The Cytolethal Distending Toxin Effects on Mammalian Cells: A DNA Damage Perspective



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. CDT-Related Pathogenicity
1.2. CDT is a Tripartite A-B Exotoxin

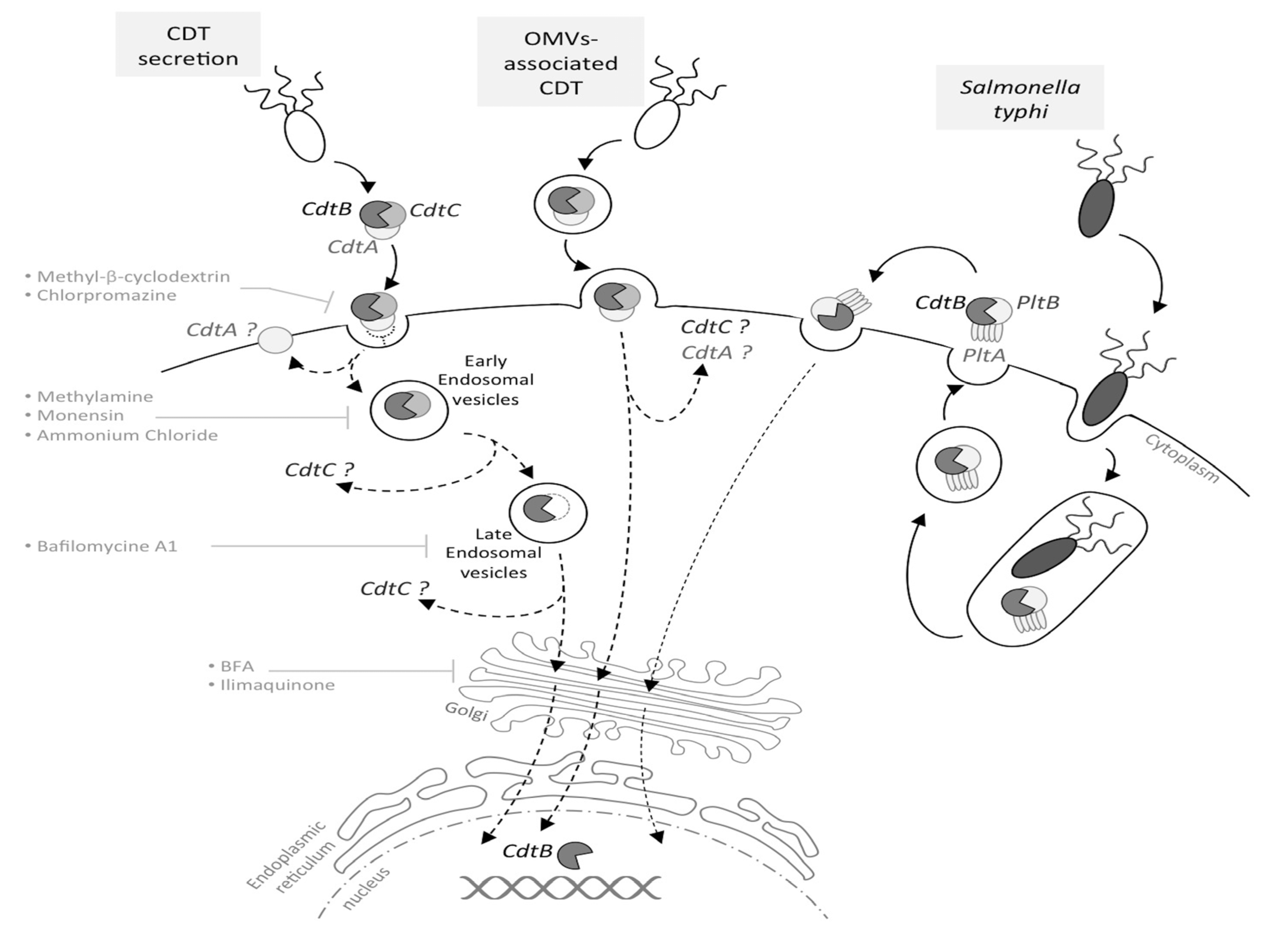
1.3. From CDT Host Cell Binding to CdtB Nuclear Localization
2. DNA Damage-Related Cellular Outcomes of CDT Intoxication
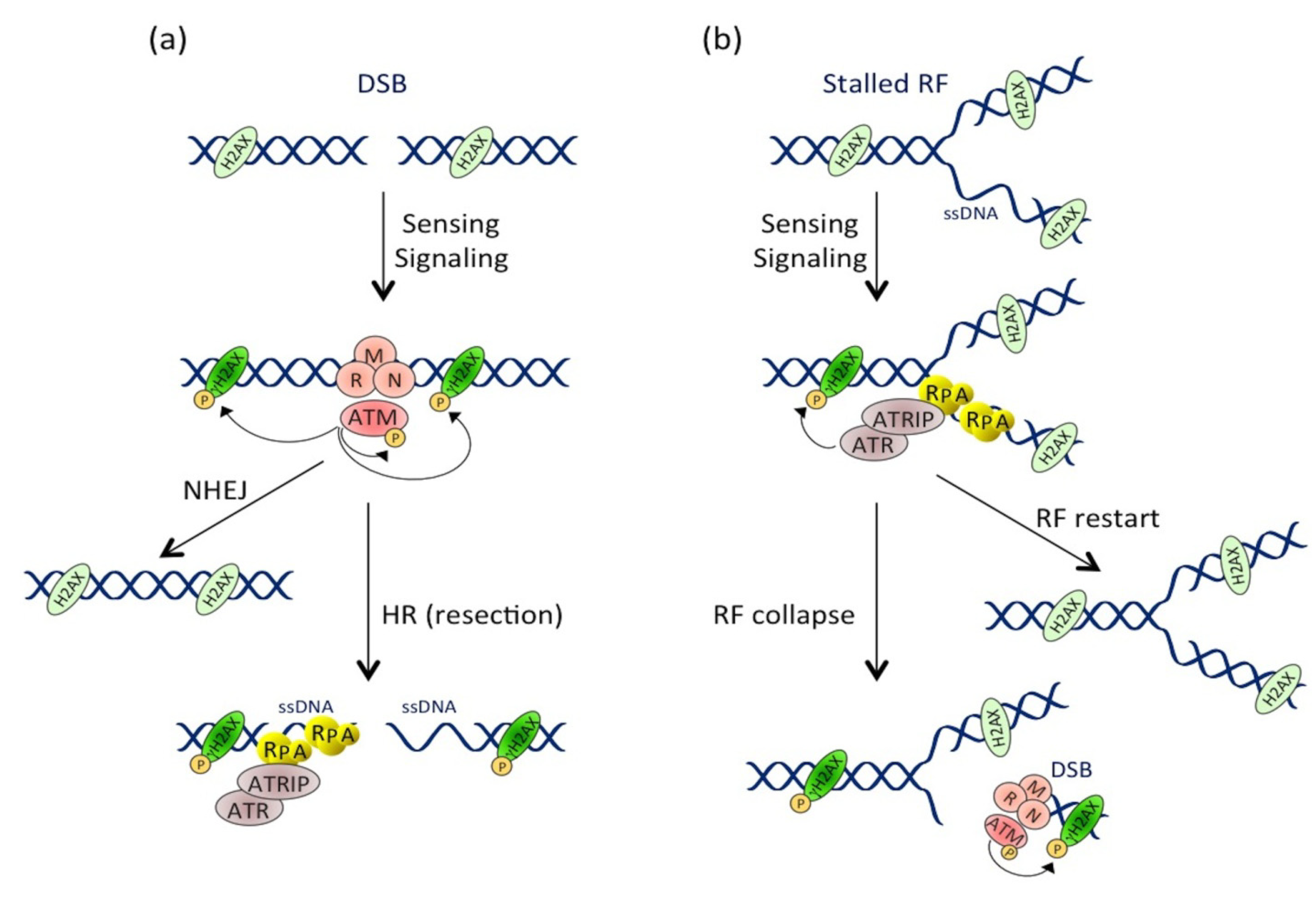
2.1. The CDT-Activated DNA Damage Response
2.1.1. Introduction to the DDR
2.1.2. CDT Activates the DNA Damage Response


2.2. Cellular Effects of the CDT-Induced DNA Damage
2.2.1. Cell Cycle Arrest
2.2.2. Cell Death or Senescence
3. Characterization of the CDT-Induced DNA Lesions
3.1. CDT Does Not Directly Induce DSBs
3.1.1. CdtB Nuclease Activity Primarily Generates SSBs
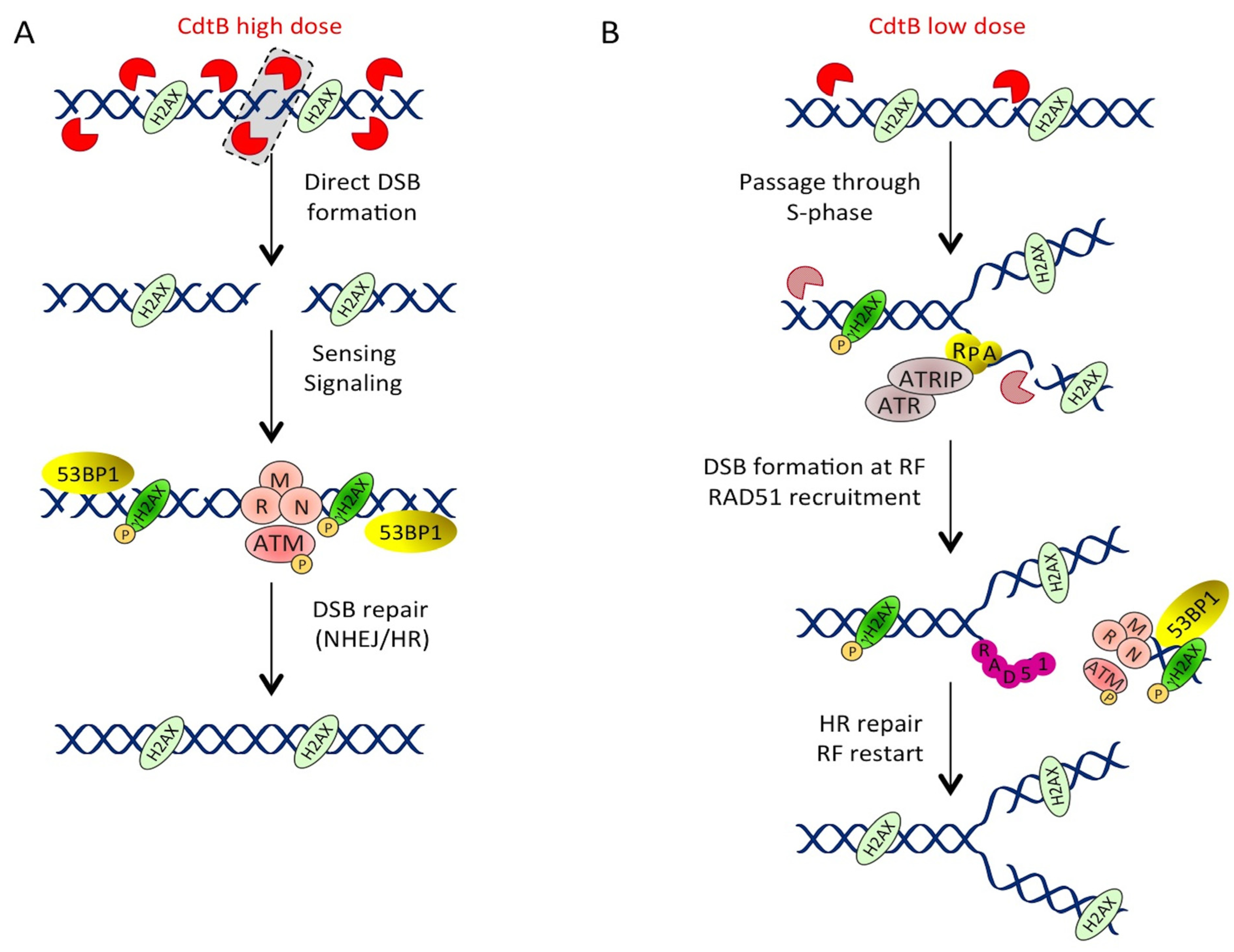
3.1.2. Different Doses, Different DNA Lesions

3.1.3. Low Doses of CDT Induce Replication-Associated DSBs
3.2. Repair of the CDT-Related DNA Damage
3.2.1. Overview of the Eukaryotic DSB Repair Mechanisms
3.2.2. Involvement of the Direct DSB Response Pathway
3.2.3. Implication of HR during the CDT-Mediated Replicative Stress Recovery
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Johnson, W.M.; Lior, H. Response of chinese hamster ovary cells to a cytolethal distending toxin (CDT) of Escherichia coli and possible misinterpretation as heat-labile (LT) enterotoxin. FEMS Microbiol. Lett. 1987, 43, 19–23. [Google Scholar] [CrossRef]
- Johnson, W.M.; Lior, H. A new heat-labile Cytolethal Distending Toxin (CLDT) produced by campylobacter spp. Microbiol. Pathol. 1988, 4, 115–126. [Google Scholar] [CrossRef]
- Cope, L.D.; Lumbley, S.; Latimer, J.L.; Klesney-Tait, J.; Stevens, M.K.; Johnson, L.S.; Purven, M.; Munson, R.S.; Lagergard, T.; Radolf, J.D.; et al. A diffusible cytotoxin of haemophilus ducreyi. Proc. Natl. Acad. Sci. 1997, 94, 4056–4061. [Google Scholar] [CrossRef]
- Sugai, M.; Kawamoto, T.; Pérès, S.Y.; Ueno, Y.; Komatsuzawa, H.; Fujiwara, T.; Kurihara, H.; Suginaka, H.; Oswald, E. The cell cycle-specific growth-inhibitory factor produced by actinobacillus actinomycetemcomitans is a cytolethal distending toxin. Infect. Immun. 1998, 66, 5008–5019. [Google Scholar]
- Shen, Z.; Feng, Y.; Rogers, A.B.; Rickman, B.; Whary, M.T.; Xu, S.; Clapp, K.M.; Boutin, S.R.; Fox, J.G. Cytolethal distending toxin promotes helicobacter cinaedi-associated typhlocolitis in interleukin-10-deficient mice. Infect. Immun. 2009, 77, 2508–2516. [Google Scholar] [CrossRef]
- Haghjoo, E.; Galán, J.E. Salmonella typhi encodes a functional cytolethal distending toxin that is delivered into host cells by a bacterial-internalization pathway. Proc. Natl. Acad. Sci. 2004, 101, 4614–4619. [Google Scholar] [CrossRef]
- Stanley, J.; Linton, D.; Burnens, A.P.; Dewhirst, F.E.; On, S.L.W.; Porter, A.; Owen, R.J.; Costas, M. Helicobacter pullorurn sp. nov.-genotype and phenotype of a new species isolated from poultry and from human patients with gastroenteritis. Microbiology 1994, 140, 3441–3449. [Google Scholar] [CrossRef]
- Chien, C.C.; Taylor, N.S.; Ge, Z.; Schauer, D.B.; Young, V.B.; Fox, J.G. Identification of cdtB homologues and cytolethal distending toxin activity in enterohepatic Helicobacter spp. J. Med. Microbiol. 2000, 49, 525–534. [Google Scholar]
- Jinadasa, R.N.; Bloom, S.E.; Weiss, R.S.; Duhamel, G.E. Cytolethal distending toxin: A conserved bacterial genotoxin that blocks cell cycle progression , leading to apoptosis of a broad range of mammalian cell lineages. Microbiology 2011, 157, 1851–1875. [Google Scholar] [CrossRef]
