Acid-Labile Surfactants Based on Poly(ethylene glycol), Carbon Dioxide and Propylene Oxide: Miniemulsion Polymerization and Degradation Studies

Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Measurements
2.3. Synthesis of (R,R)-(salcy)-CoOBzF5
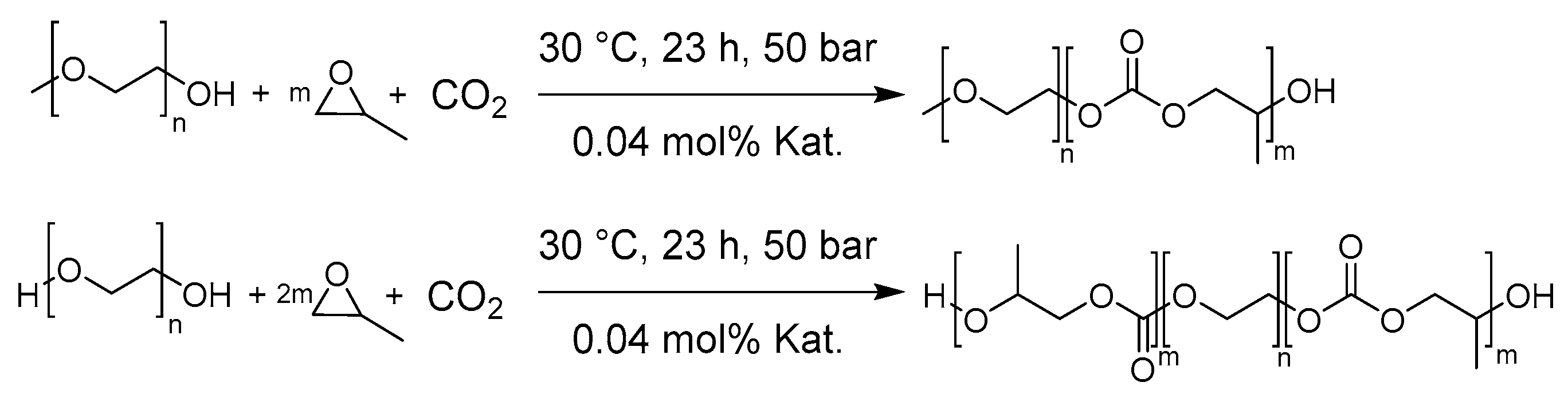
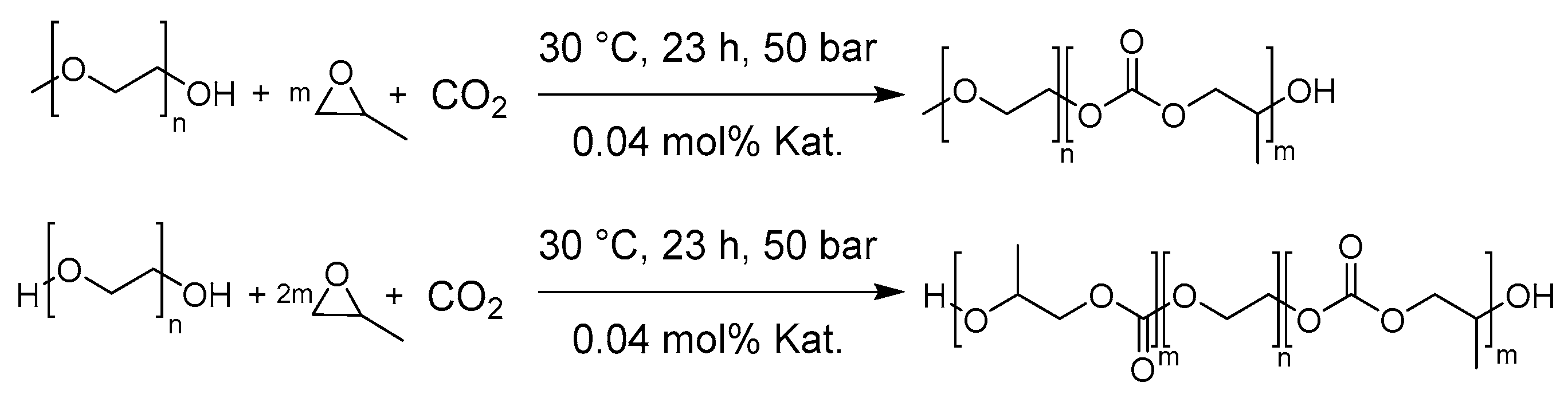
2.4. General procedure for the Synthesis of mPEG-b-PPC and PPC-b-PEG-b-PPC
2.5. General Procedure for Free-Radical Direct Miniemulsion Polymerization
2.6. General Procedure for the Destabilization of the PS Nanoparticles
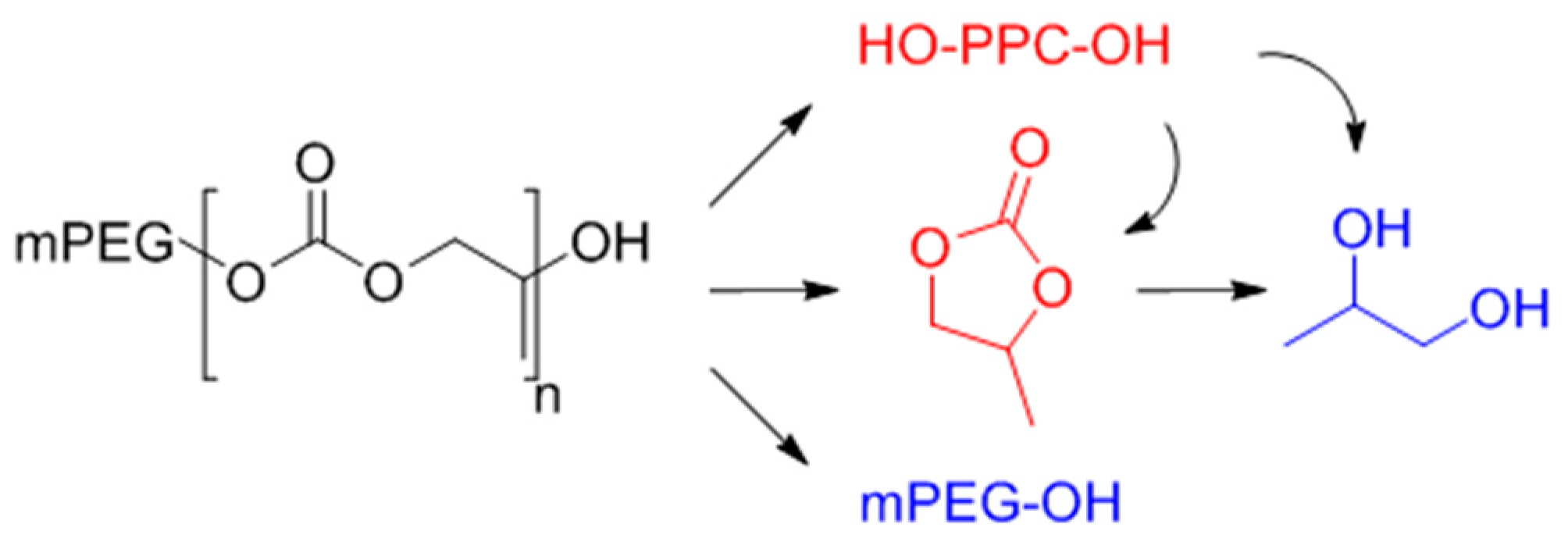
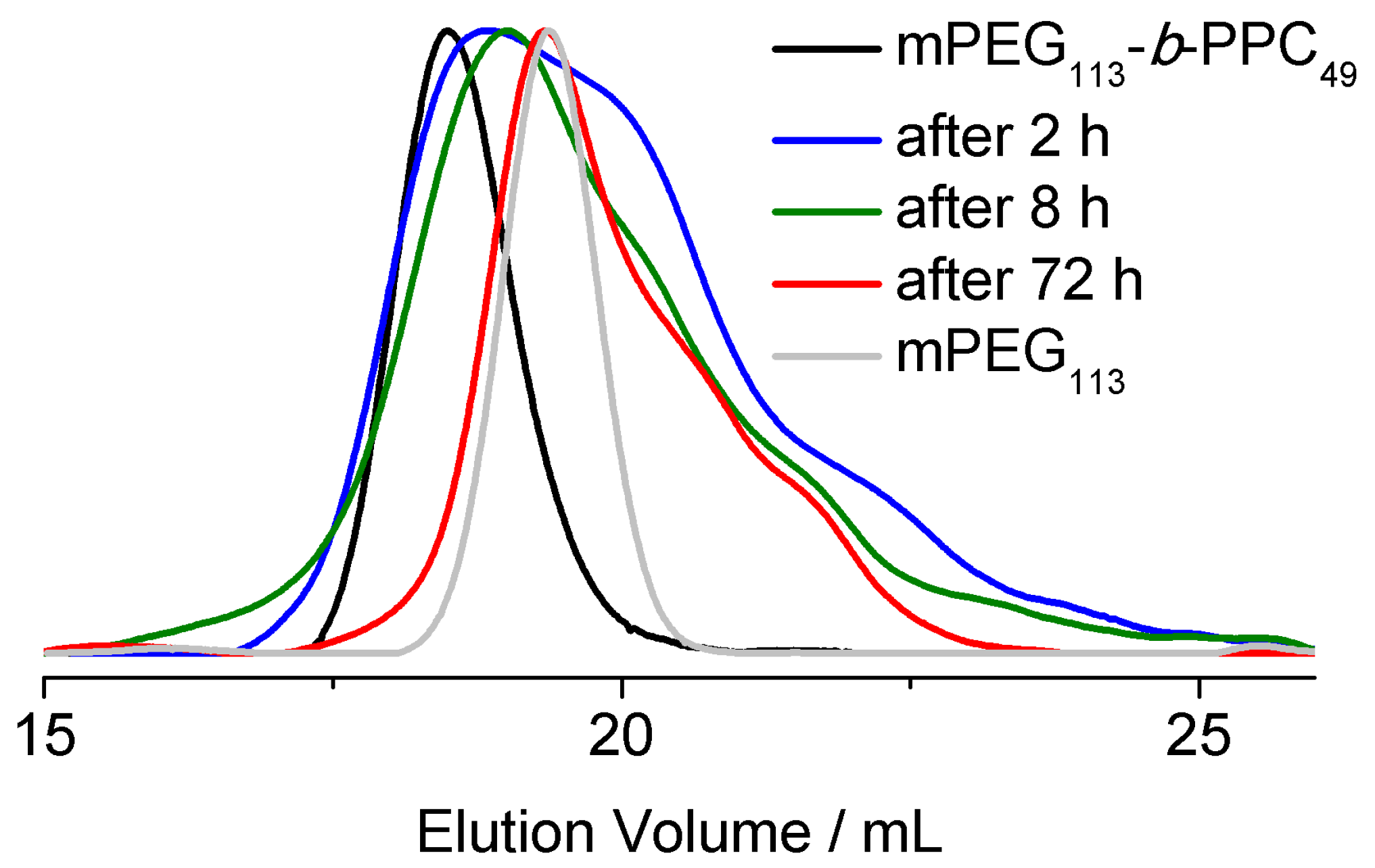
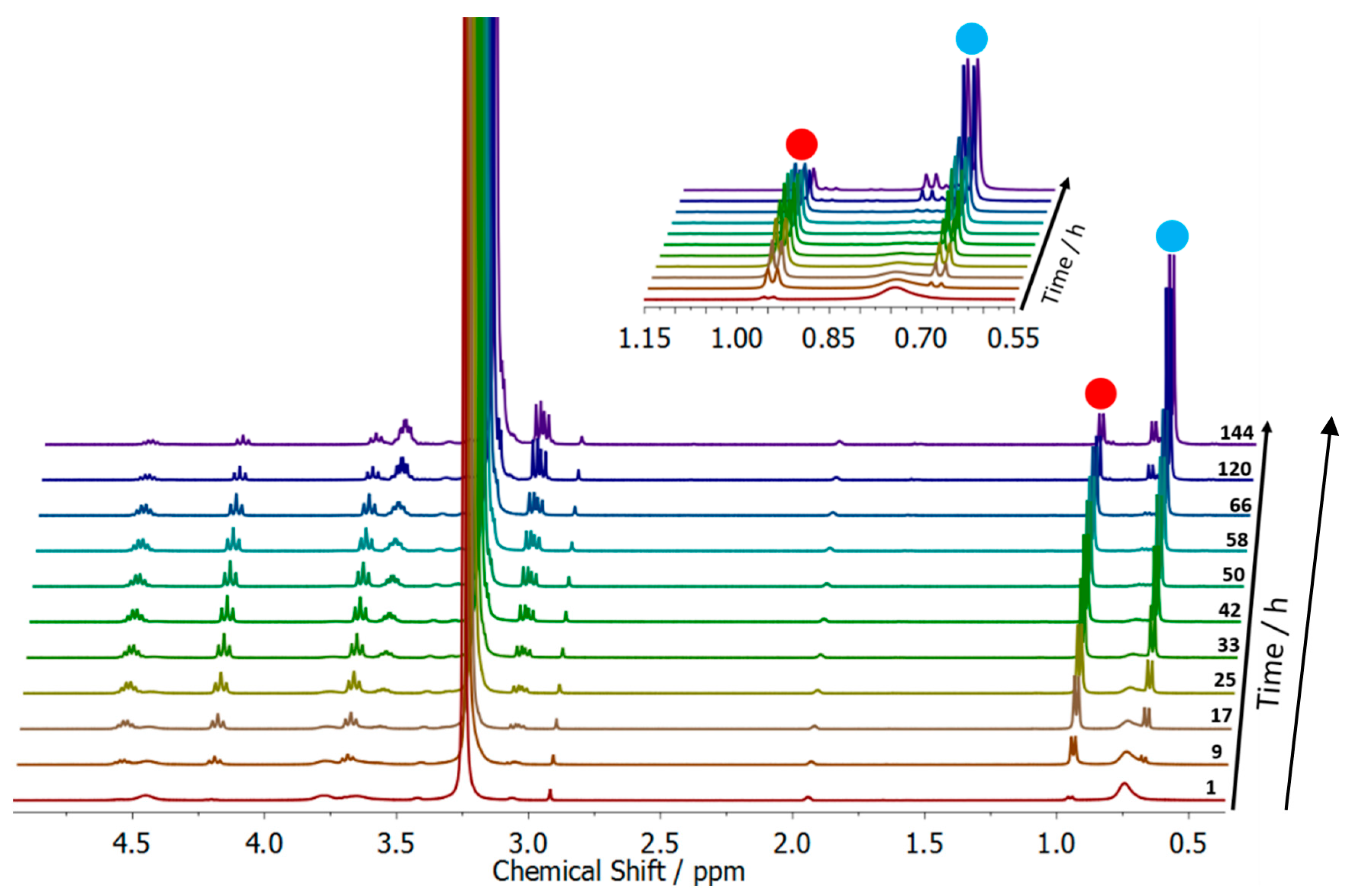
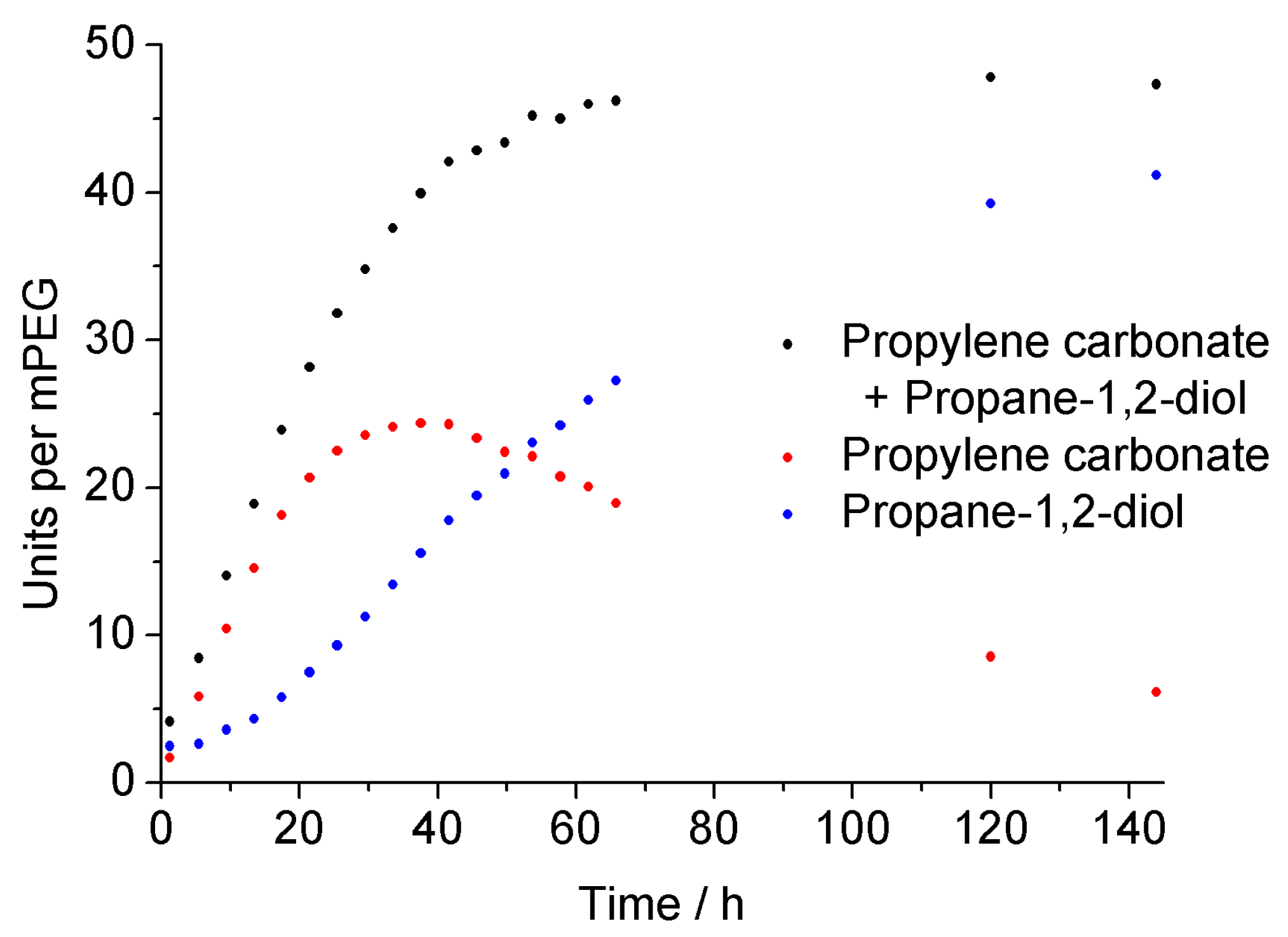
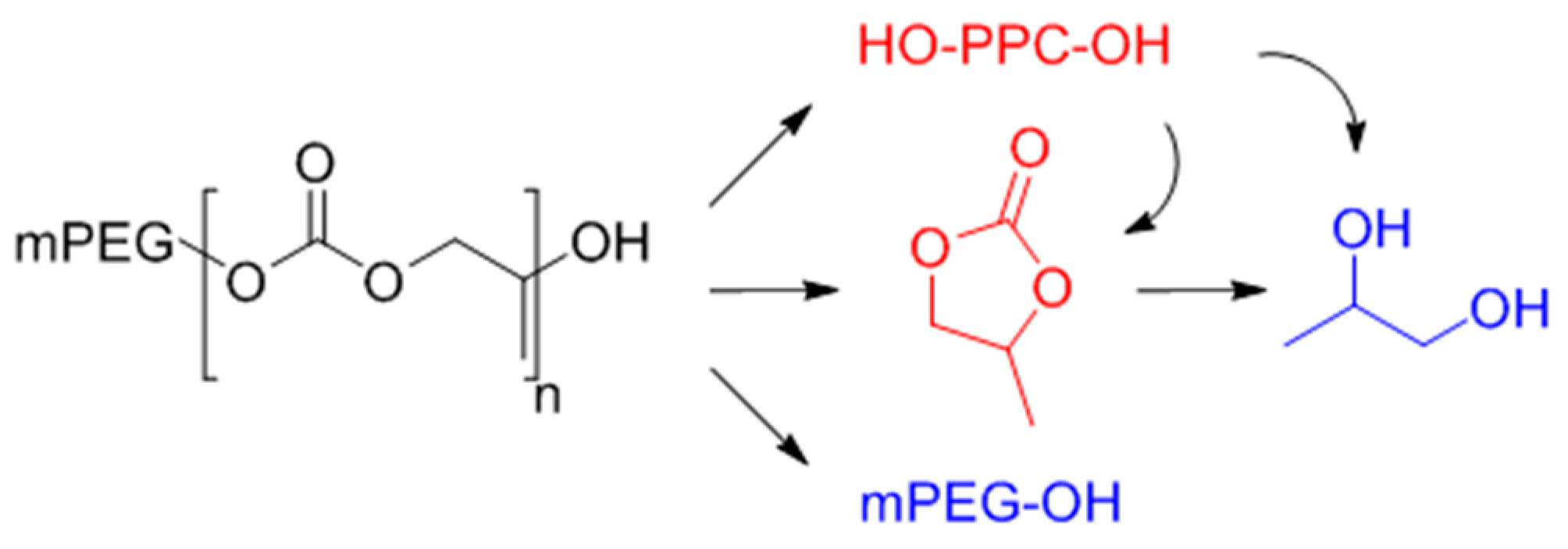
2.7. Degradation Study of the Surfactant
3. Results and Discussion
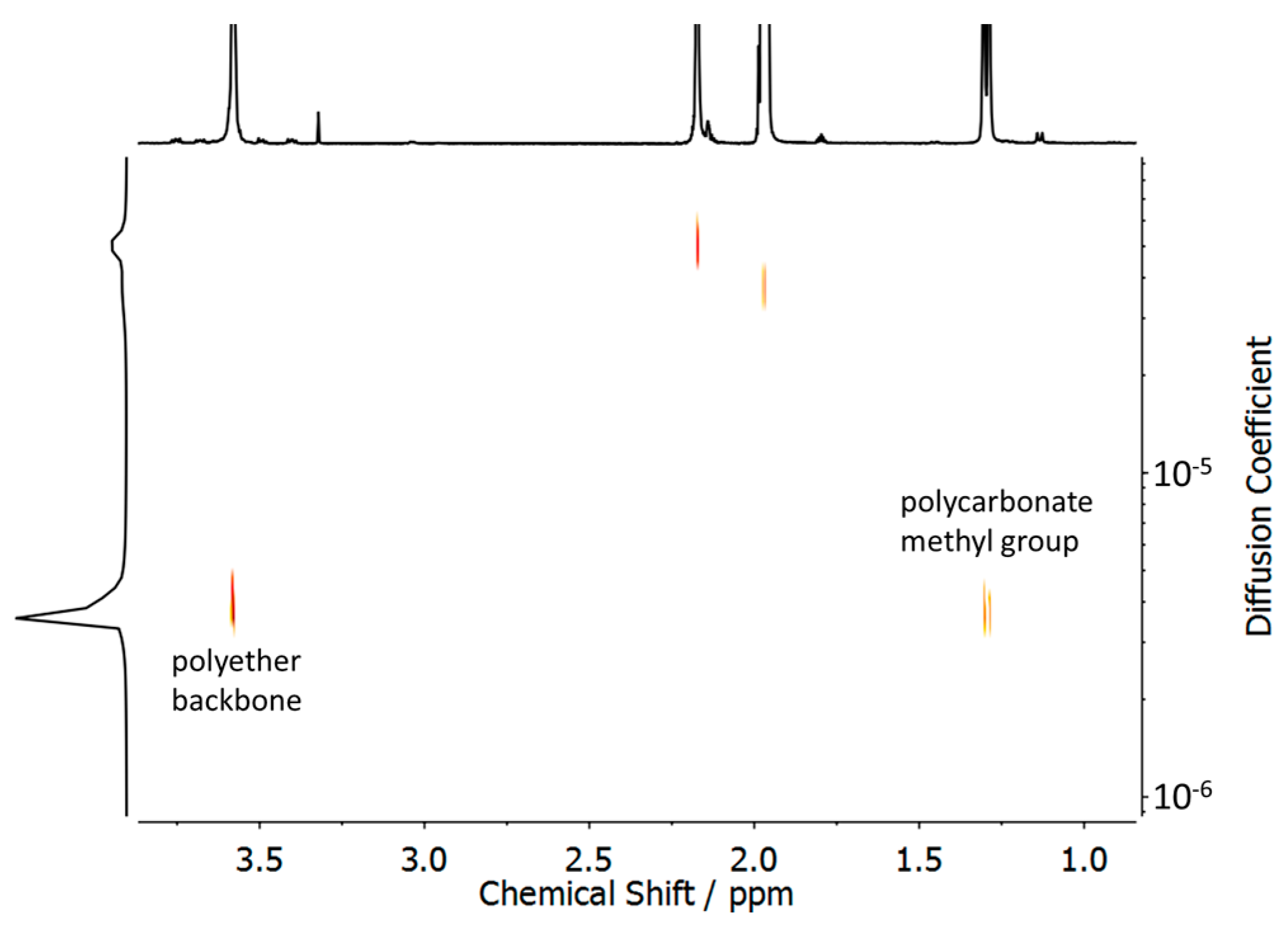
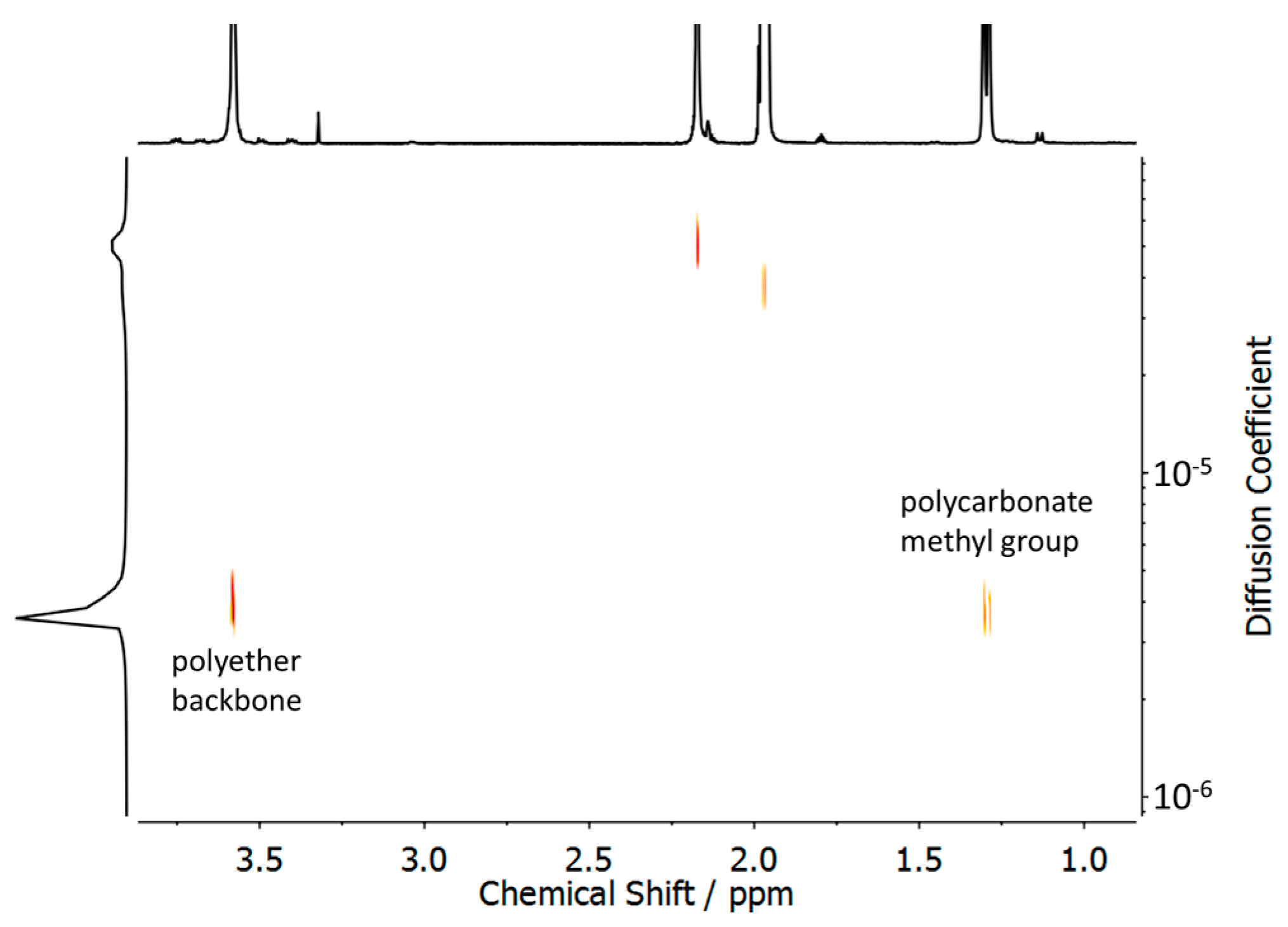
3.1. Synthesis and Characterization of Polycarbonate-Polyether Surfactants
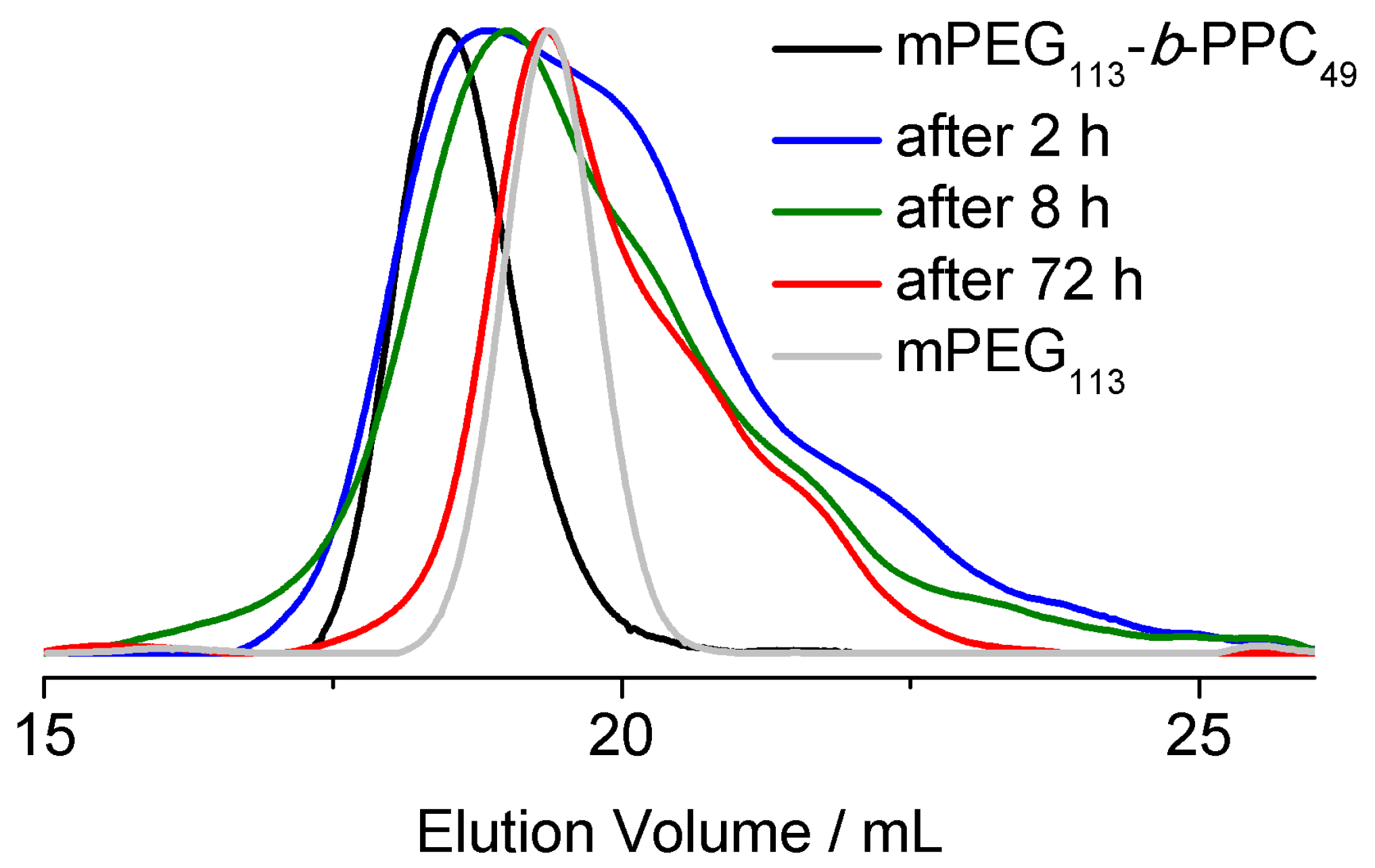
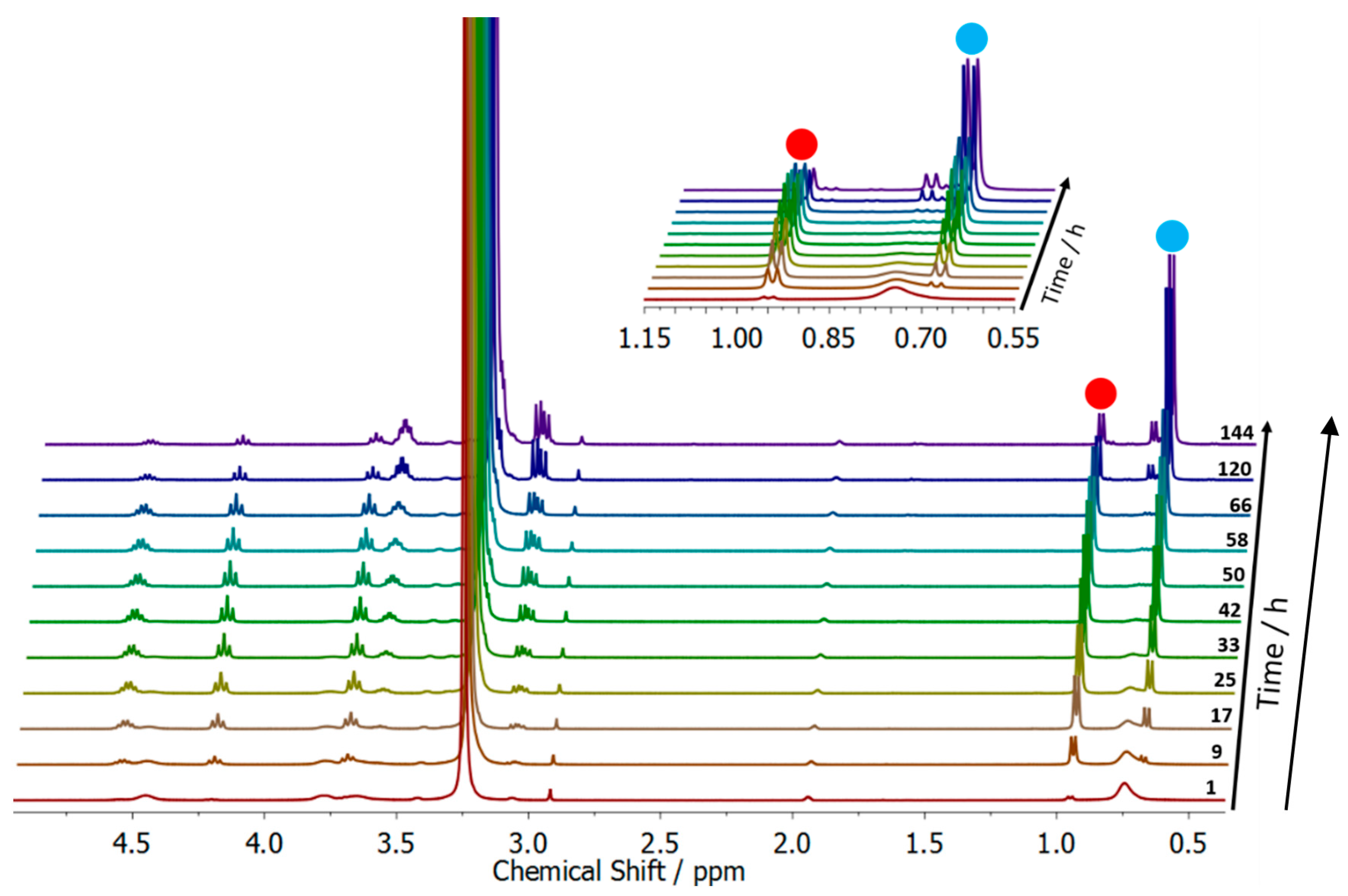
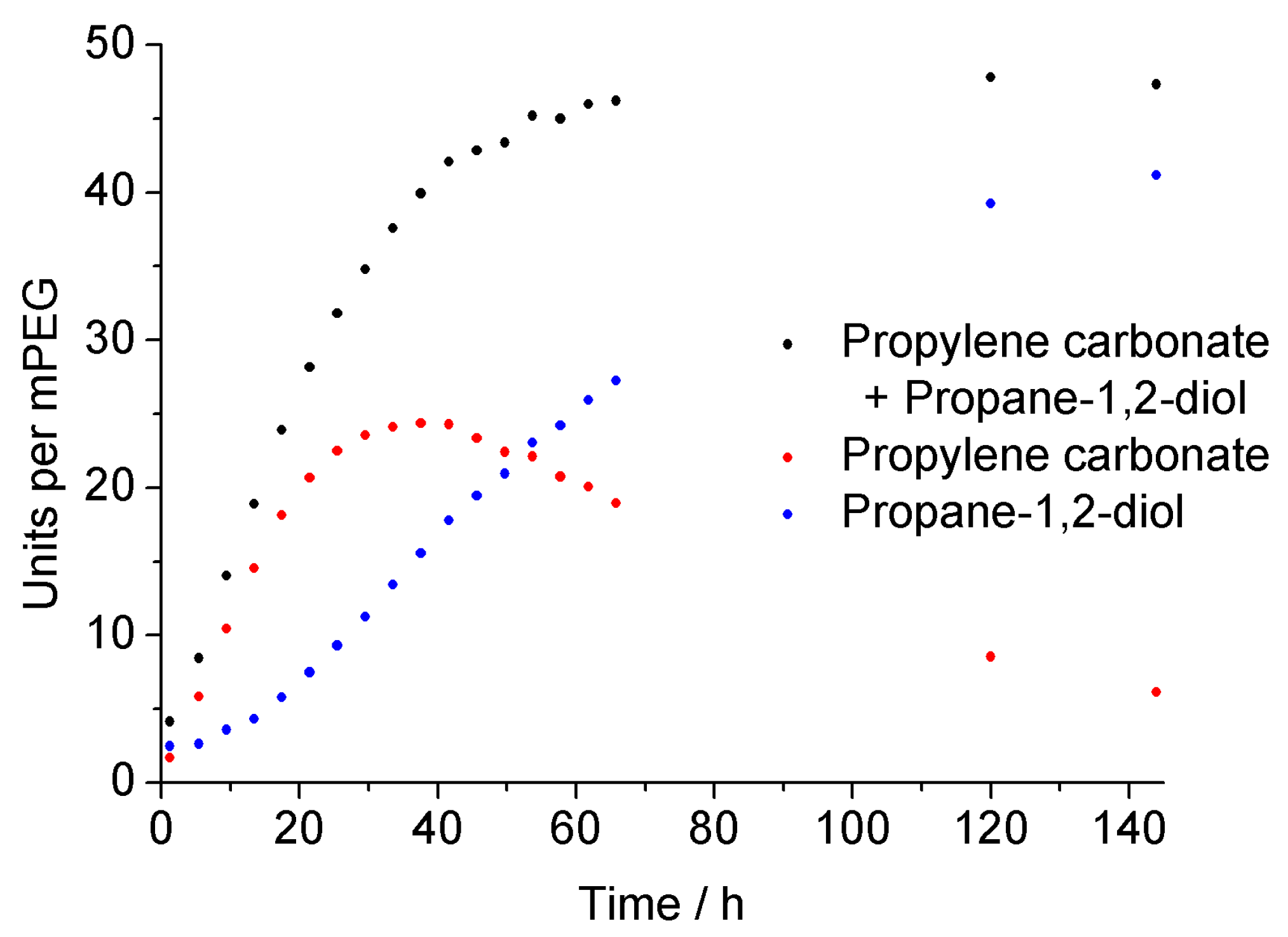
3.2. Surfactant Properties and Degradation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stolnik, S.; Illum, L.; Davis, S.S. Long circulating microparticulate drug carriers. Adv. Drug Deliv. Rev. 2012, 64, 290–301. [Google Scholar] [CrossRef]
