
Evaluation of Amphiphilic Peptide Modified Antisense Morpholino Oligonucleotides In Vitro and in Dystrophic mdx Mice

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
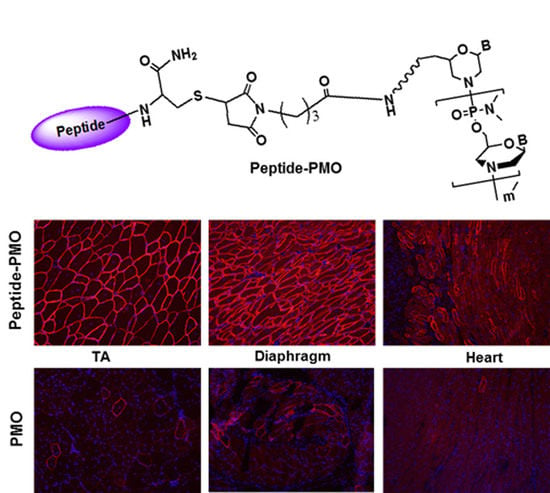
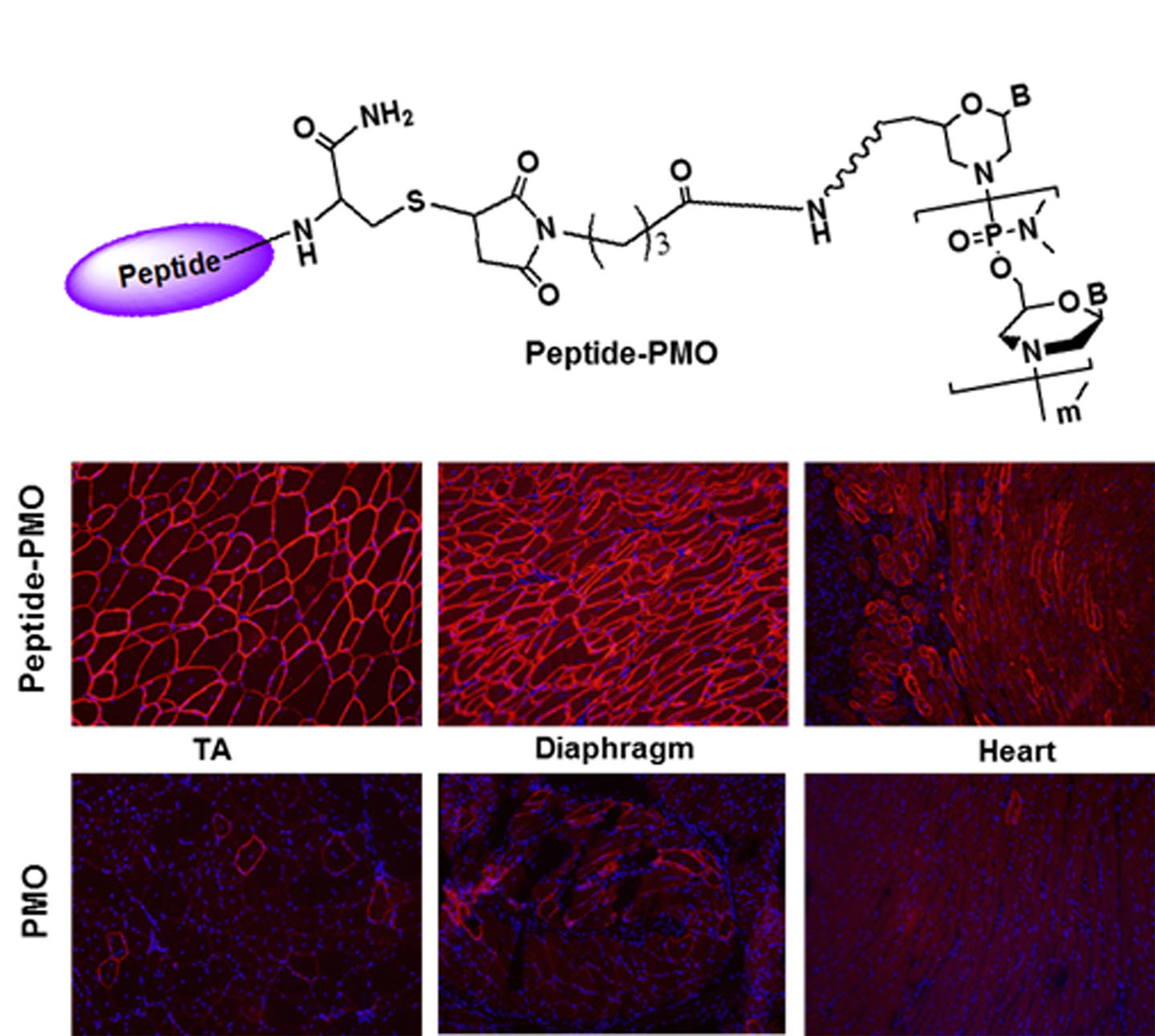
2.1. Synthesis and Characterization of Peptide-PMO Conjugates
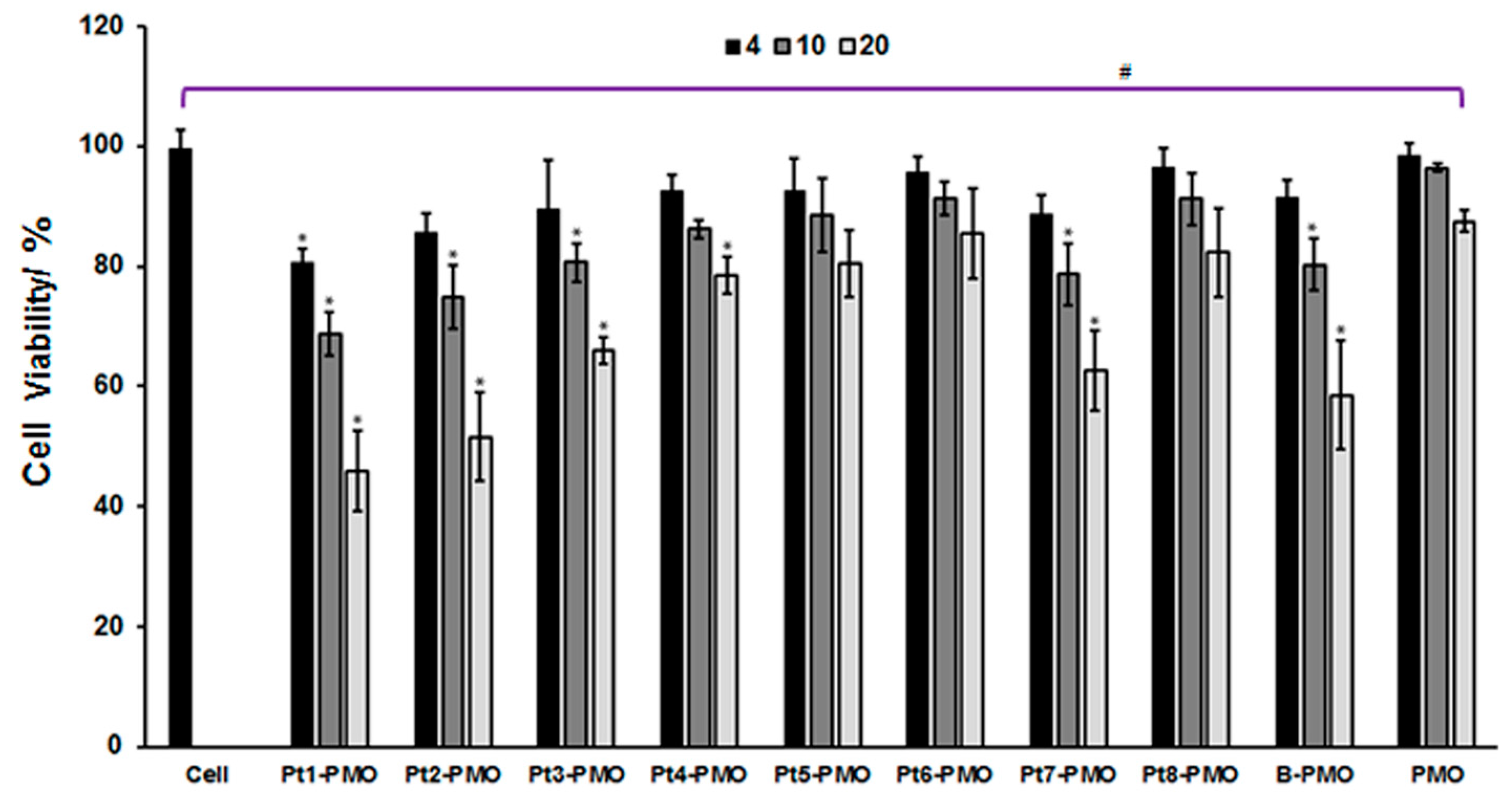
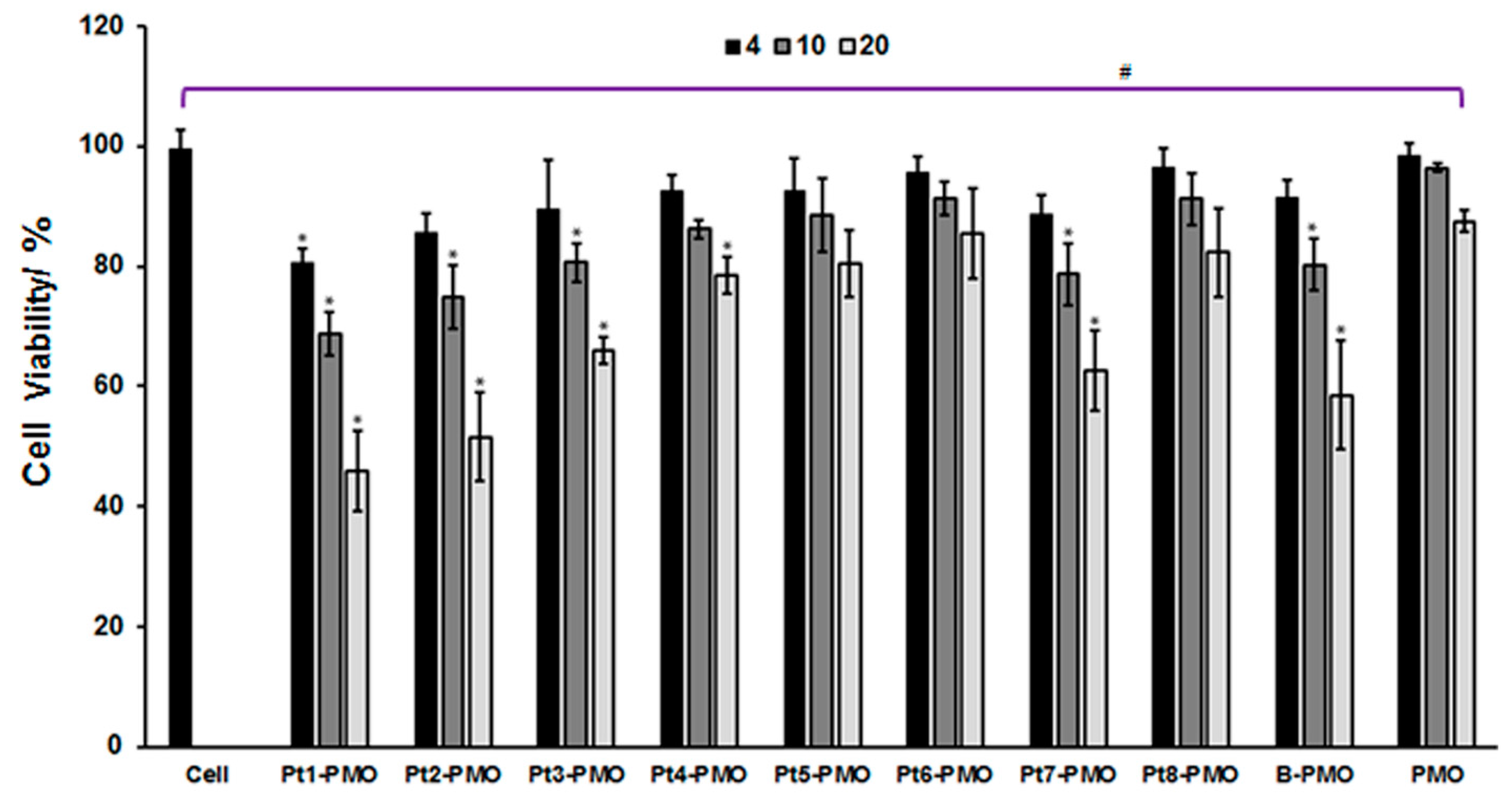
2.2. Cytotoxicity in C2C12E23 Cell Line
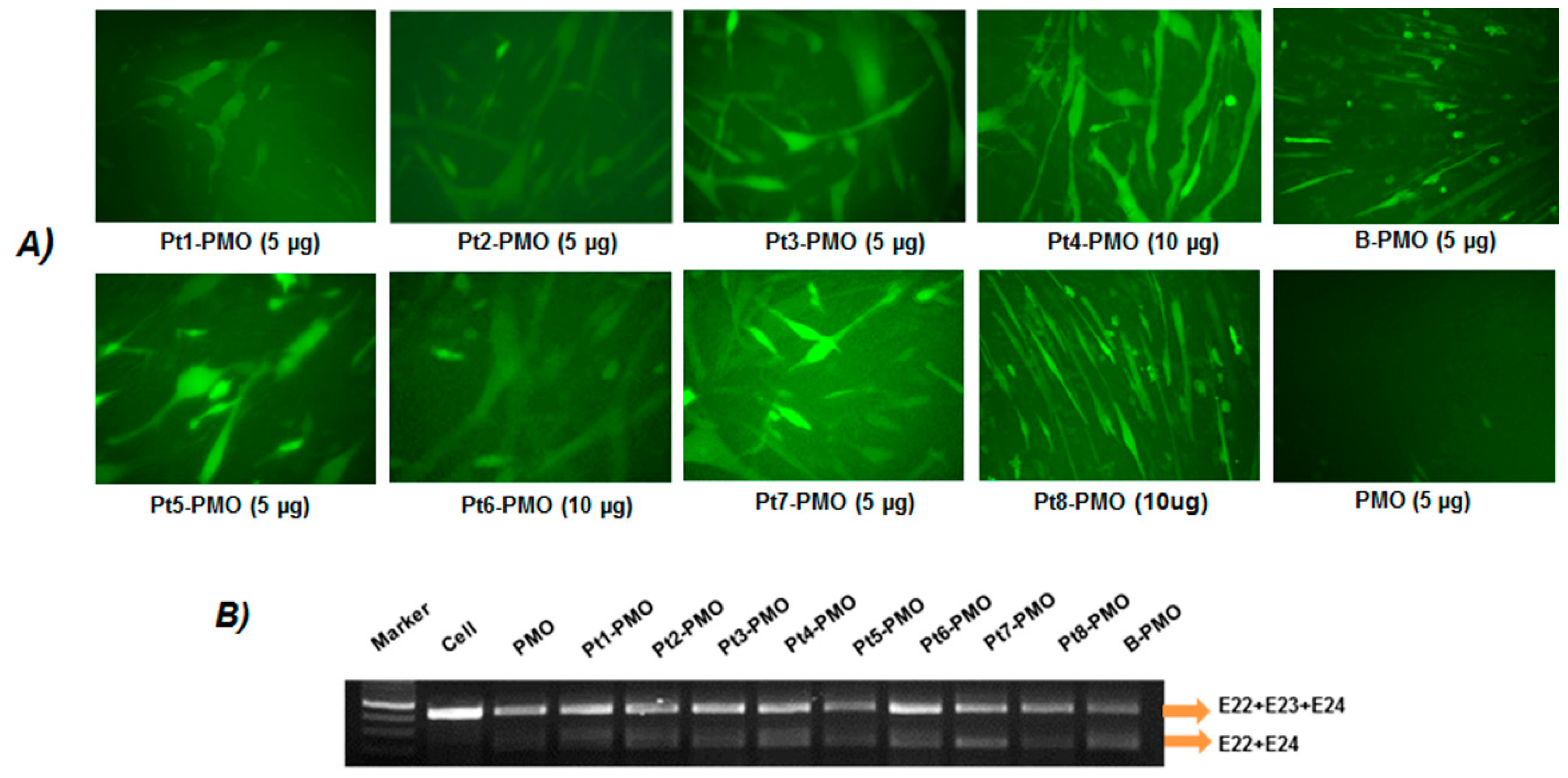
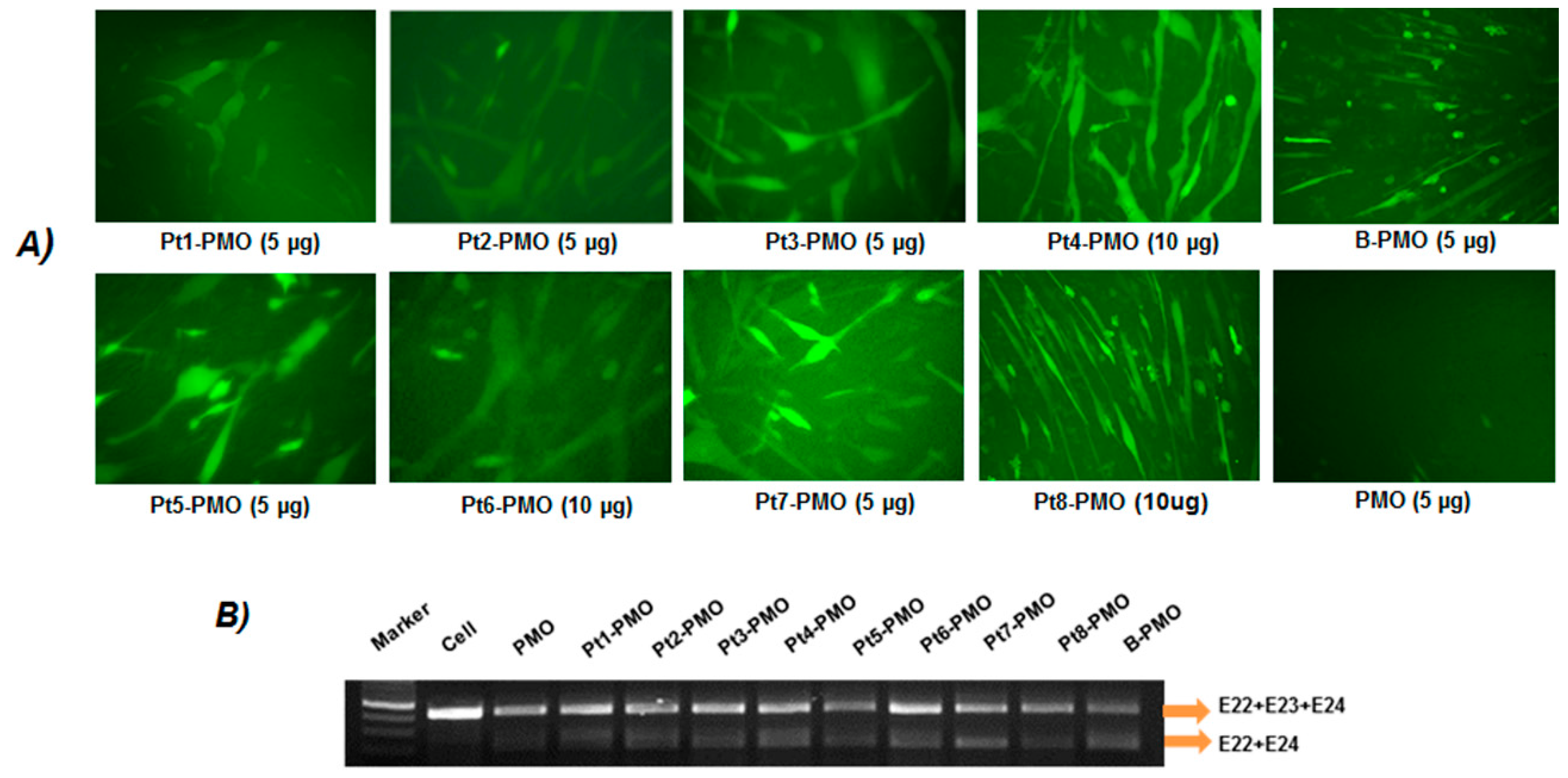
2.3. Exon-Skipping In Vitro
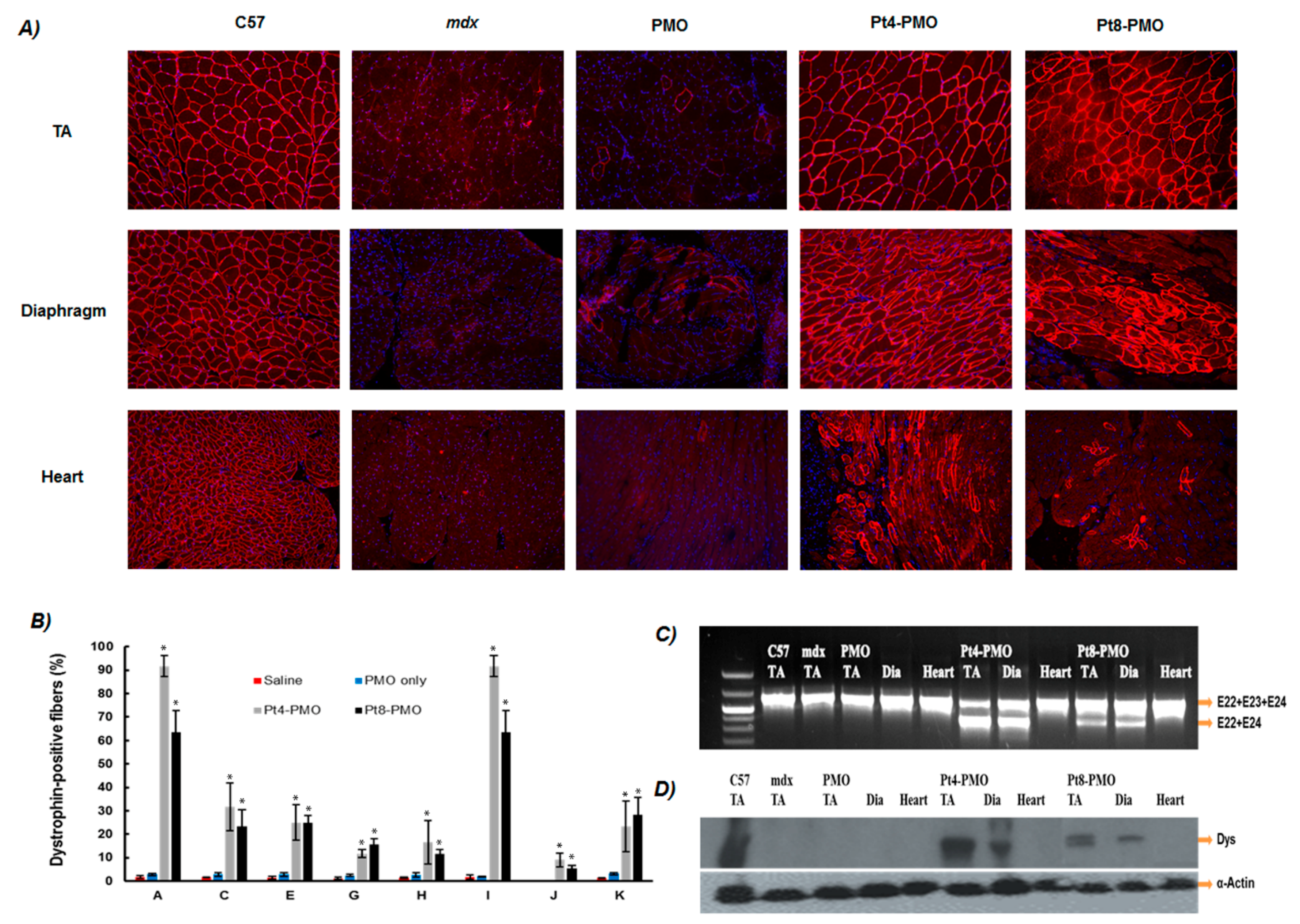
2.4. In Vivo AO Delivery and Immunohistochemistry
2.4.1. Animals and Injections
2.4.2. Immunohistochemistry and Histology
2.4.3. Western Blot and RT-PCR for In Vivo Samples
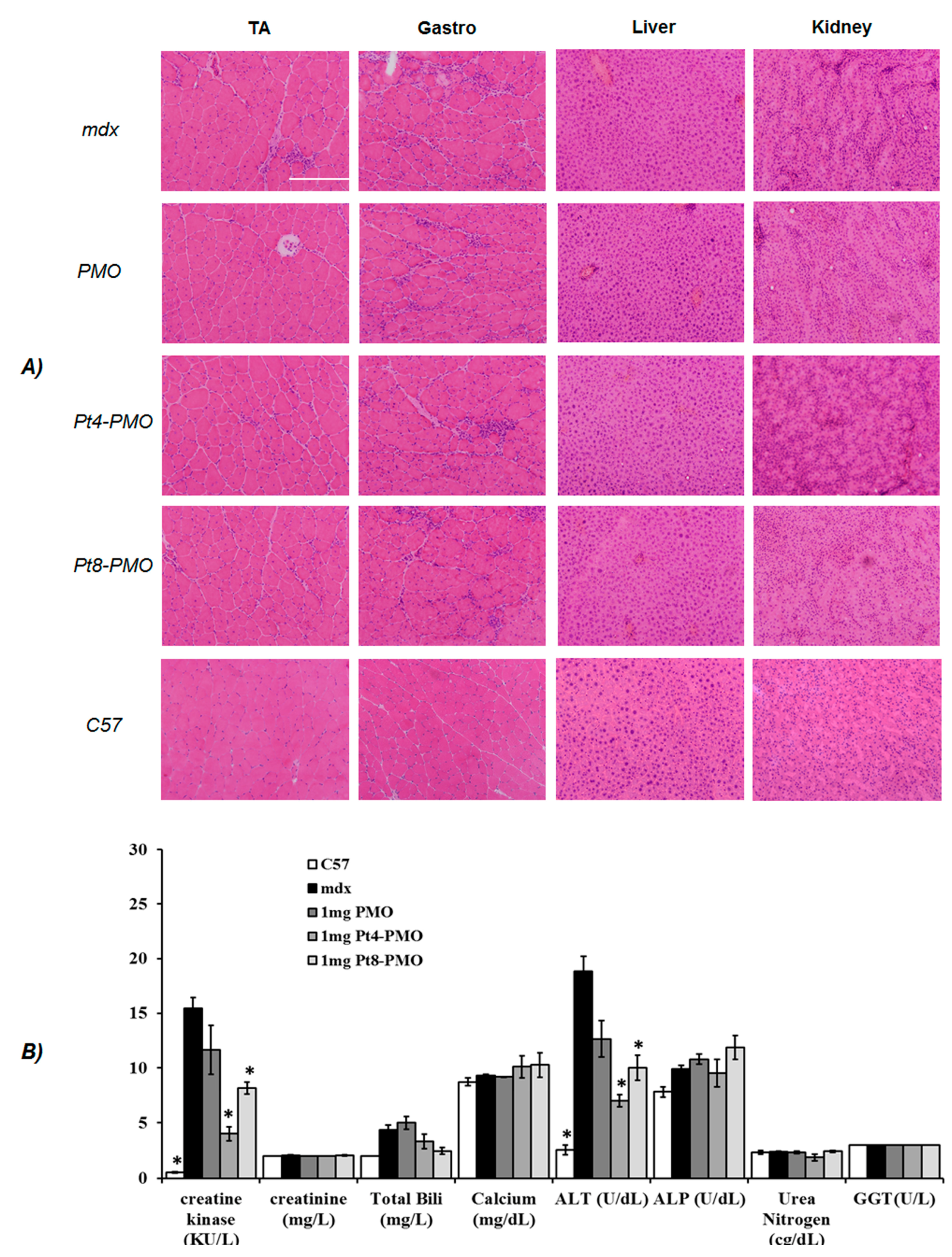
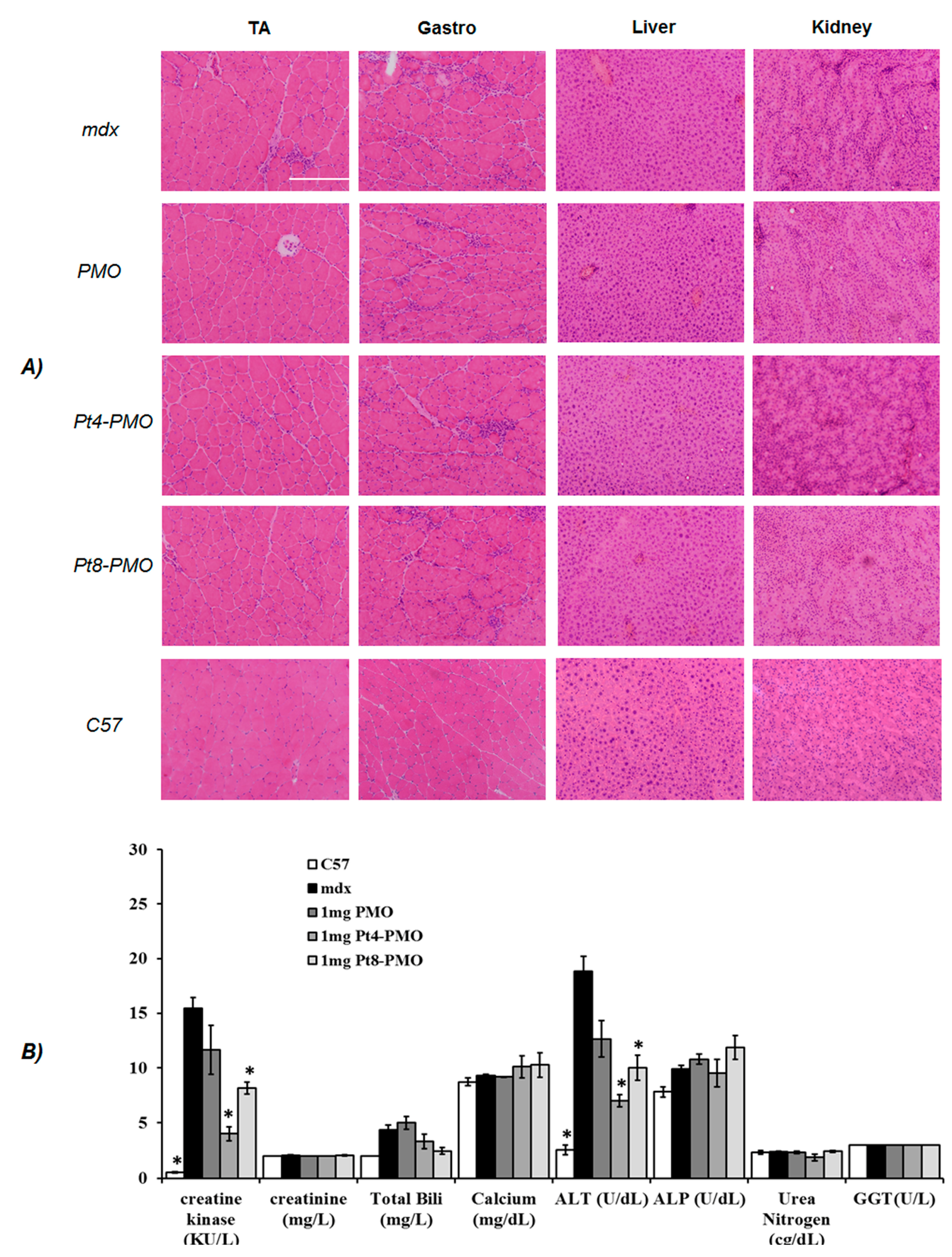
2.5. Measurement of Serum Creatine Kinase and Other Components
2.6. Transmission Electron Microscopy (TEM)
2.7. Statistical Analysis
3. Results and Discussion
3.1. Cytotoxicity
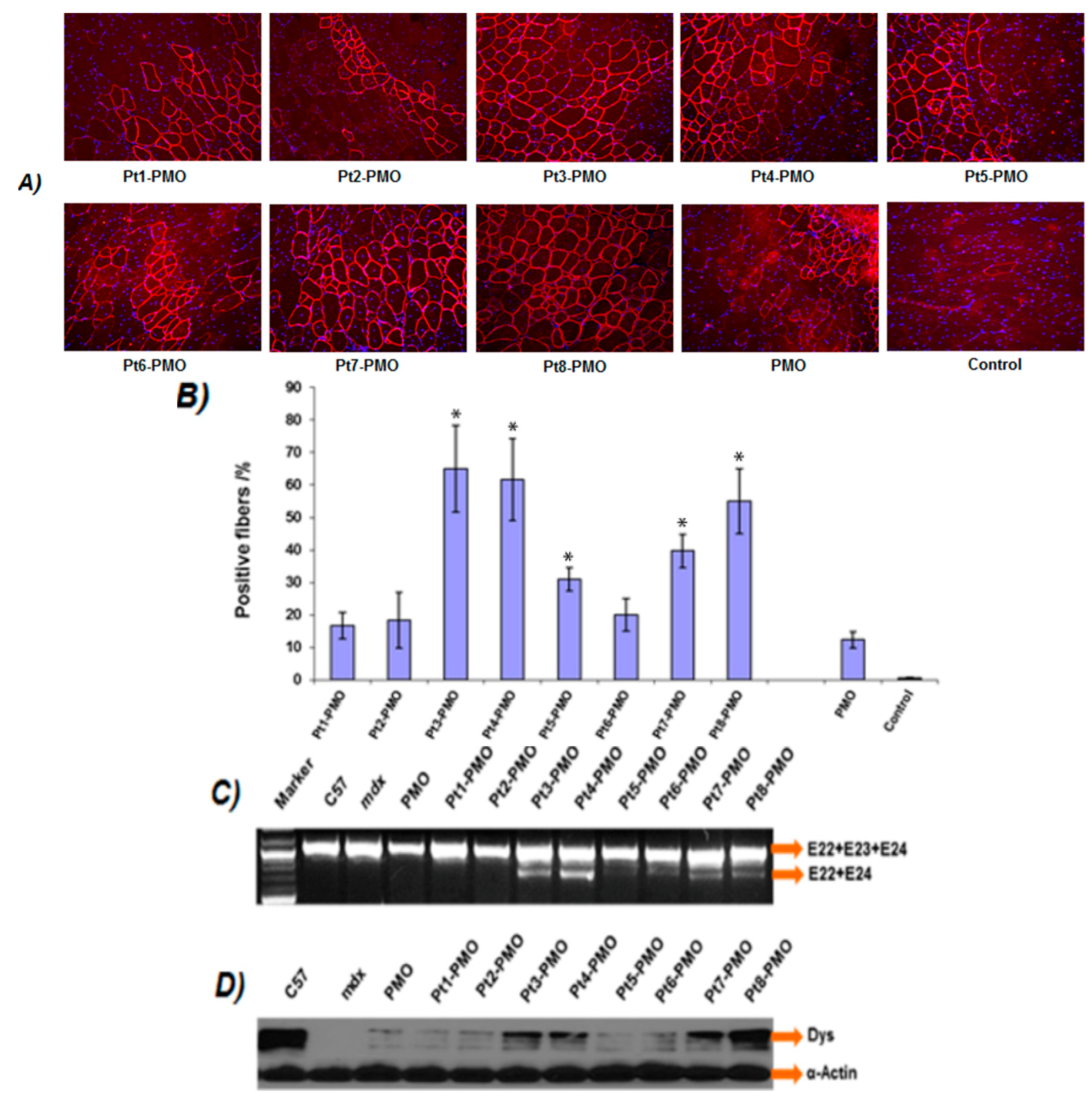
3.2. Delivery In Vitro
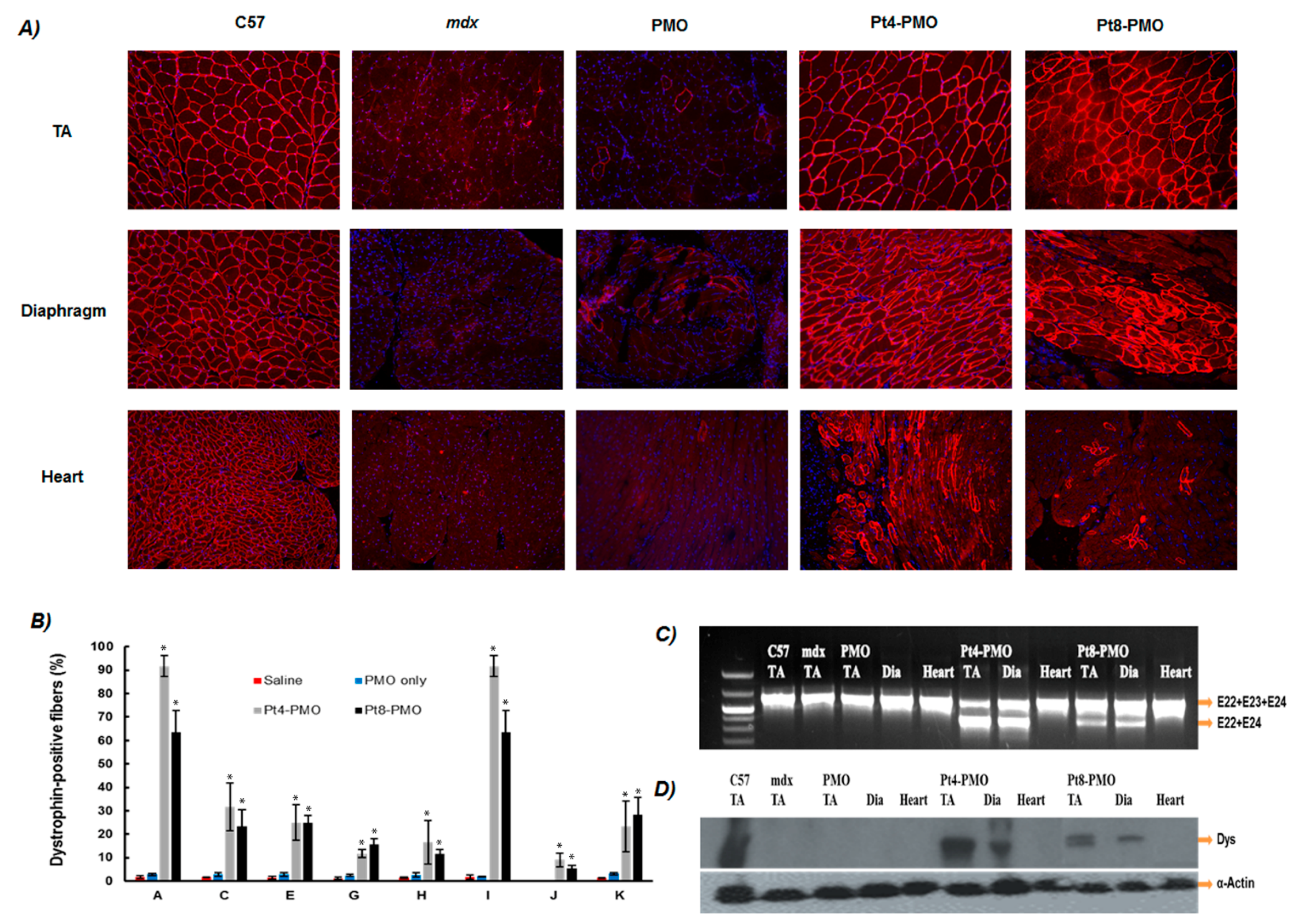
3.3. Delivery In Vivo
3.3.1. Local Delivery
3.3.2. Systemic Delivery
