
Generation and Characterisation of a Reference Transcriptome for Phalaris (Phalaris aquatica L.)

 ,
,
Abstract
:1. Introduction
2. Results
2.1. De Novo Sequence Assembly of the Phalaris Transcriptome
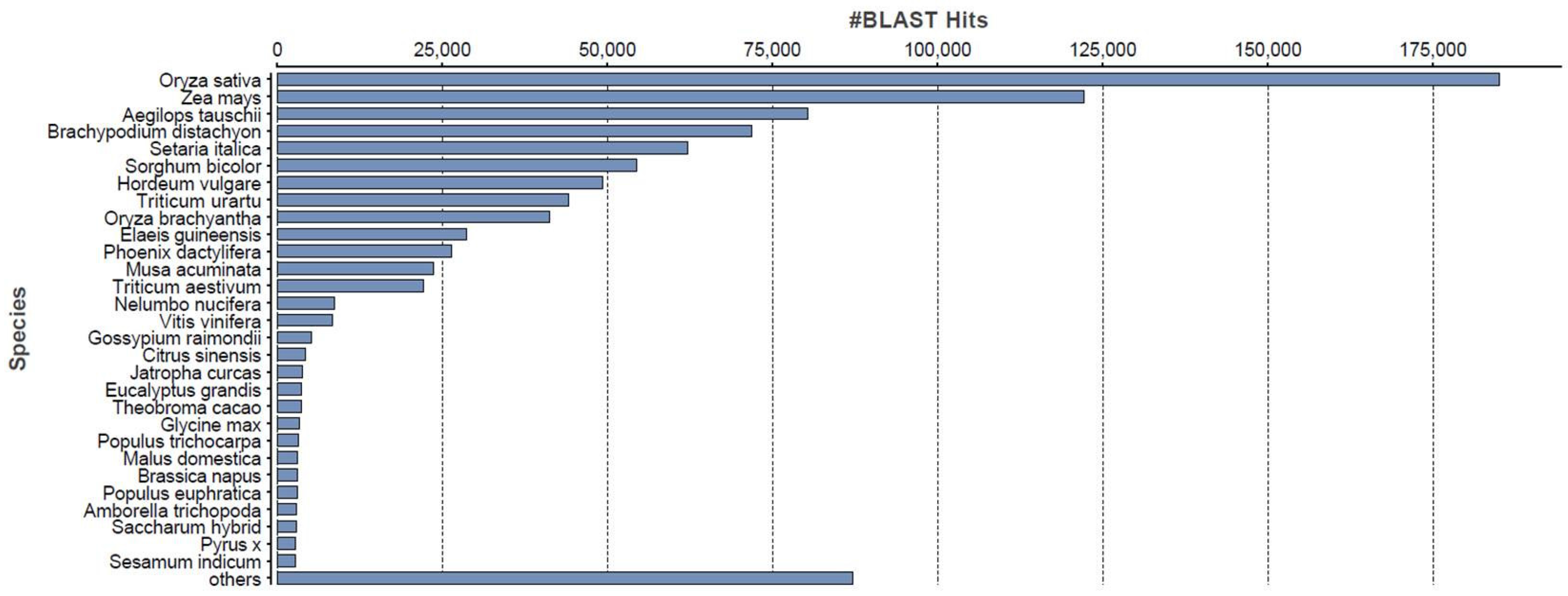
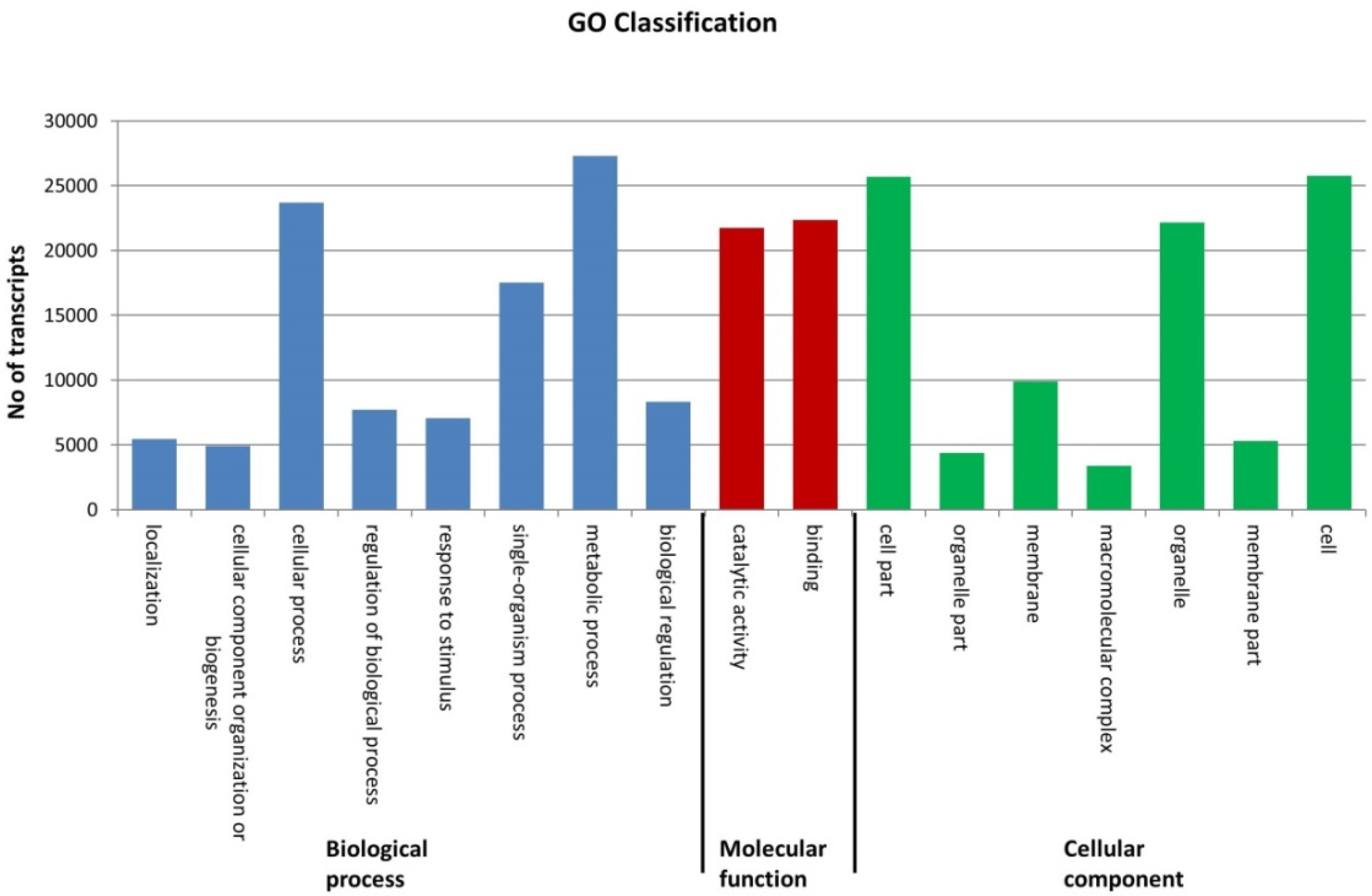
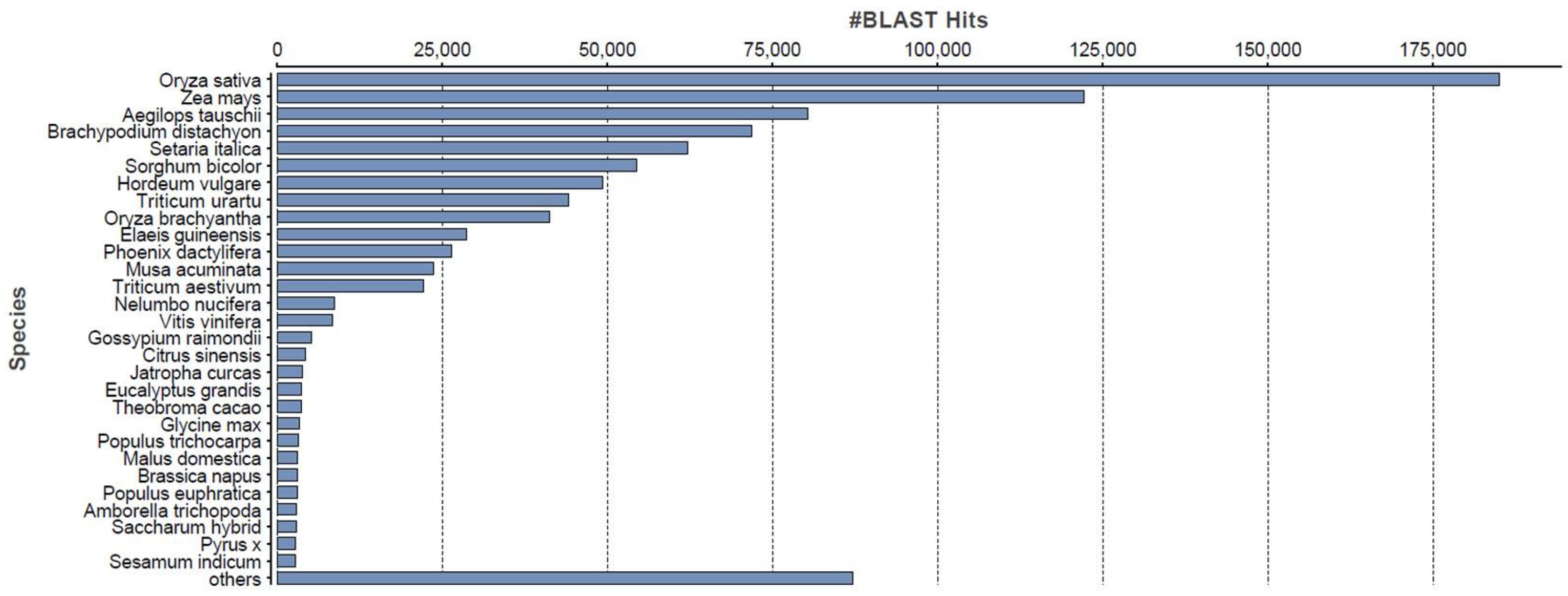
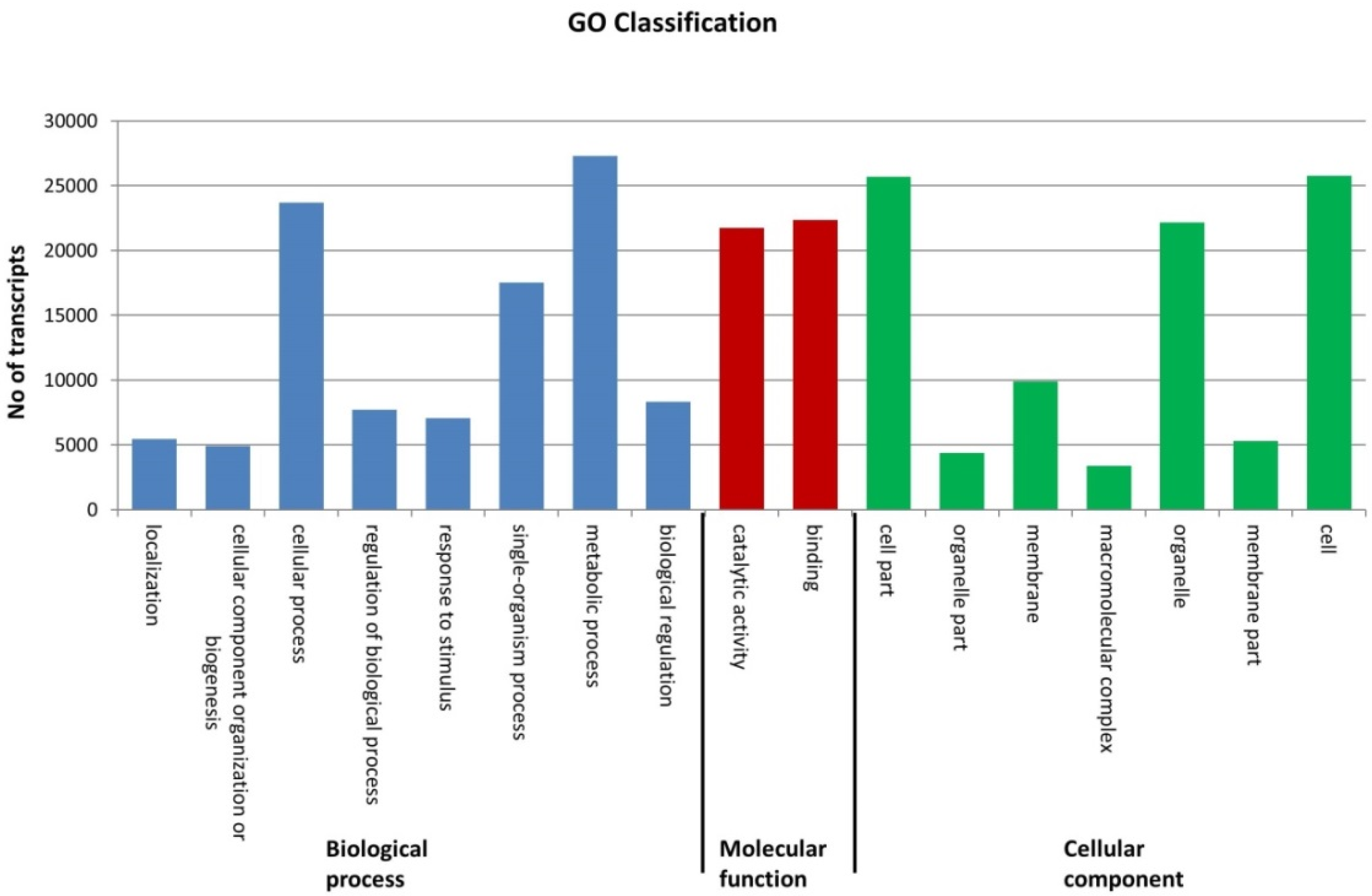
2.2. Sequence Annotation of the Phalaris Transcriptome
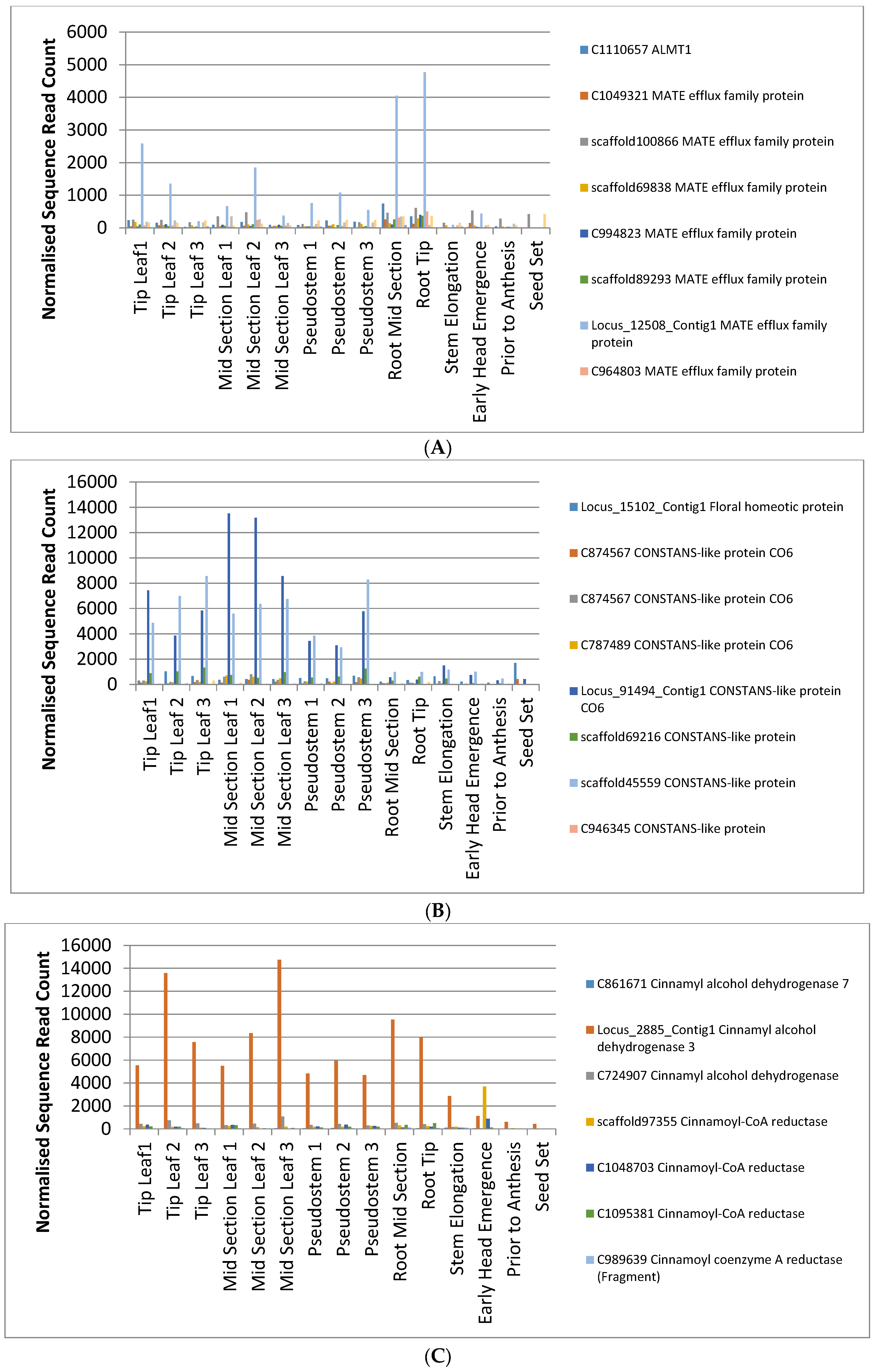
2.3. Tissue-Specific Gene Expression Analysis in Phalaris
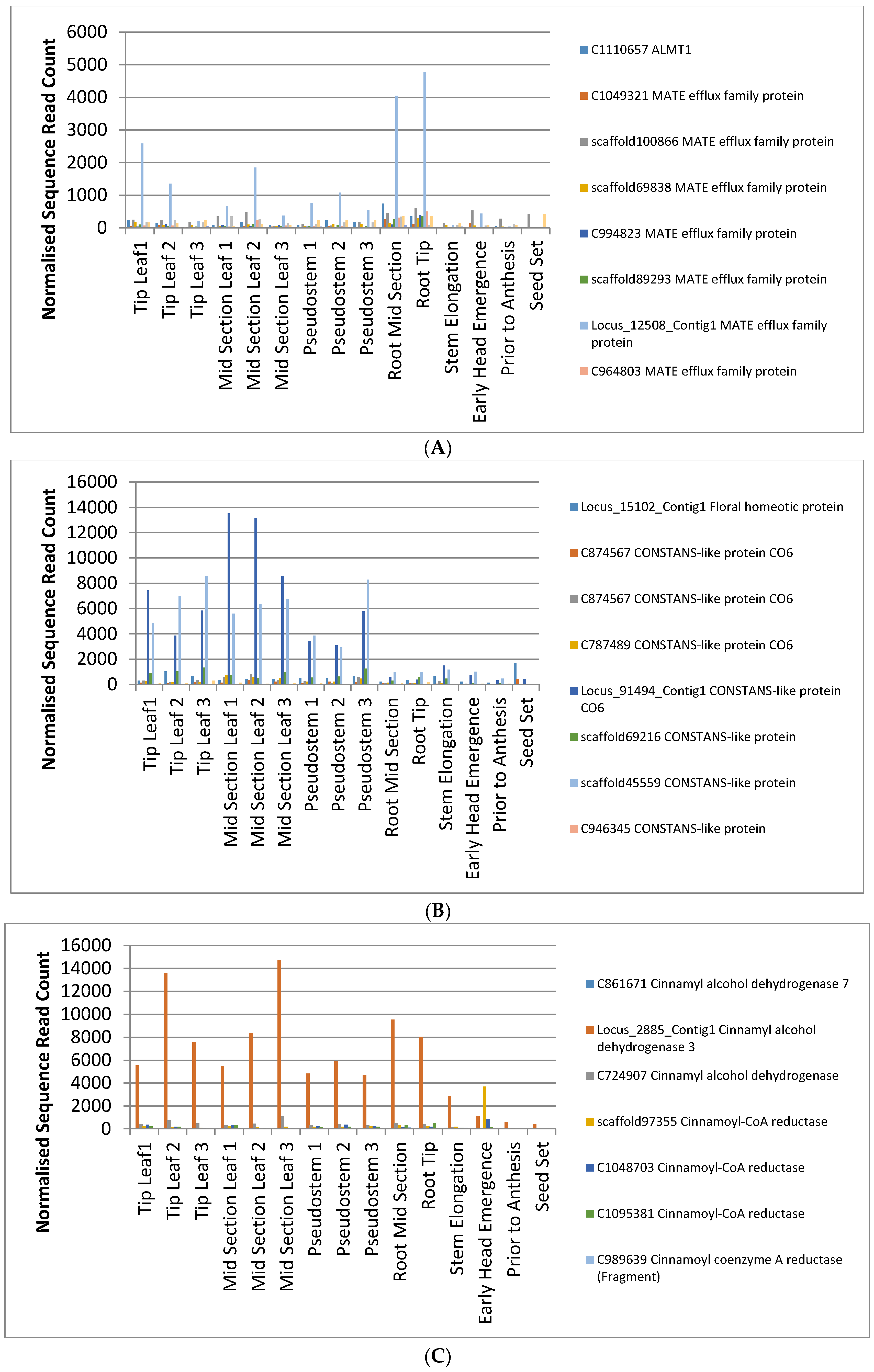
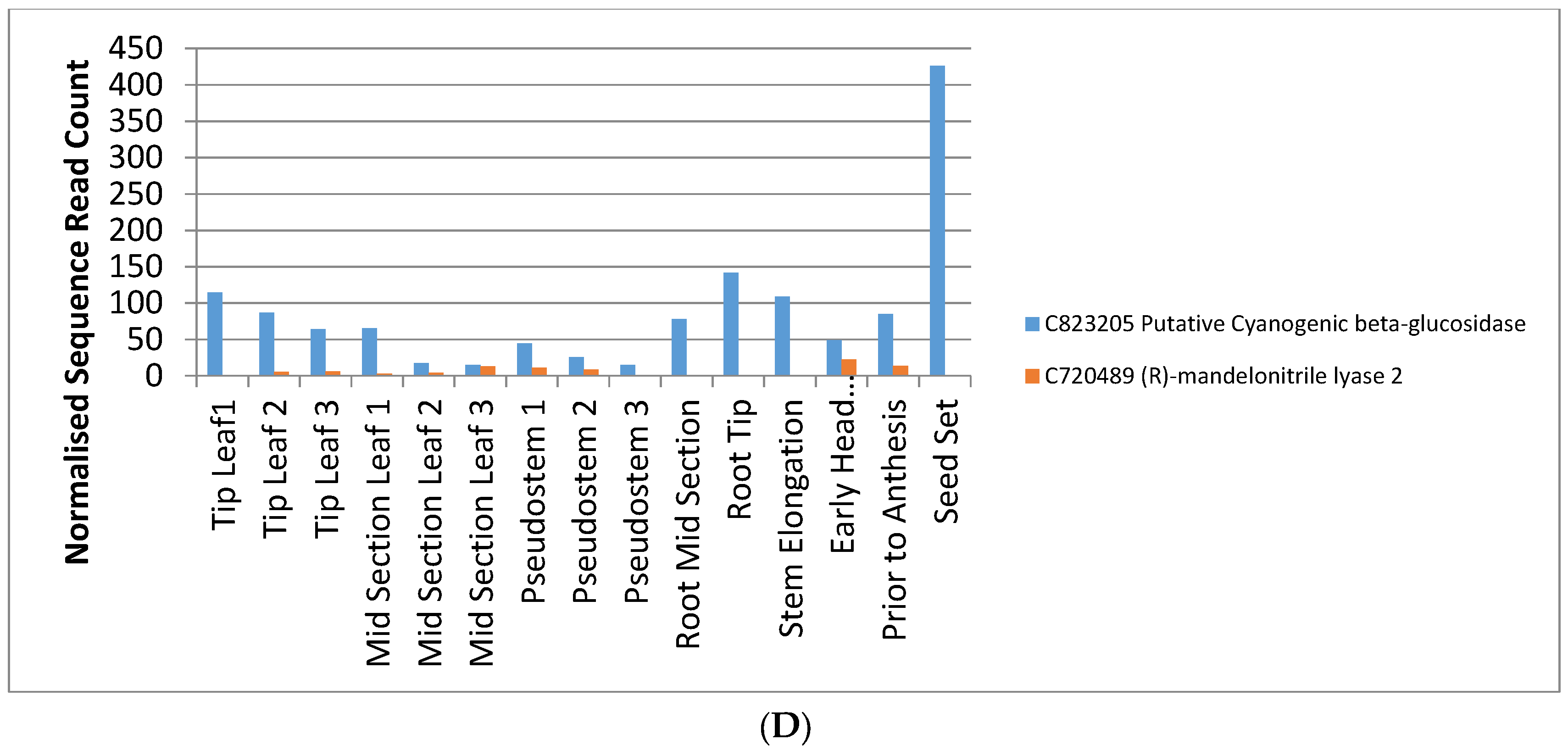
2.4. Identification of Candidate Genes for Agronomic Traits in the Phalaris Transcriptome
2.5. Identification of Long Transcripts and Molecular Phylogenetic Analysis
3. Discussion
3.1. Quality of Transcriptome Assembly
3.2. Identification and Interpretation of Candidate Genes for Agronomic Traits
3.3. Applications to Genomics-Assisted Breeding of Phalaris
4. Materials and Methods
4.1. Plant Materials
4.2. De Novo Transcriptome Sequence Assembly
4.3. De Novo Transcriptome Sequence Annotation and Tissue-Specific Expression
4.4. Identification of Long Transcripts and Molecular Phylogenetic Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Oram, R.N.; Ferreira, V.; Culvenor, R.A.; Hopkins, A.A.; Stewart, A. The first century of Phalaris aquatica L. cultivation and genetic improvement: A review. Crop Pasture Sci. 2009, 60, 1–15. [Google Scholar] [CrossRef]
- Anderson, M.W.; Cunningham, P.J.; Reed, K.F.M.; Byron, A. Perennial grasses of Mediterranean origin offer advantages for central western Victorian sheep pasture. Aust. J. Exp. Agric. 1999, 39, 275–284. [Google Scholar] [CrossRef]
- Culvenor, R.A. Breeding and use of summer-dormant grasses in Australia, with special reference to phalaris. Crop Sci. 2009, 49, 2335–2346. [Google Scholar] [CrossRef]
