microRNAs and Cardiac Cell Fate

 ,
, 
{kind=link}
Abstract
:1. Introduction
2. Biogenesis and Function of miRNAs
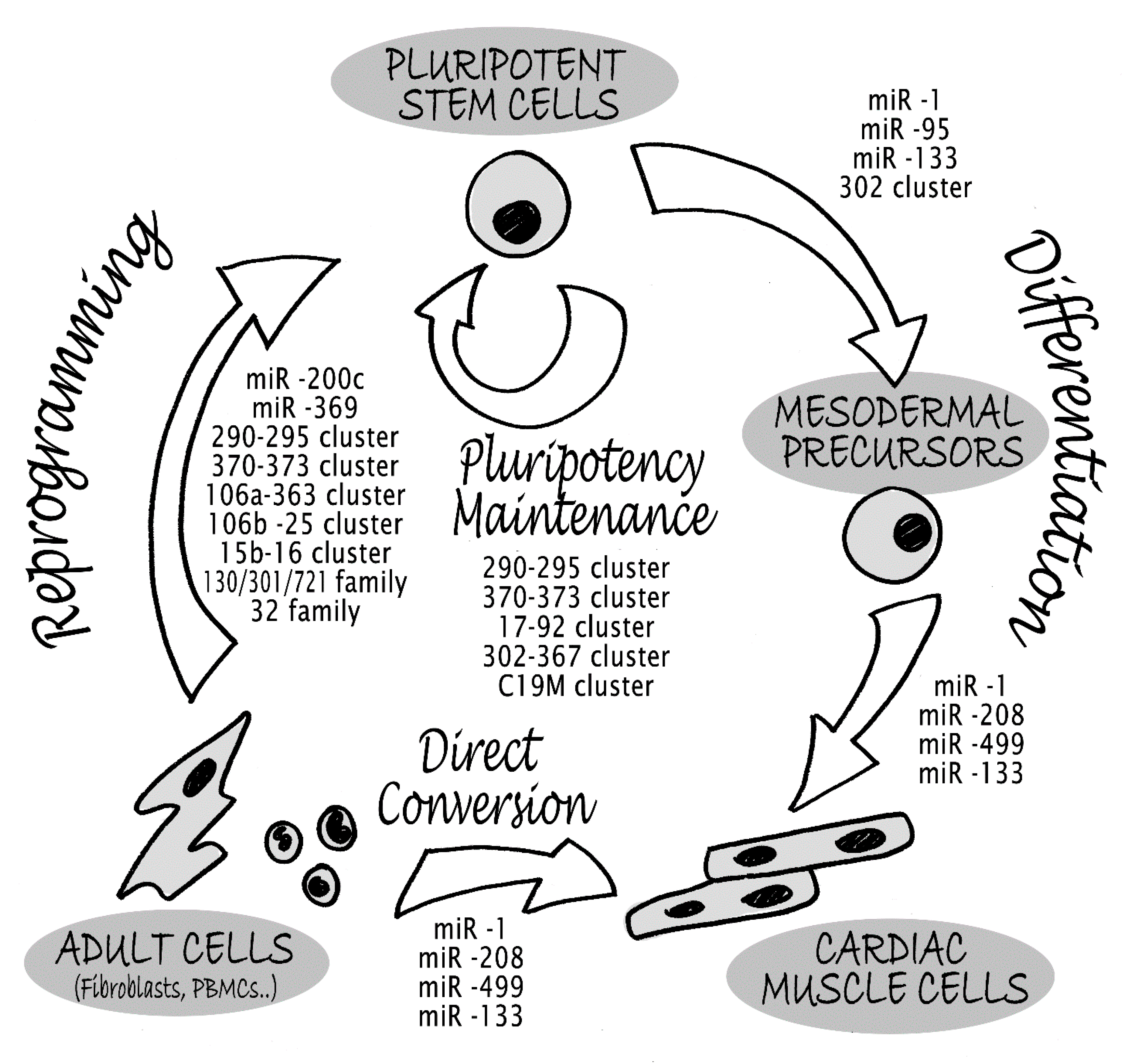
3. MicroRNAs in Pluripotency and Reprogramming Control
4. miRNAs in Cardiomyocyte Lineage Commitment and Identity
5. Direct Myocardial Reprogramming with miRNAs

6. miRNAs in Cardiac Development
7. Concluding Remarks
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Reczko, M.; Maragkakis, M.; Alexiou, P.; Papadopoulos, G.L.; Hatzigeorgiou, A.G. Accurate microRNA target prediction using detailed binding site accessibility and machine learning on proteomics data. Front. Genet. 2012, 2, 103:1–103:13. [Google Scholar]
- Kloosterman, W.P.; Wienholds, E.; Ketting, R.F.; Plasterk, R.H. Substrate requirements for let-7 function in the developing zebrafish embryo. Nucleic Acids Res. 2004, 32, 6284–6291. [Google Scholar] [CrossRef] [PubMed]
- Lytle, J.R.; Yario, T.A.; Steitz, J.A. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5' UTR as in the 3' UTR. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 9667–9672. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Olson, E.N. MicroRNA regulatory networks in cardiovascular development. Dev. Cell 2010, 18, 510–525. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Ajay, S.S.; Yook, J.I.; Kim, H.S.; Hong, S.H.; Kim, N.H.; Dhanasekaran, S.M.; Chinnaiyan, A.M.; Athey, B.D. New class of microRNA targets containing simultaneous 5'-UTR and 3'-UTR interaction sites. Genome Res. 2009, 19, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Vella, M.C.; Reinert, K.; Slack, F.J. Architecture of a validated microRNA: Target interaction. Chem. Biol. 2004, 11, 1619–1623. [Google Scholar] [CrossRef] [PubMed]
- Yekta, S.; Shih, I.H.; Bartel, D.P. MicroRNA-directed cleavage of HOXB8 mRNA. Science 2004, 304, 594–596. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell. 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Vickers, K.C.; Rye, K.A.; Tabet, F. MicroRNAs in the onset and development of cardiovascular disease. Clin. Sci. (Lond). 2014, 126, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R. microRNAs in cardiac development and regeneration. Clin. Sci. (Lond). 2013, 125, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Boettger, T.; Braun, T. A new level of complexity: The role of microRNAs in cardiovascular development. Circ. Res. 2012, 110, 1000–1013. [Google Scholar]
- Espinoza-Lewis, R.A.; Wang, D.Z. MicroRNAs in heart development. Curr. Top. Dev. Biol. 2012, 100, 279–317. [Google Scholar] [PubMed]
- Heinrich, E.M.; Dimmeler, S. MicroRNAs and stem cells: Control of pluripotency, reprogramming, and lineage commitment. Circ. Res. 2012, 110, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Hagedorn, C.H.; Cullen, B.R. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004, 10, 1957–1966. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, Y.; Yeom, K.H.; Nam, J.W.; Heo, I.; Rhee, J.K.; Sohn, S.Y.; Cho, Y.; Zhang, B.T.; Kim, V.N. Molecular basis for the recognition of primary microRNAs by the drosha-DGCR8 complex. Cell 2006, 125, 887–901. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, Y.; Yeom, K.H.; Kim, Y.K.; Jin, H.; Kim, V.N. The drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 18, 3016–3027. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Radmark, O.; Kim, S.; et al. The nuclear RNase III drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Berezikov, E.; Chung, W.J.; Willis, J.; Cuppen, E.; Lai, E.C. Mammalian mirtron genes. Mol. Cell 2007, 28, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef] [PubMed]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Small, E.M.; Frost, R.J.; Olson, E.N. MicroRNAs add a new dimension to cardiovascular disease. Circulation 2010, 121, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V.; Bartel, B.; Bartel, D.P.; Burge, C.B.; Carrington, J.C.; Chen, X.; Dreyfuss, G.; Eddy, S.R.; Griffiths-Jones, S.; Marshall, M.; et al. A uniform system for microRNA annotation. RNA 2003, 9, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Hutvagner, G.; Du, T.; Xu, Z.; Aronin, N.; Zamore, P.D. Asymmetry in the assembly of the RNAi enzyme complex. Cell 2003, 115, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Reynolds, A.; Jayasena, S.D. Functional siRNAs and miRNAs exhibit strand bias. Cell 2003, 115, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef]
- Santulli, G.; Iaccarino, G.; de Luca, N.; Trimarco, B.; Condorelli, G. Atrial fibrillation and microRNAs. Front. Physiol. 2014, 5, 15:1–15:7. [Google Scholar]
- Moradi, S.; Asgari, S.; Baharvand, H. Concise review: Harmonies played by microRNAs in cell fate reprogramming. Stem Cells 2014, 32, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.D.; Venkatasubrahmanyam, S.; Jia, F.; Sun, N.; Butte, A.J.; Wu, J.C. MicroRNA profiling of human-induced pluripotent stem cells. Stem Cells Dev. 2009, 18, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Luningschror, P.; Hauser, S.; Kaltschmidt, B.; Kaltschmidt, C. MicroRNAs in pluripotency, reprogramming and cell fate induction. Biochim. Biophys. Acta 2013, 1833, 1894–1903. [Google Scholar]
- Martinez, N.J.; Gregory, R.I. MicroRNA gene regulatory pathways in the establishment and maintenance of ESC identity. Cell. Stem. Cell 2010, 7, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Murchison, E.P.; Partridge, J.F.; Tam, O.H.; Cheloufi, S.; Hannon, G.J. Characterization of dicer-deficient murine embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 12135–12140. [Google Scholar] [CrossRef] [PubMed]
- Marson, A.; Levine, S.S.; Cole, M.F.; Frampton, G.M.; Brambrink, T.; Johnstone, S.; Guenther, M.G.; Johnston, W.K.; Wernig, M.; Newman, J.; et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 2008, 134, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Card, D.A.; Hebbar, P.B.; Li, L.; Trotter, K.W.; Komatsu, Y.; Mishina, Y.; Archer, T.K. Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol. Cell. Biol. 2008, 28, 6426–6438. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Baskerville, S.; Shenoy, A.; Babiarz, J.E.; Baehner, L.; Blelloch, R. Embryonic stem cell-specific microRNAs regulate the G1-S transition and promote rapid proliferation. Nat. Genet. 2008, 40, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microRNA polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Voorhoeve, P.M.; le Sage, C.; Schrier, M.; Gillis, A.J.; Stoop, H.; Nagel, R.; Liu, Y.P.; van Duijse, J.; Drost, J.; Griekspoor, A.; et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell 2006, 124, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Stadler, B.; Ivanovska, I.; Mehta, K.; Song, S.; Nelson, A.; Tan, Y.; Mathieu, J.; Darby, C.; Blau, C.A.; Ware, C.; et al. Characterization of microRNAs involved in embryonic stem cell states. Stem Cells Dev. 2010, 19, 935–950. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Jin, P.; Wang, E.; Marincola, F.M.; Stroncek, D.F. MicroRNA and gene expression patterns in the differentiation of human embryonic stem cells. J. Transl. Med. 2009, 7, 20:1–20:7. [Google Scholar]