- Nipic, D.; Podlesek, Z.; Budic, M.; Crnigoj, M.; Zgur-Bertok, D. Escherichia coli uropathogenic-specific protein, usp, is a bacteriocin-like genotoxin. J. Infect. Dis. 2013, 208, 1545–1552. [Google Scholar] [CrossRef]
- Nougayrède, J.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia coli induces dna double-strand breaks in eukaryotic cells. Science 2006, 313, 848–851. [Google Scholar] [CrossRef]
- Yamamoto, S.; Nakano, M.; Terai, A.; Yuri, K.; Nakata, K.; Nair, G.B.; Kurazono, H.; Ogawa, O. The presence of the virulence island containing the usp gene in uropathogenic Escherichia coli is associated with urinary tract infection in an experimental mouse model. J. Urol. 2001, 165, 1347–1351. [Google Scholar] [CrossRef]
- Arthur, J.C.; Perez-chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef]
- Finlay, B.B.; Falkow, S. Common themes in microbial pathogenicity revisited. Microbiol. Mol. Biol. Rev. 1997, 61, 136–169. [Google Scholar]
- Ge, Z.; Schauer, D.B.; Fox, J.G. In vivo virulence properties of bacterial Cytolethal Distending Toxin. Cell. Microbiol. 2008, 10, 1599–1607. [Google Scholar] [CrossRef]
- Jain, D.; Prasad, K.N.; Sinha, S.; Husain, N. Differences in virulence attributes between cytolethal distending toxin positive and negative campylobacter jejuni strains. J. Med. Microbiol. 2008, 57, 267–272. [Google Scholar] [CrossRef]
- Pokkunuri, V.; Pimentel, M.; Morales, W.; Jee, S.R.; Alpern, J.; Weitsman, S.; Marsh, Z.; Low, K.; Hwang, L.; Khoshini, R.; et al. Hole of cytolethal distending toxin in altered stool form and bowel phenotypes in a rat model of post-infectious irritable bowel syndrome. J. Neurogastroenterol. Motil. 2012, 18, 434–442. [Google Scholar] [CrossRef]
- Ge, Z.; Rogers, A.B.; Feng, Y.; Lee, A.; Xu, S.; Taylor, N.S.; Fox, J.G. Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell. Microbiol. 2007, 9, 2070–2080. [Google Scholar] [CrossRef]
- Song, J.; Gao, X.; Galán, J.E. Structure and function of the salmonella typhi chimaeric A2B5 typhoid toxin. Nature 2013, 499, 350–354. [Google Scholar]
- Scott, D.A.; Kaper, J.B. Cloning and sequencing of the genes encoding Escherichia coli cytolethal distending toxin. Infect. Immun. 1994, 62, 244–251. [Google Scholar]
- Pickett, C.L.; Cottle, D.L.; Pesci, E.C.; Bikah, G. Cloning, sequencing, and expression of the Escherichia coli cytolethal distending toxin genes. Infect. Immun. 1994, 62, 1046–1051. [Google Scholar]
- Nesić, D.; Hsu, Y.; Stebbins, C.E. Assembly and function of a bacterial genotoxin. Nature 2004, 429, 429–433. [Google Scholar]
- Yamada, T.; Komoto, J.; Saiki, K.; Konishi, K.; Takusagawa, F. Variation of loop sequence alters stability of cytolethal distending toxin (CDT): Crystal structure of CDT from actinobacillus actinomycetemcomitans. Protein Sci. 2006, 15, 362–372. [Google Scholar] [CrossRef]
- Spanò, S.; Ugalde, J.E.; Galán, J.E. Delivery of a salmonella typhi exotoxin from a host intracellular compartment. Cell Host Microbe 2008, 3, 30–38. [Google Scholar] [CrossRef]
- Kaslow, H.R.; Burns, D.L. Pertussis toxin and target eukaryotic cells: Binding, entry, and activation. FASEB J. 1992, 6, 2685–2690. [Google Scholar]
- Ueno, Y.; Ohara, M.; Kawamoto, T.; Fujiwara, T.; Komatsuzawa, H.; Oswald, E.; Sugai, M. Biogenesis of the actinobacillus actinomycetemcomitans cytolethal distending toxin holotoxin. Infect. Immun. 2006, 74, 3480–3487. [Google Scholar] [CrossRef]