- Helyar, K.R.; Anderson, A.J. Effects of lime on the growth of five species, on aluminium toxicity, and on phosphorus availability. Aust. J. Agric. Res. 1971, 22, 707–721. [Google Scholar] [CrossRef]
- McWilliam, J.R. Selection for seed retention in Phalaris tuberosa L. Aust. J. Agric. Res. 1963, 14, 755–764. [Google Scholar] [CrossRef]
- Alden, R.; Hackney, B.; Weston, L.A.; Quinn, J.C. Phalaris toxicoses in Australian livestock production systems: Prevalence, aetiology and toxicology. J. Toxins 2014, 1, 1–7. [Google Scholar]
- Gallagher, C.H.; Koch, J.H.; Moore, R.M.; Hoffmann, H. Toxicity of Phalaris tuberosa for sheep. Nature 1964, 204, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Bourke, C.A. Toxins in pasture plants—Phalaris toxicity. Anim. Prod. Aust. 1992, 19, 399–402. [Google Scholar]
- Bourke, C.A.; Carrigan, M.J. Mechanisms underlying Phalaris aquatica “sudden death” syndrome in sheep. Aust. Vet. J. 1992, 69, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Quintanar, A.; Castroviejo, S.; Catalán, P. Phylogeny of the trobe Aveneae (Pooideae, Poaceae) inferred from plastid trnT-F and nuclear ITS sequences. Am. J. Bot. 2007, 94, 1554–1569. [Google Scholar] [CrossRef] [PubMed]
- Voshell, S.M.; Baldini, R.M.; Kumar, R.; Tatalovich, N.; Hilu, K. Canary grasses (Phalaris, Poaceae): Molecular phylogenetics, polyploidy and floret evolution. Taxon 2011, 60, 1306–1316. [Google Scholar]
- Voshell, S.M.; Hilu, K.W. Canary grasses (Phalaris, Poaceae): Biogeography, molecular dating and the role of floret structure in dispersal. Mol. Ecol. 2014, 23, 212–214. [Google Scholar] [CrossRef] [PubMed]
- Starling, J.I. Cytogenetic study of interspecific hybrids between Phalaris arundinacea and P. tuberosa. Crop Sci. 1961, 1, 107–111. [Google Scholar] [CrossRef]
