling, S.B.; Ganesan, V.; Verduzco, R. Rational Design of Thermally Stable, Bicontinuous Donor/Acceptor Morphologies with Conjugated Block Copolymer Additives. ACS Macro Lett. 2015, 4, 867–871. [Google Scholar] [CrossRef]
- Mok, J.W.; Lin, Y.; Yager, K.G.; Mohite, A.D.; Nie, W.; Darling, S.B.; Lee, Y.; Gomez, E.; Gosztola, D.; Schaller, R.D.; et al. Linking Group Influences Charge Separation and Recombination in All-Conjugated Block Copolymer Photovoltaics. Adv. Funct. Mater. 2015, 25, 5578–5585. [Google Scholar] [CrossRef]
- Smith, K.A.; Lin, Y.; Mok, J.W.; Yager, K.G.; Strzalka, J.; Nie, W.; Mohite, A.D.; Verduzco, R. Molecular Origin of Photovoltaic Performance in Donor-Block-Acceptor All-Conjugated Block Copolymers. Macromolecules 2015, 48, 8346–8353. [Google Scholar] [CrossRef]
- Guo, C.; Lee, Y.; Lin, Y.; Strzalka, J.; Wang, C.; Hexemer, A.; Jaye, C.; Fischer, D.A.; Verduzco, R.; Wang, Q.; et al. Photovoltaic Performance of Block Copolymer Devices Is Independent of the Crystalline Texture in the Active Layer. Macromolecules 2016, 49, 4599–4608. [Google Scholar] [CrossRef]