- Zijnge, V.; Kieselbach, T.; Oscarsson, J. Proteomics of protein secretion by aggregatibacter actinomycetemcomitans. PLoS One 2012, 7, e41662. [Google Scholar]
- Berlanda Scorza, F.; Doro, F.; Rodríguez-Ortega, M.J.; Stella, M.; Liberatori, S.; Taddei, A.R.; Serino, L.; Gomes Moriel, D.; Nesta, B.; Fontana, M.R.; et al. Proteomics characterization of outer membrane vesicles from the extraintestinal pathogenic Escherichia coli deltatolR IHE3034 mutant. Mol. Cell. Proteomics 2008, 7, 473–485. [Google Scholar]
- Lindmark, B.; Rompikuntal, P.K.; Vaitkevicius, K.; Song, T.; Mizunoe, Y.; Uhlin, B.E.; Guerry, P.; Wai, S.N. Outer membrane vesicle-mediated release of cytolethal distending toxin (CDT) from campylobacter jejuni. BMC Microbiol. 2009, 9, 220. [Google Scholar] [CrossRef]
- Rompikuntal, P.K.; Thay, B.; Khan, M.K.; Alanko, J.; Penttinen, A.-M.; Asikainen, S.; Wai, S.N.; Oscarsson, J. Perinuclear localization of internalized outer membrane vesicles carrying active cytolethal distending toxin from aggregatibacter actinomycetemcomitans. Infect. Immun. 2012, 80, 31–42. [Google Scholar] [CrossRef]
- Gargi, A.; Reno, M.; Blanke, S.R. Bacterial toxin modulation of the eukaryotic cell cycle: Are all cytolethal distending toxins created equally? Front. Cell. Infect. Microbiol. 2012, 2, 124. [Google Scholar]
- Hu, X.; Nesic, D.; Stebbins, C.E. Comparative structure-function analysis of cytolethal distending toxins. Proteins 2006, 62, 421–434. [Google Scholar]
- Lee, R.B.; Hassane, D.C.; Cottle, D.L.; Pickett, C.L. Interactions of campylobacter jejuni cytolethal distending toxin subunits CdtA and CdtC with HeLa cells. Infect. Immun. 2003, 71, 4883–4890. [Google Scholar] [CrossRef]
- Mcsweeney, L.A.; Dreyfus, L.A. Carbohydrate-binding specificity of the Escherichia coli cytolethal distending toxin CdtA-II and CdtC-II subunits. Infect. Immun. 2005, 73, 2051–2060. [Google Scholar] [CrossRef]
- Eshraghi, A.; Maldonado-arocho, F.J.; Gargi, A.; Cardwell, M.M.; Prouty, M.G.; Blanke, S.R.; Bradley, K.A. Cytolethal distending toxin family members are differentially affected by alterations in host glycans and membrane cholesterol. Microbiology 2010, 285, 18199–18207. [Google Scholar]
- Carette, J.E.; Guimaraes, C.P.; Varadarajan, M.; Park, A.S.; Wuethrich, I.; Godarova, A.; Kotecki, M.; Cochran, B.H.; Spooner, E.; Ploegh, H.L.; et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science 2009, 326, 1231–1235. [Google Scholar] [CrossRef]
- Carette, J.E.; Guimaraes, C.P.; Wuethrich, I.; Blomen, V.A.; Sun, C.; Bell, G.; Yuan, B.; Muellner, M.K.; Nijman, M.; Ploegh, H.L.; et al. Global gene disruption in human cells to assign genes to phenotypes. Nat. Biotechnol. 2011, 29, 542–546. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Bratti, X.; Chaves-Olarte, E.; Lagergård, T.; Thelestam, M. Cellular Internalization of cytolethal distending toxin from haemophilus ducreyi. Infect. Immun. 2000, 68, 6903–6911. [Google Scholar] [CrossRef]
- Guerra, L.; Teter, K.; Lilley, B.N.; Stenerlöw, B.; Holmes, R.K.; Ploegh, H.L.; Sandvig, K.; Thelestam, M.; Frisan, T. Cellular internalization of cytolethal distending toxin: A new end to a known pathway. Cell. Microbiol. 2005, 7, 921–934. [Google Scholar] [CrossRef]
- Boesze-Battaglia, K.; Besack, D.; McKay, T.; Zekavat, A.; Otis, L.; Jordan-Sciutto, K.; Shenker, B.J. Cholesterol-rich membrane microdomains mediate cell cycle arrest induced by actinobacillus actinomycetemcomitans cytolethal-distending toxin. Cell. Microbiol. 2006, 8, 823–836. [Google Scholar] [CrossRef]
