Effect of Graphene Oxide on the Reaction Kinetics of Methyl Methacrylate In Situ Radical Polymerization via the Bulk or Solution Technique



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Graphite Oxide
2.3. Preparation of the Initial Monomer/GO Mixtures
2.4. Synthesis of PMMA/GO Nanocomposites by the In-Situ Bulk or Solution Radical Polymerization
2.5. Measurements
3. Results
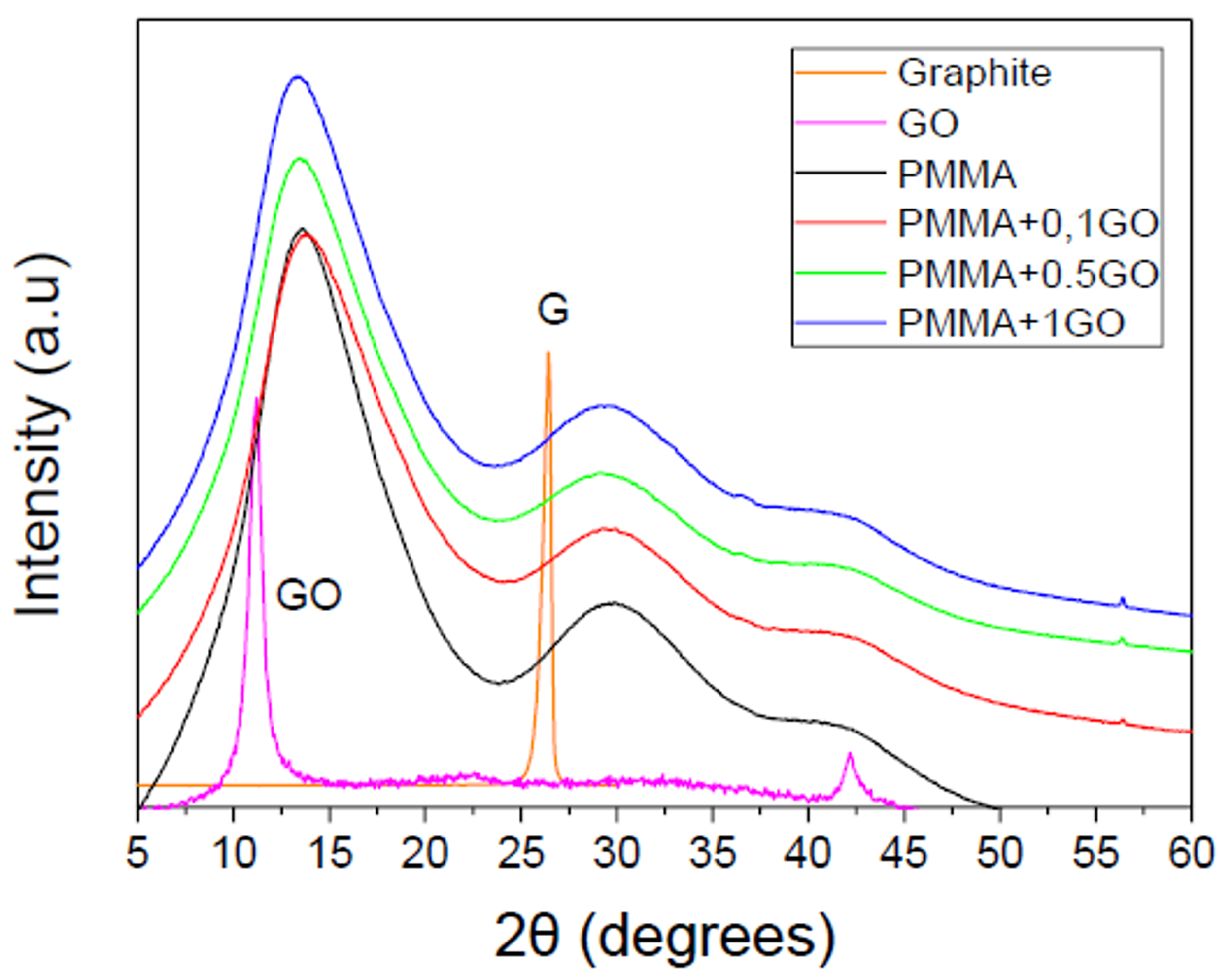
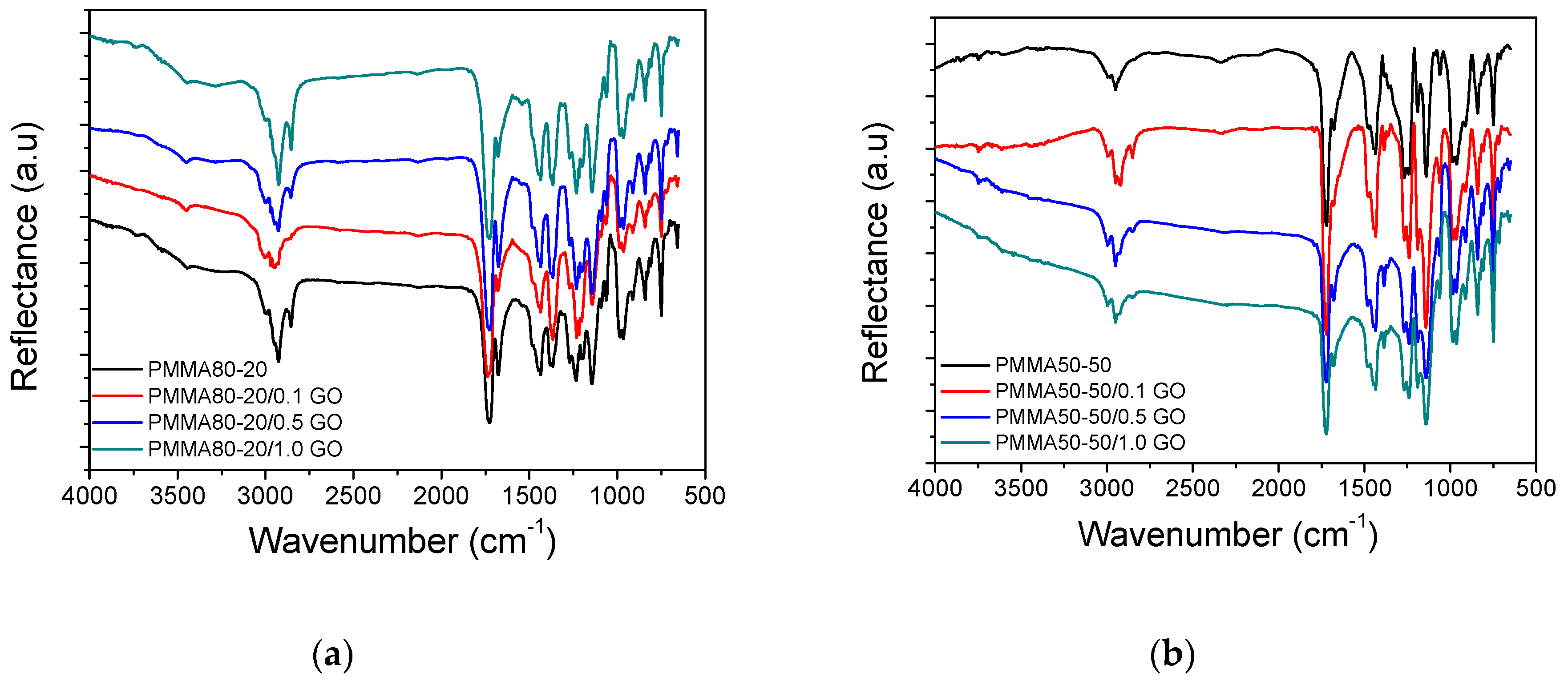
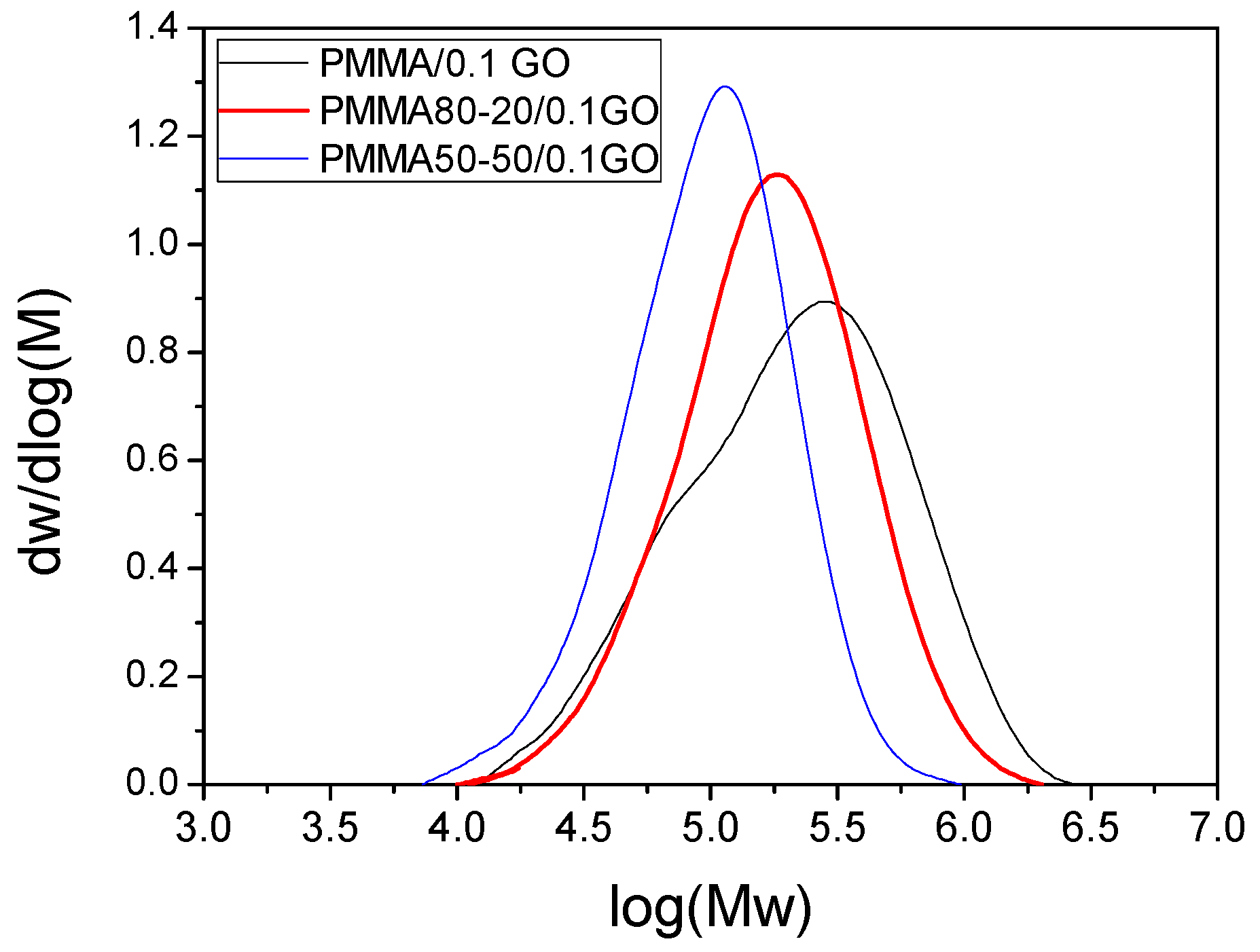
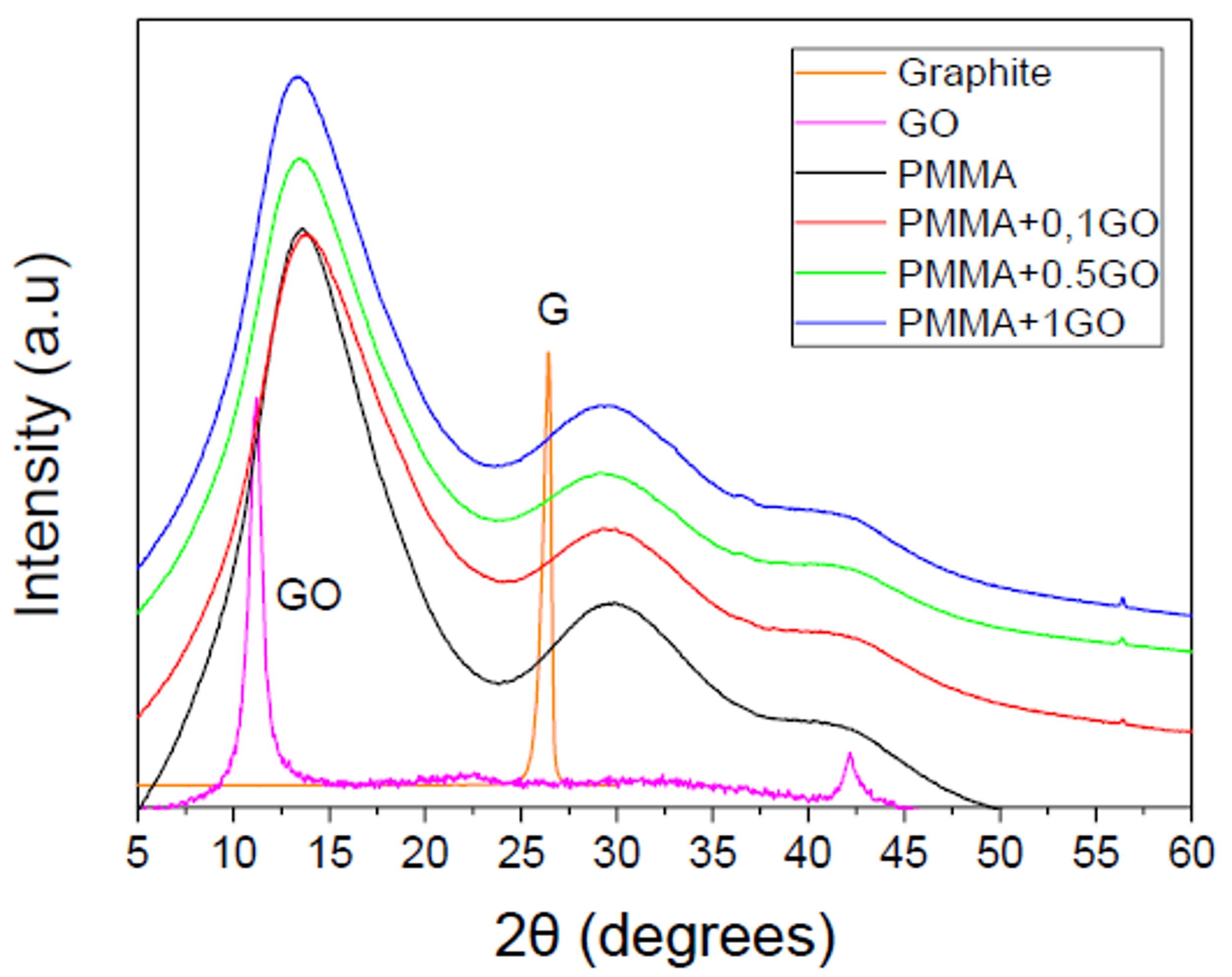
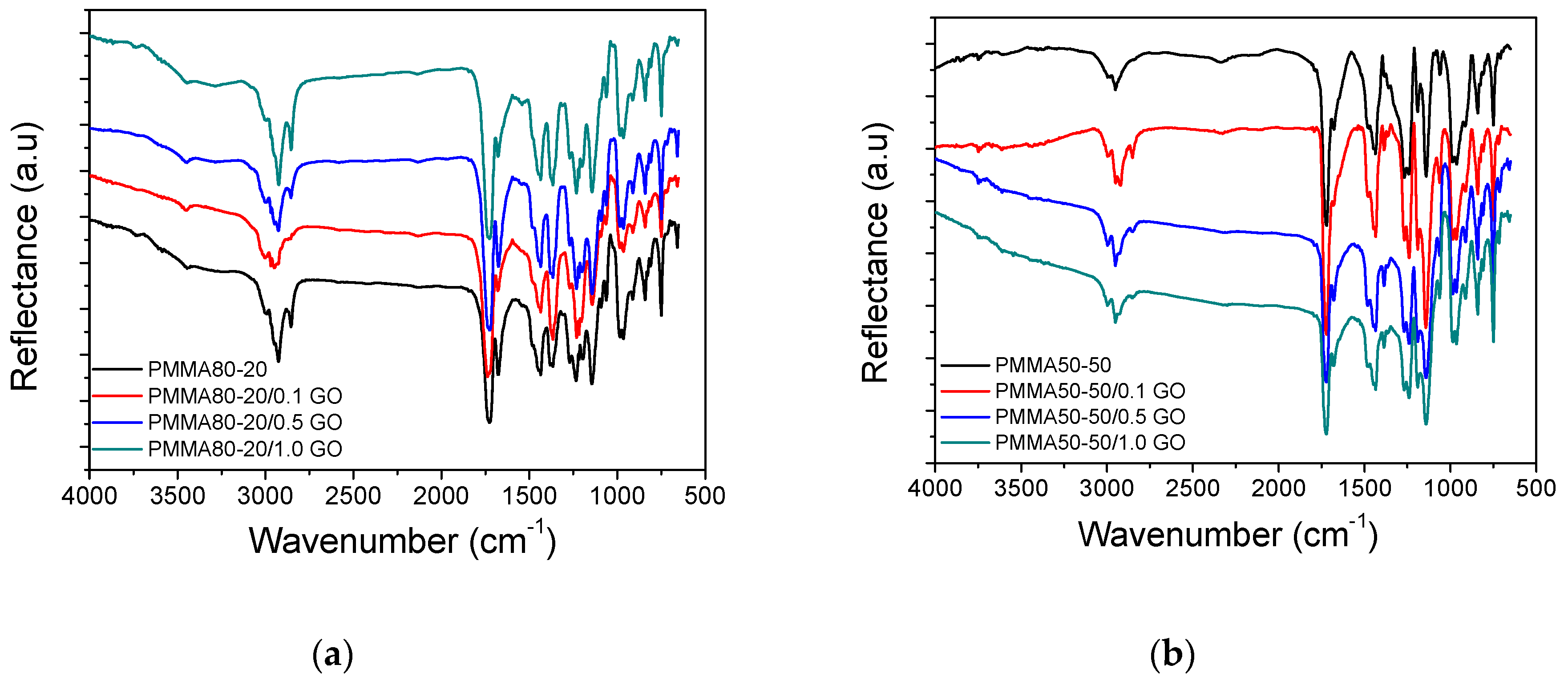
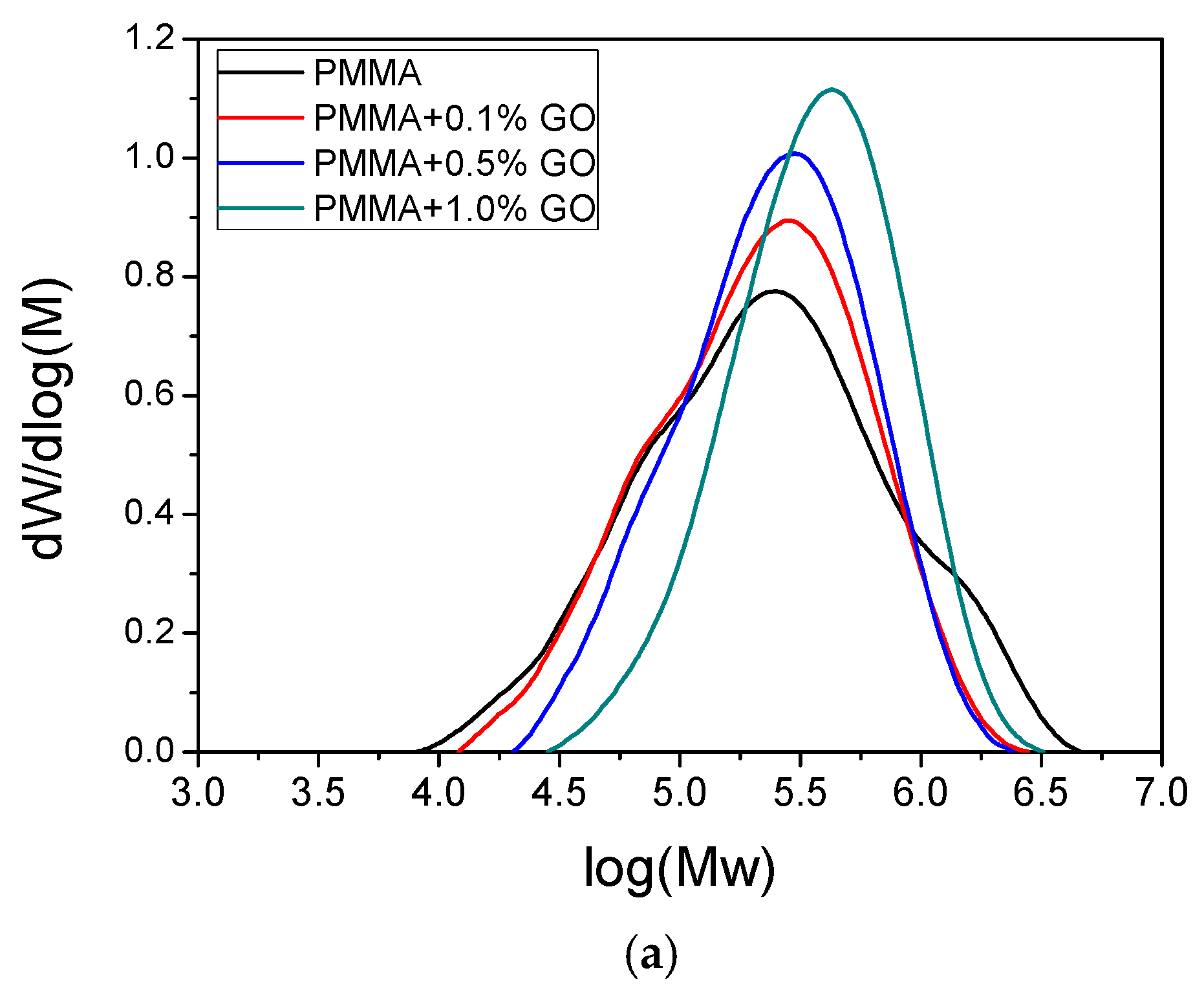
3.1. Characterization of the PMMA/GO Nanocomposites
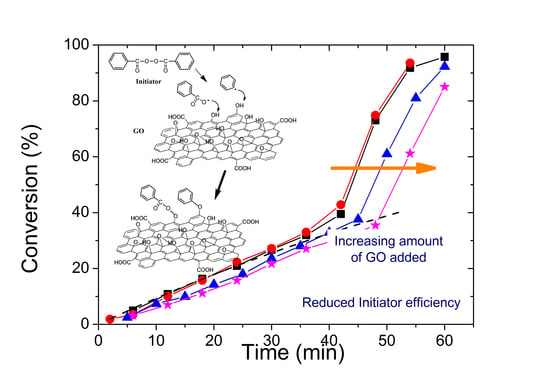
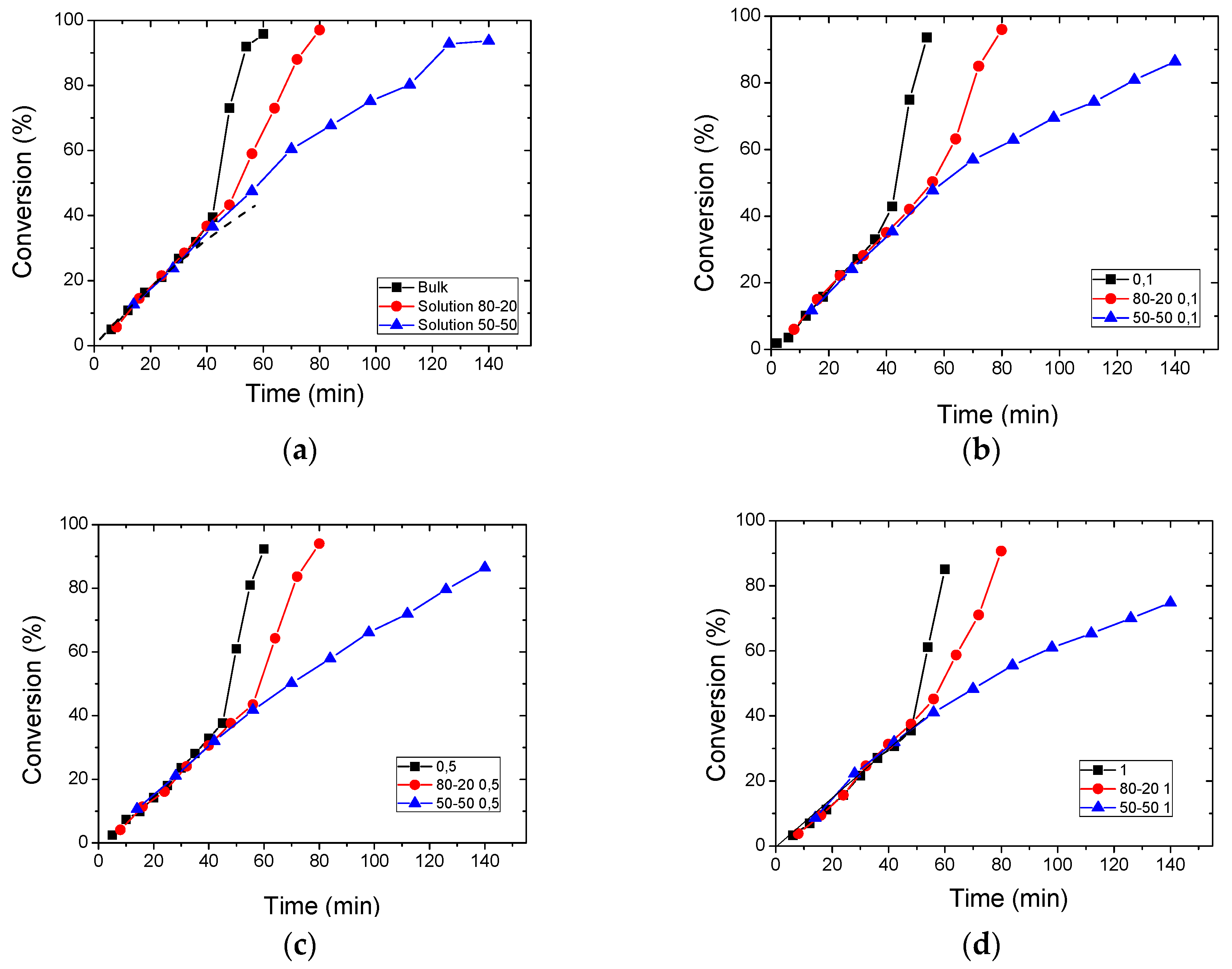
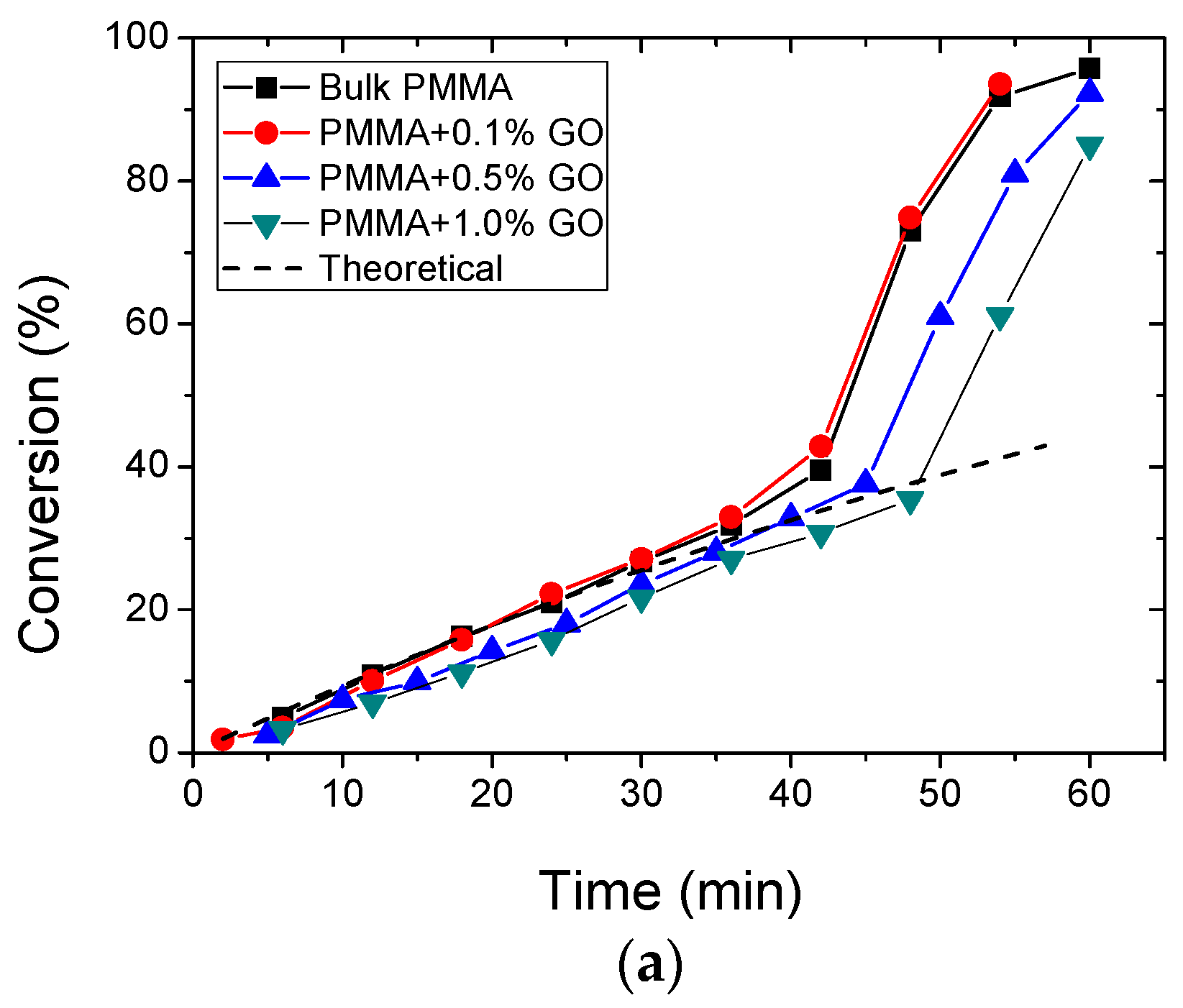
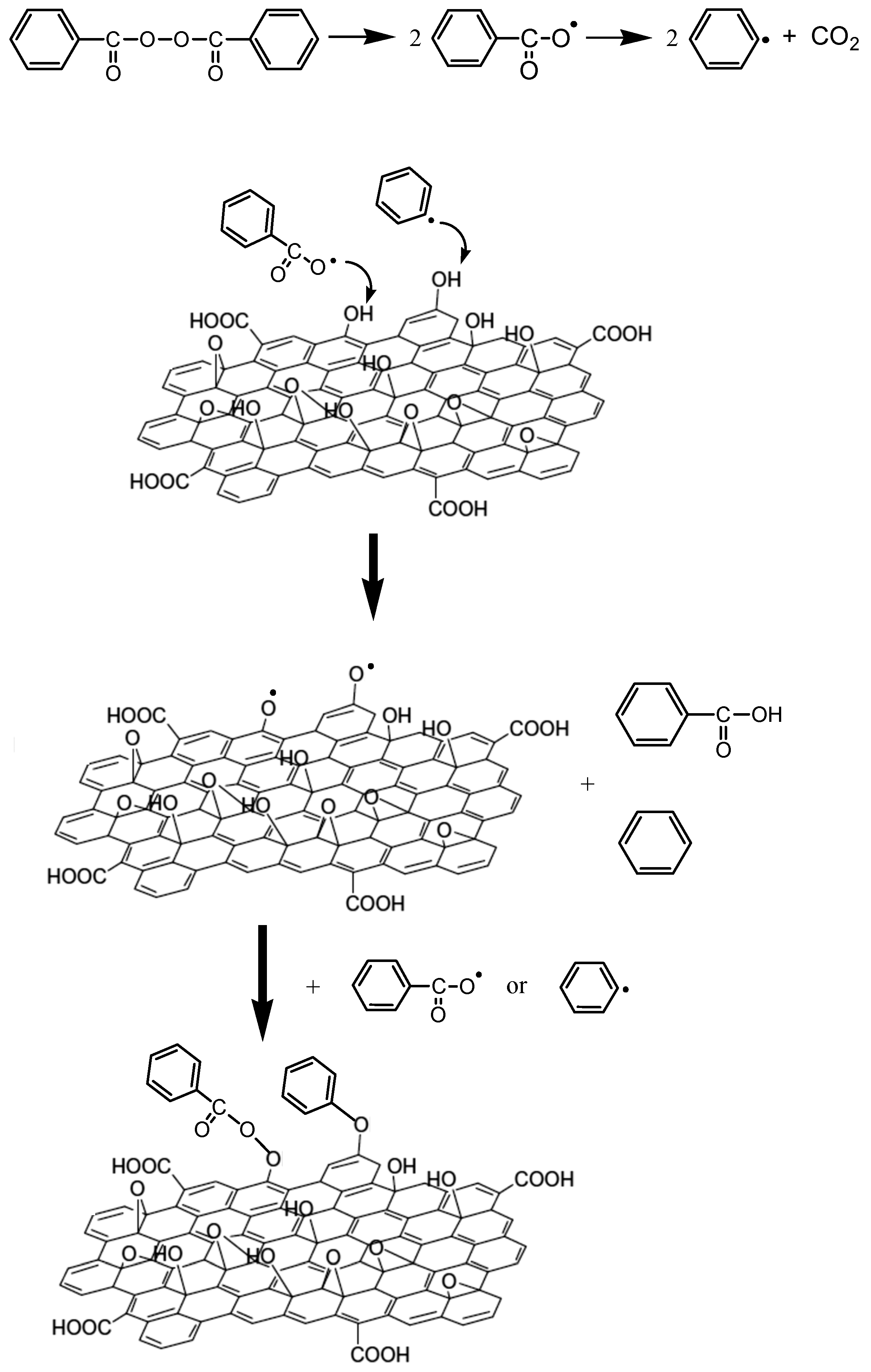
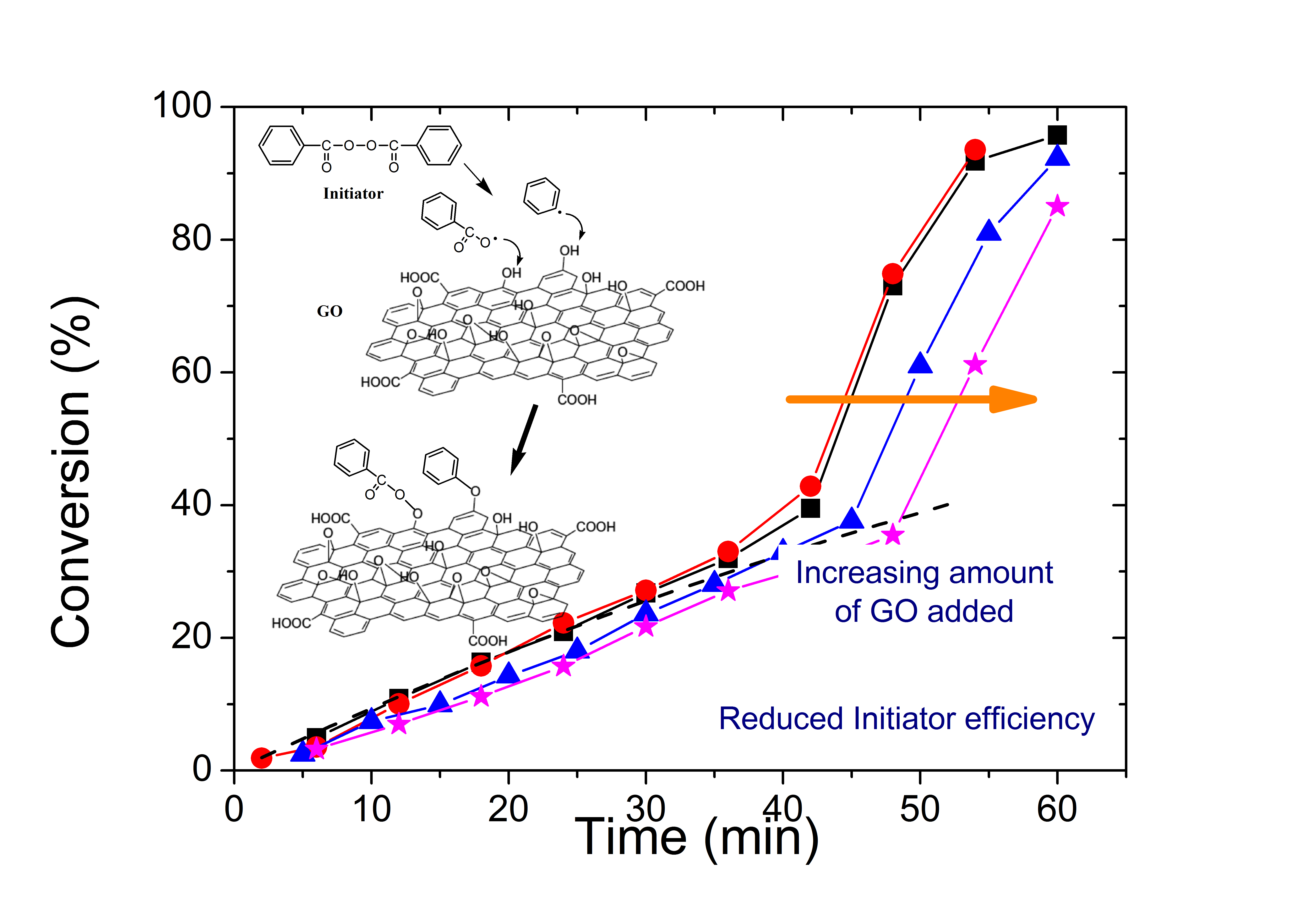
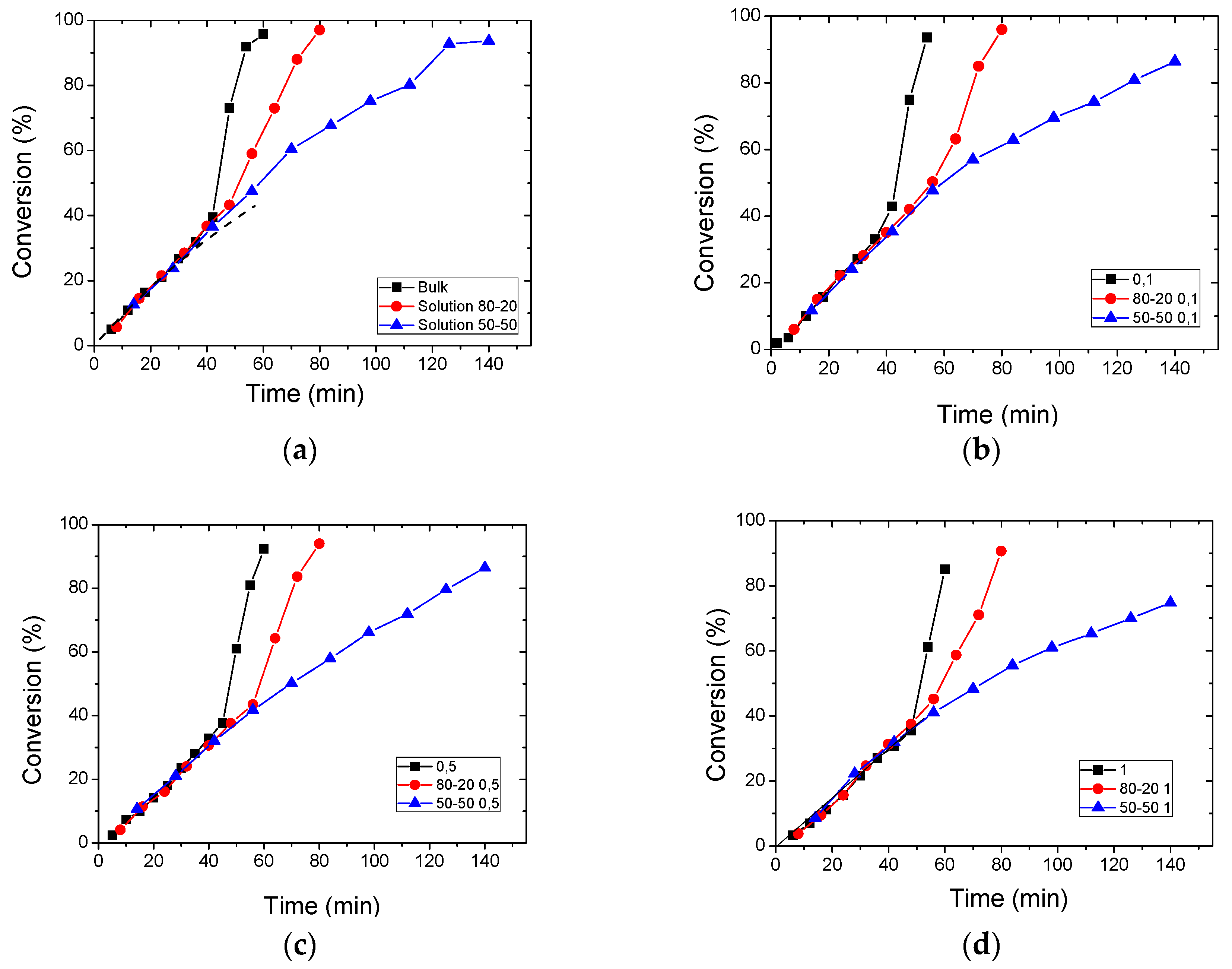
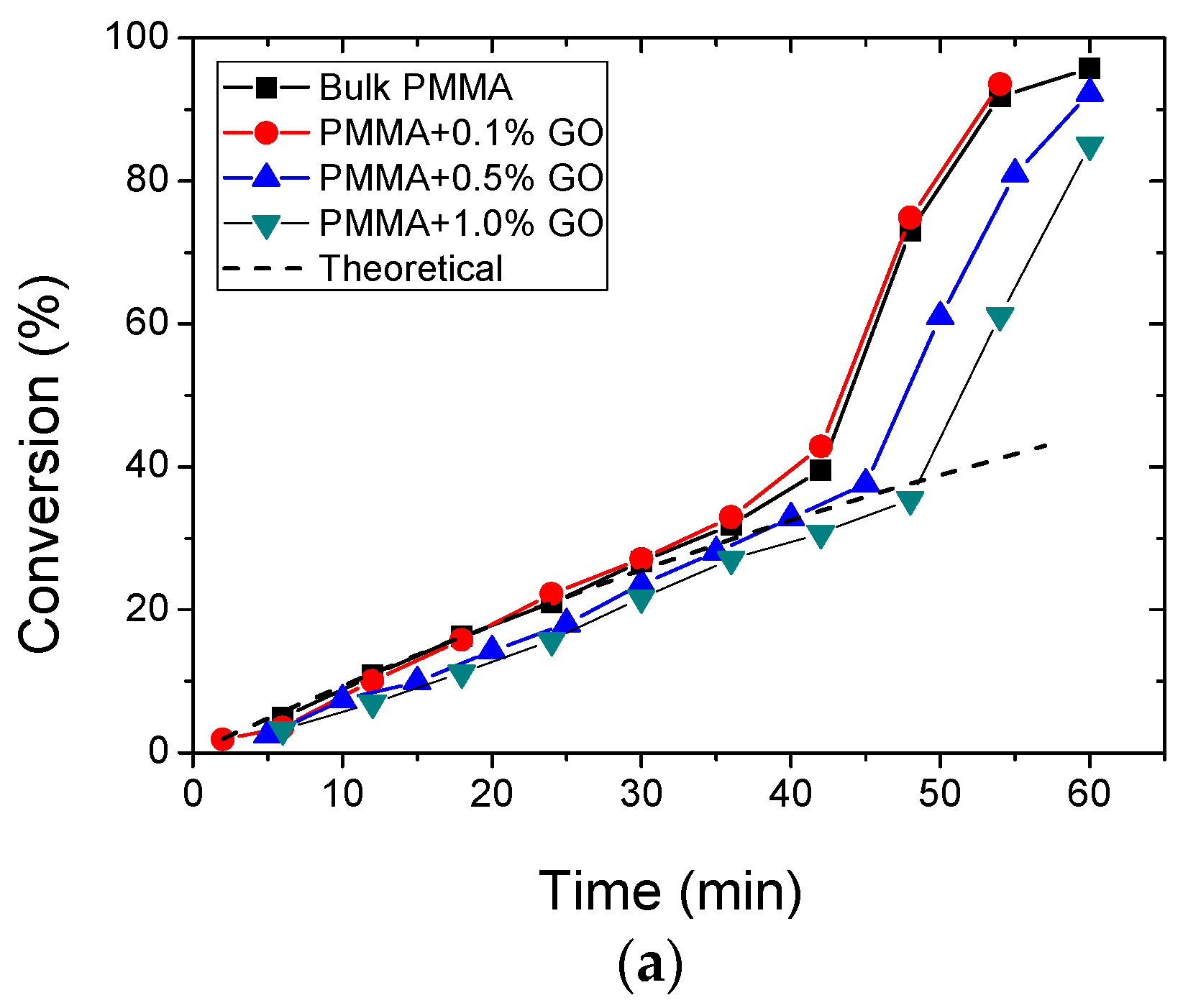
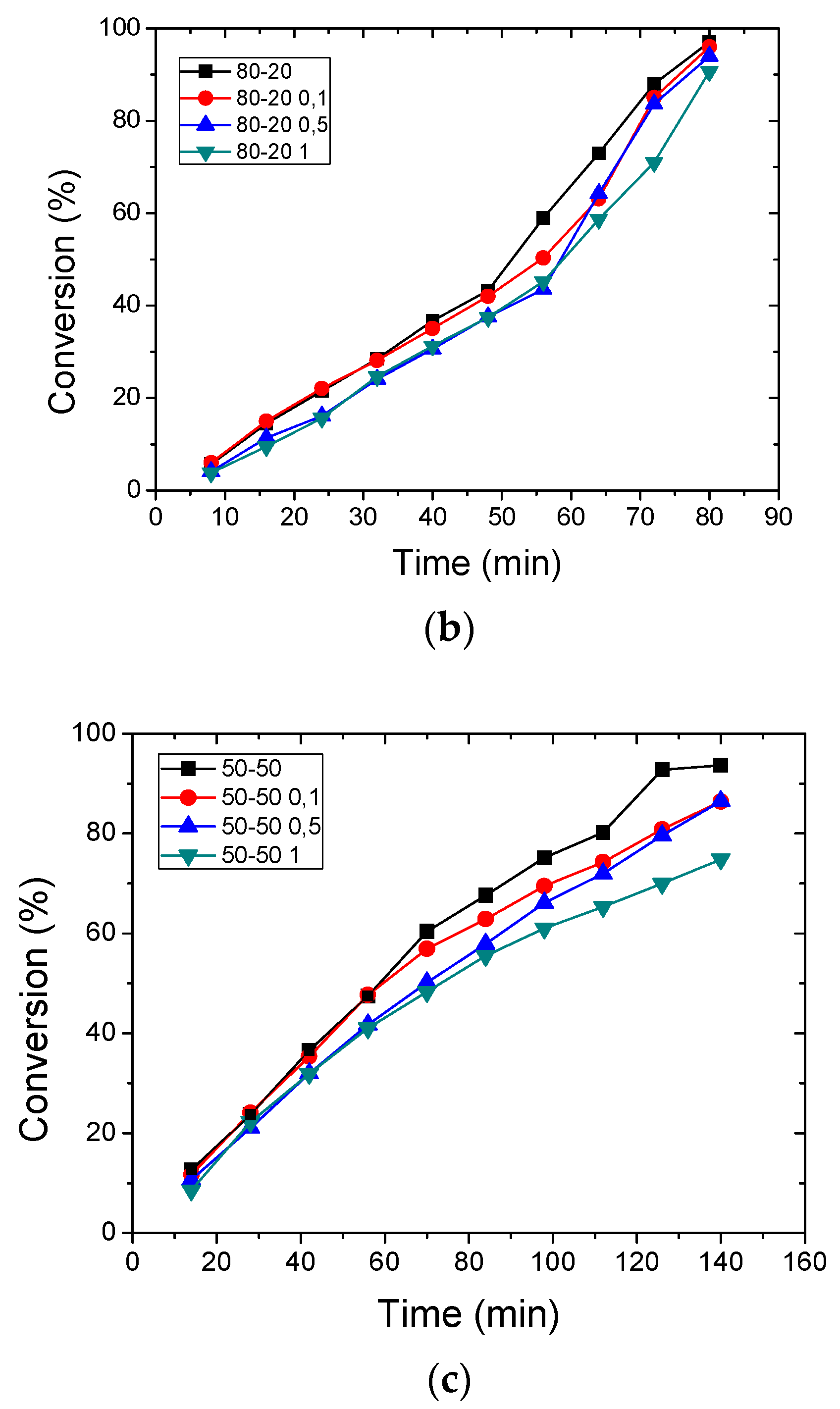
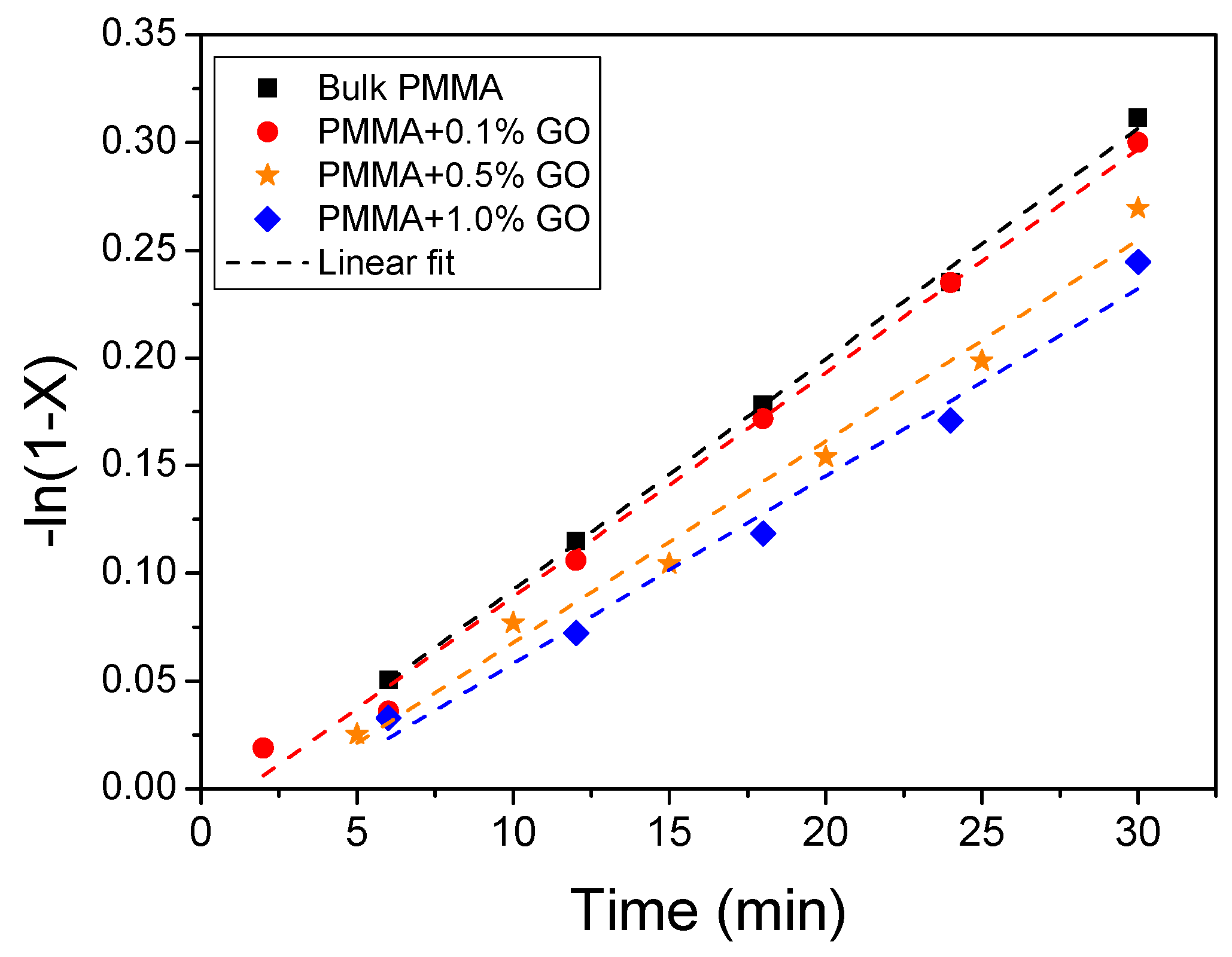
3.2. Polymerization Kinetics