- Bijlard, A.-C.; Wald, S.; Crespy, D.; Taden, A.; Wurm, F.R.; Landfester, K. Functional Colloidal Stabilization. Adv. Mater. Interfaces 2017, 4, 1600443. [Google Scholar] [CrossRef]
- Guyot, A. Advances in reactive surfactants. Adv. Colloid Interface Sci. 2004, 108–109, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Kralova, I.; Sjöblom, J. Surfactants Used in Food Industry: A Review. J. Dispers. Sci. Technol. 2009, 30, 1363–1383. [Google Scholar] [CrossRef]
- Landfester, K. Miniemulsion polymerization and the structure of polymer and hybrid nanoparticles. Angew. Chem. Int. Ed. 2009, 48, 4488–4507. [Google Scholar] [CrossRef] [PubMed]
- Ivankovic, T.; Hrenovic, J. Surfactants in the environment. Arhiv za Higijenu Rada i Toksikologiju 2010, 61, 95–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dannenfelser, R.-M.; Liu, R.; Li, S. Micellization and Drug Solubility Enhancement. In Water-Insoluble Drug Formulation, 2nd ed.; Liu, R., Ed.; CRC Press: Boca Raton, FL, USA, 2008; pp. 255–306. [Google Scholar]
- Alkan, A.; Wald, S.; Louage, B.; de Geest, B.G.; Landfester, K.; Wurm, F.R. Amphiphilic Ferrocene-Containing PEG Block Copolymers as Micellar Nanocarriers and Smart Surfactants. Langmuir 2017, 33, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Worm, M.; Kang, B.; Dingels, C.; Wurm, F.R.; Frey, H. Acid-Labile Amphiphilic PEO-b-PPO-b-PEO Copolymers: Degradable Poloxamer Analogs. Macromol. Rapid Commun. 