- Brons, I.G.; Smithers, L.E.; Trotter, M.W.; Rugg-Gunn, P.; Sun, B.; Chuva de Sousa Lopes, S.M.; Howlett, S.K.; Clarkson, A.; Ahrlund-Richter, L.; Pedersen, R.A.; et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 2007, 448, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Tesar, P.J.; Chenoweth, J.G.; Brook, F.A.; Davies, T.J.; Evans, E.P.; Mack, D.L.; Gardner, R.L.; McKay, R.D. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 2007, 448, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Osorno, R.; Tsakiridis, A.; Wilson, V. In vivo differentiation potential of epiblast stem cells revealed by chimeric embryo formation. Cell. Rep. 2012, 2, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Chenoweth, J.G.; McKay, R.D.; Tesar, P.J. Epiblast stem cells contribute new insight into pluripotency and gastrulation. Dev. Growth. Differ. 2010, 52, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Nichols, J.; Smith, A. Naive and primed pluripotent states. Cell Stem Cell 2009, 4, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Nichols, J.; Smith, A. The origin and identity of embryonic stem cells. Development 2011, 138, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Jouneau, A.; Ciaudo, C.; Sismeiro, O.; Brochard, V.; Jouneau, L.; Vandormael-Pournin, S.; Coppee, J.Y.; Zhou, Q.; Heard, E.; Antoniewski, C.; et al. Naive and primed murine pluripotent stem cells have distinct miRNA expression profiles. RNA 2012, 18, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Parchem, R.J.; Ye, J.; Judson, R.L.; LaRussa, M.F.; Krishnakumar, R.; Blelloch, A.; Oldham, M.C.; Blelloch, R. Two miRNA clusters reveal alternative paths in late-stage reprogramming. Cell Stem Cell 2014, 14, 617–631. [Google Scholar] [CrossRef] [PubMed]
- Ware, C.B.; Nelson, A.M.; Mecham, B.; Hesson, J.; Zhou, W.; Jonlin, E.C.; Jimenez-Caliani, A.J.; Deng, X.; Cavanaugh, C.; Cook, S.; et al. Derivation of naive human embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 4484–4489. [Google Scholar] [CrossRef]
- Battista, M.; Musto, A.; Navarra, A.; Minopoli, G.; Russo, T.; Parisi, S. miR-125b regulates the early steps of ESC differentiation through Dies1 in a TGF-independent manner. Int. J. Mol. Sci. 2013, 14, 13482–13496. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.G.; Langer, E.M.; Lindsley, R.C.; Cai, M.; Murphy, T.L.; Kyba, M.; Murphy, K.M. Snail and the microRNA-200 family act in opposition to regulate epithelial-to-mesenchymal transition and germ layer fate restriction in differentiating ESCs. Stem Cells 2011, 29, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Judson, R.L.; Babiarz, J.E.; Venere, M.; Blelloch, R. Embryonic stem cell-specific microRNAs promote induced pluripotency. Nat. Biotechnol. 2009, 27, 459–461. [Google Scholar] [CrossRef] [PubMed]
- Subramanyam, D.; Lamouille, S.; Judson, R.L.; Liu, J.Y.; Bucay, N.; Derynck, R.; Blelloch, R. Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.; Bao, X.; Liu, L.; Feng, S.; Zovoilis, A.; Liu, W.; Xue, Y.; Cai, J.; Guo, X.; Qin, B.; et al. MicroRNA cluster 302–367 enhances somatic cell reprogramming by accelerating a mesenchymal-to-epithelial transition. J. Biol. Chem. 2011, 286, 17359–17364. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, N.; Fiedler, J.; Holzmann, A.; Schambach, A.; Moritz, T.; Cantz, T.; Thum, T. miRNA screening reveals a new miRNA family stimulating iPS cell generation via regulation of Meox2. EMBO Rep. 2011, 12, 1153–1159. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, N.; Ishii, H.; Nagano, H.; Haraguchi, N.; Dewi, D.L.; Kano, Y.; Nishikawa, S.; Tanemura, M.; Mimori, K.; Tanaka, F.; et al. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell 2011, 8, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Chang, D.C.; Lin, C.H.; Ying, S.Y.; Leu, D.; Wu, D.T. Regulation of somatic cell reprogramming through inducible mir-302 expression. Nucleic Acids Res. 2011, 39, 1054–1065. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Chang, D.C.; Chang-Lin, S.; Lin, C.H.; Wu, D.T.; Chen, D.T.; Ying, S.Y. Mir-302 reprograms human skin cancer cells into a pluripotent ES-cell-like state. RNA 2008, 14, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Anokye-Danso, F.; Trivedi, C.M.; Juhr, D.; Gupta, M.; Cui, Z.; Tian, Y.; Zhang, Y.; Yang, W.; Gruber, P.J.; Epstein, J.A.; et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 2011, 8, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell. Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Anokye-Danso, F.; Snitow, M.; Morrisey, E.E. How microRNAs facilitate reprogramming to pluripotency. J. Cell. Sci. 2012, 125, 4179–4187. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Li, Z.; Rana, T.M. microRNAs modulate iPS cell generation. RNA 2011, 17, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, C.S.; Nakashima, K.; Rana, T.M. Small RNA-mediated regulation of iPS cell generation. EMBO J. 2011, 30, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Melton, C.; Judson, R.L.; Blelloch, R. Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. Nature 2010, 463, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 2007, 316, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Ivey, K.N.; Muth, A.; Arnold, J.; King, F.W.; Yeh, R.F.; Fish, J.E.; Hsiao, E.C.; Schwartz, R.J.; Conklin, B.R.; Bernstein, H.S.; et al. MicroRNA regulation of cell lineages in mouse and human embryonic stem cells. Cell Stem Cell 2008, 2, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.D.; Hu, S.; Venkatasubrahmanyam, S.; Fu, J.D.; Sun, N.; Abilez, O.J.; Baugh, J.J.; Jia, F.; Ghosh, Z.; Li, R.A.; et al. Dynamic microRNA expression programs during cardiac differentiation of human embryonic stem cells: Role for miR-499. Circ. Cardiovasc. Genet. 2010, 3, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Sluijter, J.P.; van Mil, A.; van Vliet, P.; Metz, C.H.; Liu, J.; Doevendans, P.A.; Goumans, M.J. MicroRNA-1 and -499 regulate differentiation and proliferation in human-derived cardiomyocyte progenitor cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Wystub, K.; Besser, J.; Bachmann, A.; Boettger, T.; Braun, T. miR-1/133a clusters cooperatively specify the cardiomyogenic lineage by adjustment of myocardin levels during embryonic heart development. PLoS Genet. 2013, 9, e1003793. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Williams, A.H.; Kim, Y.; McAnally, J.; Bezprozvannaya, S.; Sutherland, L.B.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 20844–20849. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Samal, E.; Srivastava, D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 2005, 436, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.Y.; Lin, B.; Li, Y.; Arora, A.; Han, L.; Cui, C.; Coronnello, C.; Sheng, Y.; Benos, P.V.; Yang, L. Overexpression of microRNA-1 promotes cardiomyocyte commitment from human cardiovascular progenitors via suppressing WNT and FGF signaling pathways. J. Mol. Cell. Cardiol. 