- Mok, J.W.; Kipp, D.; Hasbun, L.R.; Dolocan, A.; Strzalka, J.; Ganesan, V.; Verduzco, R. Parallel Bulk Heterojunction Photovoltaics Based on All-Conjugated Block Copolymer Additives. J. Mater. Chem. A 2016, 4, 14804–14813. [Google Scholar] [CrossRef]
- Lin, Y.; Nie, W.; Tsai, H.; Li, X.; Gupta, G.; Mohite, A.D.; Verduzco, R. Supramolecular Block Copolymer Photovoltaics through Ureido-Pyrimidinone Hydrogen Bonding Interactions. RSC Adv. 2016, 6, 51562–51568. [Google Scholar] [CrossRef]
- Shen, P.; Sang, G.; Lu, J.; Zhao, B.; Wan, M.; Zou, Y.; Li, Y.; Tan, S. Effect of 3D Pi-Pi Stacking on Photovoltaic and Electroluminescent Properties in Triphenylamine-Containing Poly(p-phenylenevinylene) Derivatives. Macromolecules 2008, 41, 5716–5722. [Google Scholar] [CrossRef]
- Yang, X.; Wang, J.; Zhang, X.; Wang, Z.; Wang, Y. STM Study on 2D Molecular Assemblies of Luminescent Quinacridone Derivatives: Structure Fine-Tuned by Introducing Bulky Substitutes and Co-Adsorption with Monofunctional/Bifunctional Acid. Langmuir 2007, 23, 1287–1291. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.; Hun, D.; Hee, M.; Won, S.; Young, J.; Yong, J.; Kyung, D. Self-Organization Polymer Consisting of Quinacridone and Quaterthiophene Units: Coplanar Structure between Benzene and Thiophene Linkage. Sol. Energy Mater. Sol. Cells 2013, 117, 285–292. [Google Scholar]