2016, 37, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Anton, P.; Heinze, J.; Laschewsky, A. Redox-active monomeric and polymeric surfactants. Langmuir 1993, 9, 77–85. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A. Utilisation of CO2 as a chemical feedstock: Opportunities and challenges. Dalton Trans. 2007, 28, 2975–2992. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of carbon dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Koinuma, H.; Tsuruta, T. Copolymerization of Carbon Dioxide and Epoxide with Organometallic Compounds. Macromol. Chem. Phys. 1969, 130, 210–220. [Google Scholar] [CrossRef]
- Inoue, S. Copolymerization of Carbon Dioxide and Epoxide: Functionality of the Copolymer. J. Macromol. Sci. Pure Appl. Chem. 1979, 13, 651–664. [Google Scholar] [CrossRef]
- Rieger, B.; Amann, M. Synthetic Biodegradable Polymers; Springer: Berlin, Germany, 2012. [Google Scholar]
- Welle, A.; Kröger, M.; Döring, M.; Niederer, K.; Pindel, E.; Chronakis, I.S. Electrospun aliphatic polycarbonates as tailored tissue scaffold materials. Biomaterials 2007, 28, 2211–2219. [Google Scholar] [CrossRef] [PubMed]
- Takanashi, M.; Nomura, Y.; Yoshida, Y.; Inoue, S. Functional Polycarbonate by Copolymerization of Carbon Dioxide and Epoxide: Synthesis and Hydrolysis. Makromol. Chem. 1982, 183, 2085–2092. [Google Scholar] [CrossRef]