- Putievsky, E.; Oram, R.N.; Malafant, K. Chromosomal differentiation among ecotypes of Phalaris aquatica L. Aust. J. Bot. 1980, 28, 645–657. [Google Scholar] [CrossRef]
- Stebbins, G.L. Variation and Evolution in Plants; Columbia University Press: New York, NY, USA; London, UK, 1950. [Google Scholar]
- Ferreira, V.; Reynoso, L.; Szpiniak, B.; Grass, E. Cytological analysis of the Phalaris arundinacea (L.) x Phalaris aquatica (L.) amphidiploid. Caryologia 2002, 55, 151–160. [Google Scholar] [CrossRef]
- Lavergne, S.; Muenke, N.J.; Molofsky, J. Genome size reduction can trigger rapid phenotypic evolution in invasive plants. Ann. Bot. 2010, 105, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Doležel, J.; Bartoš, J.; Voglmayr, H.; Greilhuber, J. Nuclear DNA content and genome size of trout and human. Cytometry A 2003, 51, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, Y.; Kimura, K.; Kubota, A.; Fujimori, M.; Yamada-Akiyama, H.; Takahara, Y.; Ueyama, Y. Comparison of genome size in reed canarygrass (Phalaris arundinacea L.) exotic and putative native Japanese genotypes by flow cytometry. Jpn. Agric. Res. Q. 2015, 49, 345–350. [Google Scholar] [CrossRef]
- Gutierrez-Gonzalez, J.J.; Tu, Z.J.; Garvin, D.F. Analysis and annotation of the hexaploid oat seed transcriptome. BMC Genom. 2013, 14, 471. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Francki, M.G.; Forster, J.W. Identification, characterisation and interpretation of single nucleotide sequence variation in allopolyploid crop plant species. Plant Biotechnol. J. 2012, 10, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Mian, M.A.R.; Zwonitzer, J.C.; Chen, Y.; Saha, M.C.; Hopkins, A.A. AFLP diversity within and among hardinggrass populations. Crop Sci. 2005, 45, 2591–2597. [Google Scholar] [CrossRef]