- Boesze-Battaglia, K.; Brown, A.; Walker, L.; Besack, D.; Zekavat, A.; Wrenn, S.; Krummenacher, C.; Shenker, B.J. cytolethal distending toxin-induced cell cycle arrest of lymphocytes is dependent upon recognition and binding to cholesterol. J. Biol. Chem. 2009, 284, 10650–10658. [Google Scholar] [CrossRef]
- Lin, C.D.; Lai, C.K.; Lin, Y.H.; Hsieh, J.T.; Sing, Y.T.; Chang, Y.C.; Chen, K.C.; Wang, W.C.; Su, H.L.; Lai, C.H. Cholesterol depletion reduces entry of Campylobacter jejuni cytolethal distending toxin and attenuates intoxication of host cells. Infect. Immun. 2011, 79, 3563–3575. [Google Scholar] [CrossRef]
- Damek-Poprawa, M.; Jang, J.Y.; Volgina, A.; Korostoff, J.; DiRienzo, J.M. Localization of Aggregatibacter actinomycetemcomitans cytolethal distending toxin subunits during intoxication of live cells. Infect. Immun. 2012, 80, 2761–2770. [Google Scholar] [CrossRef]
- Lara-Tejero, M.; Galàn, J.E. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-Like protein. Science 2000, 290, 354–357. [Google Scholar] [CrossRef]
- Nishikubo, S.; Ohara, M.; Ueno, Y.; Ikura, M.; Kurihara, H.; Komatsuzawa, H.; Oswald, E.; Sugai, M. Ann-terminal segment of the active component of the bacterial genotoxin cytolethal distending toxin B (CDTB) directs CDTB into the nucleus. J. Biol. Chem. 2003, 278, 50671–50681. [Google Scholar]
- Guerra, L.; Nemec, K.N.; Massey, S.; Tatulian, S.A.; Thelestam, M.; Frisan, T.; Teter, K. A novel mode of translocation for cytolethal distending toxin. Biochim. Biophys. Acta 2009, 1793, 489–495. [Google Scholar]
- Gargi, A.; Tamilselvam, B.; Powers, B.; Prouty, M.G.; Lincecum, T.; Eshraghi, A.; Maldonado-Arocho, F.J.; Wilson, B.A.; Bradley, K.A.; Blanke, S.R. Cellular interactions of the cytolethal distending toxins from Escherichia coli and haemophilus ducreyi. J. Biol. Chem. 2013, 288, 7492–7505. [Google Scholar]
- McSweeney, L.A.; Dreyfus, L.A. A Nuclear localization of the Escherichia coli cytolethal Distending toxin CdtB subunit. Cell. Microbiol. 2004, 6, 447–458. [Google Scholar] [CrossRef]
- Pérès, S.Y.; Marchès, O.; Daigle, F.; Nougayrède, J.P.; Herault, F.; Tasca, C.; de Rycke, J.; Oswald, E. A new cytolethal distending toxin (CDT) from Escherichia coli producing CNF2 blocks HeLa cell division in G2/M phase. Mol. Microbiol. 1997, 24, 1095–1107. [Google Scholar]
- Comayras, C.; Tasca, C.; Pe, S.Y.; Oswald, E.; Rycke, J.D.E. Escherichia coli cytolethal distending toxin blocks the HeLa cell cycle at the G2/M transition by preventing cdc2 protein kinase dephosphorylation and activation. Escherichia coli cytolethal distending toxin blocks the HeLa cell cycle at the G 2/M Tr. Infect. Immun. 1997, 65, 5088–5095. [Google Scholar]
- Ohguchi, M.; Ishisaki, A.; Okahashi, N.; Koide, M.; Koseki, T.; Yamato, K.; Noguchi, T.; Nishihara, T. Actinobacillus actinomycetemcomitans toxin induces both cell cycle arrest in the G2/M phase and apoptosis. Infect. Immun. 1998, 66, 5980–5987. [Google Scholar]
- Sert, V.; Cans, C.; Tasca, C.; Oswald, E.; Ducommun, B.; de Rycke, J. The bacterial cytolethal distending toxin (CDT) triggers a G2 cell cycle checkpoint in mammalian cells without preliminary induction of DNA strand breaks. Oncogene 1999, 18, 6296–6304. [Google Scholar] [CrossRef]
- Fahrer, J.; Huelsenbeck, J.; Jaurich, H.; Dörsam, B.; Frisan, T.; Eich, M.; Roos, W.P.; Kaina, B.; Fritz, G. Cytolethal distending toxin (CDT) is a radiomimetic agent and induces persistent levels of DNA double-strand breaks in human fibroblasts. DNA Repair (Amst) 2014, 18, 31–43. [Google Scholar] [CrossRef]