- Liao, S.; Jhuo, H.; Cheng, Y.; Chen, S. Fullerene Derivative-Doped Zinc Oxide Nanofilm as the Cathode of Inverted Polymer Solar Cells with Low-Bandgap Polymer (PTB7-Th) for High Performance. Adv. Mater. 2013, 25, 4766–4771. [Google Scholar] [CrossRef] [PubMed]
- Mori, D.; Benten, H.; Okada, I.; Ohkita, H.; Ito, S. Low-Bandgap Donor/Acceptor Polymer Blend Solar Cells with Efficiency Exceeding 4%. Adv. Energy Mater. 2013, 4, 1–6. [Google Scholar] [CrossRef]
- Kang, H.; Kim, K.; Choi, J.; Lee, C.; Kim, B.J. High-Performance All-Polymer Solar Cells Based on Face-On Stacked Polymer Blends with Low Interfacial Tension. ACS Macro Lett. 2014, 3, 1009–1014. [Google Scholar] [CrossRef]
- Schubert, M.; Dolfen, D.; Frisch, J.; Roland, S.; Steyrleuthner, R.; Stiller, B.; Chen, Z.; Scherf, U.; Koch, N.; Facchetti, A.; et al. Influence of Aggregation on the Performance of All-Polymer Solar Cells Containing Low-Bandgap Naphthalenediimide Copolymers. Adv. Energy Mater. 2012, 2, 369–380. [Google Scholar] [CrossRef]
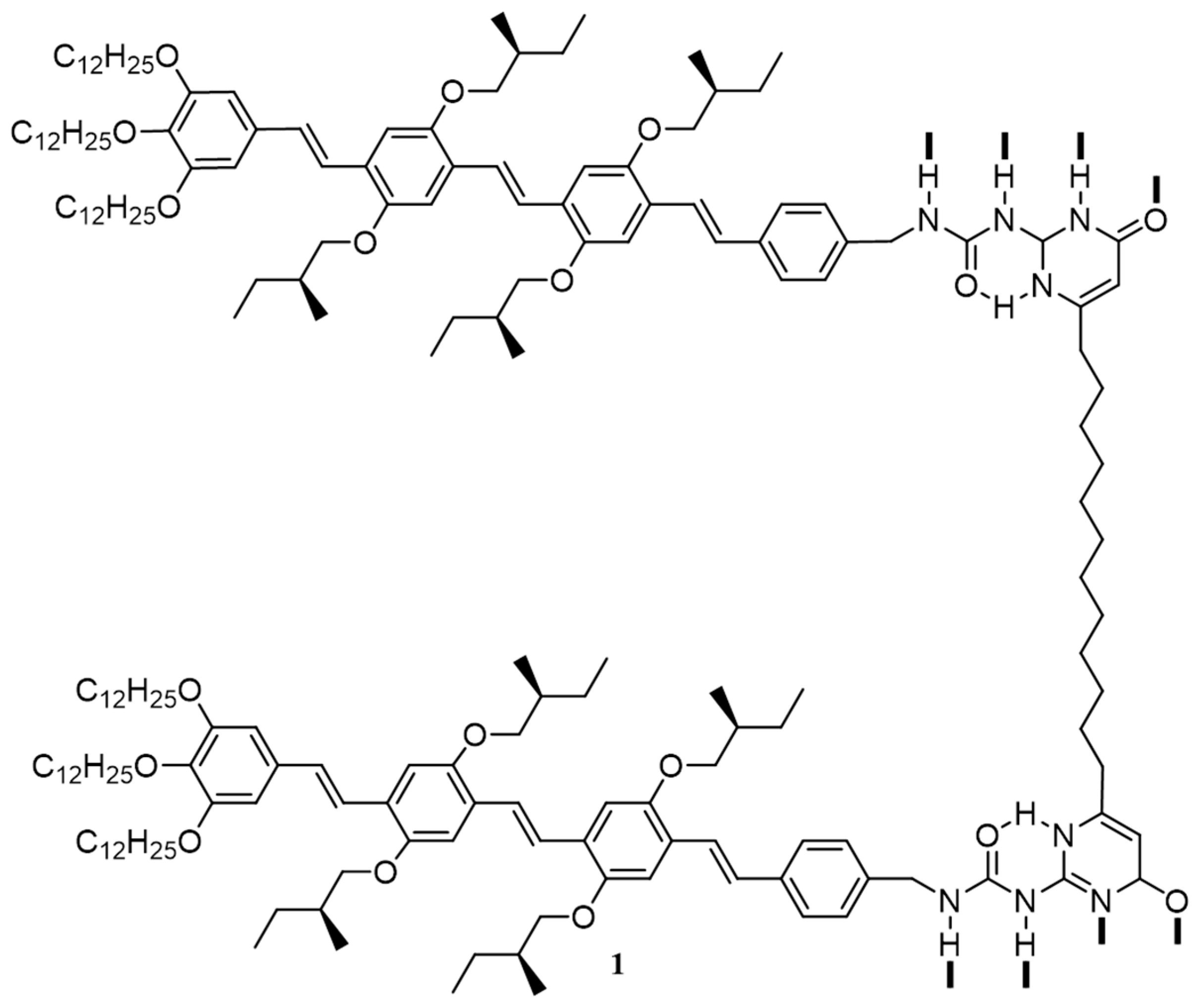