- Hayes, B.J.; Cogan, N.O.I.; Pembleton, L.W.; Goddard, M.E.; Wang, J.; Spangenberg, G.C.; Forster, J.W. Prospects for genomic selection in forage plant species. Plant Breed. 2013, 132, 133–143. [Google Scholar] [CrossRef]
- Forster, J.W.; Hand, M.L.; Cogan, N.O.I.; Hayes, B.; Spangenberg, G.C.; Smith, K.F. Resources and strategies for implementation of genomic selection in breeding of forage species. Crop Pasture Sci. 2014, 65, 1238–1247. [Google Scholar] [CrossRef]
- Garg, R.; Jain, M. RNA-Seq for transcriptome analysis in non-model plants. Methods Mol. Biol. 2013, 1069, 43–58. [Google Scholar]
- Zonneveld, B.J.M.; Leitch, I.J.; Bennett, M.D. First nuclear DNA amounts in more than 300 angiosperms. Ann. Bot. 2005, 96, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.P.; Garvin, D.F.; Mockler, T.C.; Schmutz, J.; Rokhsar, D.; Bevan, M.W.; Barry, K.; Lucas, S.; Harmon-Smith, M.; Lail, K. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 2010, 463, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Moreton, J.; Izquierdo, A.; Emes, R.D. Assembly, assessment and availability of de novo generated eukaryotic transcriptomes. Front. Genet. 2016, 6, 361. [Google Scholar] [CrossRef] [PubMed]
- Haiminen, N.; Klaas, M.; Zhou, Z.; Utro, F.; Cormican, P.; Didion, T.; Sig Jensen, C.; Mason, C.E.; Barth, S.; Parida, L. Comparative exomics of Phalaris cultivars under salt stress. BMC Genom. 2014, 15 (Suppl. 6), S18. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Jiao, C.; Zheng, Y.; Sun, H.; Liu, W.; Cai, X.; Wang, X.; Liu, S.; Xu, Y.; Mou, B.; et al. De novo and comparative transcriptome analysis of cultivated and wild spinach. Sci. Rep. 2015, 5, 17706. [Google Scholar] [CrossRef] [PubMed]