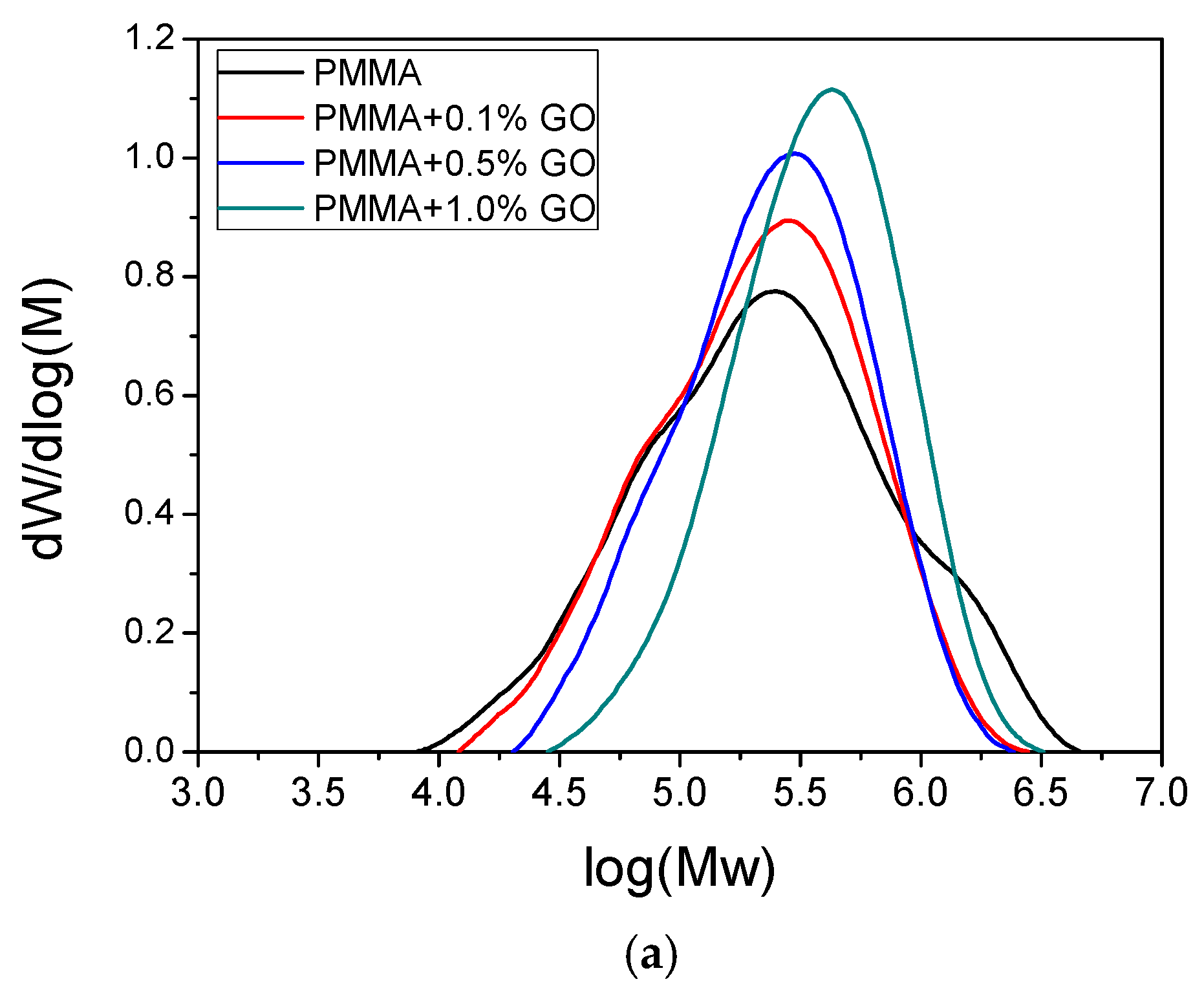
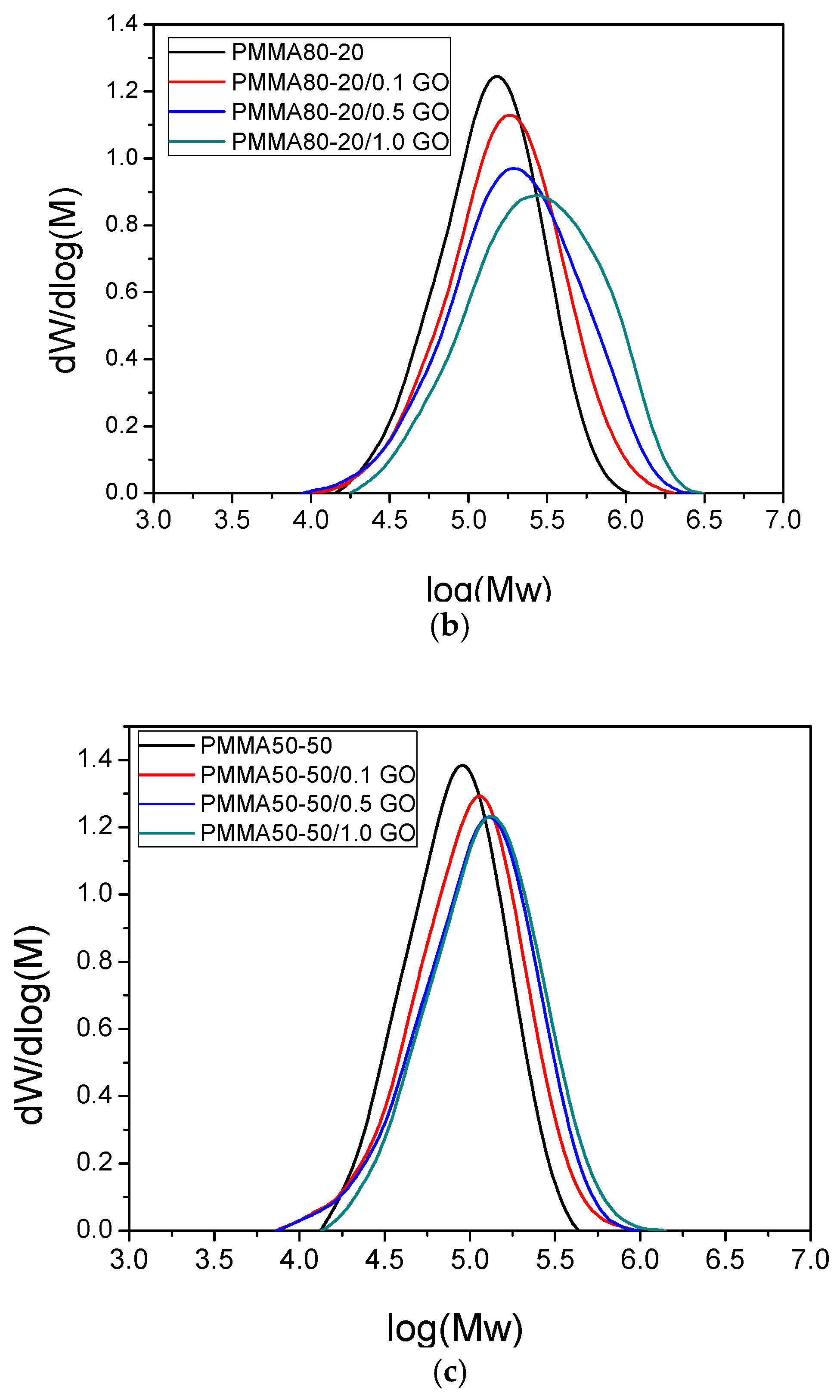
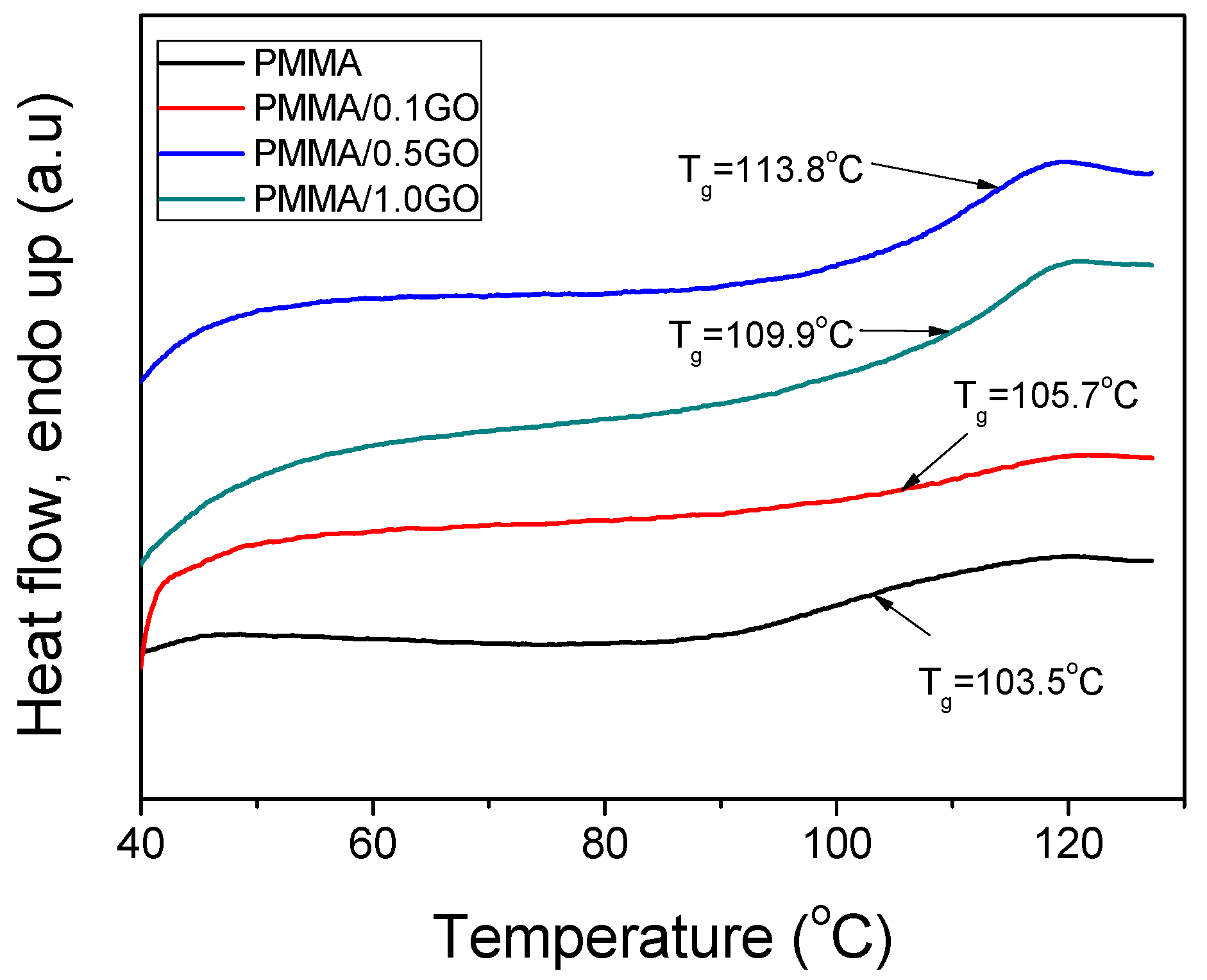
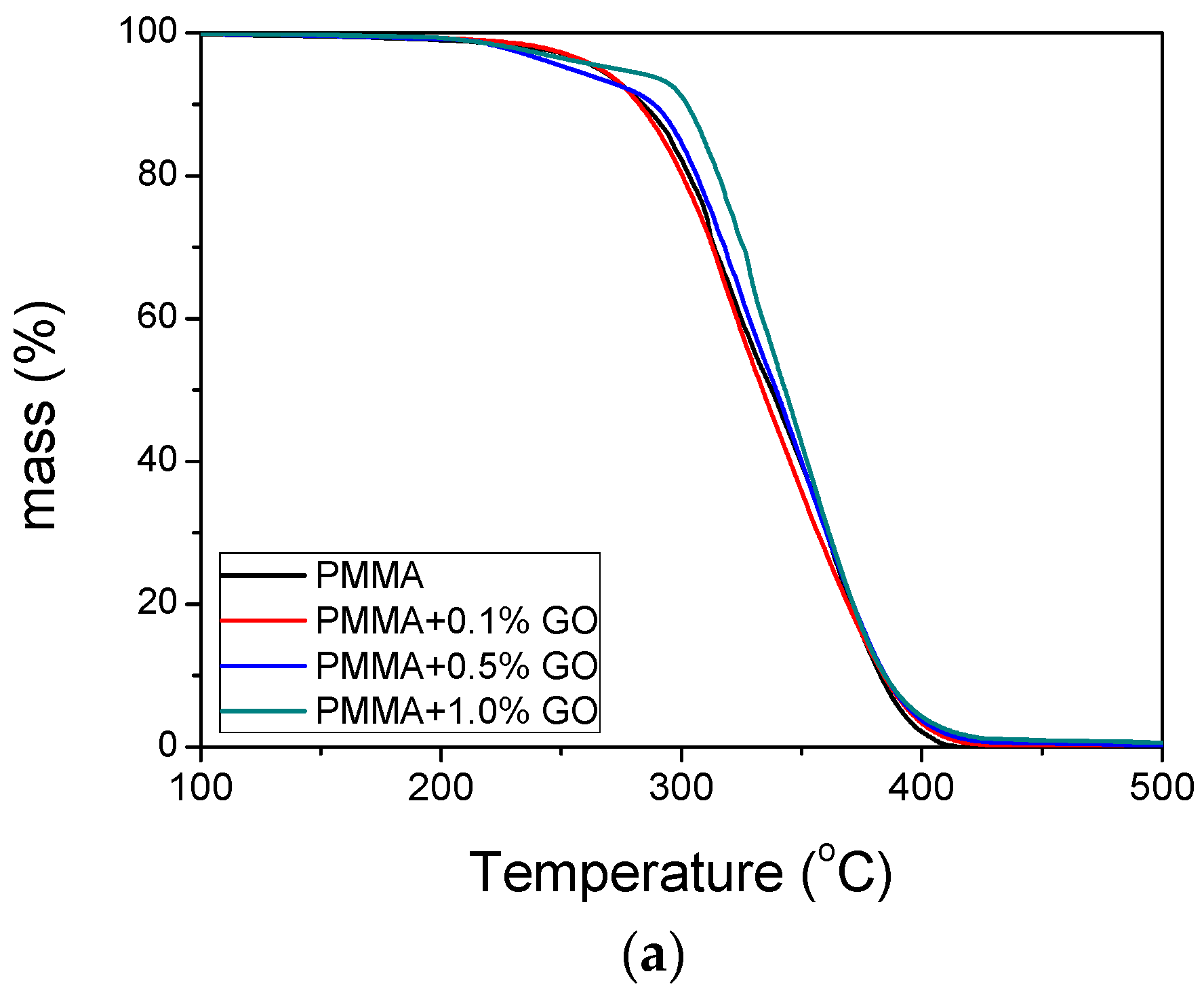
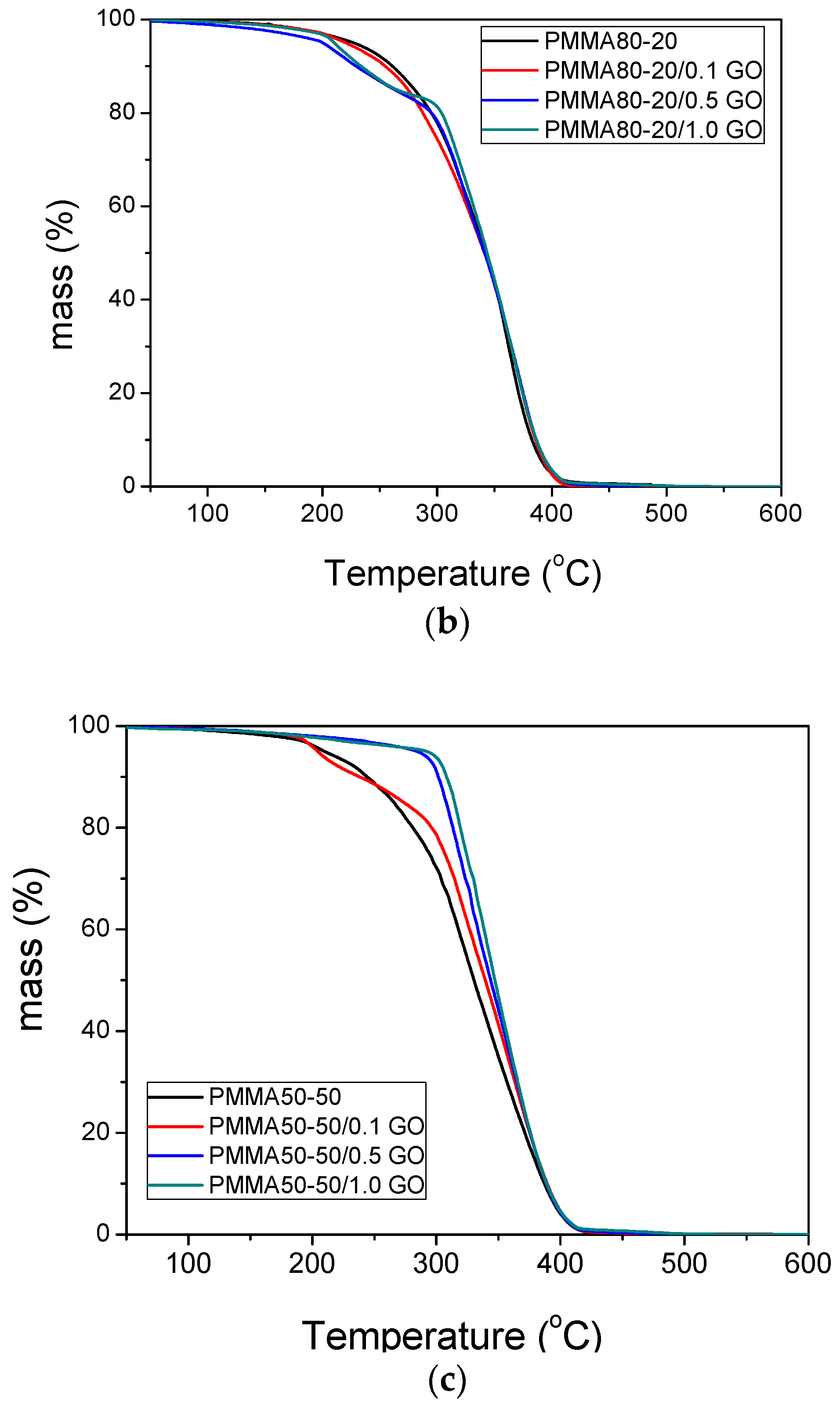
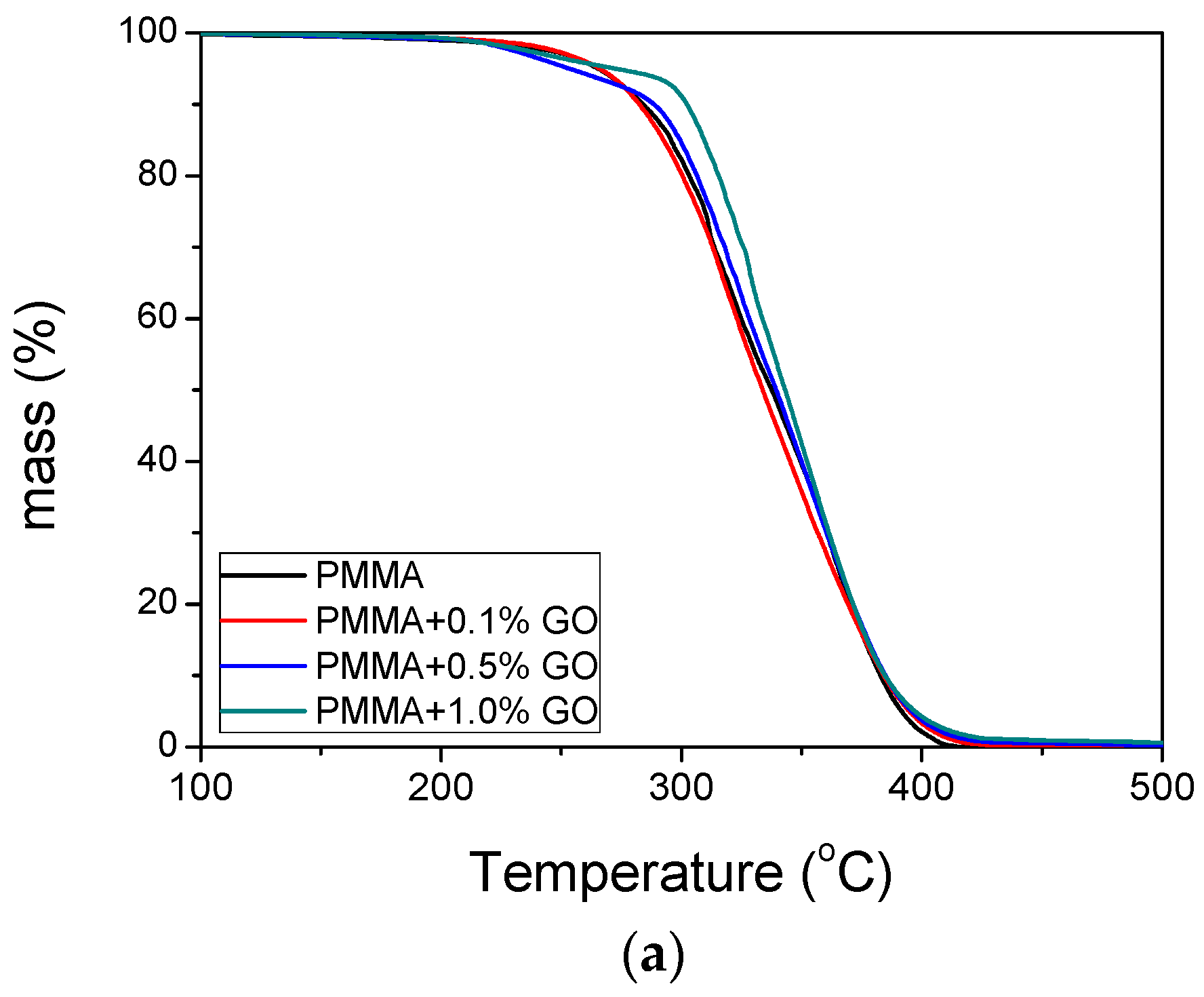
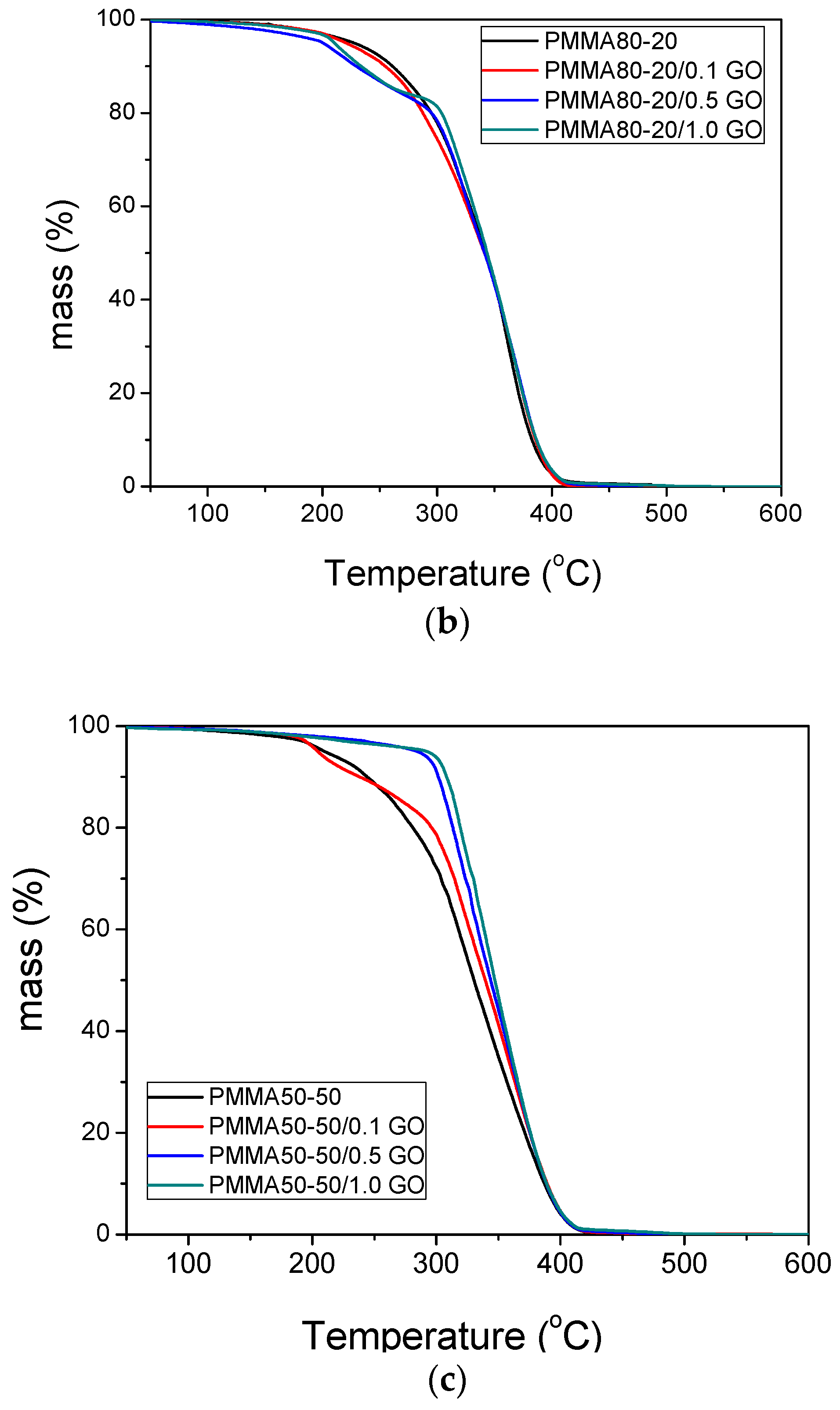
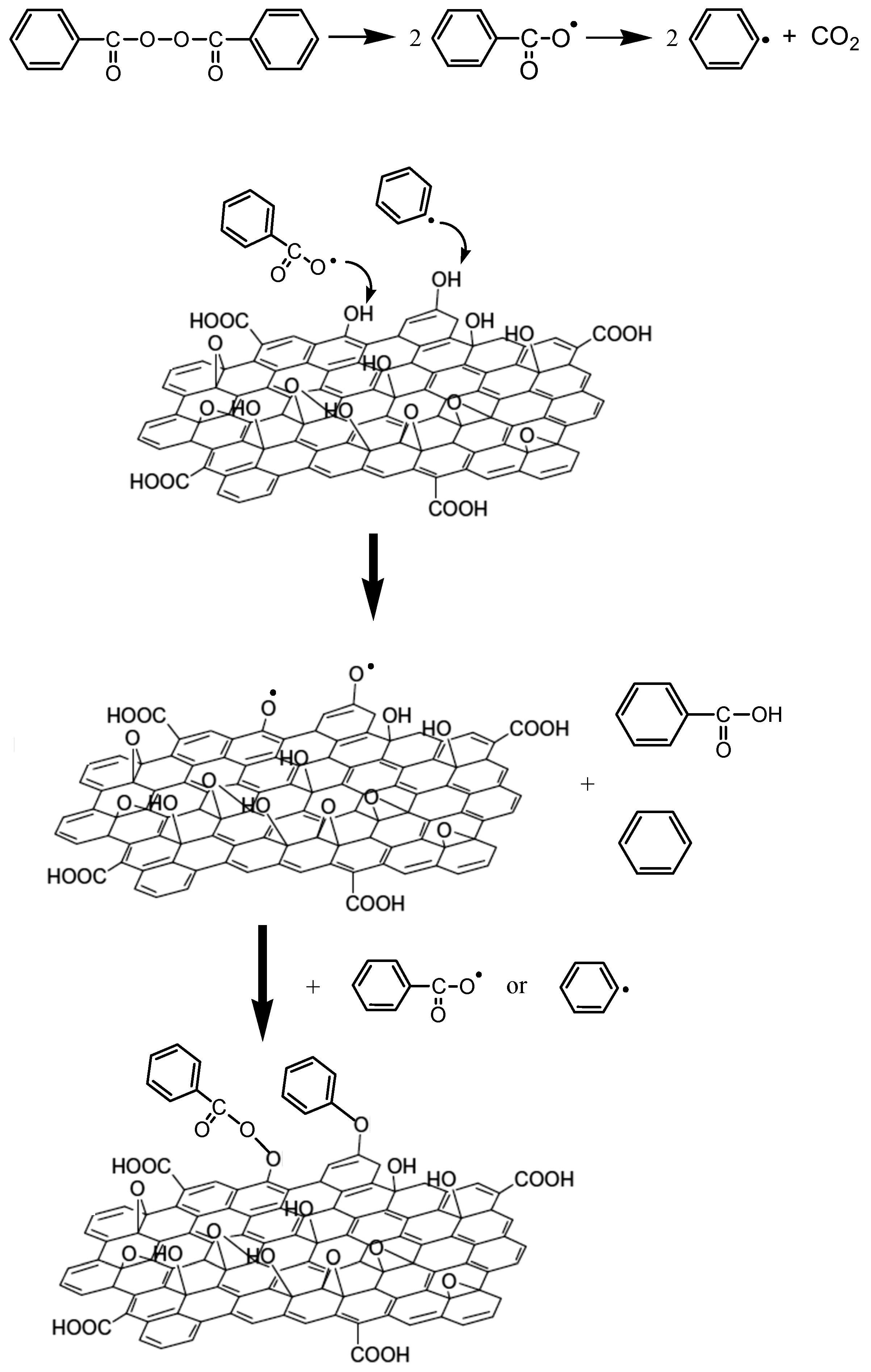
3.3. Thermal Properties of the Nanocomposites
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Kim, H.; Abdala, A.A.; Macosko, C.W. Graphene/Polymer Nanocomposites. Macromolecules 2010, 43, 6515–6530. [Google Scholar] [CrossRef]
- Park, S.; Ruoff, R.S. Chemical methods for the production of graphenes. Nat. Nano 2009, 4, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, D.R.; Park, S.; Bielawski, C.W.; Ruoff, R.S. The chemistry of graphene oxide. Chem. Soc. Rev. 2010, 39, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; An, J.; Jung, I.; Piner, R.D.; An, S.J.; Li, X.; Velamakanni, A.; Ruoff, R.S. Colloidal suspensions of highly reduced graphene oxide in a wide variety of organic solvents. Nano Lett. 2009, 9, 1593–1597. [Google Scholar] [CrossRef] [PubMed]