3.4. Morphology Study by Transmission Electron Microscopy (TEM)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Foster, K.; Foster, H.; Dickson, J.G. Gene therapy progress and prospects: Duchenne muscular dystrophy. Gene Ther. 2016, 13, 1677–1685. [Google Scholar] [CrossRef] [PubMed]
- Kole, R.; Krieg, A.M. Exon skipping therapy for Duchenne muscular dystrophy. Adv. Drug Deliv. Rev. 2015, 87, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Koo, T.; Wood, M.J. Clinical trials using antisense oligonucleotides in duchenne muscular dystrophy. Hum. Gene Ther. 2013, 24, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Cirak, S.; Partridge, T. What can we learn from clinical trials of exon skipping for DMD? Mol. Ther. Nucleic Acids 2014, 3, e152. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Lechtzin, N.; Judge, D.P. Current treatment of adult Duchenne muscular dystrophy. Biochim. Biophys. Acta 2007, 1772, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J.; et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Sipkens, J.T.; Sitsen, J.M.; et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Dickson, G.; et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009, 8, 918–928. [Google Scholar] [CrossRef]
- Malerba, A.; Sharp, P.S.; Graham, R.; Arechavala-Gomeza, V.; Foster, K.; Muntoni, F.; Wells, D.J.; Dickson, G. Chronic systemic therapy with low-dose morpholino oligomers ameliorates the pathology and normalizes locomotor behavior in mdx mice. Mol. Ther. 2011, 19, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Kota, J.; Malik, V.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Gromiha, M.M.; Sarai, A. Analysis and prediction of DNA-binding proteins and their binding residues based on composition, sequence and structural information. Bioinformatics 2004, 20, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Jirka, S.M.; Heemskerk, H.; Tanganyika-de Winter, C.L.; Muilwijk, D.; Pang, K.H.; de Visser, P.C.; Janson, A.; Vermue, R.; Karnaoukh, T.G.; Aguilera, B.; et al. Peptide conjugation of 2′-O-methyl phosphorothioate antisense oligonucleotides enhances cardiac uptake and exon skipping in mdx mice. Nucleic Acid Ther. 2014, 24, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Moulton, H.M.; Nelson, M.H.; Hatlevig, S.A.; Reddy, M.T.; Iversen, P.L. Cellular Uptake of Antisense Morpholino Oligomers Conjugated to Arginine-Rich Peptides. Bioconjug. Chem. 2004, 15, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Moulton, H.M.; Wu, B.; Jearawiriyapaisarn, N.; Sazani, P.; Lu, Q.L; Kole, R. Peptide-morpholino conjugate: A promising therapeutic for Duchenne muscular dystrophy. Ann. N. Y. Acad. Sci. 2009, 1175, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Moulton, H.M.; Moulton, J.D. Morpholinos