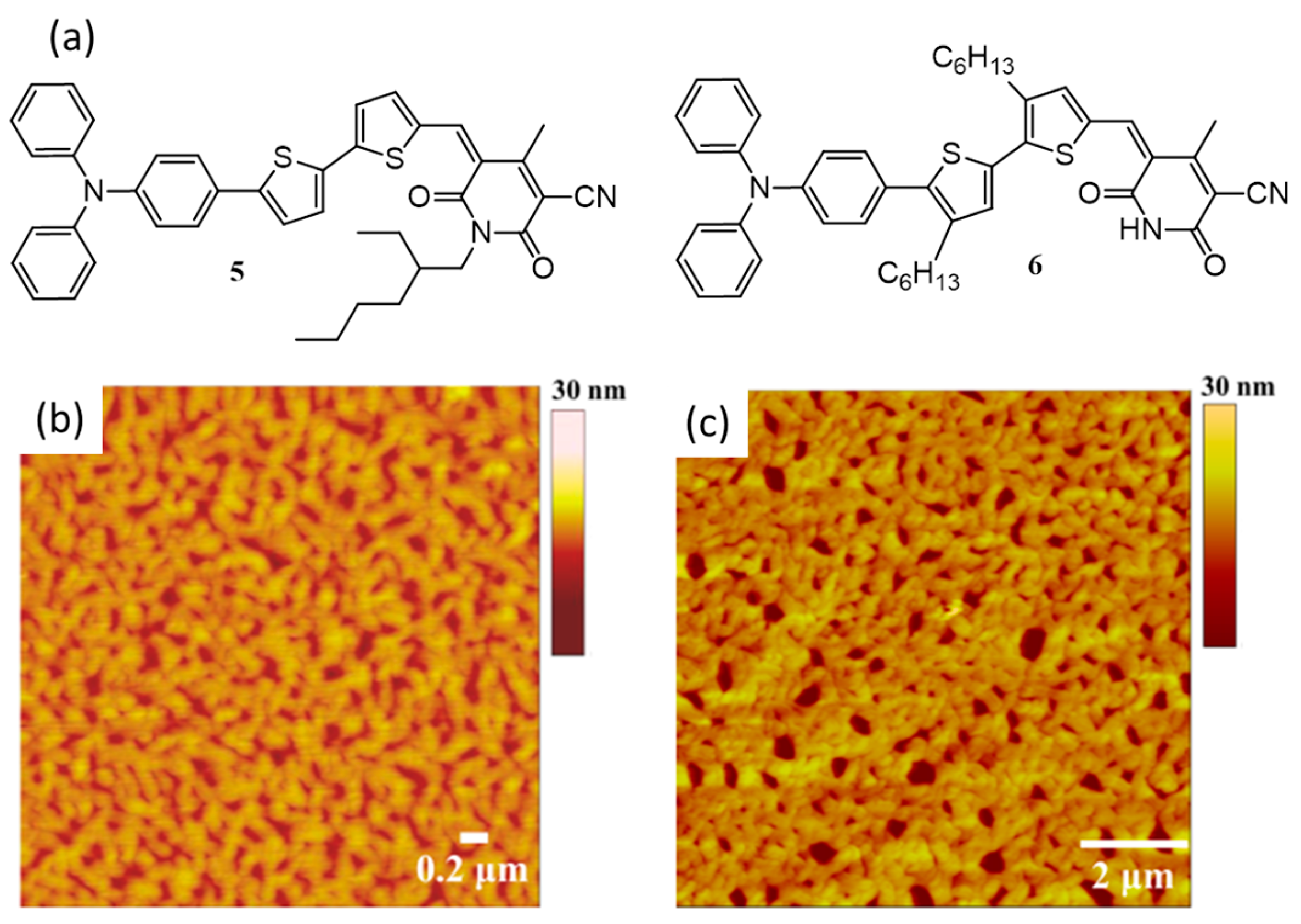
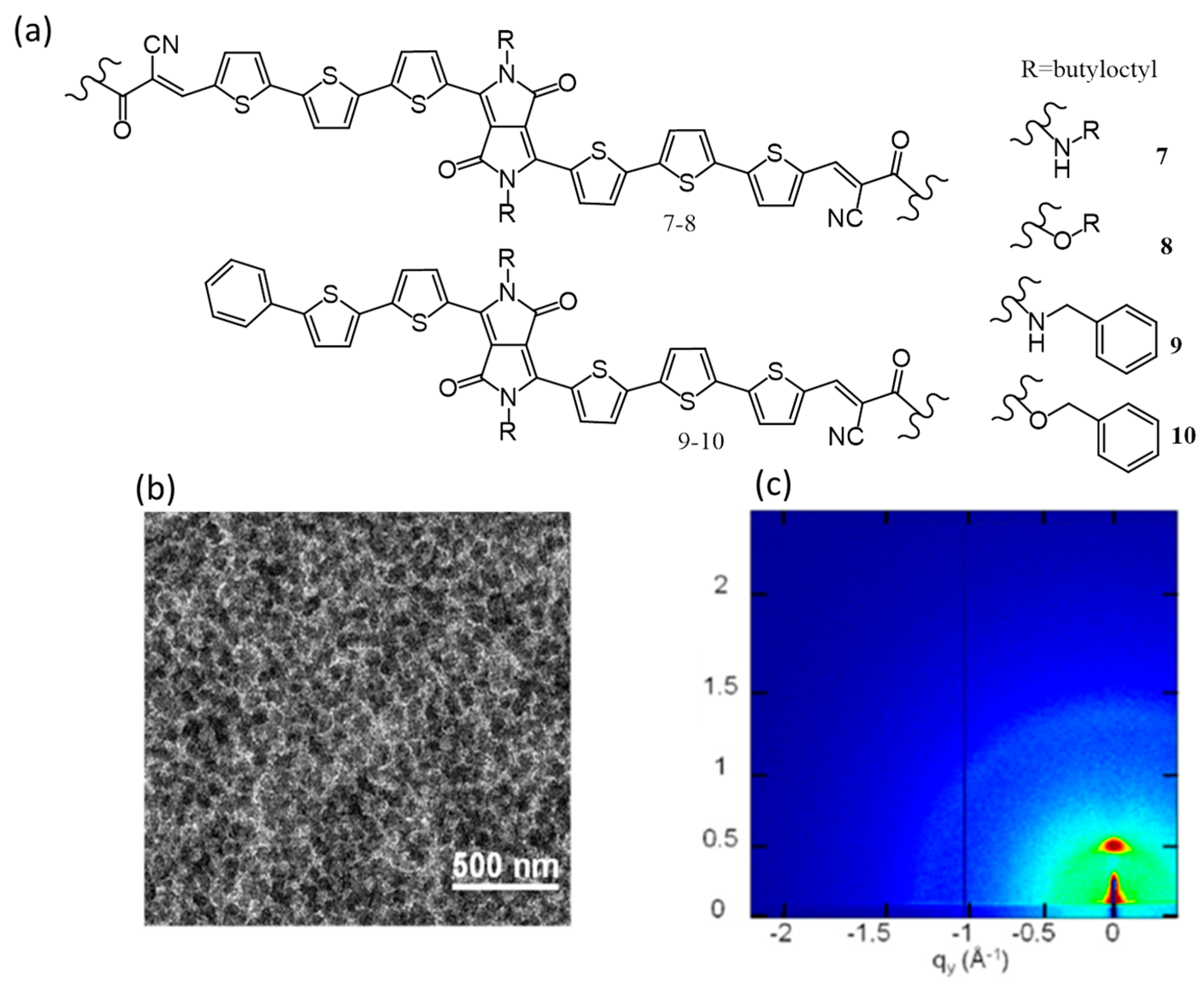
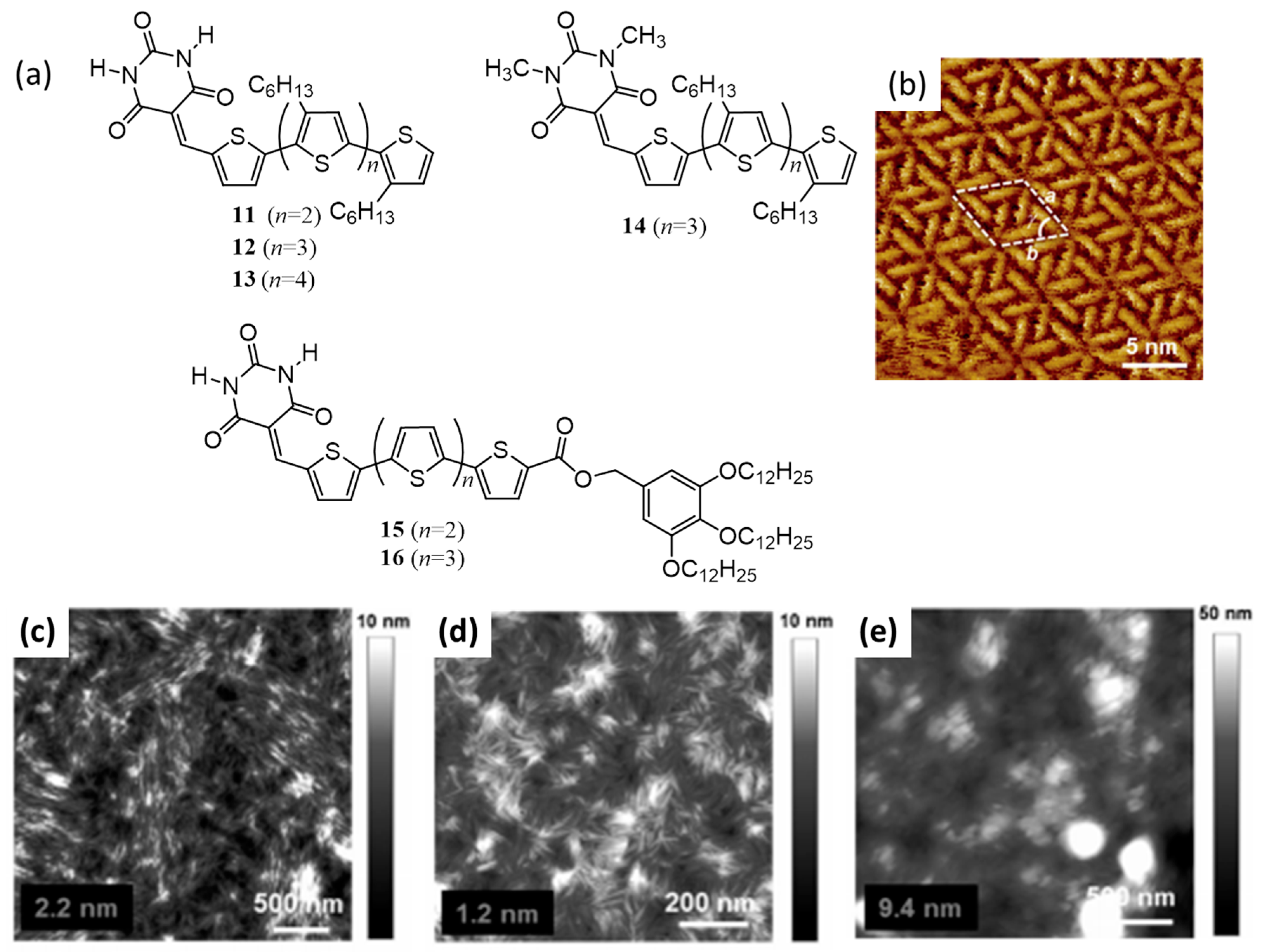
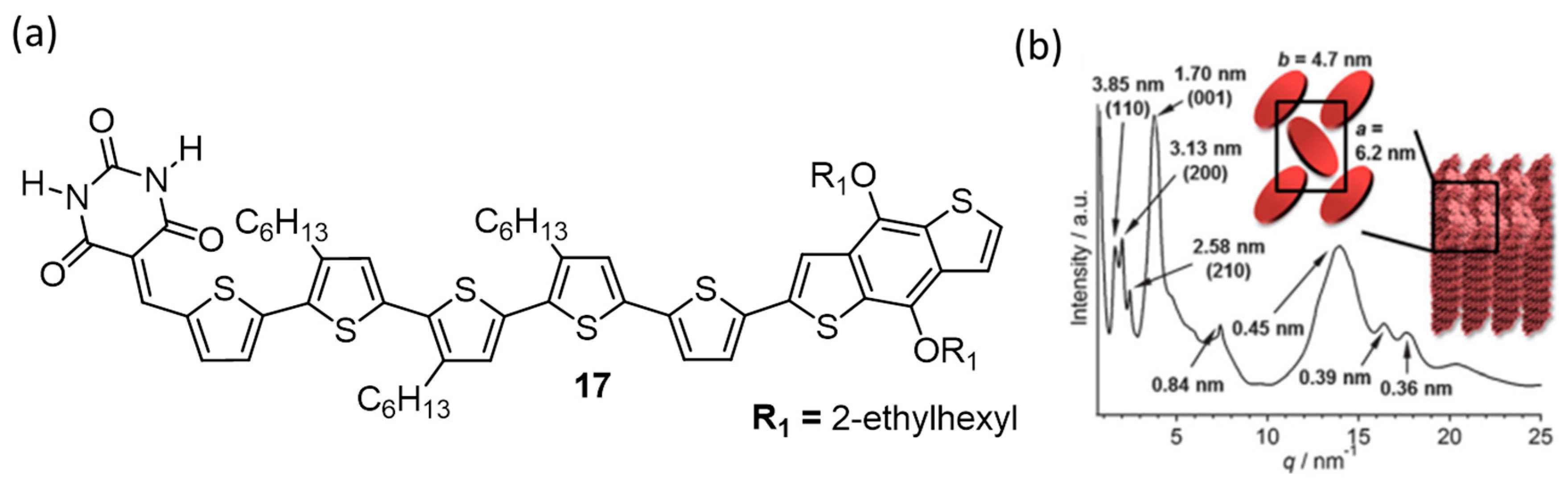

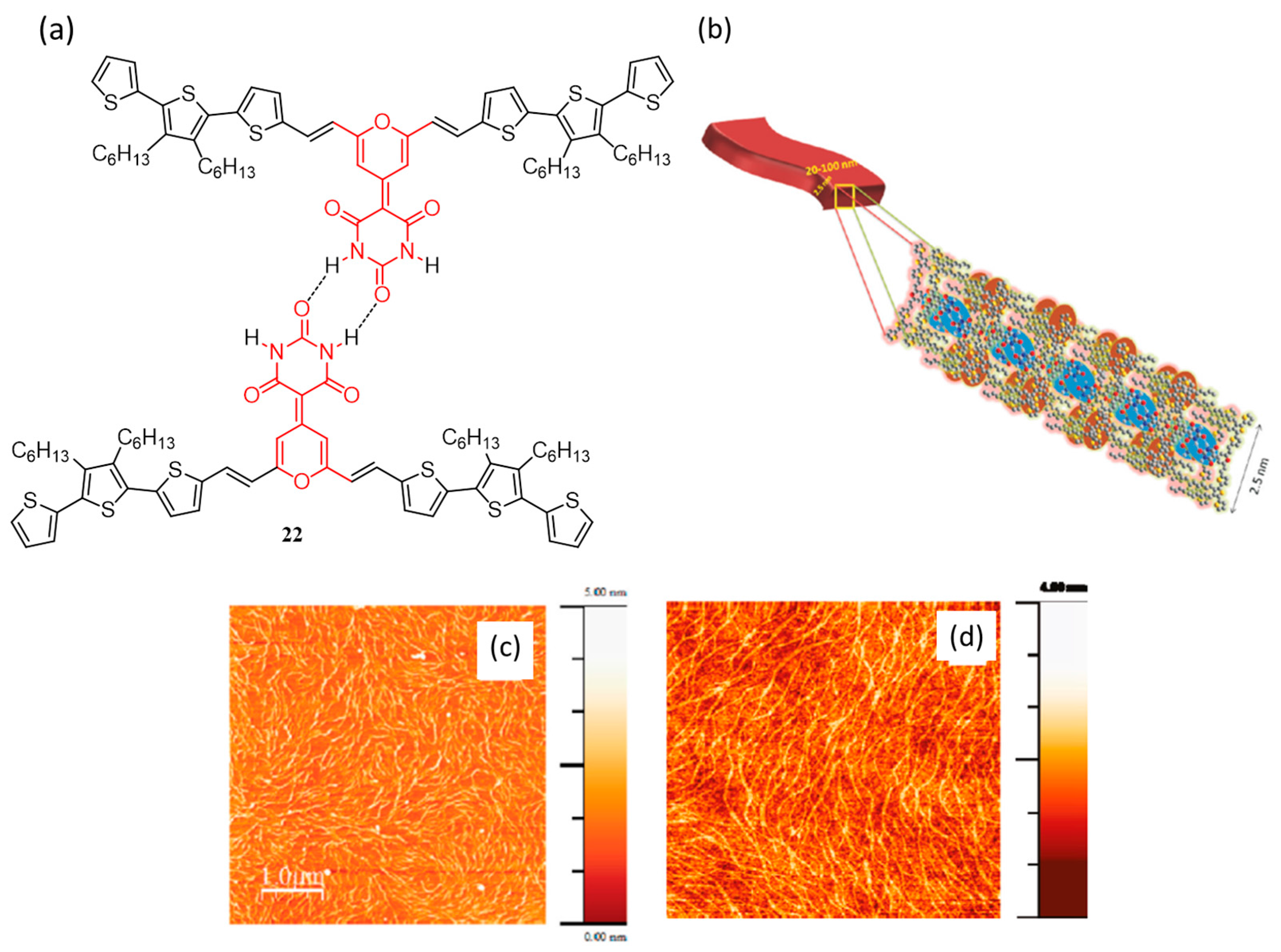



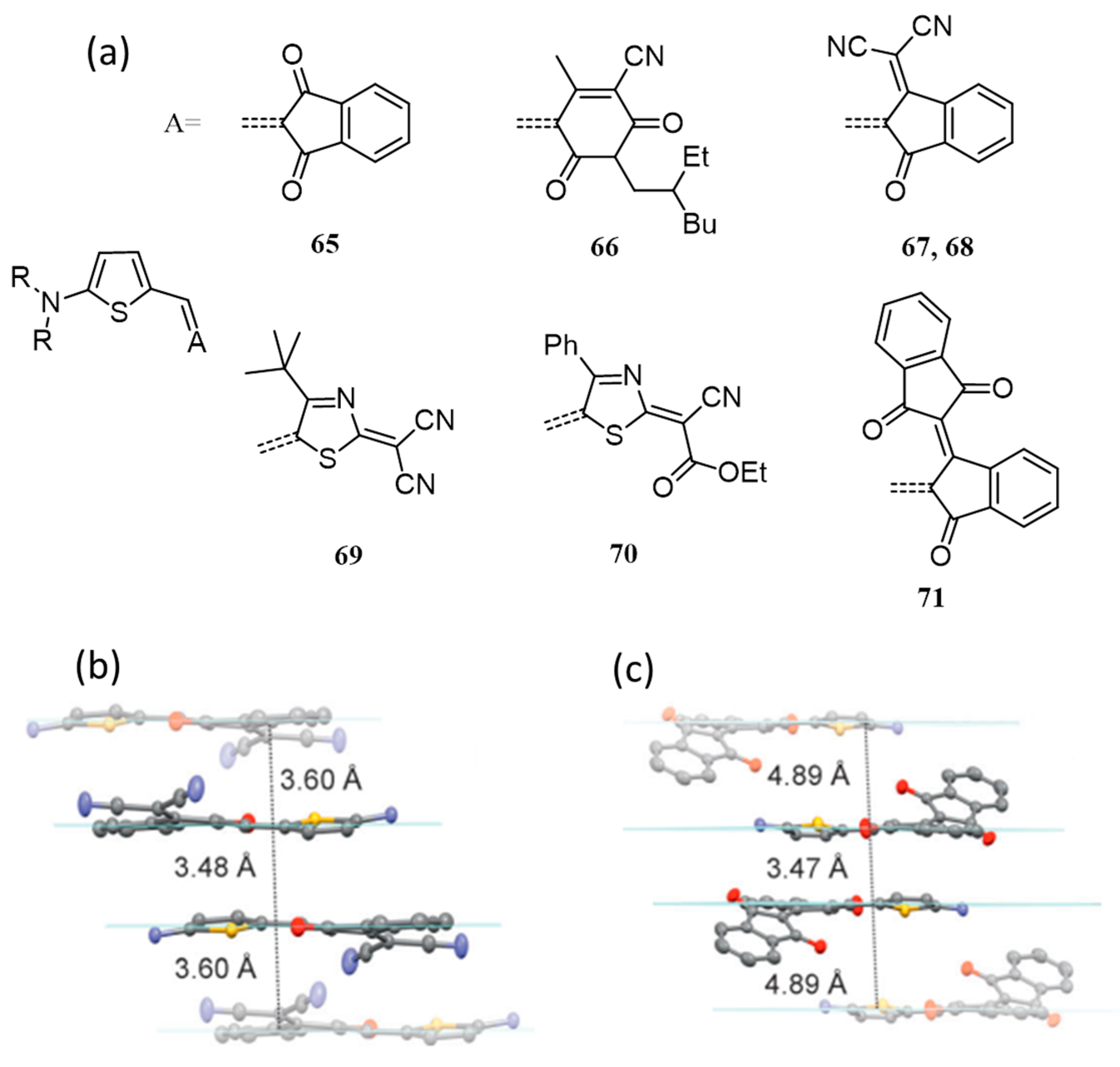
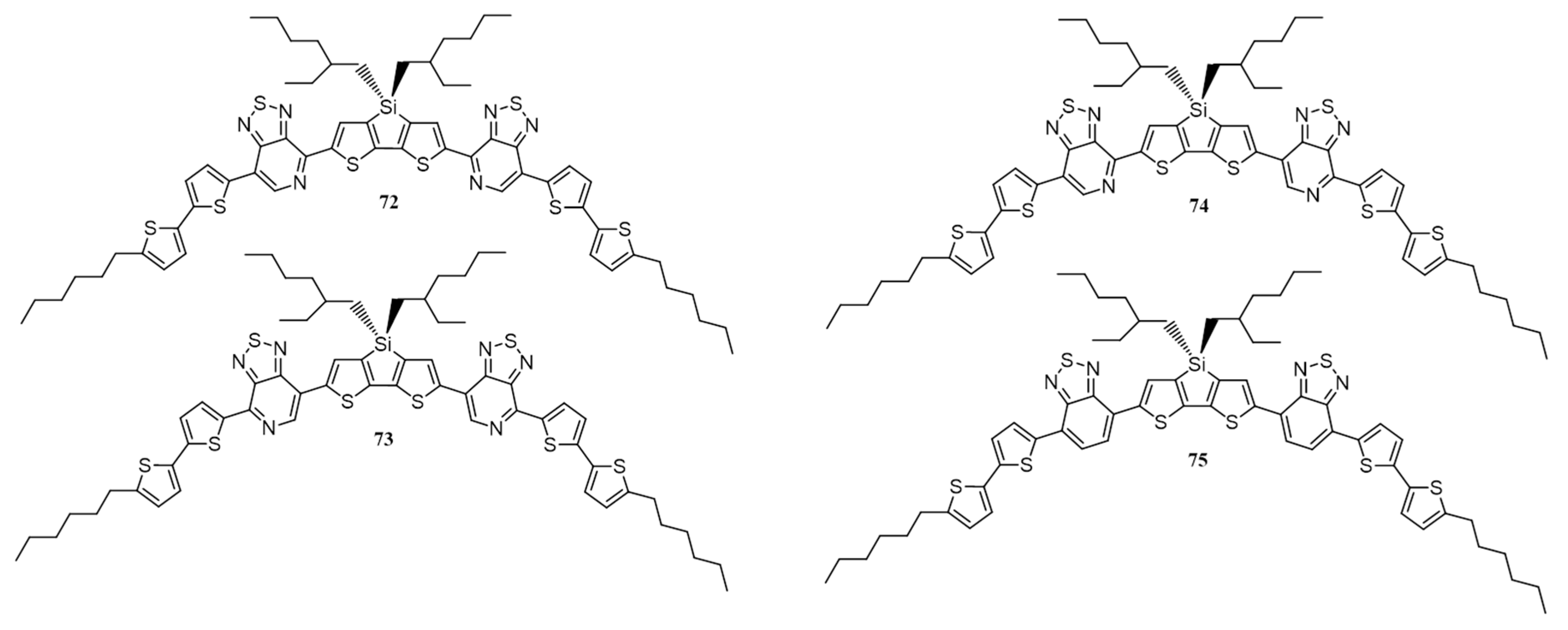
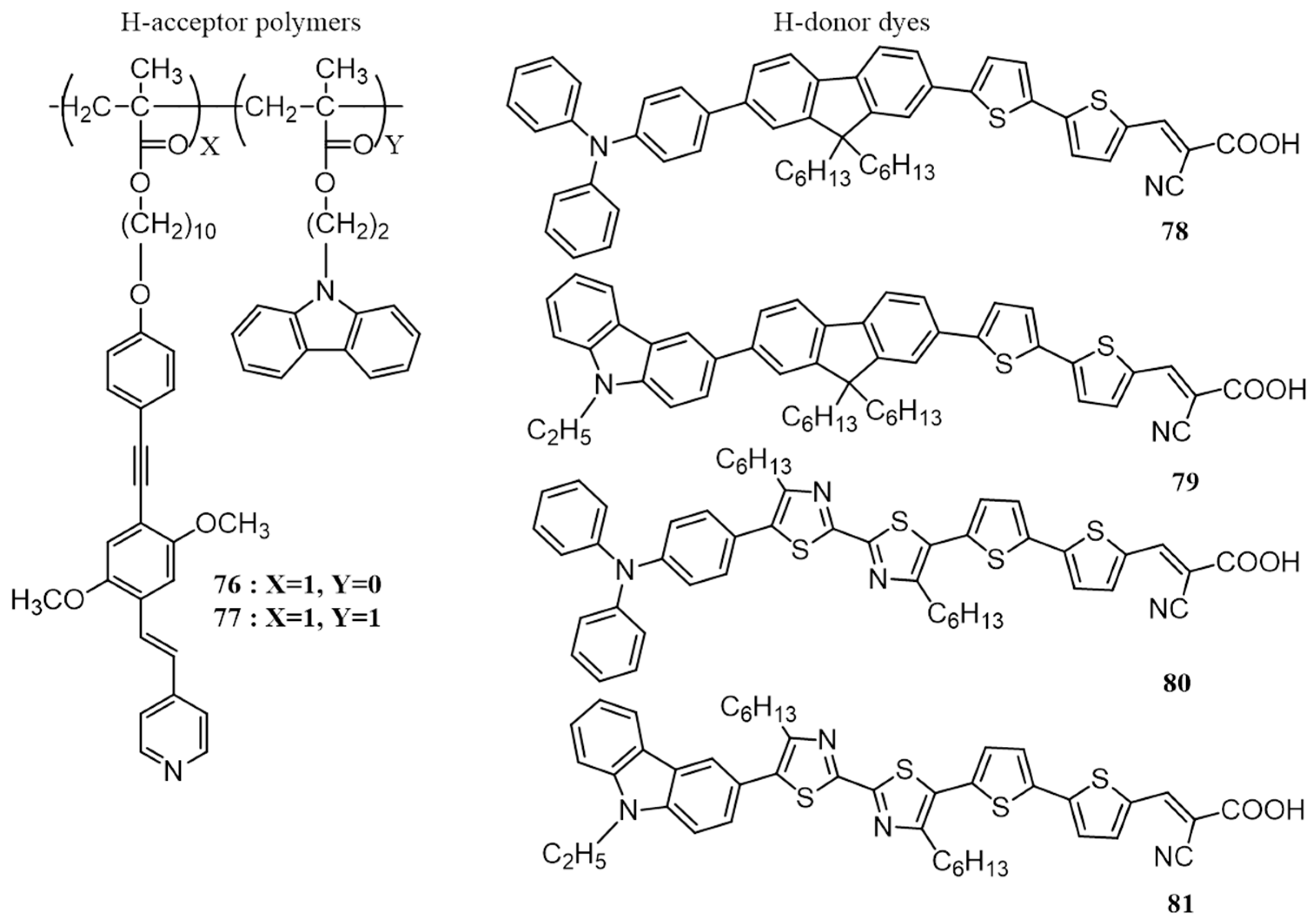
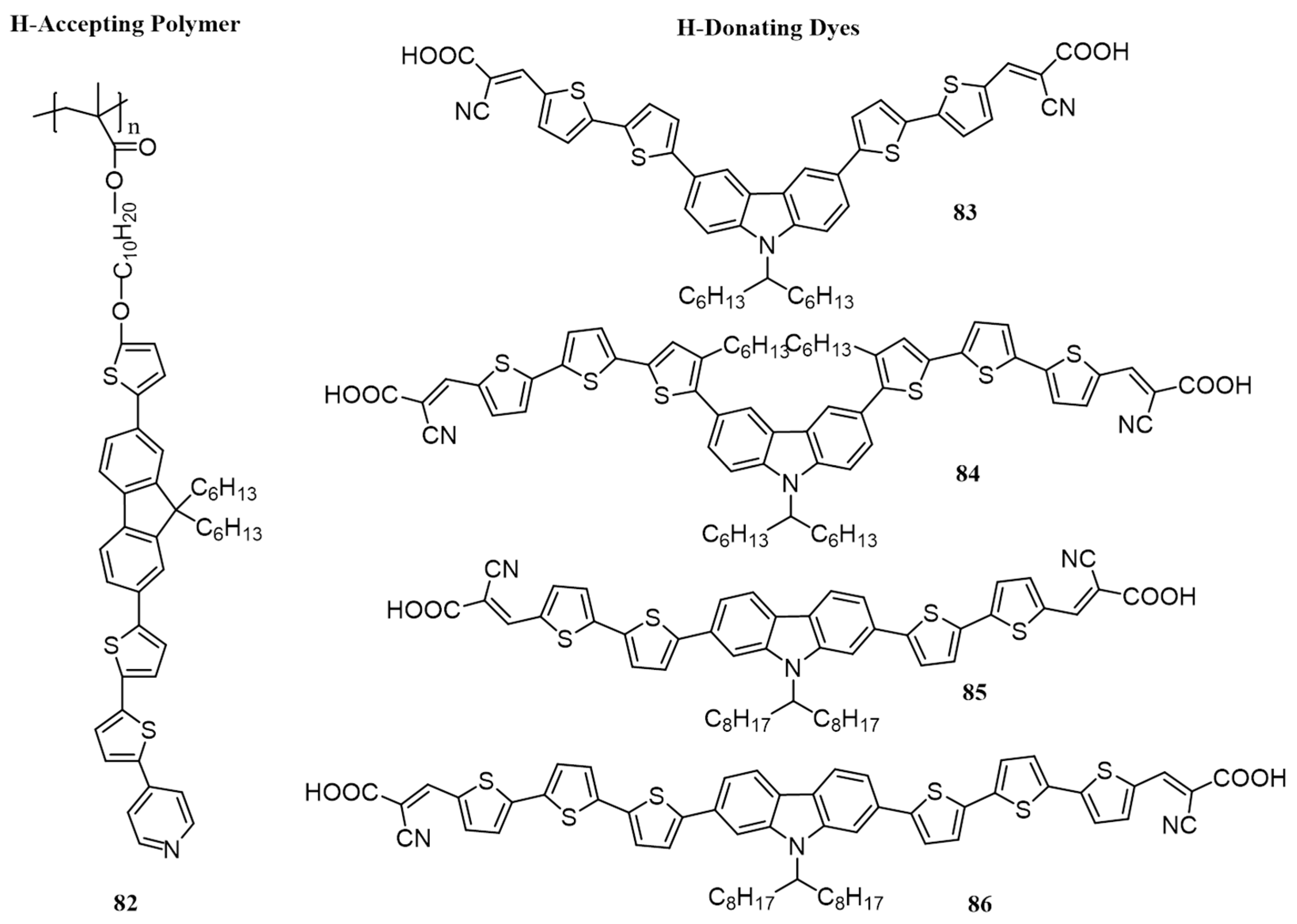
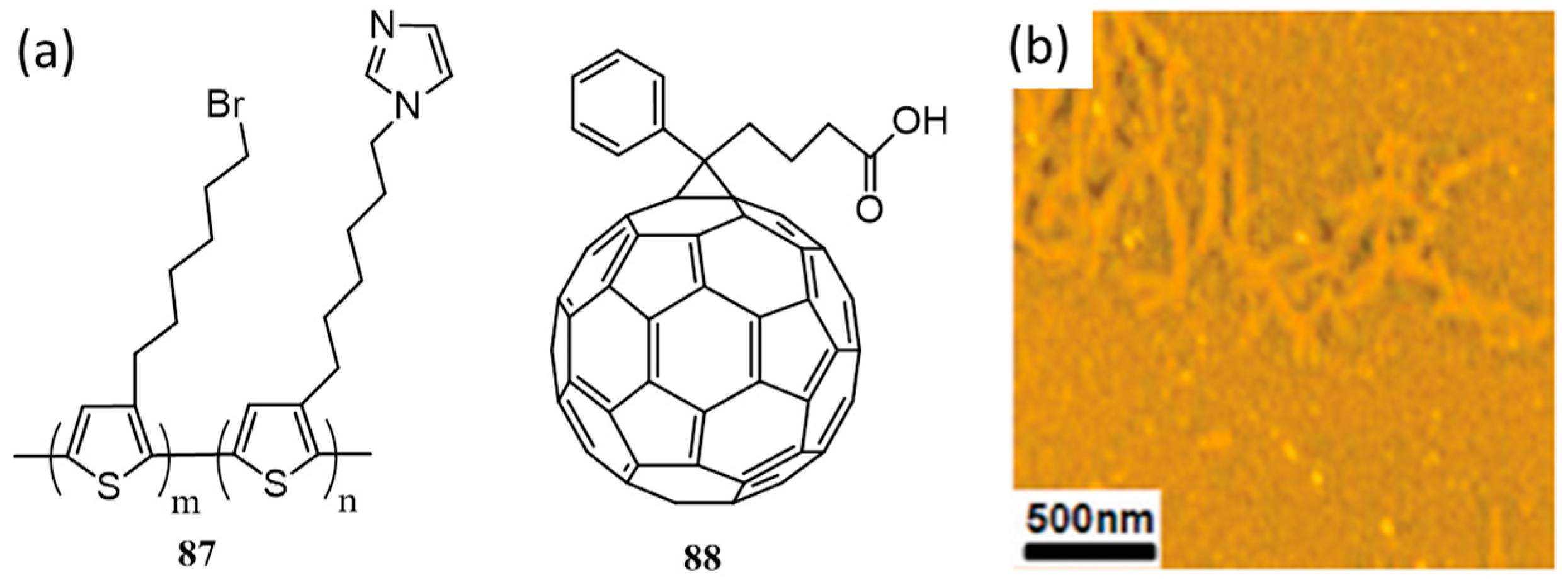
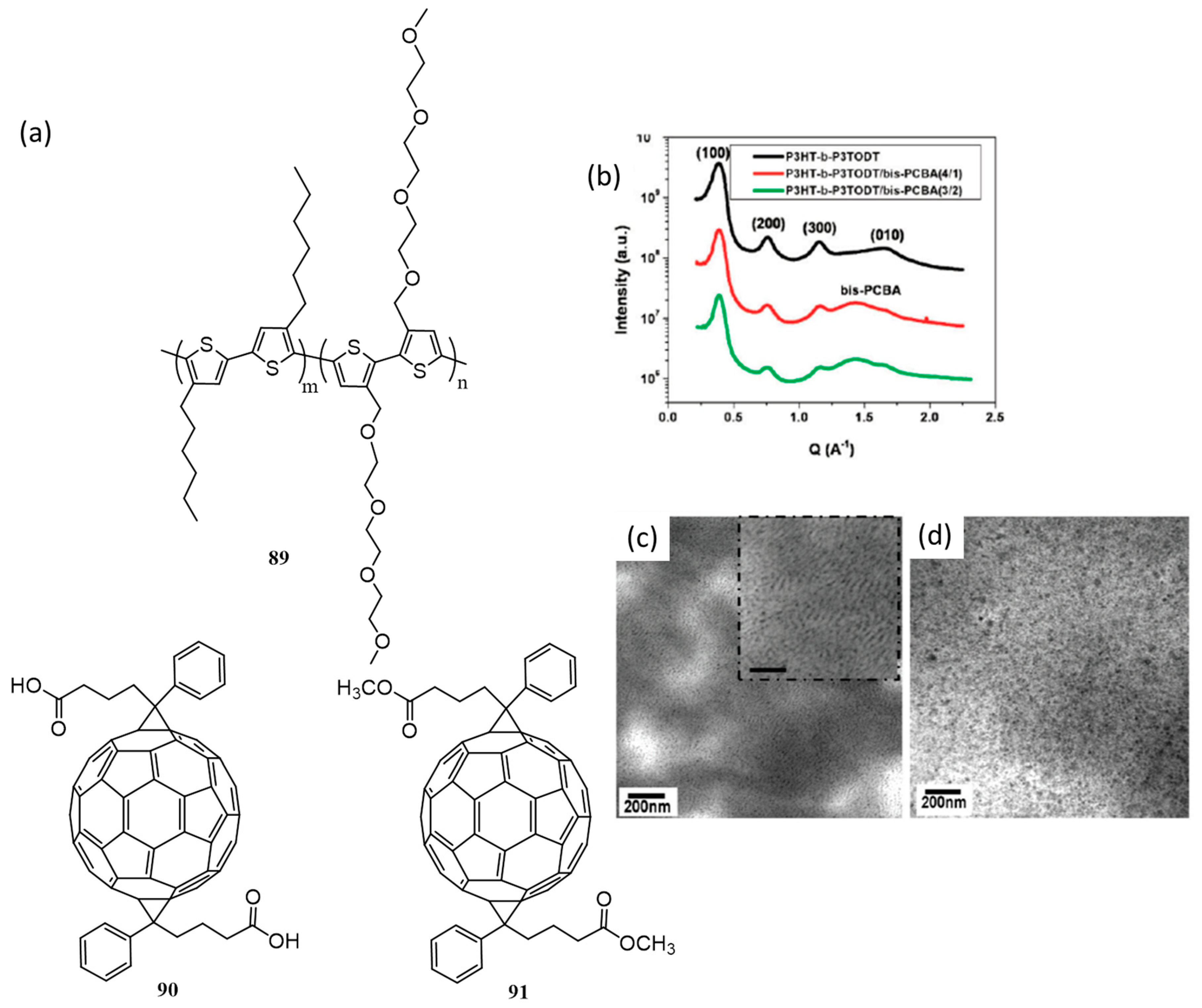

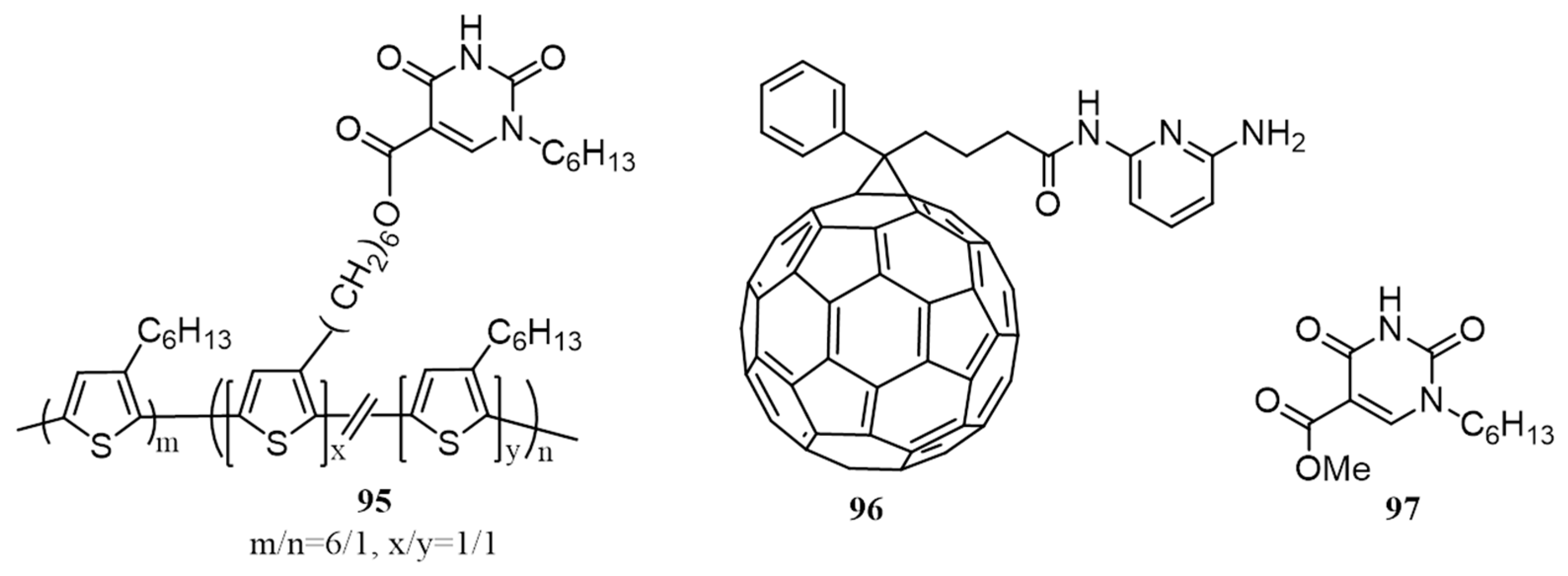
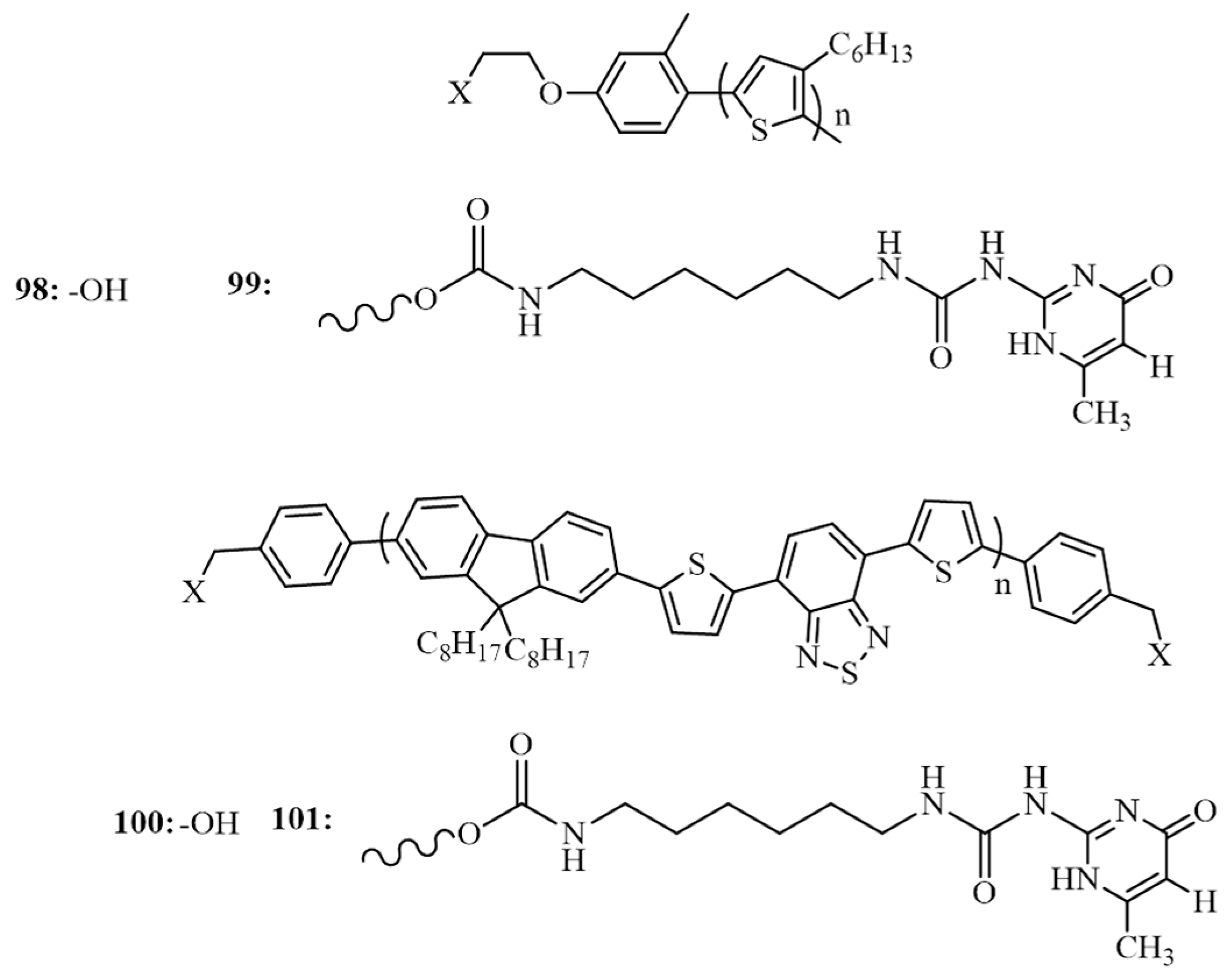


{kind=link}
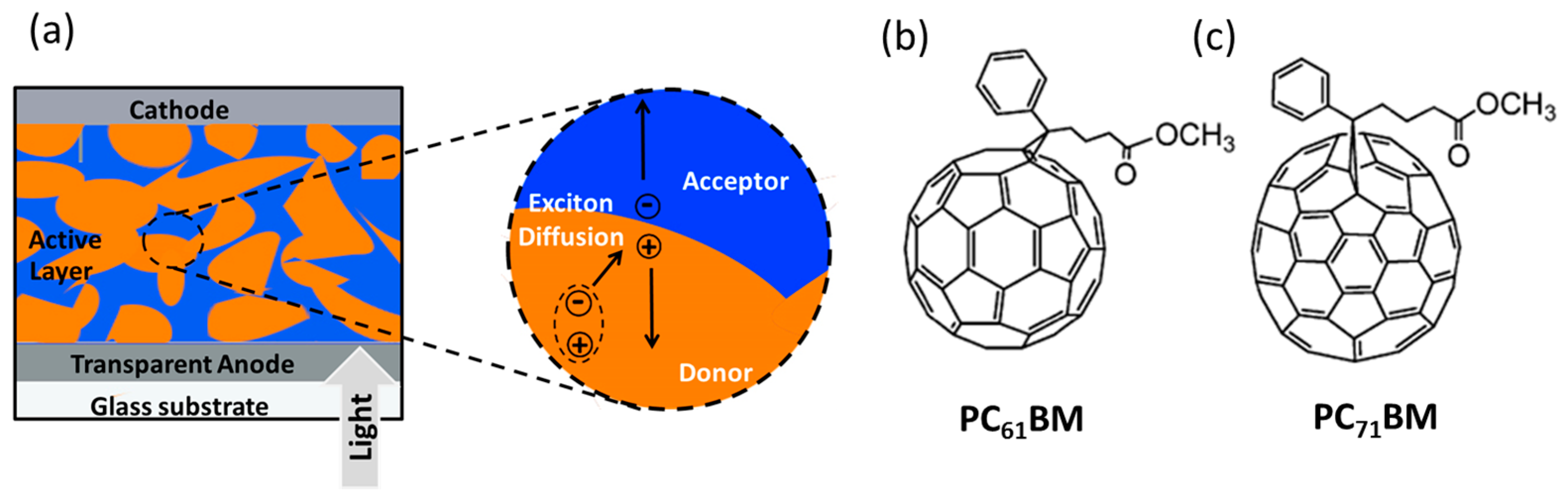{kind=link}
{kind=link}
{kind=link}
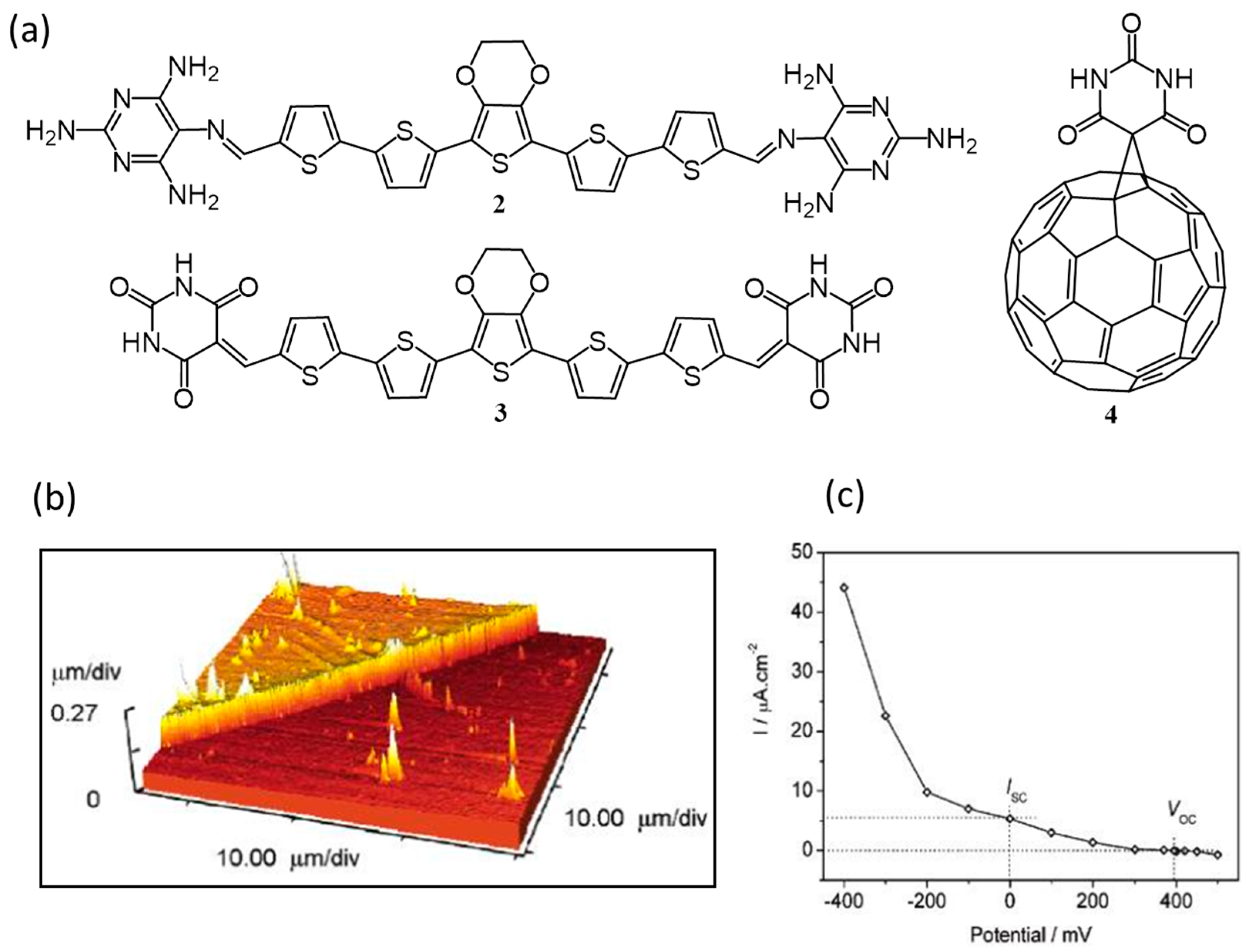
{kind=link}
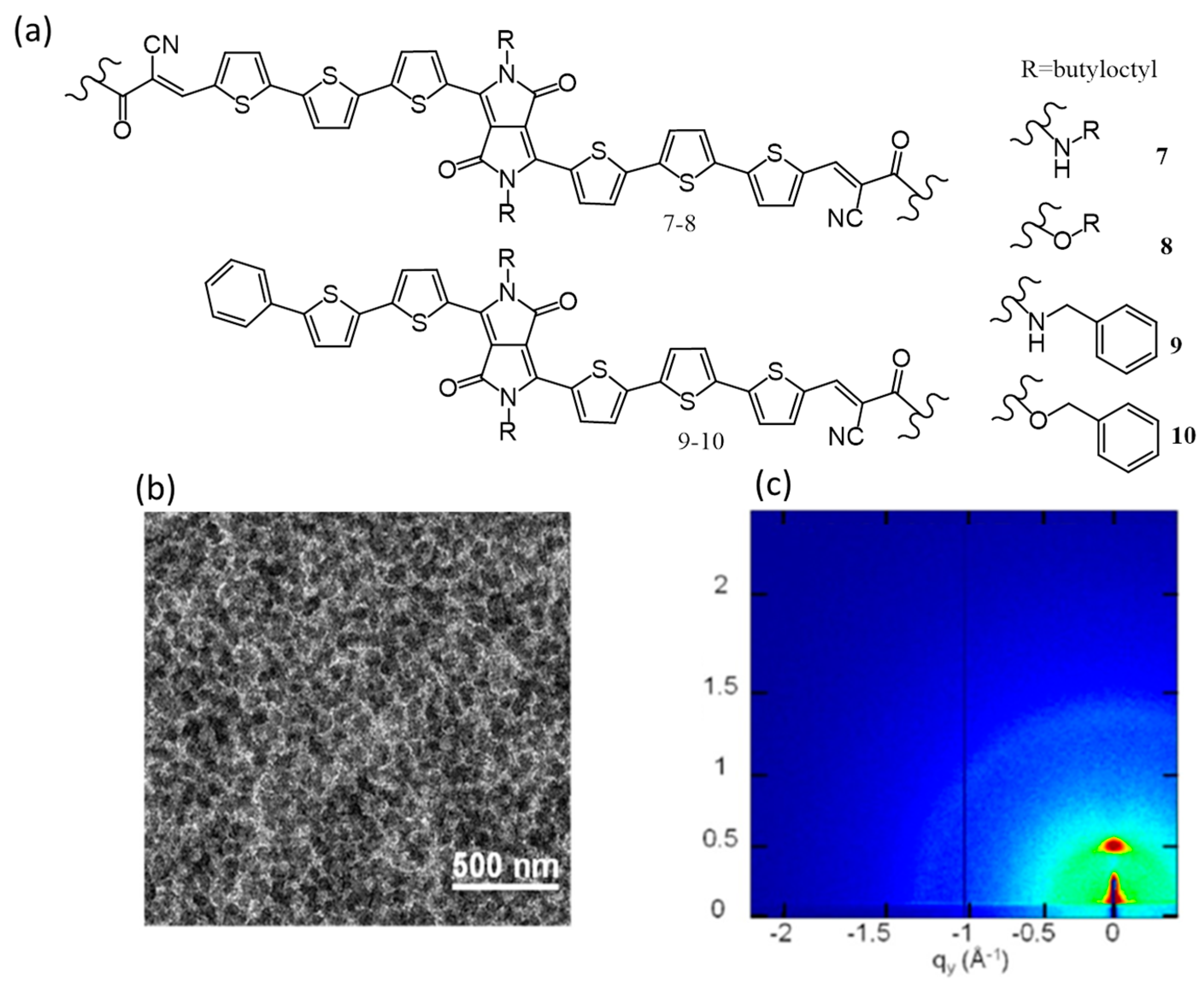{kind=link}
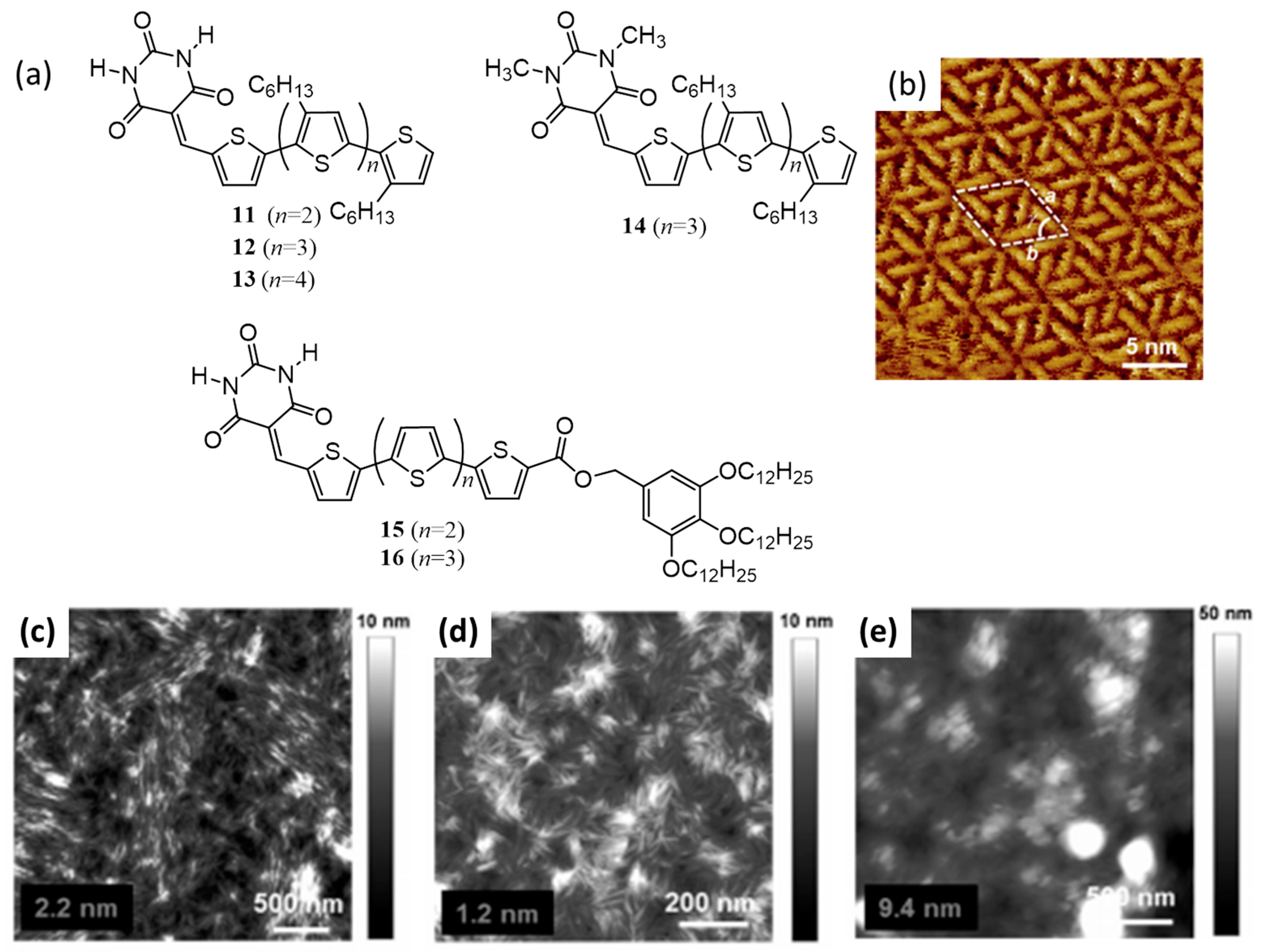{kind=link}
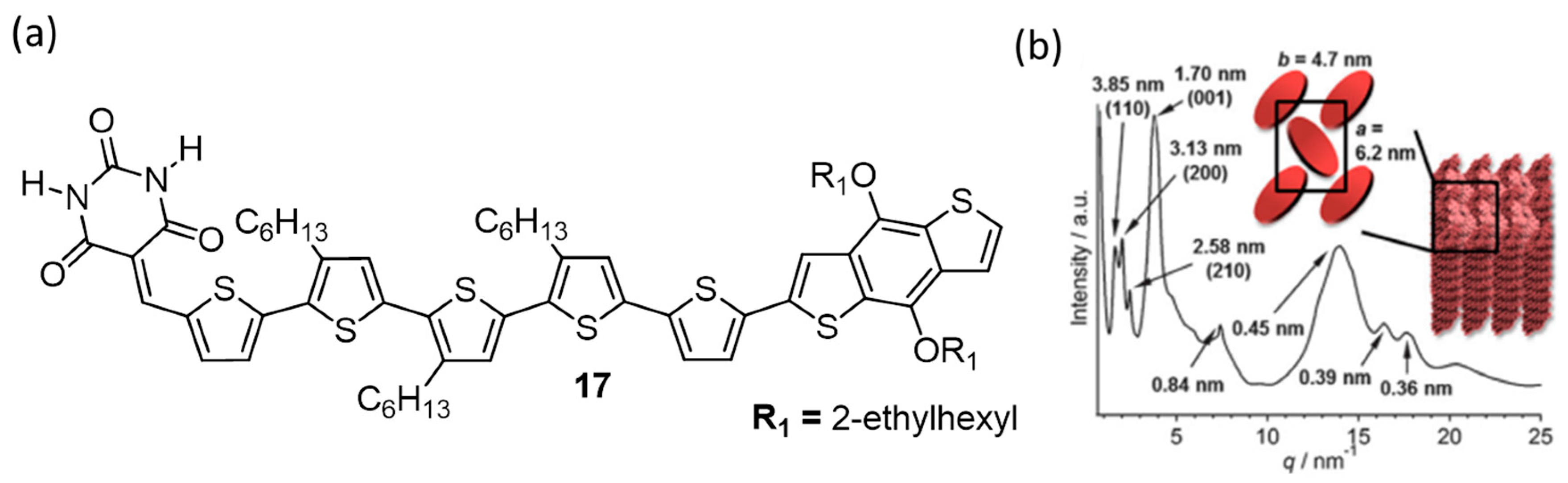{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
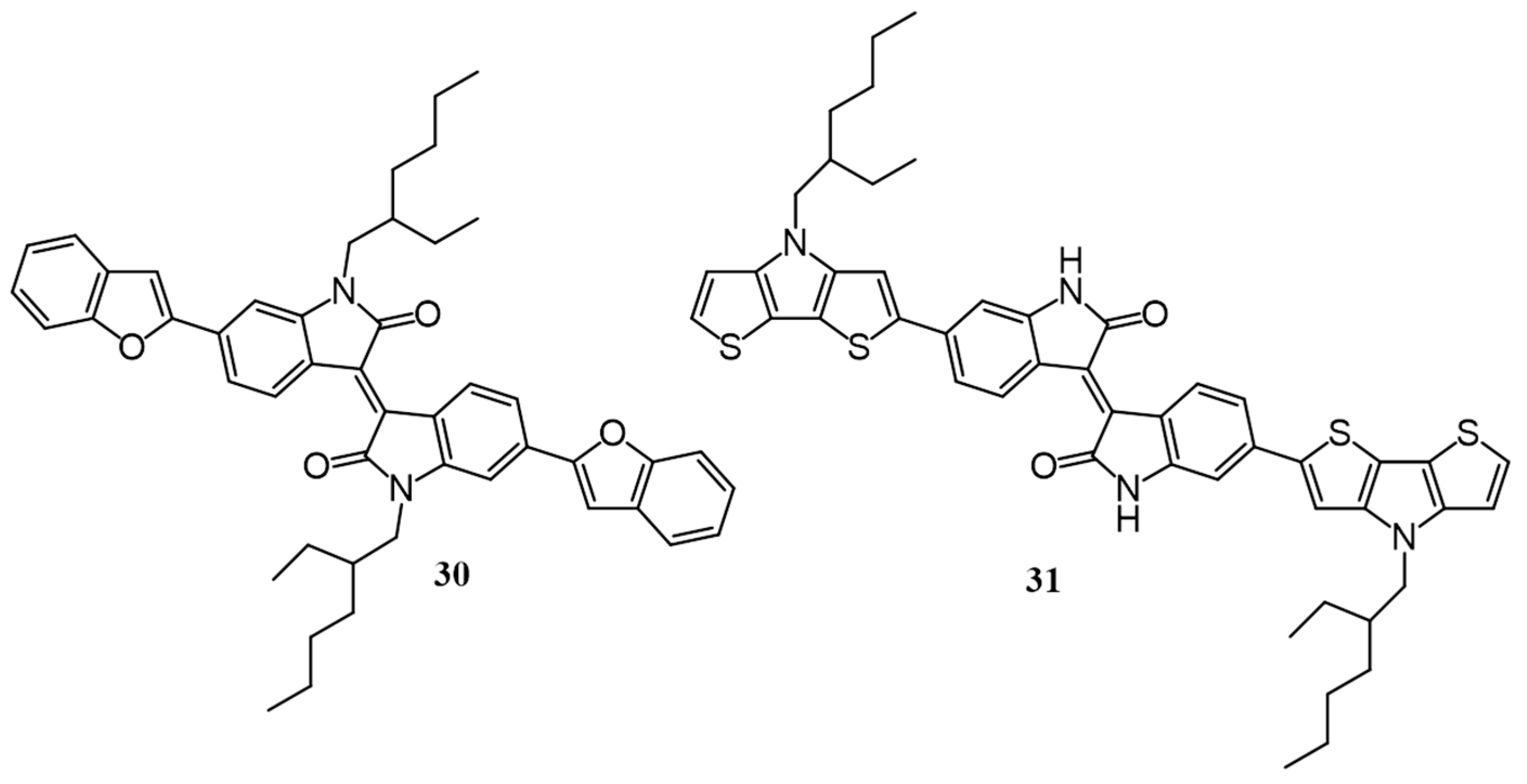{kind=link}
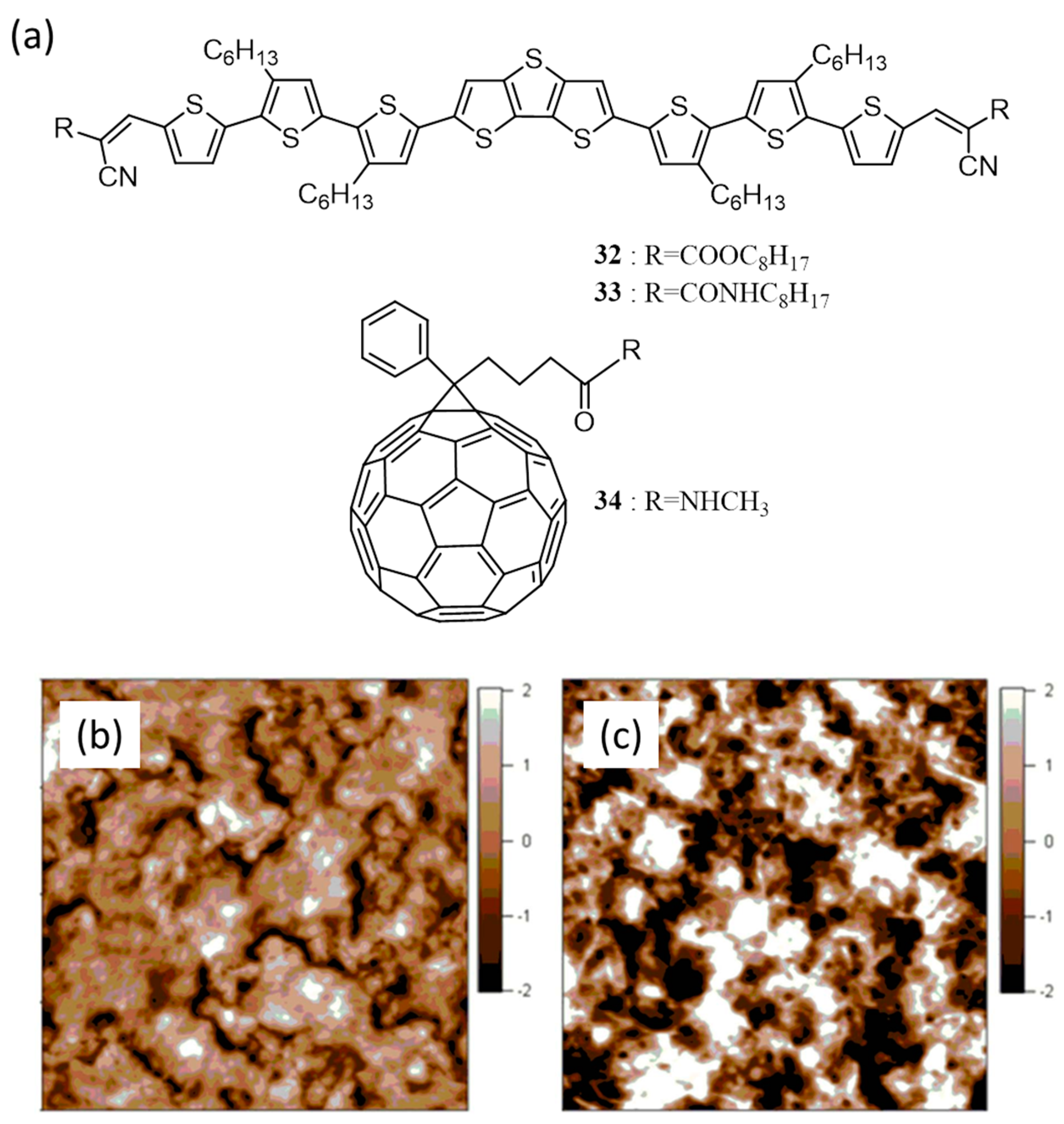{kind=link}
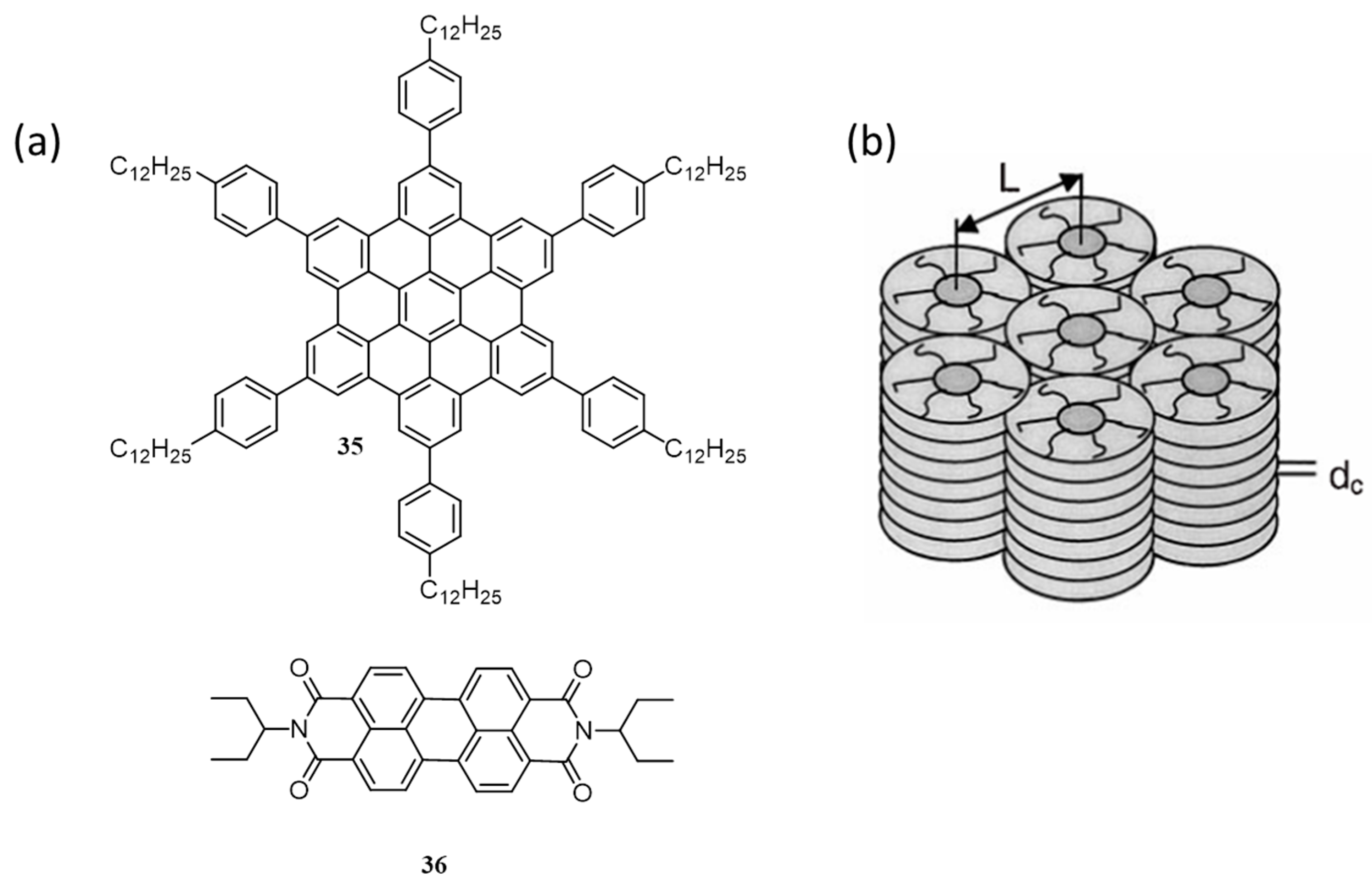{kind=link}
{kind=link}
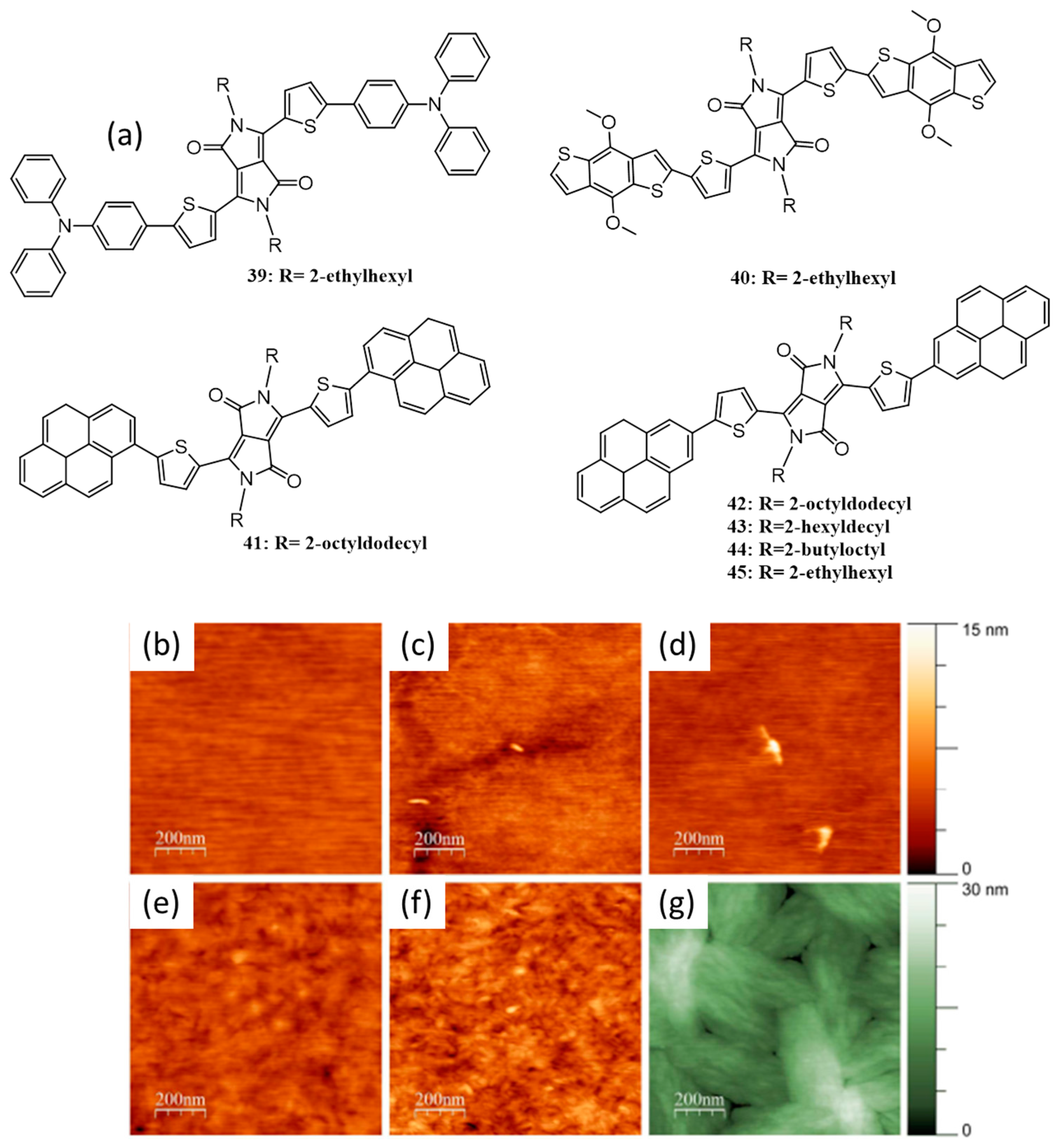{kind=link}
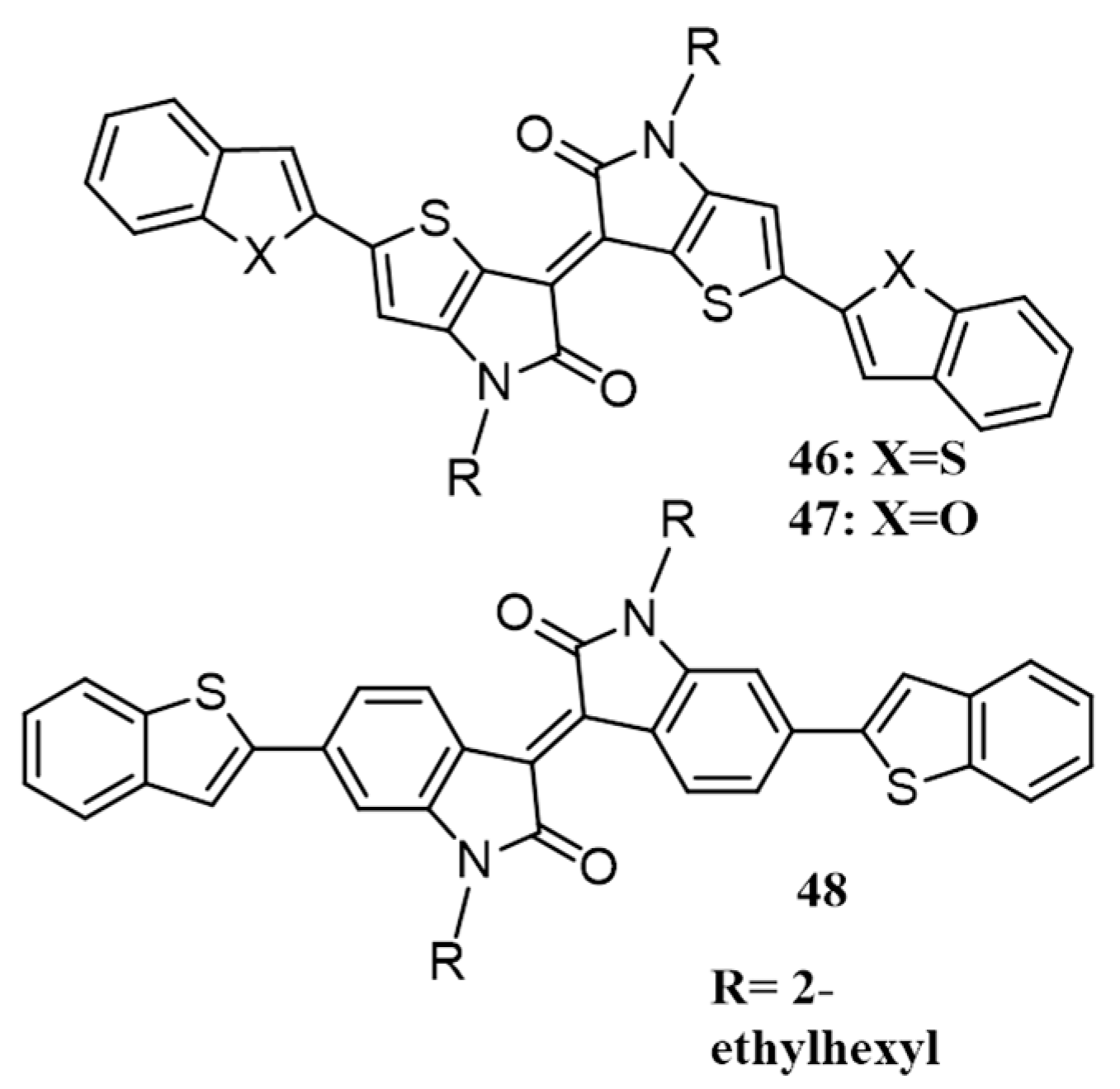{kind=link}