- Elwell, C.A.; Dreyfus, L.A. DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol. Microbiol. 2000, 37, 952–963. [Google Scholar] [CrossRef]
- Niida, H.; Nakanishi, M. DNA damage checkpoints in mammals. Mutagenesis 2006, 21, 3–9. [Google Scholar]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Lee, J.-H.; Paull, T.T. ATM activation by DNA double-Strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef]
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar]
- Matsuoka, S.; Ballif, B.A; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Gobbini, E.; Cesena, D.; Galbiati, A.; Lockhart, A.; Longhese, M.P. Interplays between ATM/Tel1 and ATR/Mec1 in sensing and signaling DNA double-strand breaks. DNA Repair (Amst) 2013, 12, 791–799. [Google Scholar] [CrossRef]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.M.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef]
- Feng, W.; di Rienzi, S.C.; Raghuraman, M.K.; Brewer, B.J. Replication stress-induced chromosome breakage is correlated with replication fork progression and is preceded by single-stranded DNA formation. G3 Genes Genomes Genet. 2011, 1, 327–335. [Google Scholar]
- Cortes-Bratti, X.; Karlsson, C.; Lagergård, T.; Thelestam, M.; Frisan, T. The haemophilus ducreyi cytolethal distending Toxin induces cell cycle arrest and apoptosis via the DNA damage checkpoint pathways. J. Biol. Chem. 2001, 276, 5296–5302. [Google Scholar]
- Li, L.; Sharipo, A.; Chaves-Olarte, E.; Masucci, M.G.; Levitsky, V.; Thelestam, M.; Frisan, T. The haemophilus ducreyi cytolethal distending toxin activates sensors of DNA damage and repair complexes in proliferating and non-proliferating cells. Cell. Microbiol. 2002, 4, 87–99. [Google Scholar] [CrossRef]
- Hassane, D.C.; Lee, R.B.; Pickett, C.L. Campylobacter jejuni cytolethal distending toxin promotes DNA repair responses in normal human cells. Infect. Immun. 2003, 71, 541–545. [Google Scholar] [CrossRef]
- Guerra, L.; Albihn, A.; Tronnersjö, S.; Yan, Q.; Guidi, R.; Stenerlöw, B.; Sterzenbach, T.; Josenhans, C.; Fox, J.G.; Schauer, D.B.; et al. Myc is required for activation of the ATM-dependent checkpoints in response to DNA damage. PLoS One 2010, 5, e8924. [Google Scholar] [CrossRef]
- Alaoui-El-Azher, M.; Mans, J.J.; Baker, H.V; Chen, C.; Progulske-Fox, A.; Lamont, R.J.; Handfield, M. Role of the ATM-checkpoint kinase 2 pathway in CDT-mediated apoptosis of gingival epithelial cells. PLoS One 2010, 5, e11714. [Google Scholar] [CrossRef]
- Bielaszewska, M.; Sinha, B.; Kuczlus, T.; Karch, H. Cytolethal distending toxin from shiga toxin-producing Escherichia coli O157 causes irreversible G2/M arrest, Inhibition of proliferation, and Death of human endothelial cells. Infect. Immun. 2005, 73, 552–562. [Google Scholar] [CrossRef]
- Taieb, F.; Nougayrède, J.; Watrin, C.; Samba-louaka, A.; Oswald, E. Escherichia coli cyclomodulin Cif induces G 2 arrest of the host cell cycle without activation of the DNA-damage checkpoint-signalling pathway. 2006, 8, 1910–1921. [Google Scholar]
- Blazkova, H.; Krejcikova, K.; Moudry, P.; Frisan, T.; Hodny, Z.; Bartek, J. Bacterial intoxication evokes cellular senescence with persistent DNA damage and cytokine signalling. J. Cell. Mol. Med. 2010, 14, 357–367. [Google Scholar] [CrossRef]
- Liyanage, N.P.M.; Manthey, K.C.; Dassanayake, R.P.; Kuszynski, C.A.; Oakley, G.G.; Duhamel, G.E. Helicobacter hepaticus cytolethal distending toxin causes cell death in Intestinal epithelial cells via mitochondrial apoptotic pathway. Helicobacter 2010, 15, 98–107. [Google Scholar] [CrossRef]