and their peptide conjugates: Therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim. Biophys. Acta 2010, 1798, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.H.; Stein, D.A.; Kroeker, A.D.; Hatlevig, S.A.; Iversen, P.L.; Moulton, H.M. Arginine-rich peptide conjugation to morpholino oligomers: Effects on antisense activity and specificity. Bioconjug. Chem. 2005, 16, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Prata, C.A.H.; Zhang, X.X.; Luo, D.; McIntosh, T.J.; Barthelemy, P.; Grinstaff, M.W. Lipophilic peptides for gene delivery. Bioconjug. Chem. 2008, 19, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, D.S.; Hatlevig, S.A.; Hassinger, J.N.; Iversen, P.L.; Moulton, H.M. Stability of cell-penetrating peptide-morpholino oligomer conjugates in human serum and in cells. Bioconjug. Chem. 2007, 18, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Moulton, H.M.; Iversen, P.L.; Jiang, J.; Li, J.; Spurney, C.F.; Sail, A.; Guerron, A.D.; Lu, P.; Xiao, X.; et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc. Natl. Acad. Sci. USA 2008, 105, 14814–14819. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Li, Y.; Morcos, P.A.; Doran, T.J.; Lu, P.; Lu, Q.L. Octa-guanidine morpholino restores dystrophin expression in cardiac and skeletal muscles and ameliorates pathology in dystrophic mdx mice. Mol. Ther. 2009, 17, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Lu, P.; Benrashid, E.; Malik, S.; Ashar, J.; Doran, T.J.; Lu, Q.L. Dose-dependent restoration of dystrophin expression in cardiac muscle of dystrophic mice by systemically delivered morpholino. Gene Ther. 2009, 17, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Cloer, C.; Shaban, M.; Moulton, H.; Lu, P.; Lu, Q.L. Long-term rescue of dystrophin expression and improvement in muscle pathology and function in dystrophic mdx mice by peptide-conjugated morpholino. Am. J. Pathol. 2012, 181, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Moulton, H.M.; Betts, C.; Seow, Y.; Boutilie, J.; Iverson, P.L.; Wood, M.J. A fusion peptide directs enhanced systemic dystrophin exon skipping and functional restoration in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2009, 18, 4405–4414. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Moulton, H.M.; Betts, C.; Merritt, T.; Seow, Y.; Ashraf, S. Functional Rescue of Dystrophin-deficient mdx Mice by a Chimeric Peptide-PMO. Mol. Ther. 2010, 18, 1822–1829. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Saleh, A.F.; Betts, C.; Camelliti, P.; Seow, Y.; Ashraf, S.; Merritt, T.; Gait, M.J.; Wood, M.J.A.; Arzumanov, A. Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol. Ther. 2011, 19, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Samoylova, T.I.; Smith, B.F. Elucidation of muscle-binding peptides by phage display screening. Muscle Nerve 1999, 22, 460–466. [Google Scholar] [CrossRef]
- Seow, Y.; Yin, H.; Wood, M.J. Identification of a novel muscle targeting peptide in mdx mice. Peptides 2010, 31, 1873–1877. [Google Scholar] [CrossRef] [PubMed]
- Moulton, J.D.; Jiang, S. Gene knockdowns in adult animals: PPMOs and Vivo-morpholinos. Molecules 2009, 14, 1304–1323. [Google Scholar] [CrossRef] [PubMed]
- Hassane, F.S.; Saleh, A.F.; Abes, R.; Gait, M.J.; Lebleu, B. Cell penetrating peptides overview and applications to the delivery of oligonucleotides. Cell. Mol. Life Sci. 2010, 67, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, D.P.; Dangott, L.J.; Lightfoot, J.T. Lessons learned from vivo-morpholinos: How to avoid vivomorpholino toxicity. BioTechniques 2014, 56, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.X.; Wu, B.; Lu, P.; Cloer, C.; Tucker, J.D.; Lu, Q.L. Polyethylenimine-modified pluronics (PCMs) improve morpholino oligomer delivery in cell culture and dystrophic mdx mice. Mol. Ther. 2013, 21, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.X.; Wu, B.; Tucker, J.D.; Lu, P.; Cloer, C.; Lu, Q.L. Evaluation