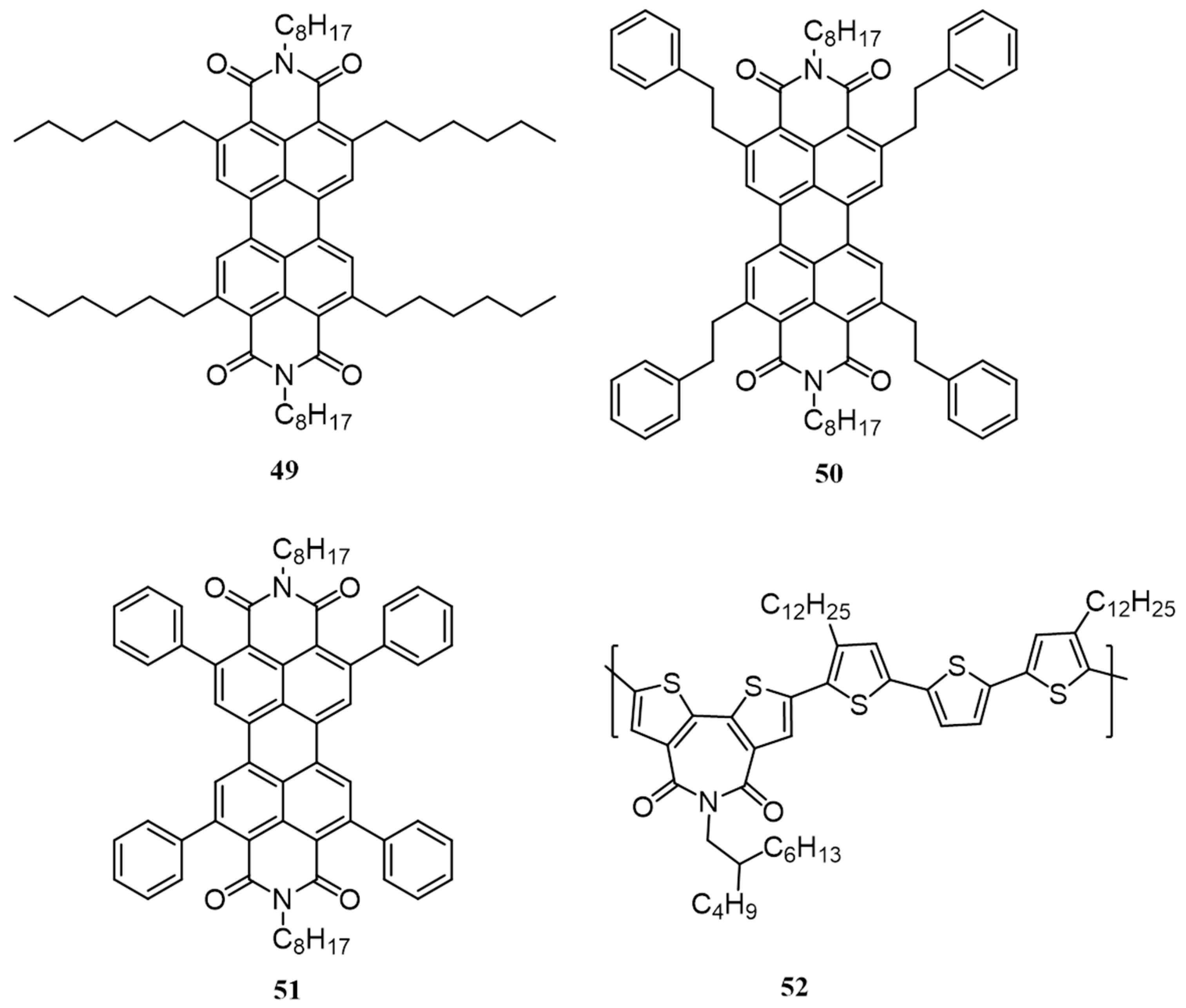{kind=link}
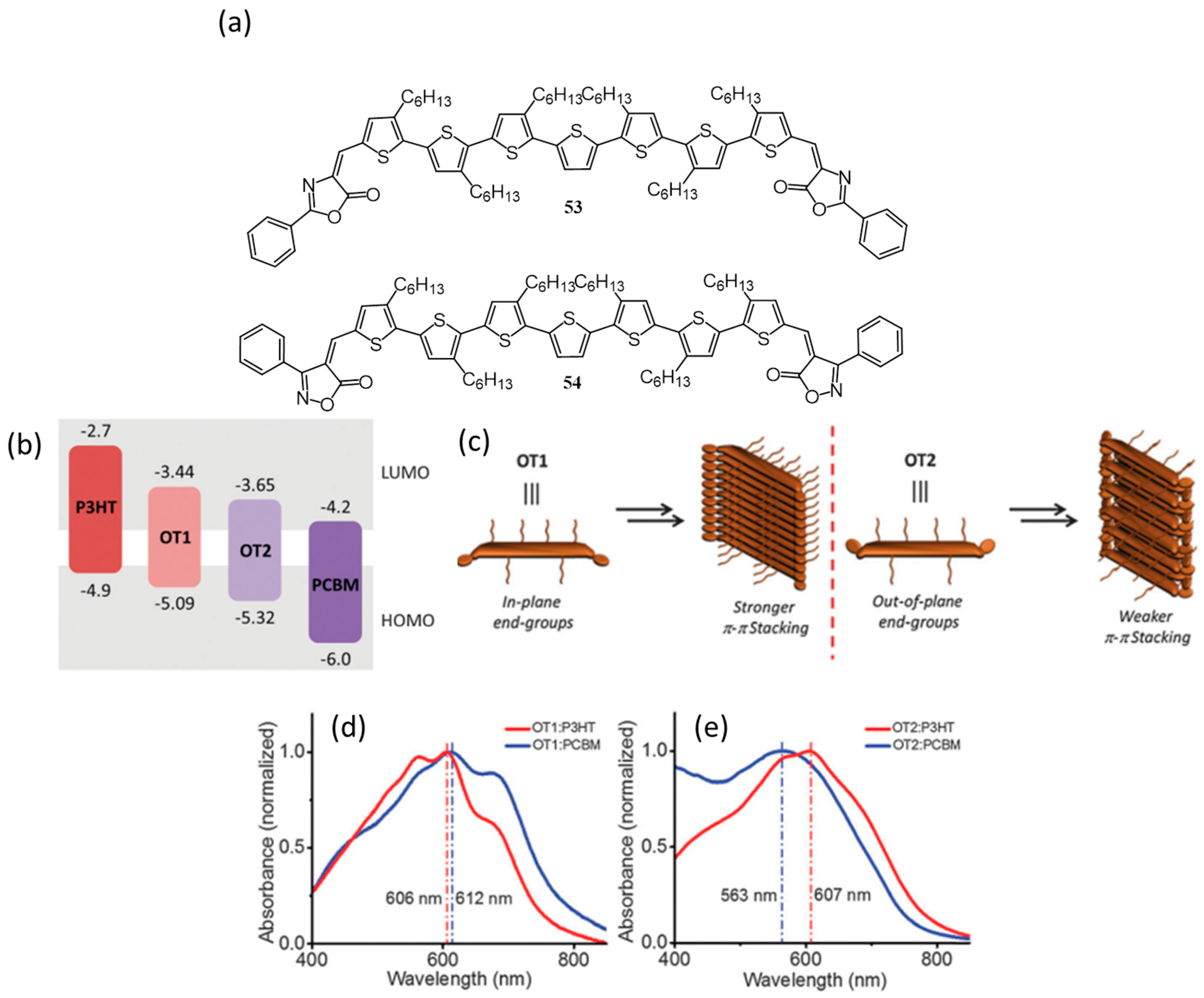{kind=link}
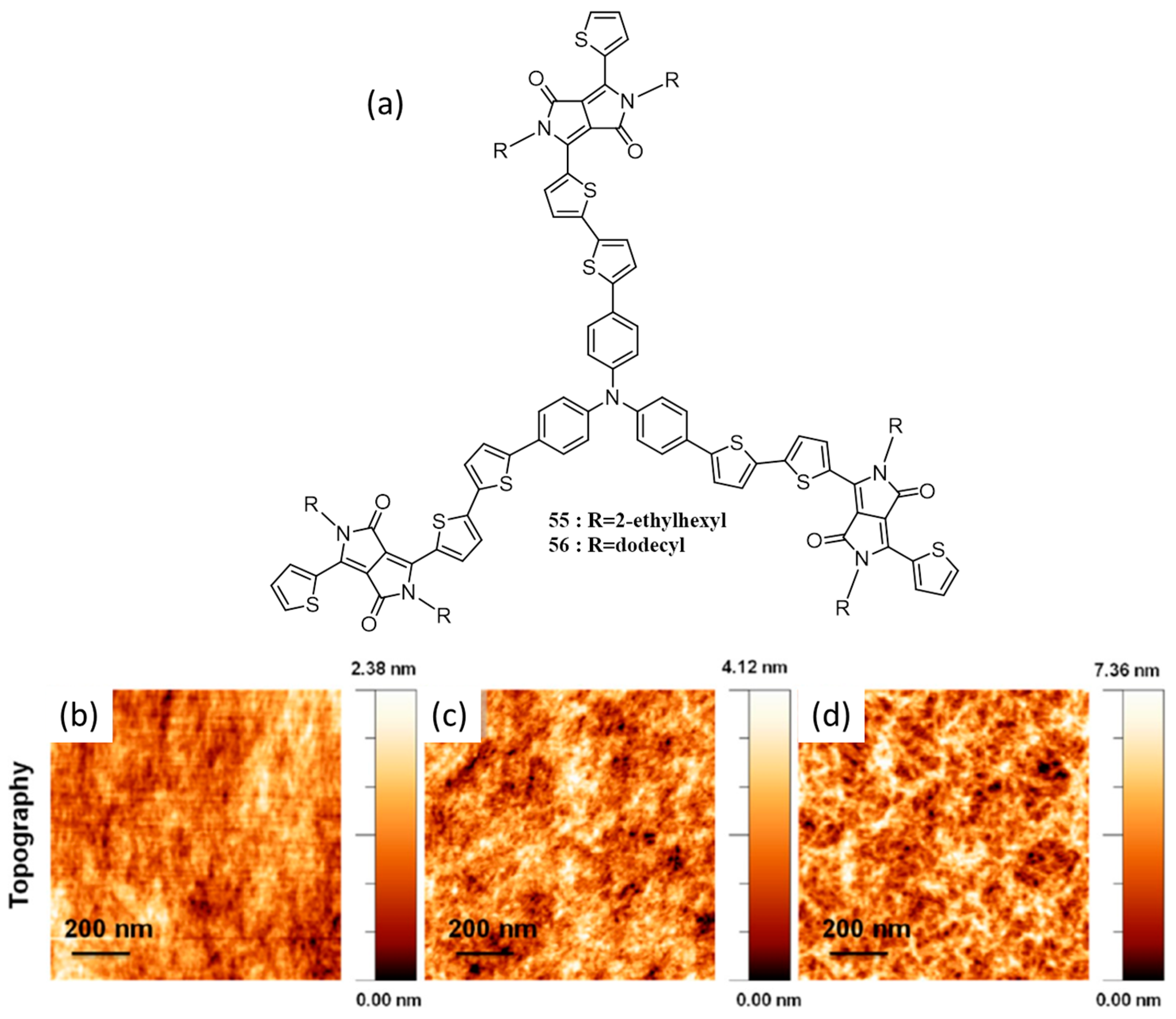{kind=link}
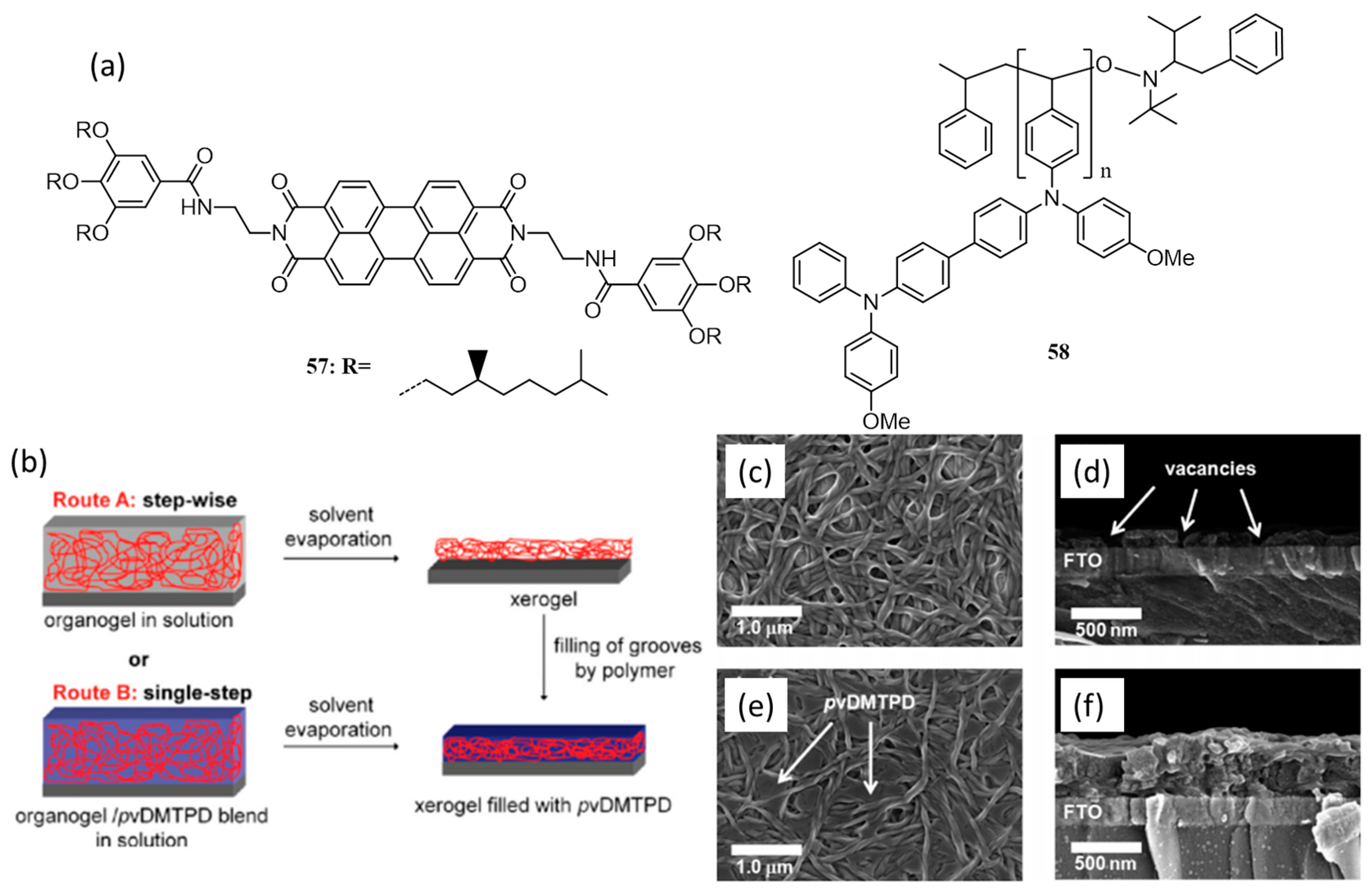{kind=link}
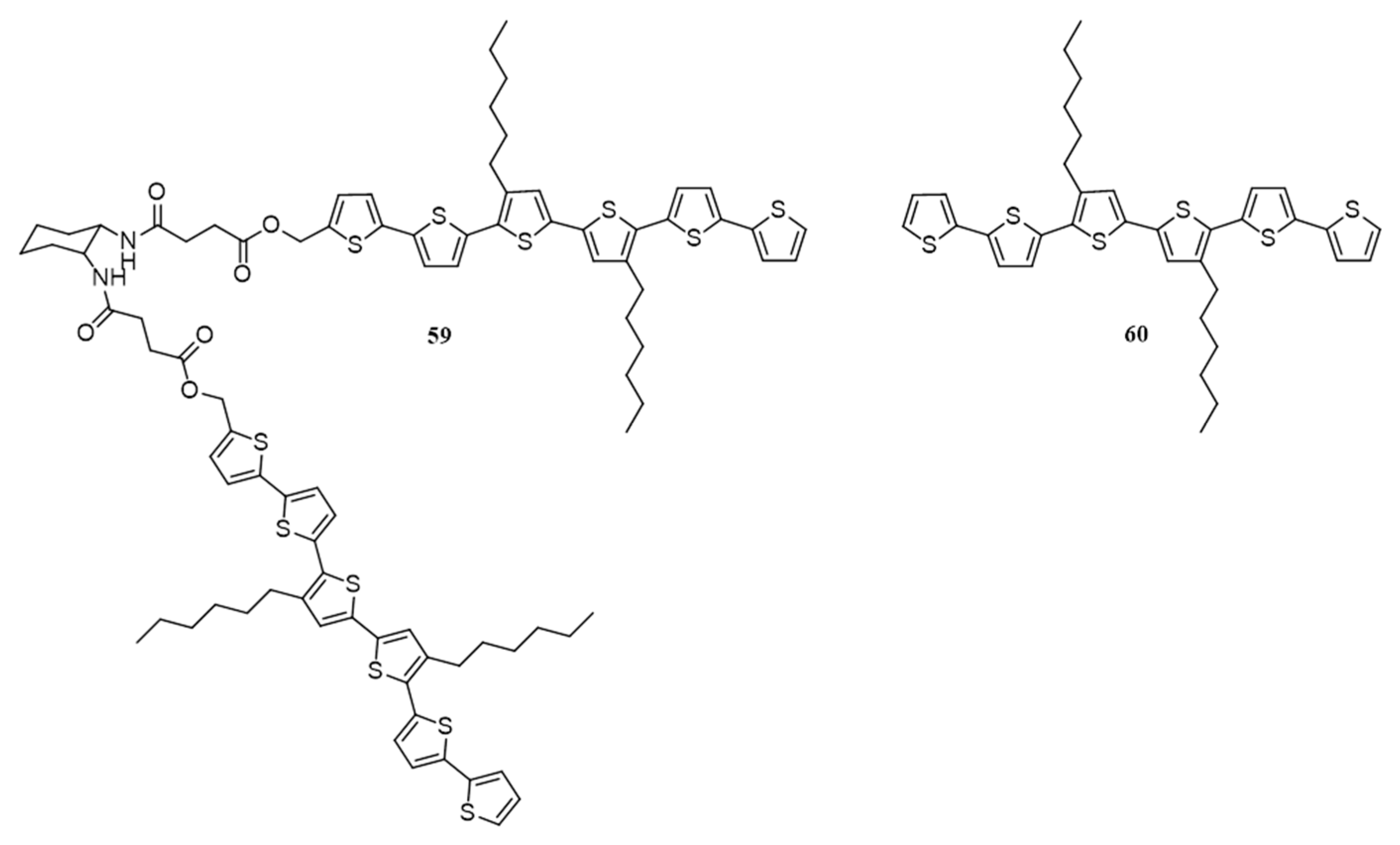{kind=link}
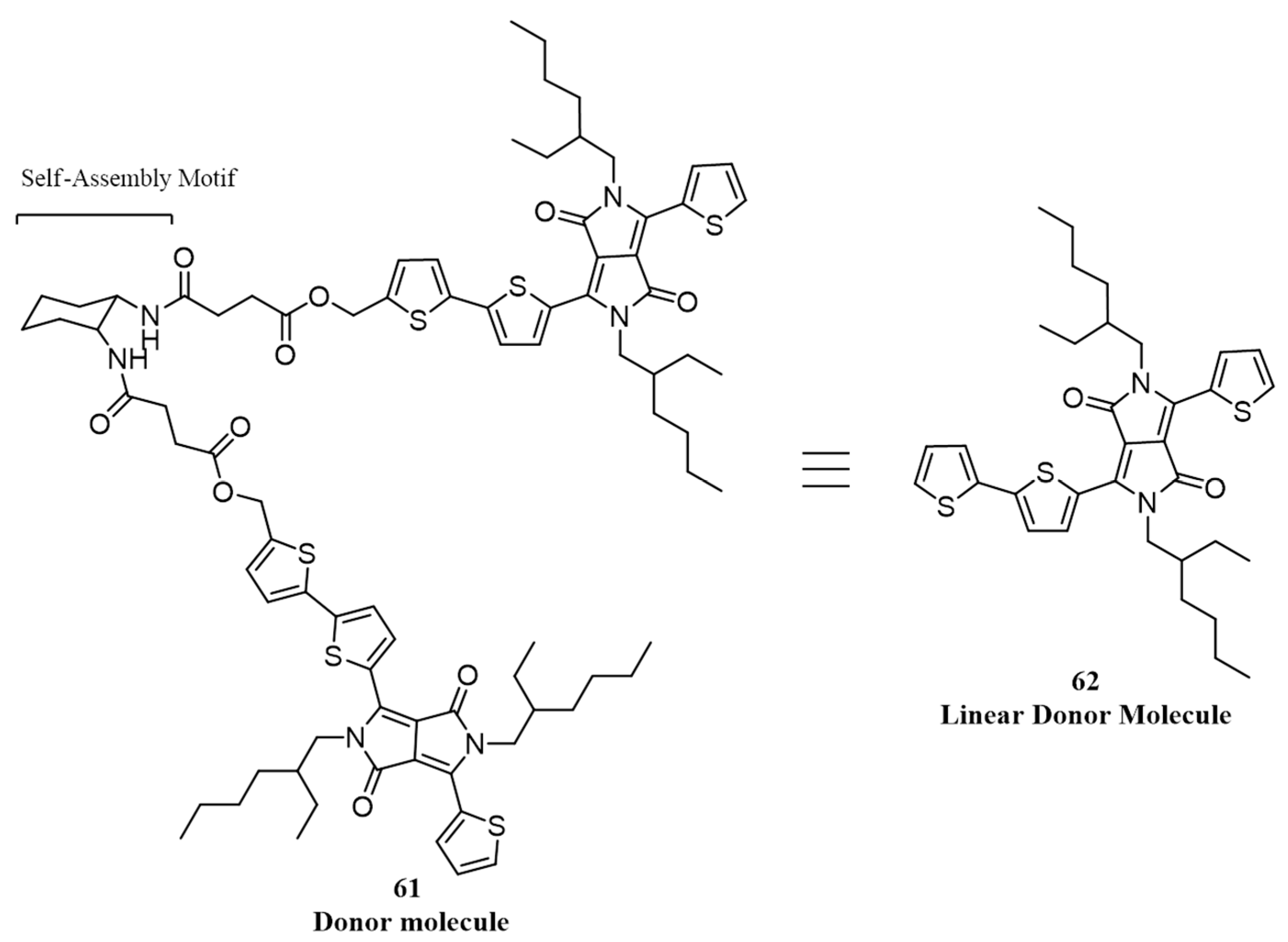{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
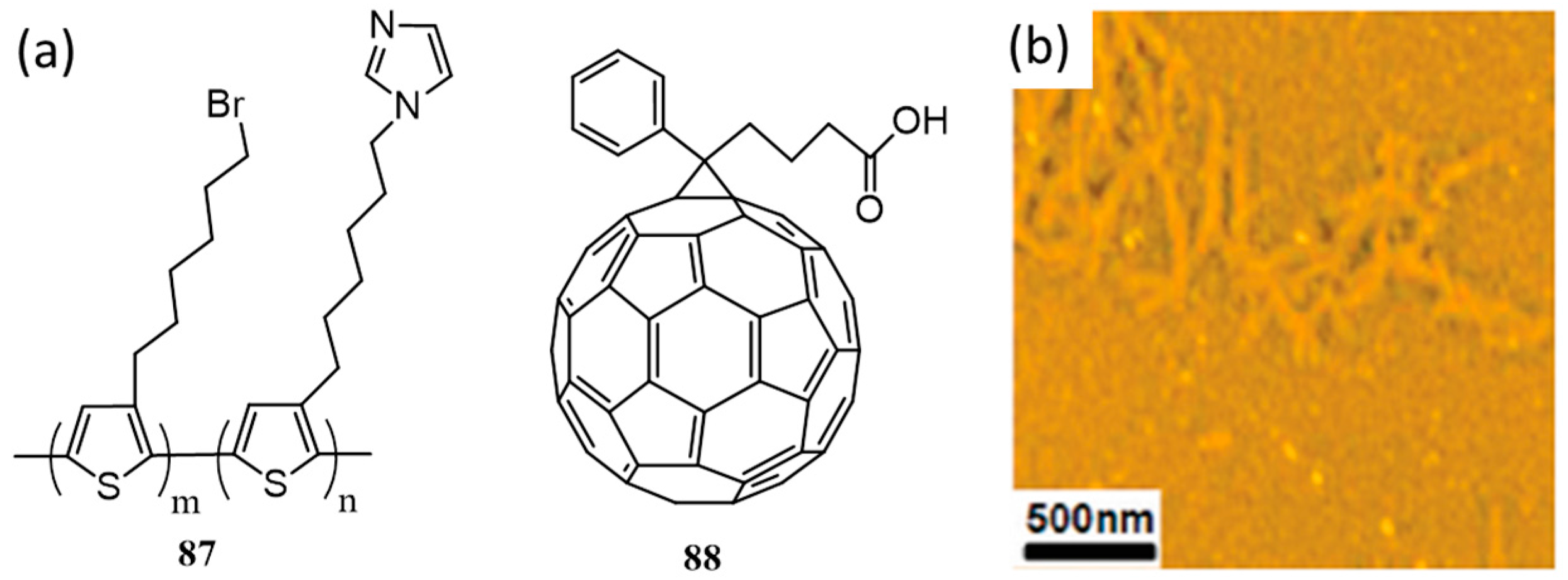{kind=link}
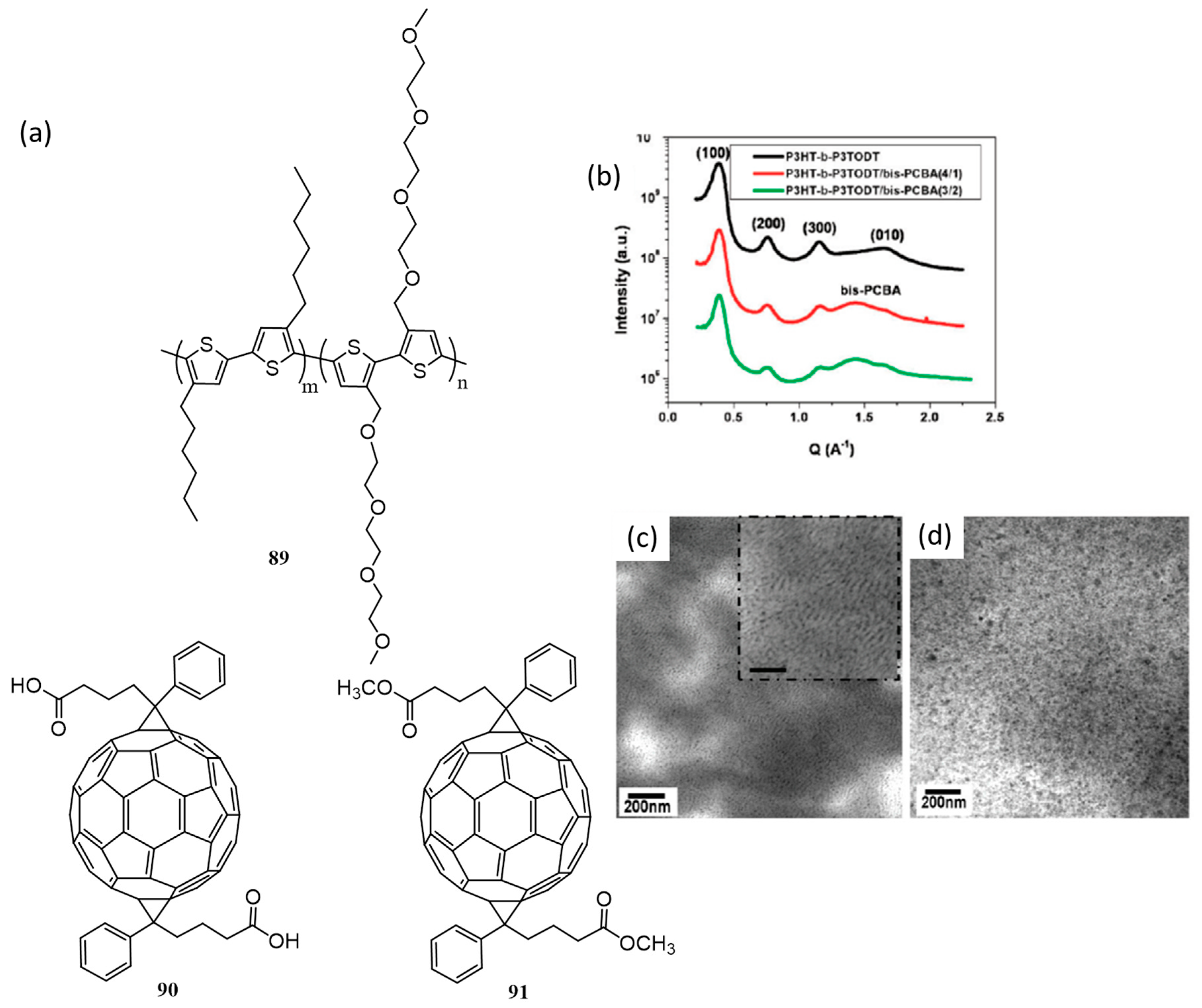{kind=link}
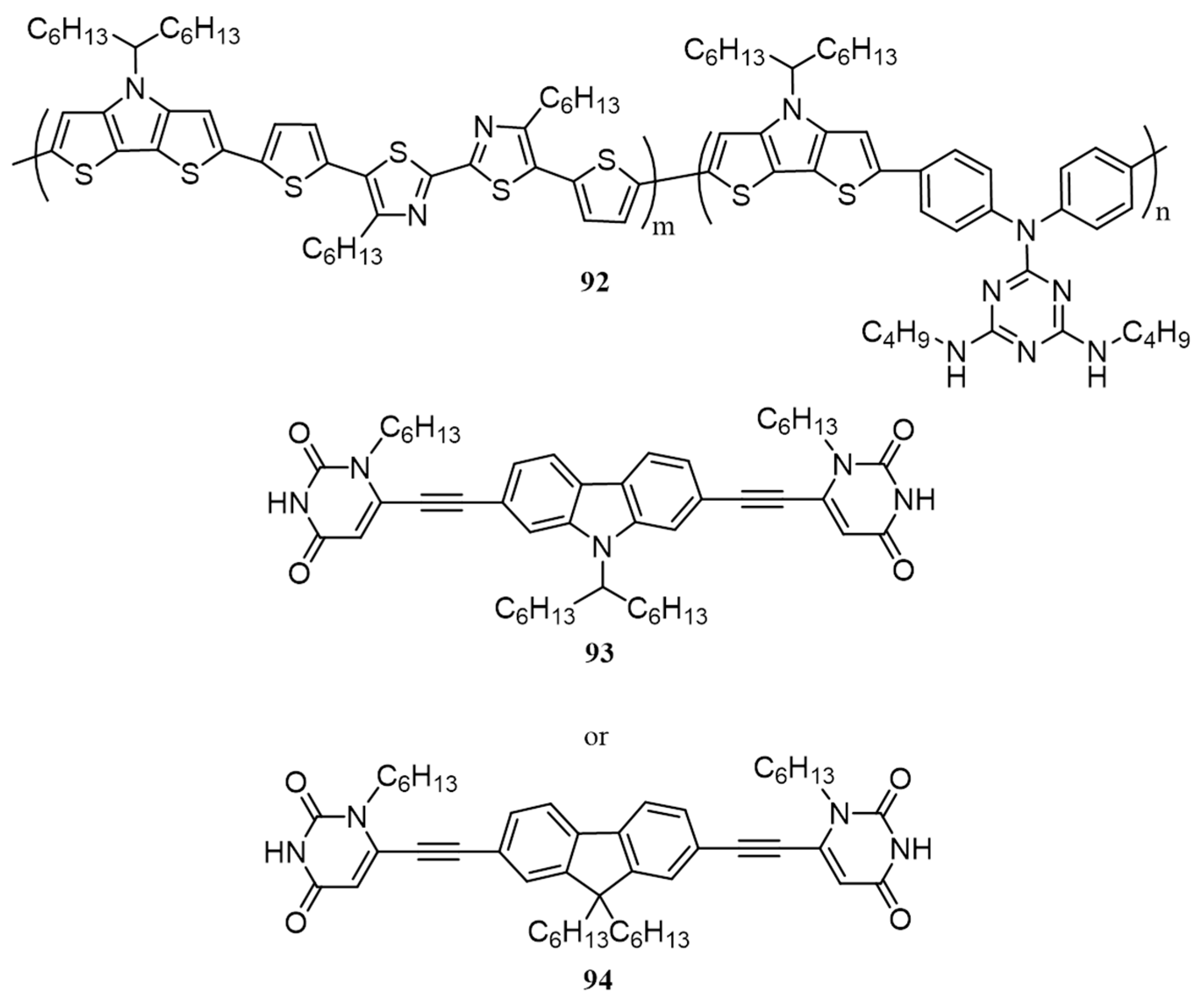{kind=link}
{kind=link}
{kind=link}
{kind=link}
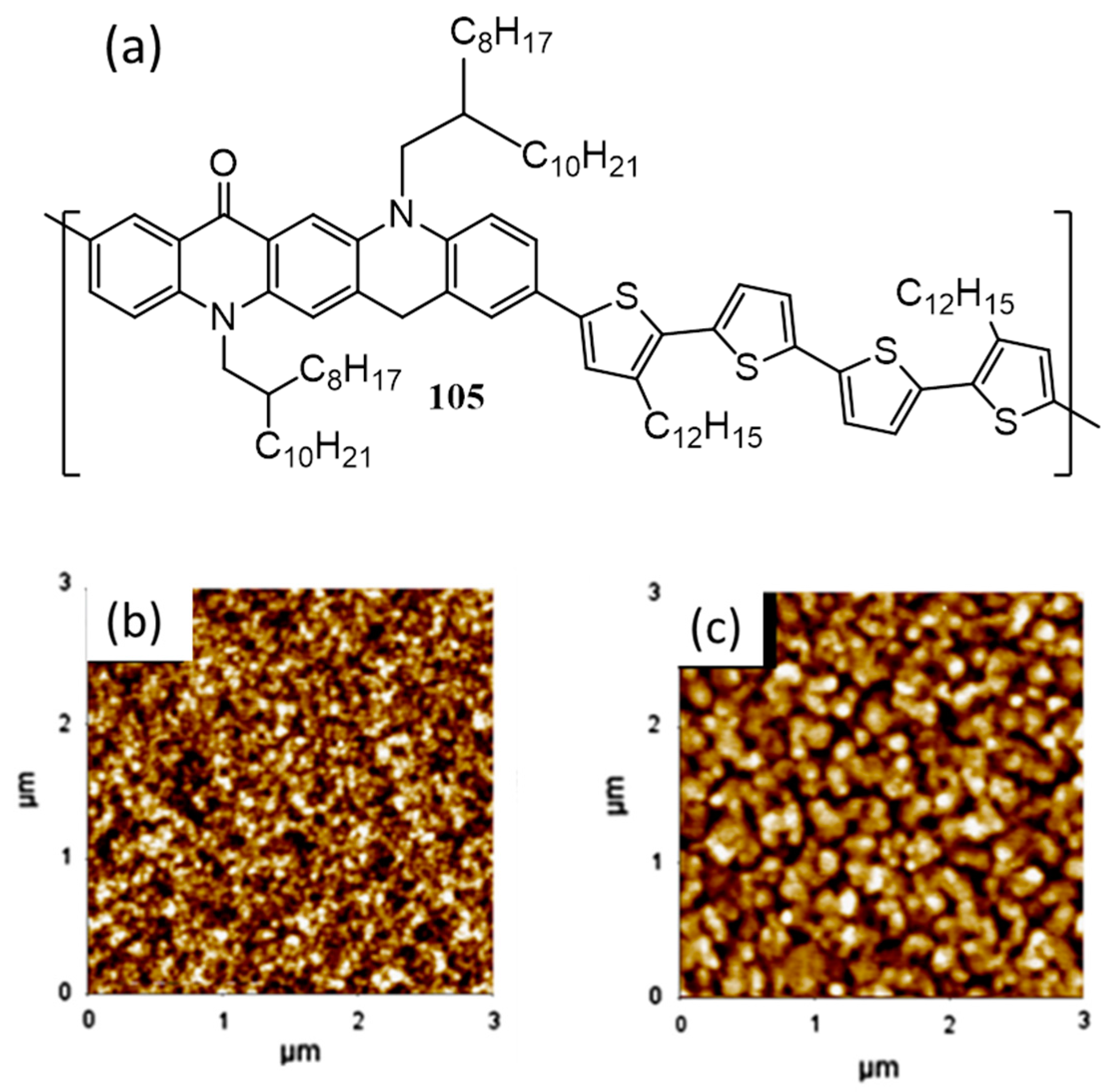{kind=link}
{kind=link}
{kind=link}
| SI No | Device Structure | JSC (mA·cm−2) | VOC (V) | FF | η (%) | Light Intensity (mW·cm−2) | Ref. |
|---|---|---|---|---|---|---|---|
| 1. | ITO/PEDOT:PSS/1:PC61BM(1:2.8)/Al | 0.32 | 0.82 | 0.39 | 0.10 | 88 | [48] |
| 2. | ITO/PEDOT:PSS/6:PC61BM(1:1)/ZnO/Al | 5.10 | 0.96 | 0.49 | 2.40 | 100 | [51] |
| 3. | ITO/MoOx/9:PC71BM(1:1)/LiF/PEDOT:PSS/Au | 12.6 | 0.83 | 0.44 | 4.57 | - | [52] |
| 4. | ITO/PEDOT:PSS/12:PC61BM(1:1)/Ca/Al | 7.00 | 0.79 | 0.54 | 3.01 | 100 | [53] |
| 5. | ITO/PEDOT:PSS/17:PC61BM(1:1)/Ca/Al | 7.74 | 0.83 | 0.46 | 2.98 | 100 | [54] |