- Yates, S.A.; Swain, M.T.; Hegarty, M.J.; Chernukin, I.; Lowe, M.; Allison, G.G.; Ruttink, T.; Abberton, M.T.; Jenkins, G.; Skøt, L. De novo assembly of red clover transcriptome based on RNA-Seq data provides insight into drought response, gene discovery and marker identification. BMC Genom. 2014, 15, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, J.D.; Byrne, S.; Paina, C.; Asp, T. De novo assembly of the perennial ryegrass transcriptome using RNA-Seq strategy. PLoS ONE 2014, 9, e103567. [Google Scholar] [CrossRef] [PubMed]
- Krasileva, K.V.; Buffalo, V.; Bailey, P.; Pearce, S.; Ayling, S.; Tabbita, F.; Soria, M.; Wang, S.; Akhunov, E.; Uauy, C.; et al. Separating homeologs by phasing in the tetraploid wheat transcriptome. Genome Biol. 2013, 14, R66. [Google Scholar] [CrossRef] [PubMed]
- Delhaize, E.; Ryan, P.R. Aluminium toxicity and tolerance in plants. Plant Phys. 1995, 107, 315–321. [Google Scholar] [CrossRef]
- Culvenor, R.A.; Oram, R.N.; Wood, J.T. Inheritance of aluminium tolerance in Phalaris aquatica L. Aust. J. Agric. Res. 1986, 37, 397–408. [Google Scholar] [CrossRef]
- Inostroza-Blancheteau, C.; Soto, B.; Ibáñez, C.; Ulloa, P.; Aquea, F.; Arce-Johnson, P.; Reyes-Diaz, M. Mapping aluminium tolerance in cereals: A tool available for crop breeding. Eur. J. Biotechnol. 2010, 13, 4. [Google Scholar]
- Sasaki, T.; Yamamoto, Y.; Ezaki, B.; Katsuhara, M.; Sung, J.U.; Ryan, P.R.; Delhaize, E.; Matsumoto, H. A wheat gene encoding an aluminium-activated malate transporter. Plant J. 2004, 37, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, J.; Yamaji, N.; Wang, H.; Mitani, N.; Murata, Y.; Sato, K.; Katsuhara, M.; Takeda, K.; Ma, J.-F. An aluminium-activated citrate transporter of barley. Plant Cell Phys. 2007, 48, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.J.; Fellers, J.P.; Trick, H.N.; Zhang, Z.; Tai, Y.