of tris[2-(acryloyloxy)ethyl] isocyanurate cross-linked polyethylenimine as antisense morpholino oligomer delivery vehicle in cell culture and dystrophic mdx mice. Hum. Gene Ther. 2014, 25, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.X.; Wu, B.; Tucker, J.D.; Lu, P.; Bollinger, L.E.; Lu, Q.L. Tween 85 grafted PEIs enhanced delivery performance of antisense 2′-O-methyl phosphorothioate oligonucleotides in vitro and in dystrophic mdx mice. J. Mater. Chem. B 2015, 3, 5330–5340. [Google Scholar] [CrossRef]
- Berdanier, C.D. Fatty acids and membrane function. In Fatty Acids in Foods and their Health Implications, 3rd ed.; Chow, C.K., Ed.; CRC Press: Boca Raton, FL, USA, 2007; pp. 693–712. [Google Scholar]
- Farazi, T.A. The biology and enzymology of protein N-myristoylation. J. Biol. Chem. 2001, 276, 39501–39504. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, K.; Aoki, Y.; Koo, T.; McClorey, G.; Benner, L.; Coenen-Stass, A.; Lehto, T.; Noedin, J.; Saleh, A.F.; Morris, J.; et al. Self-assembly into nanoparticles is essential for receptor mediated uptake of therapeutic antisense oligonucleotides. Nano Lett. 2015, 15, 4364–4373. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code of peptide-PMO | Peptides | Peptide sequence | Numbers of arginine | Numbers of myristoyl |