| 6. | ITO/PEDOT:PSS/P3HT:PC61BM(1:4)/Al | - | - | - | 1.77 | 100 | [55] |
| 7. | ITO/PEDOT:PSS/22:PC61BM(1:1)/Al | 0.84 | 0.47 | 0.46 | 0.18 | 100 | [56] |
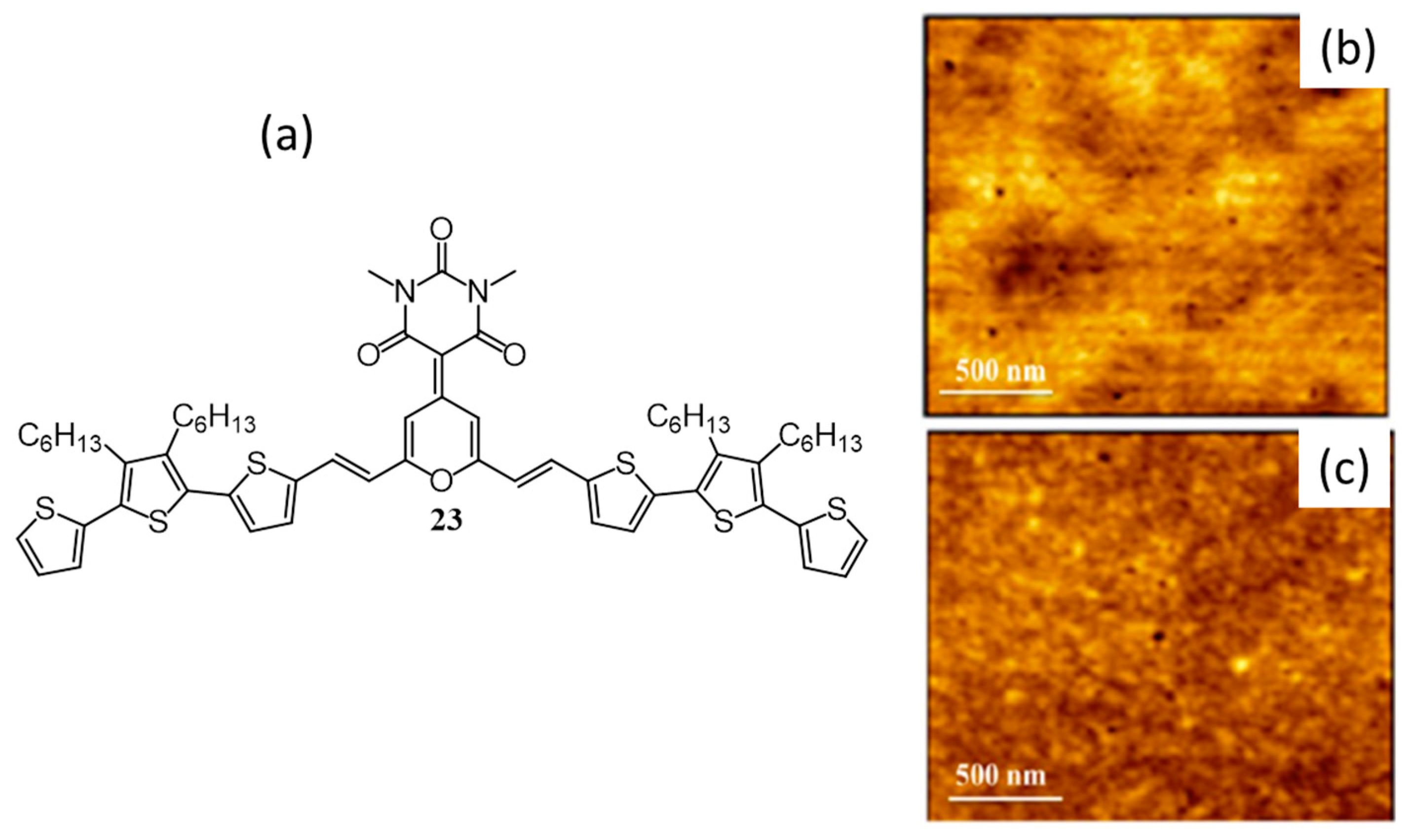
| 8. | ITO/PEDOT:PSS/23:PC61BM(1:1)/Ca/Al | 5.43 | 0.81 | 0.41 | 1.8 | 100 | [57] |
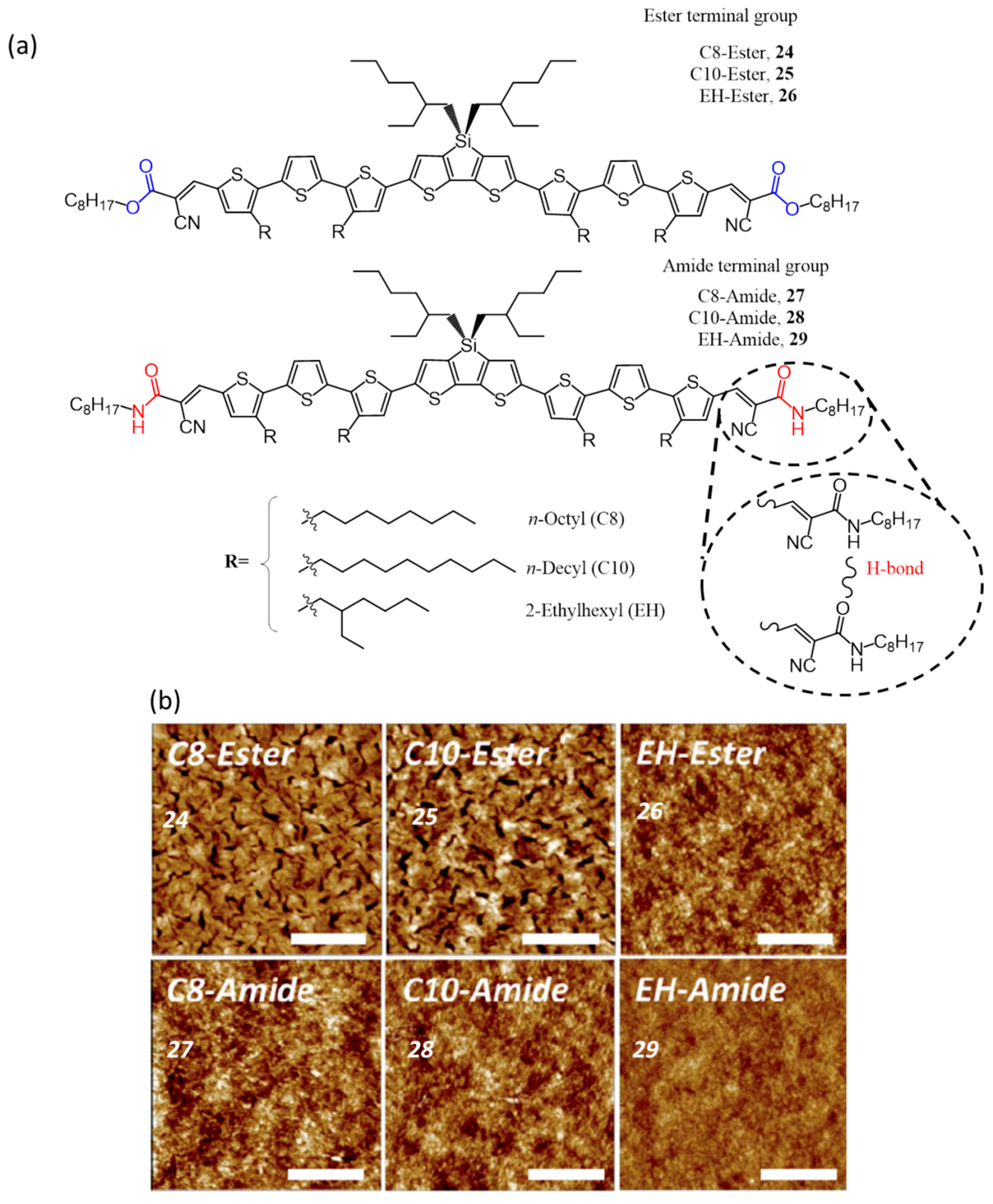
| 9. | ITO/PEDOT:PSS/24:PC61BM(1:0.7)/LiF/Al | 9.79 | 0.82 | 0.54 | 4.31 | 100 | [58] |
| 10. | ITO/PEDOT:PSS/31:PC61BM(1:2)/Al | 1 | 0.6 | 0.38 | 0.26 | 100 | [59] |
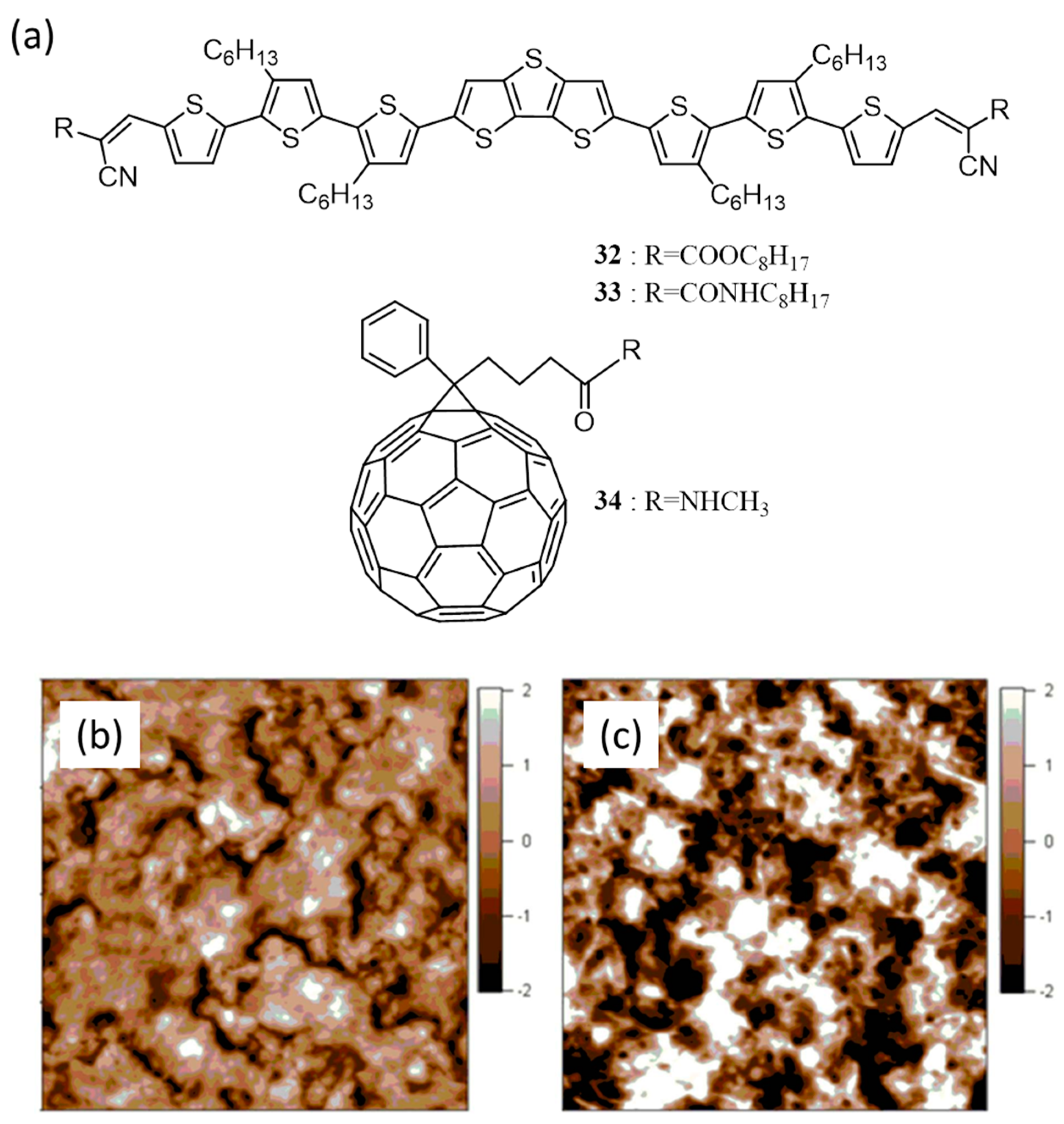
| 11. | ITO/PEDOT:PSS/32:PC61BM(1:1)/ZnO/Al | 3.90 | 0.64 | 0.47 | 1.15 | 100 | [60] |
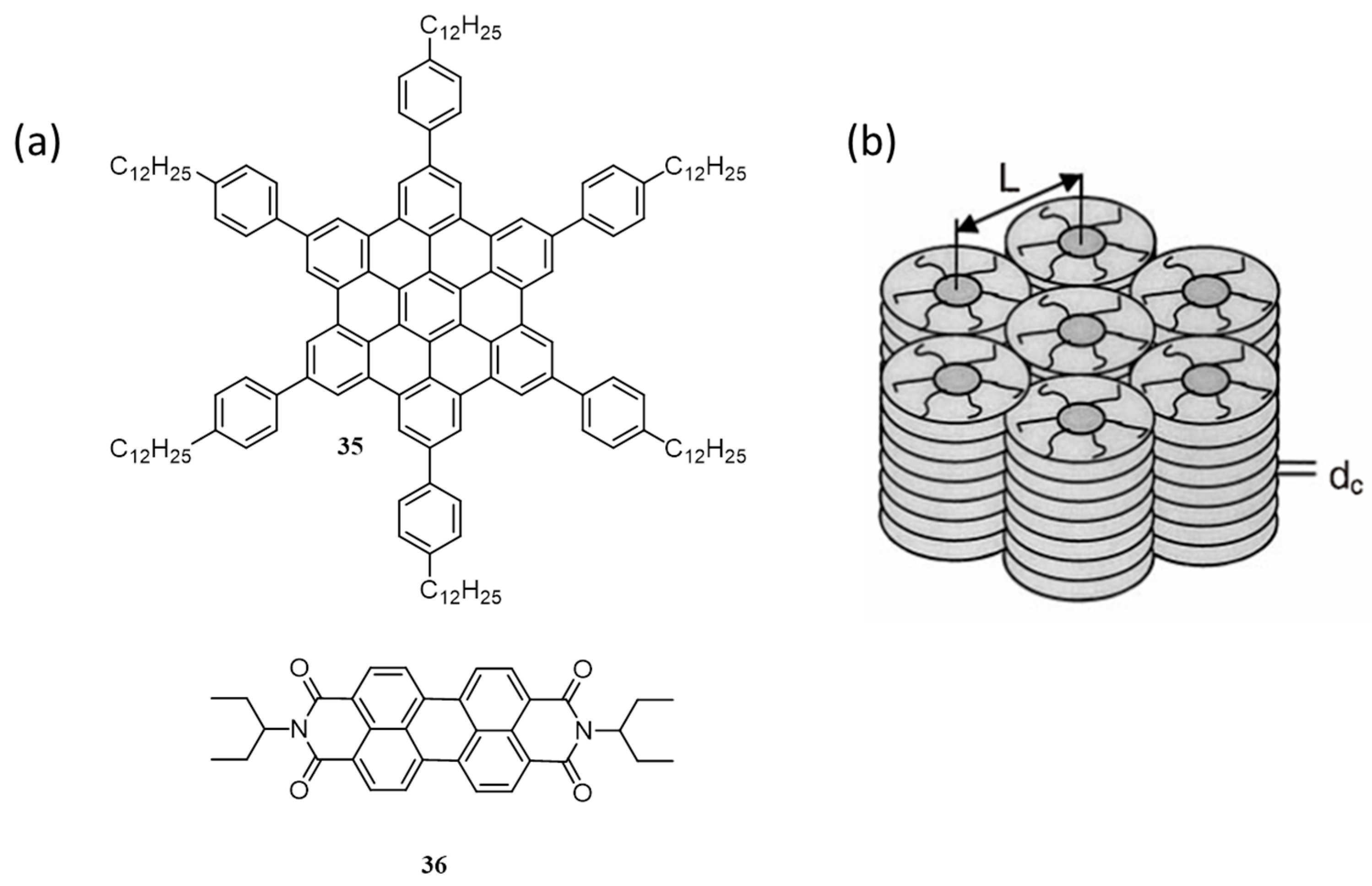
| 12. | ITO/35:36(40:60)/Al | 0.03 | 0.69 | 0.40 | 1.95 | 0.47 | [63] |
| 13. | ITO/PEDOT:PSS/38:PC71BM(1:3)/LiF/Al | 9.32 | 0.57 | 0.37 | 2.05 | 100 | [64] |
| 14. | ITO/PEDOT:PSS/43:PC71BM(2:1)/Al | 8.3 | 0.76 | 0.58 | 4.1 | 100 | [65] |
| 15. | ITO/PEDOT:PSS/46:PC71BM(3:2)/LiF/Al | 7.26 | 0.67 | 0.49 | 2.4 | 100 | [67] |
| 16. | ITO/ZnO/51:52(1:1)/MoO3/Ag | 6.56 | 1.02 | 0.54 | 3.67 | 100 | [74] |
| 17. | ITO/ZnO/53:PC61BM(1:1)/MoOx/Ag | 3.25 | 0.62 | 0.37 | 0.75 | 100 | [75] |