- Aldosari, M.A.; Othman, A.A.; Alsharaeh, E.H. Synthesis and characterization of the in situ bulk polymerization of PMMA containing graphene sheets using microwave irradiation. Molecules 2013, 18, 3152–3167. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.Y.; Zou, L.L.; Liao, C.C.; Dai, J.W. Improved properties of chemically modified graphene/ poly(methyl methacrylate) nanocomposites via a facile in-situ bulk polymerization. Express Polym. Lett. 2012, 6, 847–858. [Google Scholar] [CrossRef]
- Kuila, T.; Bose, S.; Hong, C.E.; Uddin, M.E.; Khanra, P.; Kim, N.H.; Lee, J.H. Preparation of functionalized graphene/linear low density polyethylene composites by a solution mixing method. Carbon 2011, 49, 1033–1051. [Google Scholar] [CrossRef]
- Achilias, D.S.; Nikolaidis, A.K.; Karayannidis, G.P. PMMA/organomodified montmorillonite nanocomposites prepared by in situ bulk polymerization: Study of the reaction kinetics. J. Therm. Anal. Calorim. 2010, 102, 451–460. [Google Scholar] [CrossRef]
- Achilias, D.S.; Siafaka, P.; Nikolaidis, A.K. Polymerization kinetics and thermal properties of poly(alkyl methacrylate)/organomodified montmorillonite nanocomposites. Polym. Int. 2012, 61, 1510–1518. [Google Scholar] [CrossRef]
- Siddiqui, M.N.; Redhwi, H.H.; Gkinis, K.; Achilias, D.S. Synthesis and characterization of novel nanocomposite materials based on poly(styrene-co-butyl methacrylate) copolymers and organomodified clay. Eur. Polym. J. 2013, 49, 353–365. [Google Scholar] [CrossRef]
- Siddiqui, M.N.; Redhwi, H.H.; Charitopoulou, D.; Achilias, D.S. Effect of organomodified clay on the reaction kinetics, properties and thermal degradation of nanocomposite based on poly(styrene-co-ethyl methacrylate). Polym. Int. 2014, 63, 766–777. [Google Scholar] [CrossRef]
- Nikolaidis, A.K.; Achilias, D.S.; Karayannidis, G.P. Effect of the type of organic modifier on the polymerization kinetics and the properties of poly(methyl methacrylate)/organomodified montmorillonite nanocomposites. Eur. Polym. J. 2012, 48, 240–251. [Google Scholar] [CrossRef]