- Zhou, M.; Takayanagi, M.; Yoshida, Y.; Ishii, S.; Noguchi, H. Enzyme-catalyzed degradation of aliphatic polycarbonates prepared from epoxides and carbon dioxide. Polym. Bull. 1999, 42, 419–424. [Google Scholar] [CrossRef]
- Du, L.C.; Meng, Y.Z.; Wang, S.J.; Tjong, S.C. Synthesis and Degradation Behaviour of Poly(propylene Carbonate) Derived from Carbon Dioxide and Propylene Oxide. J. Appl. Polym. Sci. 2004, 92, 1840–1846. [Google Scholar] [CrossRef]
- Jung, J.H.; Ree, M.; Kim, H. Acid- and base-catalyzed hydrolyses of aliphatic polycarbonates and polyesters. Catal. Today 2006, 115, 283–287. [Google Scholar] [CrossRef]
- Luinstra, G. Poly(Propylene Carbonate), Old Copolymers of Propylene Oxide and Carbon Dioxide with New Interests: Catalysis and Material Properties. Polym. Rev. 2008, 48, 192–219. [Google Scholar] [CrossRef]
- Luinstra, G.A.; Borchardt, E. Material properties of poly(propylene carbonates). Adv. Polym. Sci. 2012, 245, 29–48. [Google Scholar] [CrossRef]
- Zhang, H.; Grinstaff, M.W. Synthesis of atactic and isotactic poly(1,2-glycerol carbonate)s: Degradable polymers for biomedical and pharmaceutical applications. J. Am. Chem. Soc. 2013, 135, 6806–6809. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, J.; Frey, H. Poly(1,2-glycerol carbonate): A fundamental polymer structure synthesized from CO2 and glycidyl ethers. Macromolecules 2013, 46, 3280–3287. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Wei, S.-H.; Wilson, S.J. Depolymerization of poly(indene carbonate). A unique degradation pathway. Macromolecules 2013, 46, 3228–3233. [Google Scholar] [CrossRef]
- Hilf, J.; Schulze, P.; Frey, H. CO2-based non-ionic surfactants: Solvent-free synthesis of poly(ethylene glycol)-block-poly(propylene carbonate) block copolymers. Macromol. Chem. Phys. 2013, 214, 2848–2855. [Google Scholar] [CrossRef]
- Cyriac, A.; Lee, S.H.; Varghese, J.K.; Park, E.S.; Park, J.H.; Lee, B.Y. Immortal CO2/propylene oxide copolymerization: Precise control of molecular weight and architecture of various block copolymers. Macromolecules 2010, 43, 7398–7401. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, J.; Darensbourg, D.J. Construction of versatile and functional nanostructures derived from CO2-based polycarbonates. Angew. Chem. Int. Ed. 2015, 54, 10206–10210. [Google Scholar] [CrossRef] [PubMed]
- Darensbourg, D.J.; Tsai, F.-T. Postpolymerization functionalization of copolymers produced from carbon dioxide and 2-vinyloxirane: Amphiphilic/water-soluble CO2-based polycarbonates. Macromolecules 2014, 47, 3806–3813. [Google Scholar] [CrossRef]
- Gu, L.; Qin, Y.; Gao, Y.; Wang, X.; Wang, F. Hydrophilic CO2-based biodegradable polycarbonates: Synthesis and rapid thermo-responsive behavior. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 2834–2840. [Google Scholar] [CrossRef]