| 18. | ITO/MoOx/56:PC71BM(1:2)/LiF/Al | 10.17 | 0.75 | 0.58 | 4.39 | 100 | [78] |
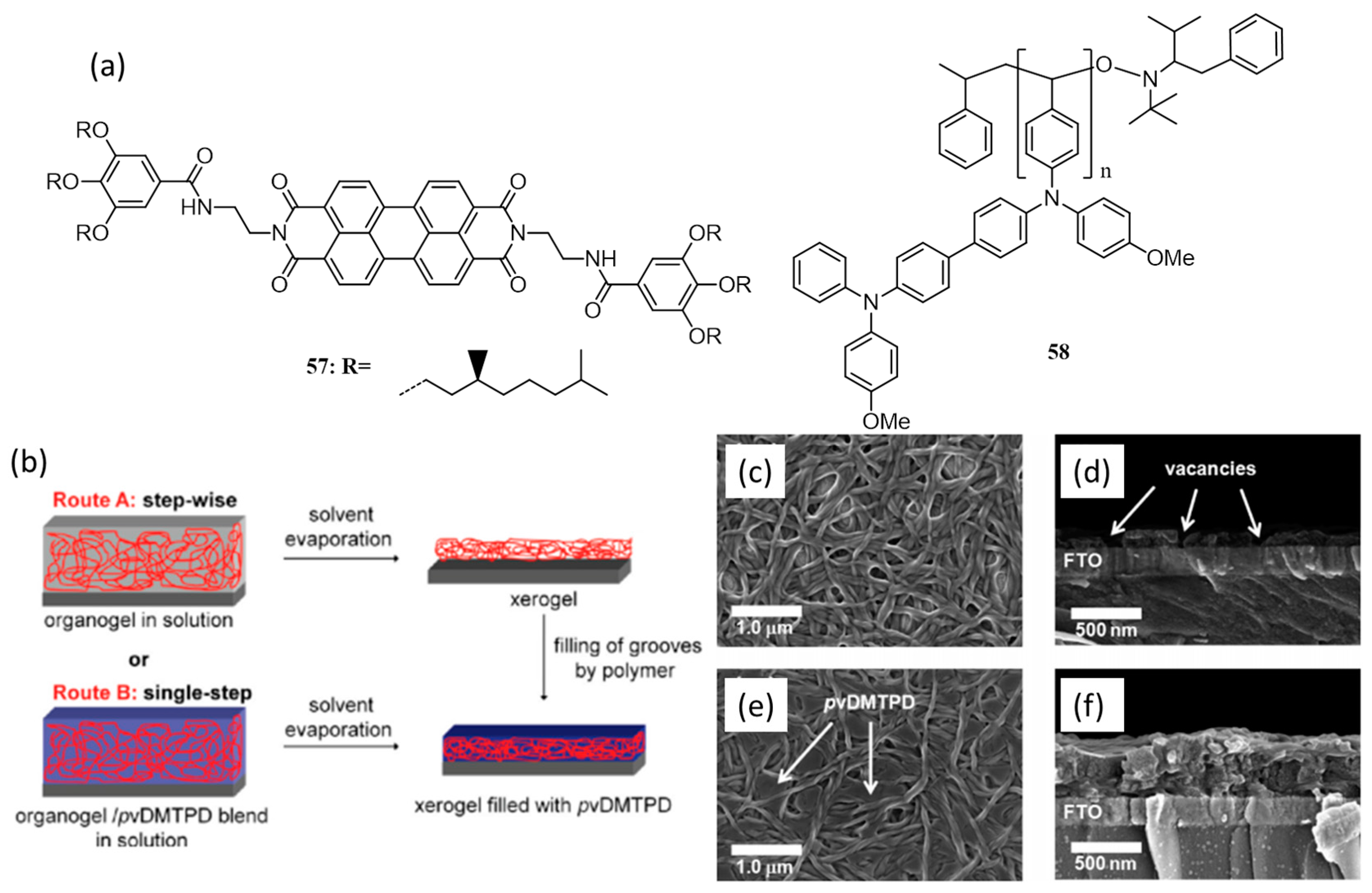
| 19. | FTO/bl-TiO2/57:58(3:1)/52/PEDOT:PSS/Au | 0.28 | 0.39 | 0.38 | 0.04 | 100 | [81] |
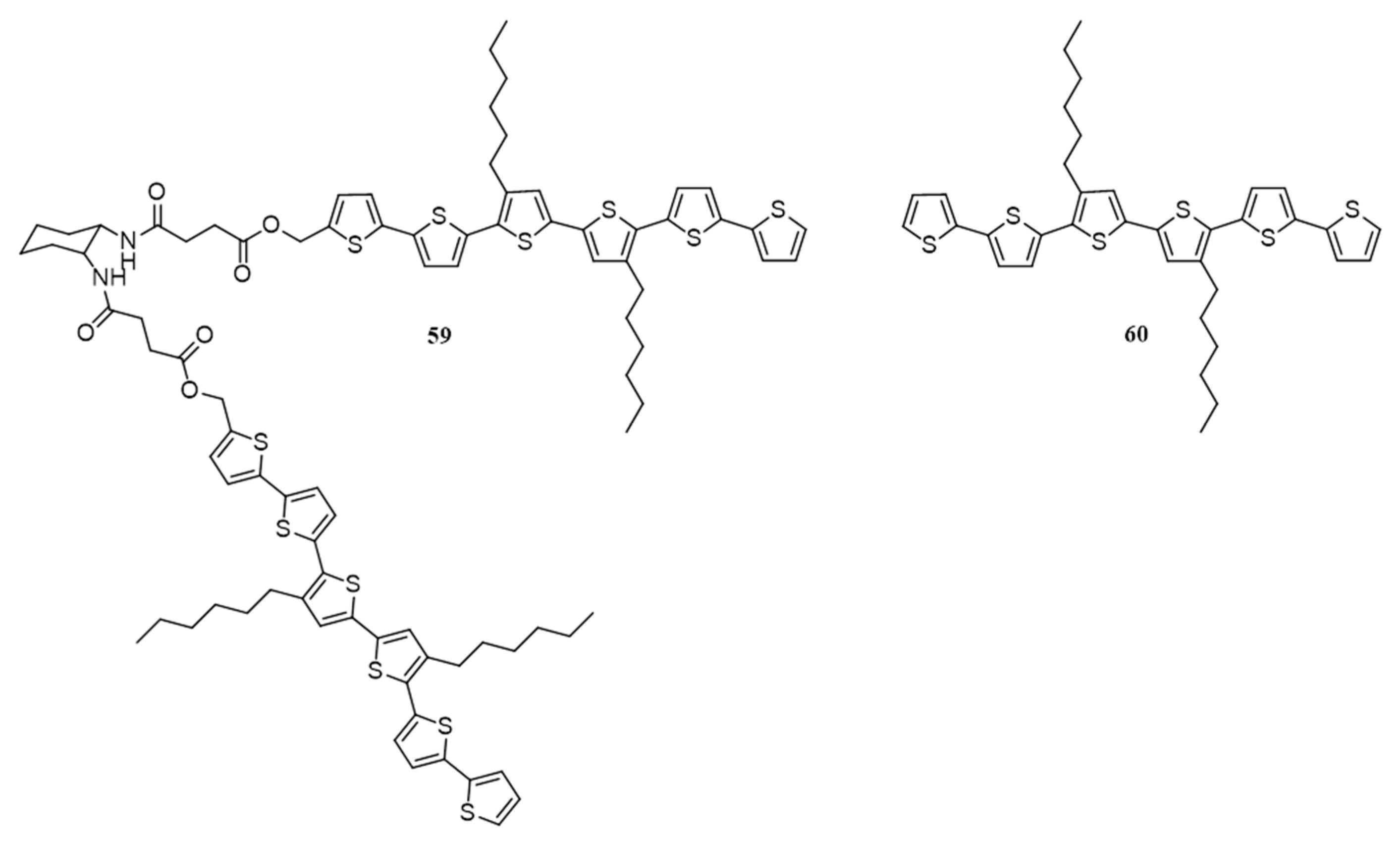
| 20. | ITO/PEDOT:PSS/59:PC71BM(40:60)/LiF/Al | 1.79 | 0.62 | 0.35 | 0.48 | 80 | [83] |
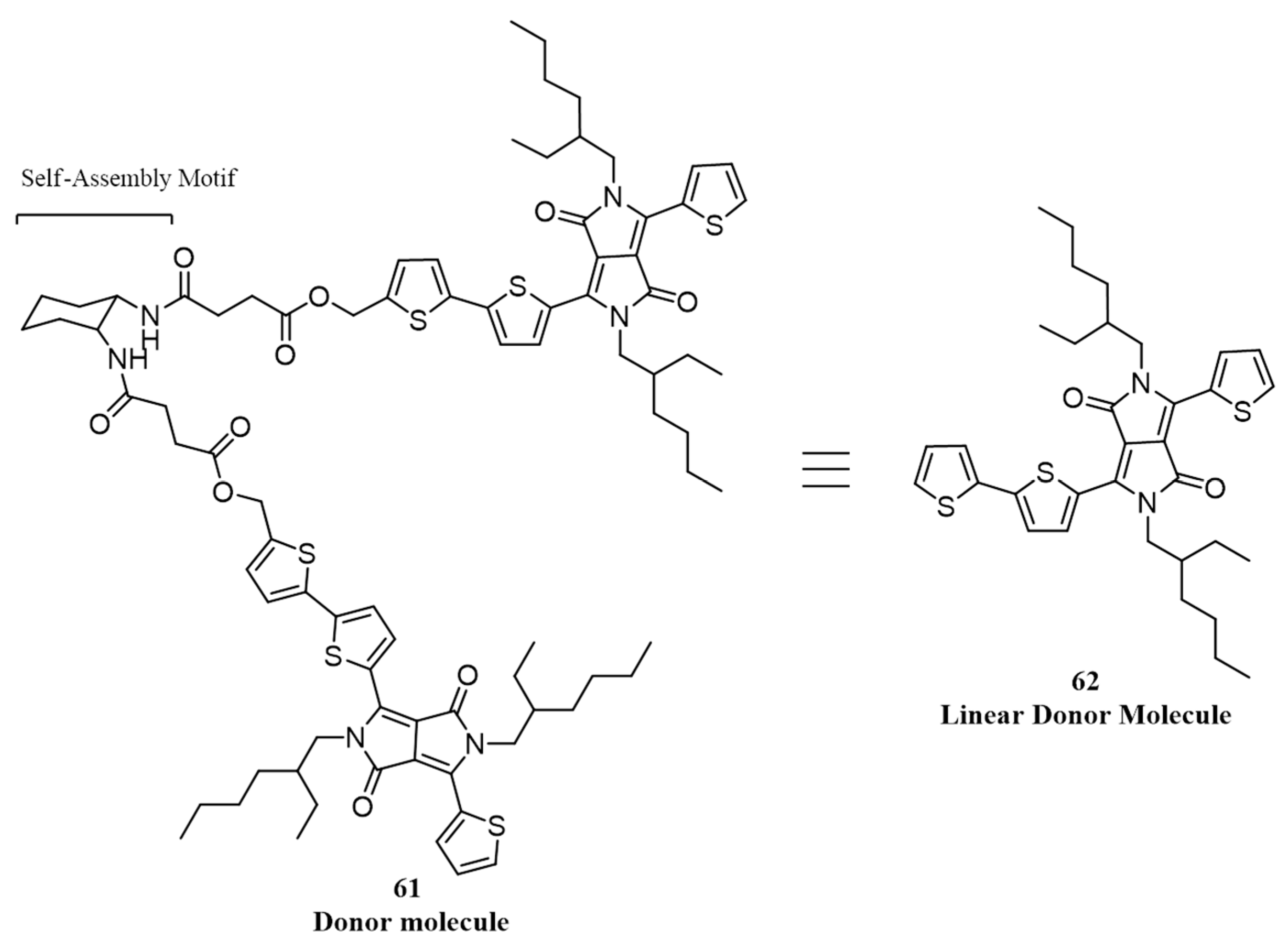
| 21. | ITO/PEDOT:PSS/61:PC71BM(1:1)/LiF/Al | 2.98 | 0.66 | 0.27 | 0.53 | 100 | [84] |
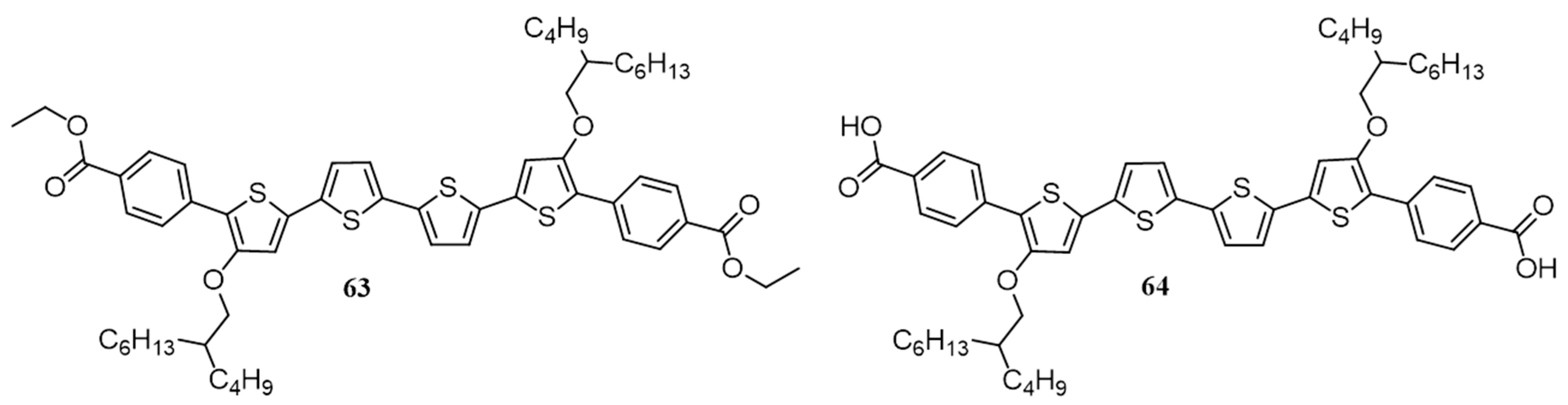
| 22. | ITO/PEDOT:PSS/64:PC61BM(1:1)/Al | 2.86 | 0.71 | 0.45 | 0.91 | 100 | [85] |
| 23. | ITO/MoO3/68:PC71BM(45:55)/Ba/Ag | 10.5 | 0.99 | 0.49 | 5.10 | 23 | [89] |
| 24. | ITO/MoOx/72:PC70BM(70:30)/Al | 0.78 | 14.4 | 0.59 | 6.70 | 100 | [90] |
| 25. | ITO/PEDOT:PSS/76/80:PC61BM(1:1)/Ca/Al | 3.17 | 0.47 | 0.34 | 0.50 | 100 | [95] |
| 26. | ITO/PEDOT:PSS/82/84:PC61BM(1:1)Ca/Al | 1.90 | 0.55 | 0.29 | 0.31 | 100 | [96] |
| 27. | ITO/PEDOT:PSS/87:88(1:0.8)/LiF/Al | 9.11 | 0.67 | 0.51 | 3.16 | 100 | [97] |
| 28. | ITO/PEDOT:PSS/89/90(3:2)/LiF/Al | 6.32 | 0.58 | 0.55 | 2.04 | 100 | [106] |
| 29. | ITO/PEDOT:PSS/92/93:PC71BM(1:2)/Ca/Al | 7.16 | 0.60 | 0.36 | 1.56 | 100 | [107] |