- Paredes, J.I.; Villar-Rodil, S.; Martínez-Alonso, A.; Tascón, J.M.D. Graphene Oxide Dispersions in Organic Solvents. Langmuir 2008, 24, 10560–10564. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.N.; Redhwi, H.H.; Verros, G.D.; Achilias, D.S. Evaluating the role of nanomontmorillonite in bulk in situ radical polymerization kinetics of butyl methacrylate through a simulation model. Ind. Eng. Chem. Res. 2014, 53, 11303–11311. [Google Scholar] [CrossRef]
- Verros, G.D.; Achilias, D.S. Towards the development of a mathematical model for the bulk in situ radical polymerization of methyl methacrylate in the presence of nano-additives. Can. J. Chem. Eng. 2016, 94, 1783–1791. [Google Scholar] [CrossRef]
- Siddiqui, M.N.; Redhwi, H.H.; Vakalopoulou, E.; Tsagkalias, I.; Ioannidou, M.D.; Achilias, D.S. Synthesis, characterization and reaction kinetics of PMMA/silver nanocomposites prepared via in situ radical polymerization. Eur. Polym. J. 2015, 72, 256–269. [Google Scholar] [CrossRef]
- Verros, G.D.; Latsos, T.; Achilias, D.S. Development of a unified framework fr calculating molecular weight distribution in diffusion controlled free radical bulk homo-polymerization. Polymer 2005, 46, 539–552. [Google Scholar] [CrossRef]