- Hauenstein, O.; Agarwal, S.; Greiner, A. Bio-based polycarbonate as synthetic toolbox. Nat. Commun. 2016, 7, 11862. [Google Scholar] [CrossRef] [PubMed]
- Tsai, F.-T.; Wang, Y.; Darensbourg, D.J. Environmentally benign CO2-based copolymers: Degradable polycarbonates derived from dihydroxybutyric acid and their platinum-polymer conjugates. J. Am. Chem. Soc. 2016, 138, 4626–4633. [Google Scholar] [CrossRef] [PubMed]
- Trott, G.; Saini, P.K.; Williams, C.K. Catalysts for CO2/epoxide ring-opening copolymerization. Philos. Trans. R. Soc. A 2016, 374. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.T.; Chu, T.; Coates, G.W. Cobalt catalysts for the alternating copolymerization of propylene oxide and carbon dioxide: Combining high activity and selectivity. J. Am. Chem. Soc. 2005, 127, 10869–10878. [Google Scholar] [CrossRef] [PubMed]
- Alakhov, V.Y.; Kabanov, A.V. Block copolymeric biotransport carriers as versatile vehicles for drug delivery. Expert Opin. Investig. Drugs 1998, 7, 1453–1473. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Zhao, C.L.; Wang, Y.; Xu, R.; Winnik, M.A. Poly(styrene-ethylene oxide) block copolymer micelle formation in water: A fluorescence probe study. Macromolecules 1991, 24, 1003–1040. [Google Scholar] [CrossRef]