- Fedor, Y.; Vignard, J.; Nicolau-Travers, M.-L.; Boutet-Robinet, E.; Watrin, C.; Salles, B.; Mirey, G. From single-strand breaks to double-strand breaks during S-phase: A new mode of action of the Escherichia coli cytolethal distending toxin. Cell. Microbiol. 2013, 15, 1–15. [Google Scholar] [CrossRef]
- Vignard, J.; Mirey, G.; Salles, B. Ionizing-radiation induced DNA double-strand breaks: A direct and indirect lighting up. Radiother. Oncol. 2013, 108, 362–369. [Google Scholar] [CrossRef]
- Ahn, J.; Urist, M.; Prives, C. The Chk2 protein kinase. DNA Repair (Amst) 2004, 3, 1039–1047. [Google Scholar] [CrossRef]
- Sato, T.; Koseki, T.; Yamato, K.; Saiki, K.; Konishi, K.; Yoshikawa, M.; Ishikawa, I.; Nishihara, T. p53-Independent Expression of p21 CIP1/WAF1 in plasmacytic cells during G2 cell cycle arrest induced by actinobacillus actinomycetemcomitans cytolethal distending toxin. Infect. Immun. 2002, 70, 528–534. [Google Scholar] [CrossRef]
- Hochegger, H.; Takeda, S.; Hunt, T. Cyclin-dependent kinases and cell-cycle transitions: Does one fit all? Nat. Rev. Mol. Cell Biol. 2008, 9, 910–916. [Google Scholar] [CrossRef]
- Langerak, P.; Russell, P. Regulatory networks integrating cell cycle control with DNA damage checkpoints and double-strand break repair. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3562–3571. [Google Scholar] [CrossRef]
- Alby, F.; Mazars, R.; de Rycke, J.; Guillou, E.; Baldin, V.; Darbon, J.M.; Ducommun, B. Study of the cytolethal distending toxin (CDT)-activated cell cycle checkpoint. Involvement of the CHK2 kinase. FEBS Lett. 2001, 491, 261–265. [Google Scholar] [CrossRef]
- De Rycke, J.; Sert, V.; Comayras, C.; Tasca, C. Sequence of lethal events in HeLa cells exposed to the G2 blocking cytolethal distending toxin. Eur. J. Cell Biol. 2000, 79, 192–201. [Google Scholar] [CrossRef]
- Escalas, N.; Davezac, N.; de Rycke, J.; Baldin, V.; Mazars, R.; Ducommun, B. Study of the cytolethal distending toxin-induced cell cycle arrest in HeLa cells: Involvement of the CDC25 phosphatase. Exp. Cell Res. 2000, 257, 206–212. [Google Scholar] [CrossRef]
- Bielaszewska, M.; Fell, M.; Greune, L.; Prager, R.; Fruth, A.; Tschäpe, H.; Schmidt, M.A.; Karch, H.; Tscha, H. Characterization of cytolethal distending toxin genes and expression in shiga toxin-producing Escherichia coli strains of non-O157 serogroups. Infect. Immun. 2004, 72, 1812–1816. [Google Scholar] [CrossRef]
- Cortes-Bratti, X.; Frisan, T.; Thelestam, M. The cytolethal distending toxins induce DNA damage and cell cycle arrest. Toxicon 2001, 39, 1729–1736. [Google Scholar] [CrossRef]
- Frisan, T.; Cortes-bratti, X.; Chaves-olarte, E.; Stenerlöw, B.; Thelestam, M.; Rica, U.D.C.; José, S.; Rica, C. The Haemophilus ducreyi cytolethal distending toxin induces DNA double-strand breaks and promotes ATM-dependent activation of RhoA. Cell. Microbiol. 2003, 5, 695–707. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. P21 in Cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Shenker, B.J.; Hoffmaster, R.H.; Zekavat, A.; Yamaguchi, N.; Lally, E.T.; Demuth, D.R. Induction of apoptosis in human T cells by actinobacillus actinomycetemcomitans cytolethal distending toxin Is a consequence of G2 arrest of the cell cycle. J. Immunol. 2001, 167, 435–441. [Google Scholar] [CrossRef]