- Achilias, D.S. A review of modelling of diffusion controlled polymerization reactions. Macromol. Theory Simul. 2007, 16, 319–347. [Google Scholar] [CrossRef]
- Verros, G.D.; Achilias, D.S. Modeling gel effect in branched polymer systems: Free radical solution homopolymerization of vinyl acetate. J. Appl. Polym. Sci. 2009, 111, 2171–2185. [Google Scholar] [CrossRef]
- Achilias, D.S.; Verros, G.D. Modelling of diffusion controlled reactions in free radical solution and bulk polymerization: Model validation by DSC experiments. J. Appl. Polym. Sci. 2010, 116, 1842–1856. [Google Scholar]
- Zoller, A.; Gigmes, D.; Guillaneuf, Y. Simulation of “cold” free radical polymerization of methyl methacrylate using a tertiary amine/BPO initiating system. Polym. Chem. 2015, 6, 5719–5727. [Google Scholar] [CrossRef]
- Michailidis, M.; Verros, G.D.; Deliyanni, E.A.; Andriotis, E.G.; Achilias, D.S. An Experimental and Theoretical Study of Butyl Methacrylate In Situ Radical Polymerization Kinetics in the Presence of Graphene Oxide Nanoadditive. J. Polym. Sci. A 2017, 55, 1433–1441. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | keff (min−1) | Standard Error | R2 | f |
|---|---|---|---|---|
| PMMA | 0.0107 | 2.68 × 10−4 | 0.9975 | 0.47 |
| PMMA/0.1GO | 0.01038 | 3.75 × 10−4 | 0.9935 | 0.44 |
| PMMA/0.5GO | 0.00935 | 5.65 × 10−4 | 0.9820 | 0.36 |
| PMMA/1.0GO | 0.0087 | 6.24 × 10−4 | 0.9797 | 0.31 |
| Sample | PD | Tg | |||
|---|---|---|---|---|---|
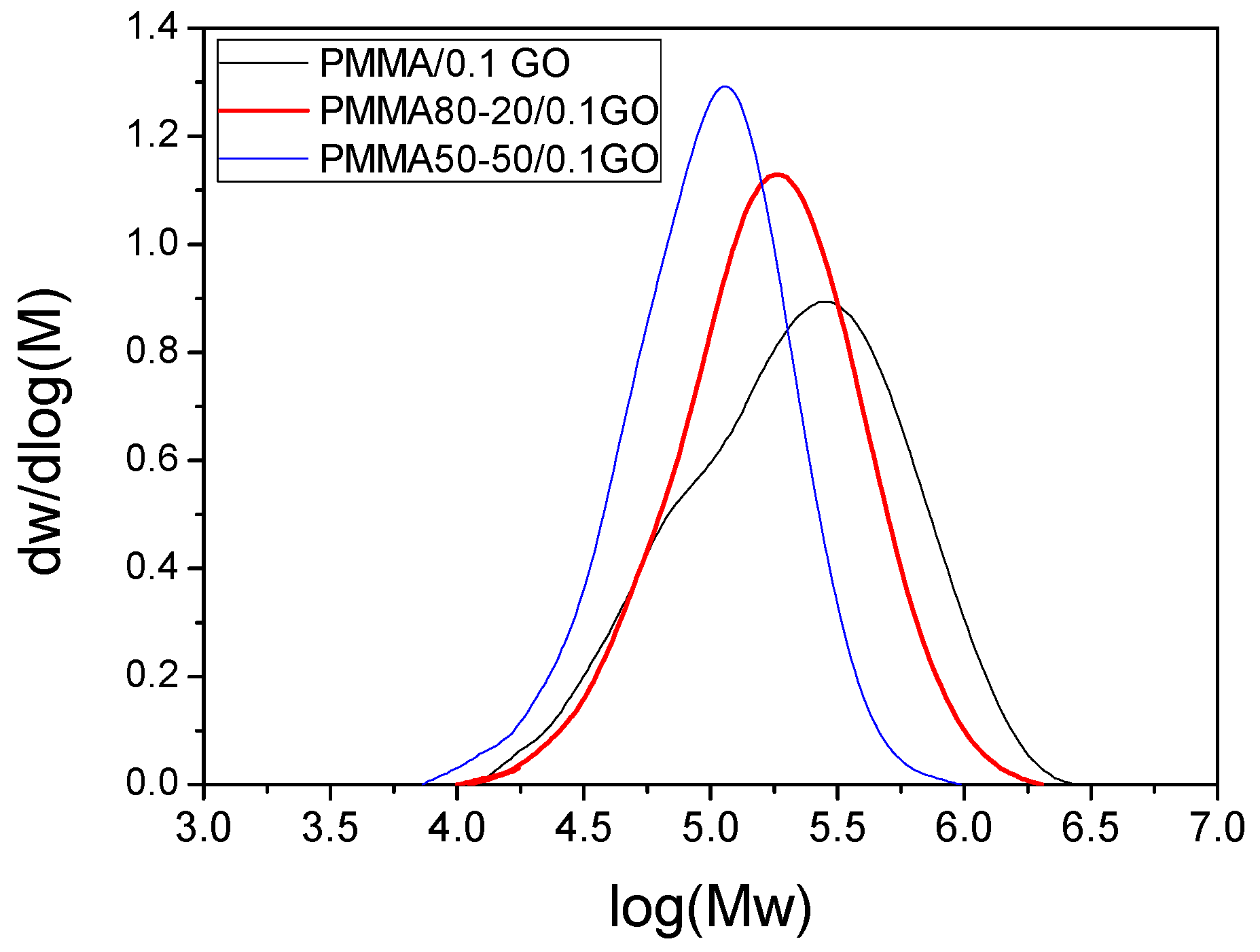
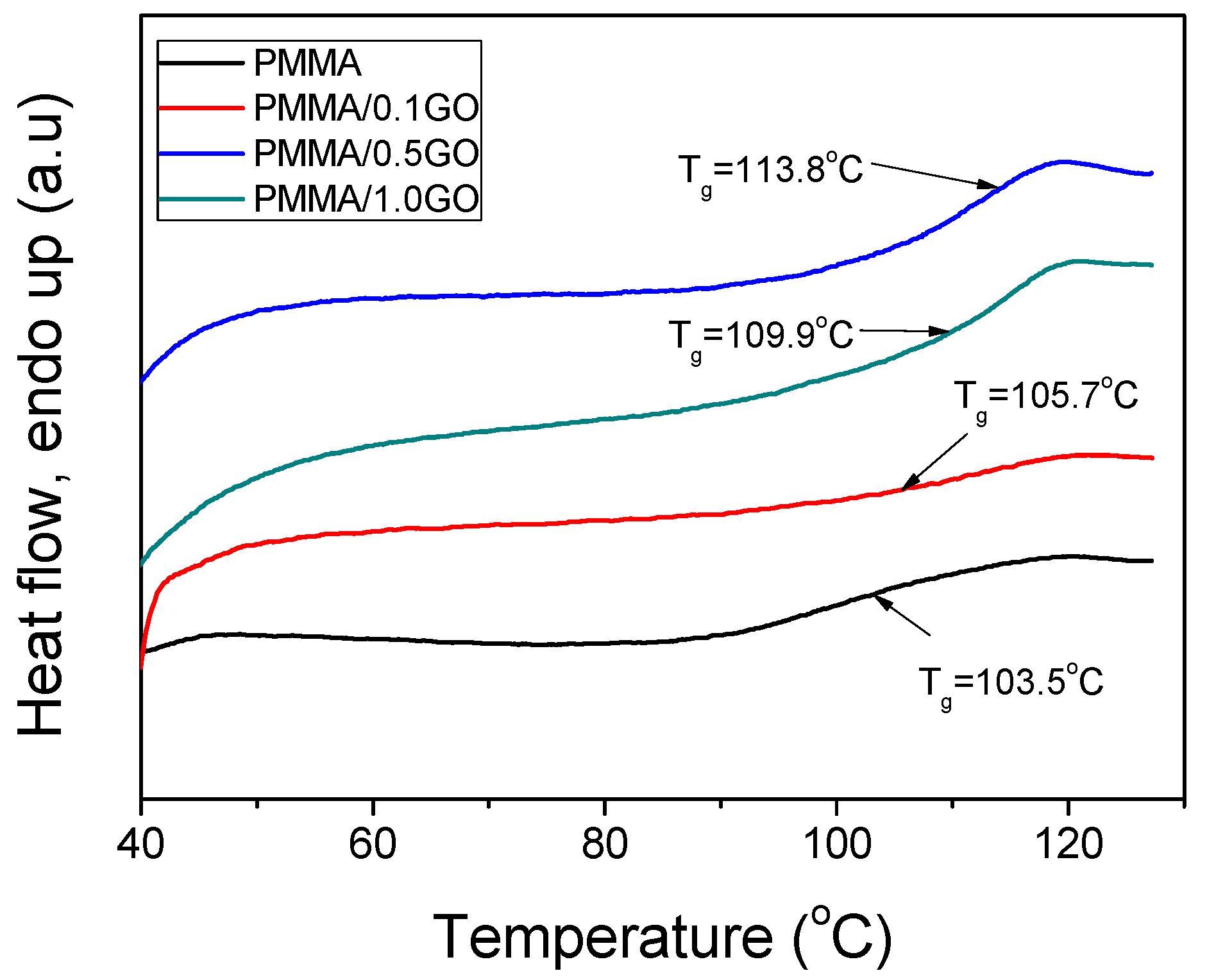
| PMMA | 114,570 | 427,250 | 1,121,690 | 3.73 | 103.5 |
| PMMA/0.1GO | 124,024 | 321,516 | 626,860 | 2.59 | 105.7 |
| PMMA/0.5GO | 160,872 | 339,196 | 592,744 | 2.11 | 109.9 |
| PMMA/1.0GO | 255,553 | 480,359 | 781,407 | 1.88 | 113.8 |
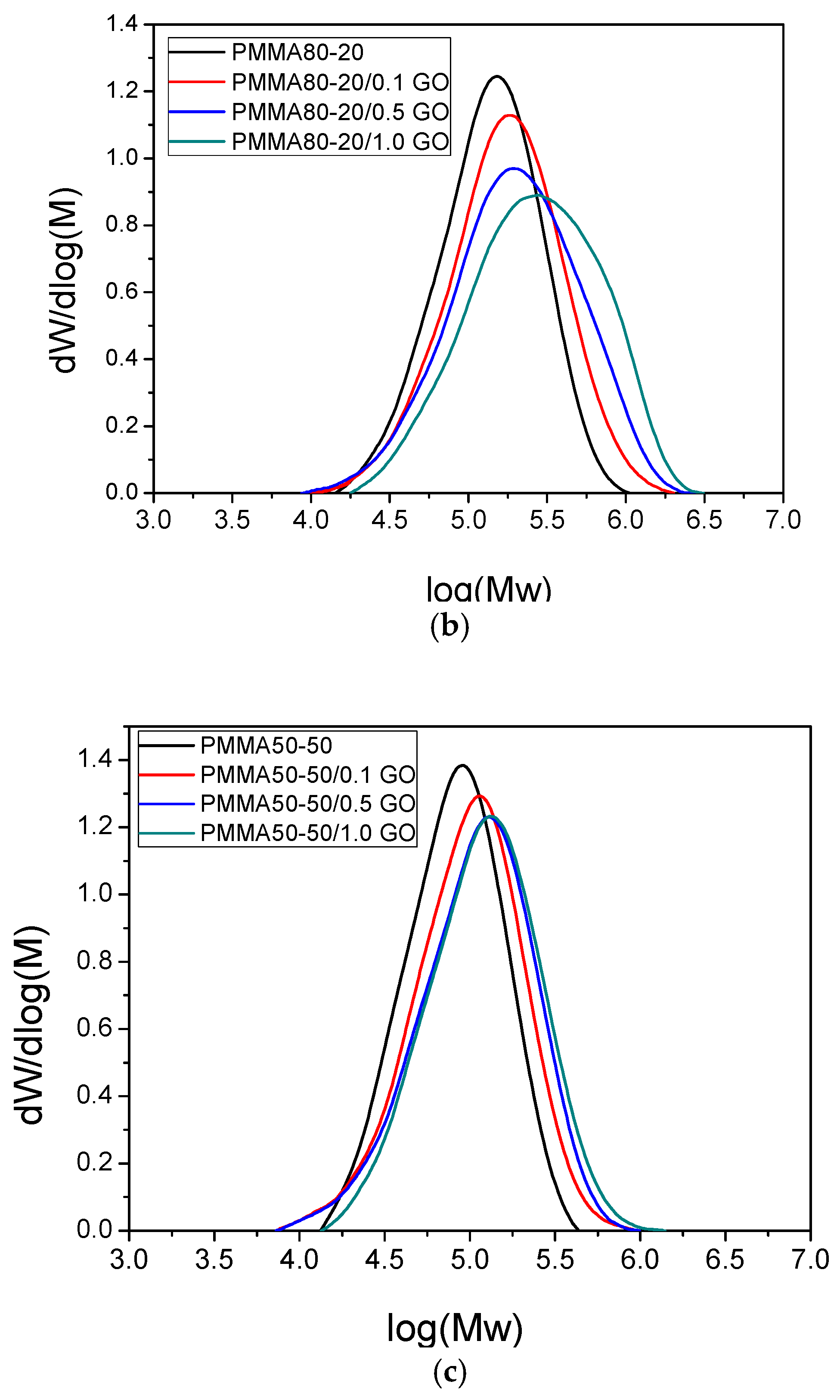
| PMMA80–20 | 102,673 | 173,944 | 267,436 | 1.69 | 86.6 |
| PMMA80–20/0.1GO | 119,534 | 233,037 | 409,315 | 1.95 | 88.0 |
| PMMA80–20/0.5GO | 144,446 | 333,565 | 634,007 | 2.31 | 91.1 |
| PMMA80–20/1.0GO | 168,209 | 390,492 | 723,507 | 2.32 | 92.9 |