2013, 63, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Takaya, T.; Ono, K.; Kawamura, T.; Takanabe, R.; Kaichi, S.; Morimoto, T.; Wada, H.; Kita, T.; Shimatsu, A.; Hasegawa, K. MicroRNA-1 and MicroRNA-133 in spontaneous myocardial differentiation of mouse embryonic stem cells. Circ. J. 2009, 73, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Bezprozvannaya, S.; Williams, A.H.; Qi, X.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008, 22, 3242–3254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, X.; Chen, C.; Li, Y.; Zhao, L.; Jing, Y.; Liu, W.; Wang, X.; Zhang, Y.; Xia, H.; et al. Attenuation of p38-mediated miR-1/133 expression facilitates myoblast proliferation during the early stage of muscle regeneration. PLoS One. 2012, 7, e41478. [Google Scholar] [CrossRef] [PubMed]
- Mishima, Y.; Abreu-Goodger, C.; Staton, A.A.; Stahlhut, C.; Shou, C.; Cheng, C.; Gerstein, M.; Enright, A.J.; Giraldez, A.J. Zebrafish miR-1 and miR-133 shape muscle gene expression and regulate sarcomeric actin organization. Genes Dev. 2009, 23, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Rossini, A.; Frati, C.; Lagrasta, C.; Graiani, G.; Scopece, A.; Cavalli, S.; Musso, E.; Baccarin, M.; di Segni, M.; Fagnoni, F.; et al. Human cardiac and bone marrow stromal cells exhibit distinctive properties related to their origin. Cardiovasc.Res. 2011, 89, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Vecellio, M.; Meraviglia, V.; Nanni, S.; Barbuti, A.; Scavone, A.; DiFrancesco, D.; Farsetti, A.; Pompilio, G.; Colombo, G.I.; Capogrossi, M.C.; Gaetano, C.; et al. In vitro epigenetic reprogramming of human cardiac mesenchymal stromal cells into functionally competent cardiovascular precursors. PLoS One. 2012, 7, e51694. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, T.; Zheng, H.; Cabral-da-Silva, M.; Sanada, F.; Ide-Iwata, N.; Ogorek, B.; Ferreira-Martins, J.; Arranto, C.; D'Amario, D.; del Monte, F.; et al. Human cardiac stem cell differentiation is regulated by a mircrine mechanism. Circulation 2011, 123, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Quiat, D.; Johnson, B.A.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Kelm, R.J., Jr.; Olson, E.N. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev. Cell 2009, 17, 662–673. [Google Scholar]
- Fu, J.D.; Rushing, S.N.; Lieu, D.K.; Chan, C.W.; Kong, C.W.; Geng, L.; Wilson, K.D.; Chiamvimonvat, N.; Boheler, K.R.; Wu, J.C.; et al. Distinct roles of microRNA-1 and -499 in ventricular specification and functional maturation of human embryonic stem cell-derived cardiomyocytes. PLoS One. 2011, 6, e27417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoda, T. The mircrine mechanism controlling cardiac stem cell fate. Front. Genet. 2013, 4, 204:1–204:3. [Google Scholar]
- Berardi, E.; Pues, M.; Thorrez, L.; Sampaolesi, M. miRNAs in ESC differentiation. Am. J. Physiol. Heart. Circ. Physiol. 2012, 303, H931–H939. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Spagnoli, F.M.; Brivanlou, A.H. The miR-430/427/302 family controls mesendodermal fate specification via species-specific target selection. Dev. Cell. 2009, 16, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Foshay, K.M.; Gallicano, G.I. miR-17 family miRNAs are expressed during early mammalian development and regulate stem cell differentiation. Dev. Biol. 2009, 326, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Papagiannakopoulos, T.; Pan, G.; Thomson, J.A.; Kosik, K.S. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell 2009, 137, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Pournasr, B.; Khaloughi, K.; Salekdeh, G.H.; Totonchi, M.; Shahbazi, E.; Baharvand, H. Concise review: Alchemy of biology: Generating desired cell types from abundant and accessible cells. Stem Cells. 2011, 29, 1933–1941. [Google Scholar] [CrossRef] [PubMed]