- Allen, C.; Maysinger, D.; Eisenberg, A. Nano-engineering block copolymer aggregates for drug delivery. Colloids Surf. B 1999, 16, 3–27. [Google Scholar] [CrossRef]
- Griffin, W.C. Classification of surface active agents by HLB. J. Soc. Cosmet. Chem. 1949, 1, 311–326. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Yarbrough, J.C.; Ortiz, C.; Fang, C.C. Comparative kinetic studies of the copolymerization of cyclohexene oxide and propylene oxide with carbon dioxide in the presence of chromium salen derivatives. In situ FTIR measurements of copolymer vs cyclic carbonate production. J. Am. Chem. Soc. 2003, 125, 7586–7591. [Google Scholar] [CrossRef] [PubMed]
- Hecht, L.L.; Schoth, A.; Muñoz-Espí, R.; Javadi, A.; Köhler, K.; Miller, R.; Landfester, K.; Schuchmann, H.P. Determination of the ideal surfactant concentration in miniemulsion polymerization. Macromol. Chem. Phys. 2013, 214, 812–823. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Composition a | Mn/g·mol−1 b | Đ b | Mn/g·mol−1 a | CMC/mg·L−1 c | HLB d |
|---|---|---|---|---|---|---|
| 1 | mPEG113-b-PPC10 | 5000 | 1.05 | 6000 | 2 | 16.6 |
| 2 | mPEG113-b-PPC20 | 5300 | 1.08 | 7000 | 1 | 14.2 |
| 3 | mPEG113-b-PPC49 | 6500 | 1.09 | 10,000 | 2 | 10.0 |
| 4 | mPEG45-b-PPC8 | 2500 | 1.04 | 2800 | 9 | 14.2 |
| 5 | mPEG45-b-PPC10 | 2600 | 1.03 | 3000 | 3 | 13.2 |
| 6 | mPEG45-b-PPC26 | 3600 | 1.04 | 4600 | 3 | 8.5 |