-S.; Gill, B.S.; Faris, J.D. Molecular characterisation of the major wheat domestication gene Q. Genetics 2006, 172, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.P.; McWilliam, J.R. Climatic variation in forage grasses. II. Germination, flowering and leaf development in Mediterranean populations of Phalaris tuberosa. J. Appl. Ecol. 1966, 3, 191–212. [Google Scholar] [CrossRef]
- Suárez-López, P.; Wheatley, K.; Robson, F.; Onouichi, H.; Valverde, F.; Coupland, G. CONSTANS mediates between the circadian clock and the control of flowering in Arabidopsis. Nature 2001, 410, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Busk, P.K.; Møller, B.L. Dhurrin synthesis in sorghum is regulated at the transcriptional level and induced by nitrogen fertilisation in older plants. Plant Phys. 2002, 129, 1222–1231. [Google Scholar] [CrossRef] [PubMed]
- Cheeke, P.R. Endogenous toxins and mycotoxins in forage grasses and their effects on livestock. J. Anim. Sci. 1995, 73, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Oram, R.N.; Edlington, J.P. Breeding non-toxic phalaris (Phalaris aquatica L.). In Proceedings of the 8th Australian Agronomy Conference, University of Southern Queensland, Toowoomba, Queensland, Australia, 30 January–2 February 1996; Asghar, M., Ed.; Australian Society of Agronomy Carlton: Victoria, Australia, 1996. Available online: http://www.regional.org.au/au/asa/1996/contributed/450oram.htm (accessed on 13 February 2017). [Google Scholar]
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.-X.; Liu, H.; Li, Z. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 2014, 5, 484. [Google Scholar] [CrossRef] [PubMed]
- Harper, A.L.; Trick, M.; Higgins, J.; Fraser, F.; Clissold, L.; Wells, R.; Hattori, C.; Werner, P.; Bancroft, I. Associative transcriptomics of traits in the polyploidy crop species Brassica napus. Nat. Biotechnol. 2012, 30, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Wang, J. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013. [Google Scholar]