- Jayawardena, T.M.; Egemnazarov, B.; Finch, E.A.; Zhang, L.; Payne, J.A.; Pandya, K.; Zhang, Z.; Rosenberg, P.; Mirotsou, M.; Dzau, V.J. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ. Res. 2012, 110, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Efe, J.A.; Hilcove, S.; Kim, J.; Zhou, H.; Ouyang, K.; Wang, G.; Chen, J.; Ding, S. Conversion of mouse fibroblasts into cardiomyocytes using a direct reprogramming strategy. Nat.Cell. Biol. 2011, 13, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.J.; Song, K.; Olson, E.N. Heart repair by cardiac reprogramming. Nat. Med. 2013, 19, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Nam, Y.J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G.; Hill, J.A.; et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1–2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Murchison, E.P.; Tang, R.; Callis, T.E.; Tatsuguchi, M.; Deng, Z.; Rojas, M.; Hammond, S.M.; Schneider, M.D.; Selzman, C.H.; Meissner, G.; et al. Targeted deletion of dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 2111–2116. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.K.; Toyama, Y.; Chiang, H.R.; Gupta, S.; Bauer, M.; Medvid, R.; Reinhardt, F.; Liao, R.; Krieger, M.; Jaenisch, R.; et al. Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circ. Res. 2009, 105, 585–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Costa Martins, P.A.; Bourajjaj, M.; Gladka, M.; Kortland, M.; van Oort, R.J.; Pinto, Y.M.; Molkentin, J.D.; de Windt, L.J. Conditional dicer gene deletion in the postnatal myocardium provokes spontaneous cardiac remodeling. Circulation 2008, 118, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.; Huang, Y.; Maher, S.E.; Kim, R.W.; Giordano, F.J.; Tellides, G.; Geirsson, A. miR-1 mediated suppression of sorcin regulates myocardial contractility through modulation of Ca2+ signaling. J. Mol. Cell Cardiol. 2012, 52, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Tritsch, E.; Mallat, Y.; Lefebvre, F.; Diguet, N.; Escoubet, B.; Blanc, J.; de Windt, L.J.; Catalucci, D.; Vandecasteele, G.; Li, Z.; Mericskay, M. An SRF/miR-1 axis regulates NCX1 and annexin A5 protein levels in the normal and failing heart. Cardiovasc. Res. 2013, 98, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Heidersbach, A.; Saxby, C.; Carver-Moore, K.; Huang, Y.; Ang, Y.S.; de Jong, P.J.; Ivey, K.N.; Srivastava, D. microRNA-1 regulates sarcomere formation and suppresses smooth muscle gene expression in the mammalian heart. Elife 2013, 2, e01323. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.; Han, Z.; Olson, E.N.; Srivastava, D. MicroRNA1 influences cardiac differentiation in drosophila and regulates notch signaling. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 18986–18991. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkeland, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008, 132, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Novotny, G.W.; Sonne, S.B.; Nielsen, J.E.; Jonstrup, S.P.; Hansen, M.A.; Skakkebaek, N.E.; Rajpert-De Meyts, E.; Kjems, J.; Leffers, H. Translational repression of E2F1 mRNA in carcinoma in situ and normal testis correlates with expression of the miR-17–92 cluster. Cell. Death Differ. 2007, 14, 879–882. [Google Scholar] [CrossRef] [PubMed]