- Ohara, M.; Hayashi, T.; Kusunoki, Y.; Miyauchi, M.; Takata, T.; Sugai, M.; Mmun, I.N.I. Caspase-2 and Caspase-7 are involved in cytolethal distending toxin-induced apoptosis in jurkat and MOLT-4 T-cell lines. Infect. Immun. 2004, 72, 871–879. [Google Scholar] [CrossRef]
- Hickey, T.E.; Majam, G.; Guerry, P. Intracellular survival of campylobacter jejuni in human monocytic cells and induction of apoptotic death by cytholethal distending toxin. Infect. Immun. 2005, 73, 5194–5197. [Google Scholar] [CrossRef]
- Guidi, R.; Guerra, L.; Levi, L.; Stenerlöw, B.; Fox, J.G.; Josenhans, C.; Masucci, M.G.; Frisan, T. Chronic exposure to the cytolethal distending toxins of Gram-negative bacteria promotes genomic instability and altered DNA damage response. Cell. Microbiol. 2013, 15, 98–113. [Google Scholar] [CrossRef]
- Gelfanova, V.; Hansen, E.J.; Spinola, S.M. Cytolethal distending toxin of haemophilus ducreyi induces apoptotic death of jurkat T cells. Infect. Immun. 1999, 67, 6394–6402. [Google Scholar]
- Wising, C.; Azem, J.; Zetterberg, M.; Svensson, L.A.; Ahlman, K.; Lagerga, T. Induction of apoptosis/necrosis in various human cell lineages by haemophilus ducreyi cytolethal distending toxin. Toxicon 2005, 45, 767–776. [Google Scholar] [CrossRef]
- Shenker, B.J.; Dlakic, M.; Walker, L.P.; Besack, D.; Jaffe, E.; LaBelle, E.; Boesze-Battaglia, K. A novel mode of action for a microbial-derivated immunotoxin: The cytolethal distending toxin subunit B exhibits phosphatidylinositol3,4,5-triphosphate phosphatase activity. J. Immunol. 2007, 178, 5099–5108. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Guerra, L.; Guidi, R.; Frisan, T. Do bacterial genotoxins contribute to chronic inflammation, genomic instability and tumor progression? FEBS J. 2011, 278, 4577–4588. [Google Scholar] [CrossRef]
- Mao, X.; DiRienzo, J.M. Functional studies of the recombinant subunits of a cytolethal distending holotoxin. Cell. Microbiol. 2002, 4, 245–255. [Google Scholar] [CrossRef]
- Hontz, J.S.; Villar-Lecumberri, M.T.; Dreyfus, L.A; Yoder, M.D. Crystallization of Escherichia coli CdtB, the biologically active subunit of cytolethal distending toxin. Acta Crystallogr. Sect. F: Struct. Biol. Cryst. Commun. 2006, 62, 192–195. [Google Scholar] [CrossRef]
- Campbell, V.W.; Jackson, D.A. The effect of divalent cations on the mode of action of DNase I. J. Biol. Chem. 1980, 255, 3726–3735. [Google Scholar]
- Pan, C.Q.; Ulmer, J.S.; Herzka, A.; Lazarus, R.A. Mutational analysis of human DNase I at the DNA binding interface: Implications for DNA recognition, catalysis, and metal ion dependence. Protein Sci. 1998, 7, 628–636. [Google Scholar] [CrossRef]
- Dassanayake, R.P.; Griep, M.A.; Duhamel, G.E. The cytolethal distending toxin B sub-unit of Helicobacter hepaticus is a Ca2+—and Mg2+ -dependent neutral nuclease. FEMS Microbiol. Lett. 2005, 251, 219–225. [Google Scholar] [CrossRef]
- DiRienzo, J.M.; Cao, L.; Volgina, A.; Bandelac, G.; Korostoff, J. Functional and structural characterization of chimeras of a bacterial genotoxin and human type I DNAse. FEMS Microbiol. Lett. 2009, 291, 222–231. [Google Scholar] [CrossRef]
- Roots, R.; Holley, W.; Chatterjee, A.; Rachal, E.; Kraft, G. The influence of radiation quality on the formation of DNA breaks. Adv. Space Res. 1989, 9, 45–55. [Google Scholar]
- Averbeck, D. Mécanismes de réparation et mutagenèse radio-induite chez les eucaryotes supérieurs. Cancer Radiother. 2000, 4, 335–354. [Google Scholar] [CrossRef]