- Sudheesh, S.; Sawbridge, T.I.; Cogan, N.O.I.; Kennedy, P.; Forster, J.W.; Kaur, S. De novo assembly and characterisation of the field pea transcriptome using RNA-Seq. BMC Genom. 2015, 16, 611. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. ClustalW and ClustalX Version 2. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Assembly—SOAPdenovo-Trans | |
| Total number of filtered reads | 553,566,274 |
| Total number of contigs | 437,776 |
| N50 length | 1304 |
| Total base pairs | 233,054,090 |
| Secondary Assembly—CAP3 and Filtering | |
| Total number of scaffolds and contigs | 56,873 |
| N50 length | 1558 |
| Total base pairs | 58,174,765 |
| Trait Category/Common Gene Name | Uniref90 Description | Brachypodium BLAST Match | Rice BLAST Match |
|---|---|---|---|
| Flowering | |||
| Q gene | Floral homeotic protein | Bradi1g03880.1 | Os07g0235800 |
| CONSTANS-like protein CO6 | |||
| CONSTANS-like protein CO6 | Bradi3g05800.1 | Os06g0654900 | |
| CONSTANS-like protein CO6 | |||
| CONSTANS-like protein CO6 | Bradi1g31280.1 | Os04g0497700 | |
| CONSTANS-like protein | Bradi5g14600.1 | Os04g0497700 | |
| CONSTANS-like protein | Bradi5g14600.1 | Os04g0497700 | |
| CONSTANS-like protein | Bradi1g43670.1 | ||
| CONSTANS-like protein | |||
| CONSTANS | Bradi1g43670.1 | ||
| Aluminium Tolerance | |||
| ALMT1 | ALMT1 | Bradi5g09690.1 | Os04g0417000 |
| MATE efflux family protein | Bradi2g17260.1 | ||
| MATE efflux family protein | Bradi1g69120.1 | Os03g0227966 | |
| MATE efflux family protein | |||
| MATE efflux family protein | |||
| MATE efflux family protein | Bradi1g69120.1 | Os03g0227966 | |