| 7 | mPEG45-b-PPC35 | 3900 | 1.08 | 5600 | 14 | 7.1 |
| 8 | mPEG45-b-PPC47 | 4400 | 1.07 | 6800 | n.s. e | 5.8 |
| 9 | PPC10-b-PEG45-b-PPC10 | 3300 | 1.06 | 4000 | n.s. e | 9.9 |
| 10 | PPC25-b-PEG45-b-PPC25 | 4700 | 1.08 | 7100 | n.s. e | 5.6 |
| Sample a | Surfactant | wt % PPC of Surfactant c | Particle Size d/nm d | PDI d |
|---|---|---|---|---|
| NP2 | 2 | 29 | 330 | 0.12 |
| NP3 | 3 | 50 | 270 | 0.08 |
| NP4 | 4 | 29 | 940 | 0.08 |
| NP6 | 6 | 57 | 140–>1000 | 0.57 |
| NPL | Lut. AT50 b | 0 | 160 | 0.06 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scharfenberg, M.; Wald, S.; Wurm, F.R.; Frey, H. Acid-Labile Surfactants Based on Poly(ethylene glycol), Carbon Dioxide and Propylene Oxide: Miniemulsion Polymerization and Degradation Studies. Polymers 2017, 9, 422. https://doi.org/10.3390/polym9090422
Scharfenberg M, Wald S, Wurm FR, Frey H. Acid-Labile Surfactants Based on Poly(ethylene glycol), Carbon Dioxide and Propylene Oxide: Miniemulsion Polymerization and Degradation Studies. Polymers. 2017; 9(9):422. https://doi.org/10.3390/polym9090422
Chicago/Turabian StyleScharfenberg, Markus, Sarah Wald, Frederik R. Wurm, and Holger Frey. 2017. "Acid-Labile Surfactants Based on Poly(ethylene glycol), Carbon Dioxide and Propylene Oxide: Miniemulsion Polymerization and Degradation Studies" Polymers 9, no. 9: 422. https://doi.org/10.3390/polym9090422