- Collins, A.R.; Oscoz, A.A.; Brunborg, G.; Gaivão, I.; Giovannelli, L.; Kruszewski, M.; Smith, C.C.; Stetina, R. The comet assay: Topical issues. Mutagenesis 2008, 23, 143–151. [Google Scholar] [CrossRef]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Jeggo, P.A. The repair and signaling responses to DNA double-strand breaks. Adv. Genet. 2013, 82, 1–45. [Google Scholar] [CrossRef]
- Bétermier, M.; Bertrand, P.; Lopez, B.S. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 2014, 10, e1004086. [Google Scholar]
- Aze, A.; Zhou, J.C.; Costa, A.; Costanzo, V. DNA replication and homologous recombination factors: Acting together to maintain genome stability. Chromosoma 2013, 122, 401–413. [Google Scholar] [CrossRef]
- Amunugama, R.; Fishel, R. Homologous Recombination in Eukaryotes, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2012; Volume 110, pp. 155–206. [Google Scholar]
- Barlow, C.; Hirotsune, S.; Paylor, R.; Liyanage, M.; Eckhaus, M.; Collins, F.; Shiloh, Y.; Crawley, J.N.; Ried, T.; Tagle, D.; et al. Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell 1996, 86, 159–171. [Google Scholar]
- Takai, H.; Naka, K.; Okada, Y.; Watanabe, M.; Harada, N.; Saito, S.; Anderson, C.W.; Appella, E.; Nakanishi, M.; Suzuki, H.; et al. Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. EMBO J. 2002, 21, 5195–5205. [Google Scholar] [CrossRef]
- Matangkasombut, O.; Wattanawaraporn, R.; Tsuruda, K.; Ohara, M.; Sugai, M.; Mongkolsuk, S. Cytolethal distending toxin from aggregatibacter actinomycetemcomitans induces DNA damage, S/G2 cell cycle arrest, and caspase-independent death in a saccharomyces cerevisiae model. Infect. Immun. 2010, 78, 783–792. [Google Scholar] [CrossRef]
- Kitagawa, T.; Hoshida, H.; Akada, R. Genome-wide analysis of cellular response to bacterial genotoxin CdtB in yeast. Infect. Immun. 2007, 75, 1393–1402. [Google Scholar] [CrossRef]
- Janssen, A.; Medema, R.H. Genetic instability: Tipping the balance. Oncogene 2013, 32, 4459–4470. [Google Scholar]
- Morrison, W.B. Inflammation and cancer: A comparative view. J. Vet. Intern. Med. 2012, 26, 18–31. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Polk, D.B.; Peek, R.M., Jr. Helicobacter pylori: Gastric cancer and beyond. Nat. Rev. Cancer 2010, 10, 403–414. [Google Scholar] [CrossRef]
- Machado, A.M.; Fiqueiredo, C.; Seruca, R.; Rasmussen, L. Helicobacter pylori infection generates genetic instability in gastric cells. Biochim. Biophys. Acta 2010, 1806, 58–65. [Google Scholar]
- Touati, E.; Michel, V.; Thiberge, J.M.; Wuscher, N.; Huerre, M.; Labigne, A. Chronic helicobacter pylori infections induce gastric mutations in mice. Gastroenterology 2003, 124, 1408–1419. [Google Scholar]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef]
- Samaras, V.; Rafailidis, P.I.; Mourtzoukou, E.G.; Peppas, G.; Falagas, M.E. Chronic bacteria and parasitic infections and cancer: A review. J. Infect. Dev. Ctries. 2010, 4, 267–281. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bezine, E.; Vignard, J.; Mirey, G. The Cytolethal Distending Toxin Effects on Mammalian Cells: A DNA Damage Perspective. Cells 2014, 3, 592-615. https://doi.org/10.3390/cells3020592
Bezine E, Vignard J, Mirey G. The Cytolethal Distending Toxin Effects on Mammalian Cells: A DNA Damage Perspective. Cells. 2014; 3(2):592-615. https://doi.org/10.3390/cells3020592
Chicago/Turabian StyleBezine, Elisabeth, Julien Vignard, and Gladys Mirey. 2014. "The Cytolethal Distending Toxin Effects on Mammalian Cells: A DNA Damage Perspective" Cells 3, no. 2: 592-615. https://doi.org/10.3390/cells3020592