| MATE efflux family protein | Bradi2g17260.1 | Os05g0554000 | |
| MATE efflux family protein | Os02g0676400 | ||
| Aluminum-activated malate transporter 12 | Bradi3g33980.1 | Os10g0572100 | |
| Aluminum resistance transcription factor 1 | |||
| Aluminum resistance transcription factor 1 | |||
| Toxin Biosynthesis | |||
| Putative Cyanogenic beta-glucosidase | |||
| (R)-mandelonitrile lyase 2 | Bradi1g31250.1 | ||
| Herbage Digestibility | |||
| Cinnamyl alcohol dehydrogenase 7 | |||
| Cinnamyl alcohol dehydrogenase 3 | Bradi4g29770.1 | Os09g0400400 | |
| Cinnamyl alcohol dehydrogenase | Bradi4g29770.1 | Os09g0399800 | |
| Cinnamoyl-CoA reductase | Bradi3g36890.1 | ||
| Cinnamoyl-CoA reductase | Bradi3g36890.1 | Os08g0441500 | |
| Cinnamoyl-CoA reductase | Bradi3g36890.1 | Os08g0441500 | |
| Cinnamoyl coenzyme A reductase (Fragment) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baillie, R.C.; Drayton, M.C.; Pembleton, L.W.; Kaur, S.; Culvenor, R.A.; Smith, K.F.; Spangenberg, G.C.; Forster, J.W.; Cogan, N.O.I. Generation and Characterisation of a Reference Transcriptome for Phalaris (Phalaris aquatica L.). Agronomy 2017, 7, 14. https://doi.org/10.3390/agronomy7010014
Baillie RC, Drayton MC, Pembleton LW, Kaur S, Culvenor RA, Smith KF, Spangenberg GC, Forster JW, Cogan NOI. Generation and Characterisation of a Reference Transcriptome for Phalaris (Phalaris aquatica L.). Agronomy. 2017; 7(1):14. https://doi.org/10.3390/agronomy7010014
Chicago/Turabian StyleBaillie, Rebecca C., Michelle C. Drayton, Luke W. Pembleton, Sukhjiwan Kaur, Richard A. Culvenor, Kevin F. Smith, German C. Spangenberg, John W. Forster, and Noel O. I. Cogan. 2017. "Generation and Characterisation of a Reference Transcriptome for Phalaris (Phalaris aquatica L.)" Agronomy 7, no. 1: 14. https://doi.org/10.3390/agronomy7010014