- O'Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. C-myc-regulated microRNAs modulate E2F1 expression. Nature 2005, 435, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Shan, S.W.; Lee, D.Y.; Deng, Z.; Shatseva, T.; Jeyapalan, Z.; Du, W.W.; Zhang, Y.; Xuan, J.W.; Yee, S.P.; Siragam, V.; Yang, B.B. MicroRNA MiR-17 retards tissue growth and represses fibronectin expression. Nat. Cell. Biol. 2009, 11, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Danielson, L.S.; Park, D.S.; Rotllan, N.; Chamorro-Jorganes, A.; Guijarro, M.V.; Fernandez-Hernando, C.; Fishman, G.I.; Phoon, C.K.; Hernando, E. Cardiovascular dysregulation of miR-17–92 causes a lethal hypertrophic cardiomyopathy and arrhythmogenesis. FASEB J. 2013, 27, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Johnson, B.A.; Aurora, A.B.; Simpson, E.; Nam, Y.J.; Matkovich, S.J.; Dorn, G.W.; van Rooij, E.; Olson, E.N. MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ. Res. 2011, 109, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Mano, M.; Dal Ferro, M.; Zentilin, L.; Sinagra, G.; Zacchigna, S.; Giacca, M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012, 492, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Hullinger, T.G.; Montgomery, R.L.; Seto, A.G.; Dickinson, B.A.; Semus, H.M.; Lynch, J.M.; Dalby, C.M.; Robinson, K.; Stack, C.; Latimer, P.A.; et al. Inhibition of miR-15 protects against cardiac ischemic injury. Circ. Res. 2012, 110, 71–81. [Google Scholar]
- Zhu, H.; Yang, Y.; Wang, Y.; Li, J.; Schiller, P.W.; Peng, T. MicroRNA-195 promotes palmitate-induced apoptosis in cardiomyocytes by down-regulating Sirt1. Cardiovasc. Res. 2011, 92, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Callis, T.E.; Pandya, K.; Seok, H.Y.; Tang, R.H.; Tatsuguchi, M.; Huang, Z.P.; Chen, J.F.; Deng, Z.; Gunn, B.; Shumate, J.; et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clin. Invest. 2009, 119, 2772–2786. [Google Scholar] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Liu, N.; Williams, A.H.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 18255–18260. [Google Scholar] [CrossRef] [PubMed]
- Morton, S.U.; Scherz, P.J.; Cordes, K.R.; Ivey, K.N.; Stainier, D.Y.; Srivastava, D. microRNA-138 modulates cardiac patterning during embryonic development. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 17830–17835. [Google Scholar] [CrossRef] [PubMed]
- Chiavacci, E.; Dolfi, L.; Verduci, L.; Meghini, F.; Gestri, G.; Evangelista, A.M.; Wilson, S.W.; Cremisi, F.; Pitto, L. MicroRNA 218 mediates the effects of Tbx5a over-expression on zebrafish heart development. PLoS One 2012, 7, e50536. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Piubelli, C.; Meraviglia, V.; Pompilio, G.; D'Alessandra, Y.; Colombo, G.I.; Rossini, A. microRNAs and Cardiac Cell Fate. Cells 2014, 3, 802-823. https://doi.org/10.3390/cells3030802
Piubelli C, Meraviglia V, Pompilio G, D'Alessandra Y, Colombo GI, Rossini A. microRNAs and Cardiac Cell Fate. Cells. 2014; 3(3):802-823. https://doi.org/10.3390/cells3030802
Chicago/Turabian StylePiubelli, Chiara, Viviana Meraviglia, Giulio Pompilio, Yuri D'Alessandra, Gualtiero I. Colombo, and Alessandra Rossini. 2014. "microRNAs and Cardiac Cell Fate" Cells 3, no. 3: 802-823. https://doi.org/10.3390/cells3030802