| 30. | ITO/MoO3/95:96(10:8)/Al | 8.59 | 0.59 | 0.52 | 2.65 | 100 | [108] |
| 31. | ITO/PEDOT:PSS/99:101(1:1)/Al | 3.18 | 0.96 | 0.25 | 0.77 | 100 | [115] |
| 32. | ITO/PEDOT:PSS/103:PC61BM(1:1)/LiF/Al | 1.20 | 0.89 | 0.38 | 0.45 | 90 | [116] |
| 33. | ITO/PEDOT:PSS/105:PC71BM(1:2)/Al | 7.90 | 0.59 | 0.49 | 2.3 | 100 | [118] |
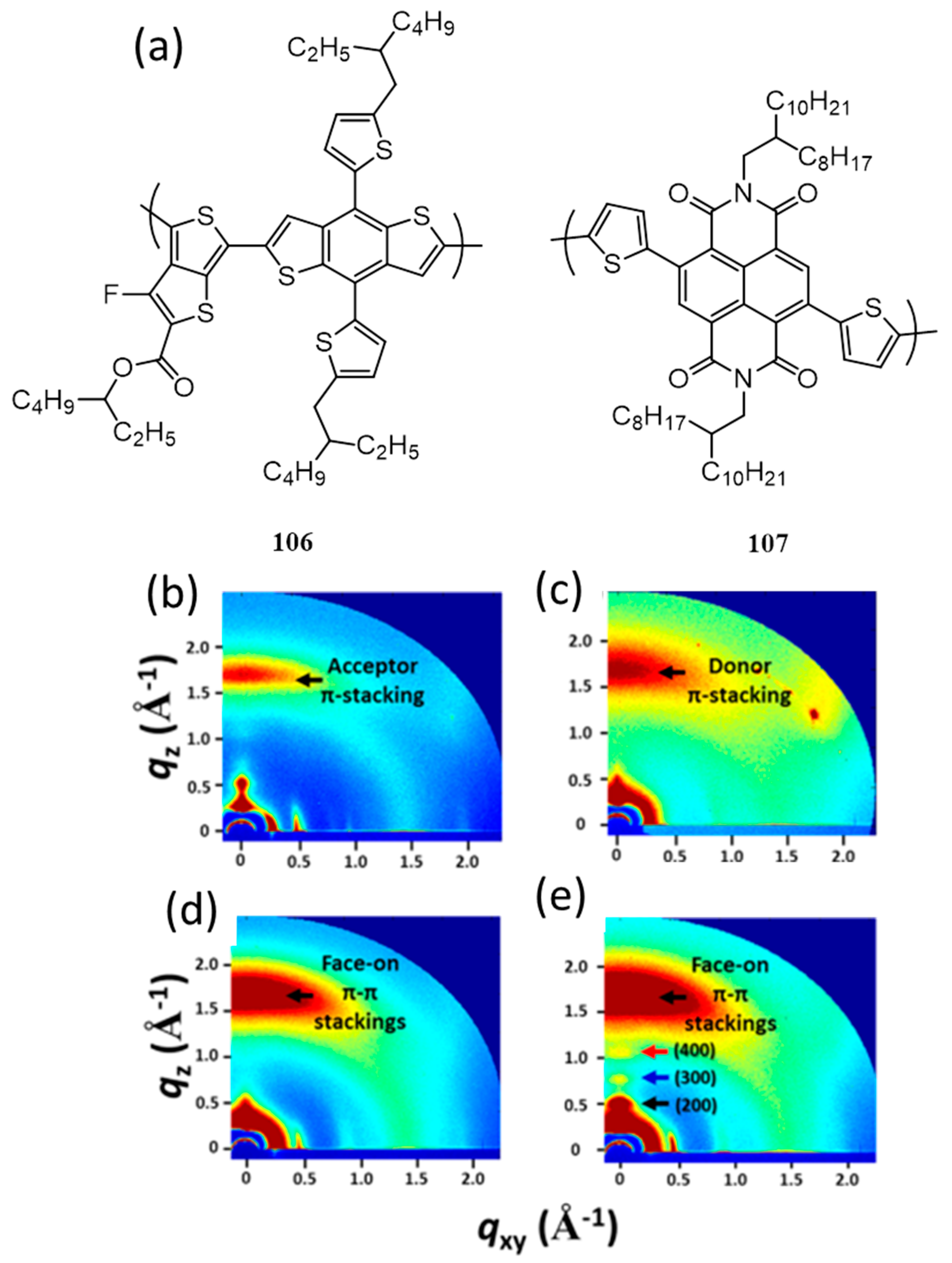
| 34. | ITO/PEDOT:PSS/106:107(1.3:1)/LiF/Al | 10.61 | 0.82 | 0.53 | 4.60 | 100 | [120] |
| 35. | ITO/PEDOT:PSS/18:108(1:0.75)/Sm/Al | 3.77 | 0.56 | 0.65 | 1.4 | 100 | [121] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosh, T.; Panicker, J.S.; Nair, V.C. Self-Assembled Organic Materials for Photovoltaic Application. Polymers 2017, 9, 112. https://doi.org/10.3390/polym9030112
Ghosh T, Panicker JS, Nair VC. Self-Assembled Organic Materials for Photovoltaic Application. Polymers. 2017; 9(3):112. https://doi.org/10.3390/polym9030112
Chicago/Turabian StyleGhosh, Tanwistha, Jayanthy S. Panicker, and Vijayakumar C. Nair. 2017. "Self-Assembled Organic Materials for Photovoltaic Application" Polymers 9, no. 3: 112. https://doi.org/10.3390/polym9030112