| PMMA50–50 | 65,699 | 98,390 | 138,636 | 1.50 | 82.2 |
| PMMA50–50/0.1GO | 71,766 | 123,872 | 193,358 | 1.73 | 85.8 |
| PMMA50–50/0.5GO | 77,709 | 140,321 | 218,256 | 1.81 | 88.0 |
| PMMA50–50/1.0GO | 91,579 | 156,504 | 250,996 | 1.71 | 90.9 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsagkalias, I.S.; Manios, T.K.; Achilias, D.S. Effect of Graphene Oxide on the Reaction Kinetics of Methyl Methacrylate In Situ Radical Polymerization via the Bulk or Solution Technique. Polymers 2017, 9, 432. https://doi.org/10.3390/polym9090432
Tsagkalias IS, Manios TK, Achilias DS. Effect of Graphene Oxide on the Reaction Kinetics of Methyl Methacrylate In Situ Radical Polymerization via the Bulk or Solution Technique. Polymers. 2017; 9(9):432. https://doi.org/10.3390/polym9090432
Chicago/Turabian StyleTsagkalias, Ioannis S., Triantafyllos K. Manios, and Dimitris S. Achilias. 2017. "Effect of Graphene Oxide on the Reaction Kinetics of Methyl Methacrylate In Situ Radical Polymerization via the Bulk or Solution Technique" Polymers 9, no. 9: 432. https://doi.org/10.3390/polym9090432