|---|---|---|---|---|
| Pt1-PMO | Pt1 | RXRRBRK(Myr)BRK(Myr)RXRRB | 8 | 2 |
| Pt2-PMO | Pt2 | RXRRBRK(Myr)K(Myr)RXRRB | 7 | 2 |
| Pt3-PMO | Pt3 | RXRRBRK(Myr)RRBRXB | 7 | 1 |
| Pt4-PMO | Pt4 | RBRRK(Myr)RRBRXB | 6 | 1 |
| Pt5-PMO | Pt5 | RGDRK(Myr)RRBRXB | 5 | 1 |
| Pt6-PMO | Pt6 | [(RGD)2K]2K-X | 4 | 0 |
| Pt7-PMO | Pt7 | CKWKS(Myr)S(Myr) | 0 | 2 |
| Pt8-PMO | Pt8 | RXRRBRRXRRBRXE(Cys)BE(Cys)BE(Cys)B | 8 | 0 |
| B-PMO | B | RXRRBRRXRRBRXB | 8 | 0 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Wu, B.; Lu, P.; Shah, S.N.; Tucker, J.D.; Bollinger, L.E.; Lu, Q. Evaluation of Amphiphilic Peptide Modified Antisense Morpholino Oligonucleotides In Vitro and in Dystrophic mdx Mice. Polymers 2017, 9, 177. https://doi.org/10.3390/polym9050177
Wang M, Wu B, Lu P, Shah SN, Tucker JD, Bollinger LE, Lu Q. Evaluation of Amphiphilic Peptide Modified Antisense Morpholino Oligonucleotides In Vitro and in Dystrophic mdx Mice. Polymers. 2017; 9(5):177. https://doi.org/10.3390/polym9050177
Chicago/Turabian StyleWang, Mingxing, Bo Wu, Peijuan Lu, Sapana N. Shah, Jason D. Tucker, Lauren E. Bollinger, and Qilong Lu. 2017. "Evaluation of Amphiphilic Peptide Modified Antisense Morpholino Oligonucleotides In Vitro and in Dystrophic mdx Mice" Polymers 9, no. 5: 177. https://doi.org/10.3390/polym9050177