Bacterial Effectors and Their Functions in the Ubiquitin-Proteasome System: Insight from the Modes of Substrate Recognition

Abstract
:1. Introduction
2. Exploitation of the Ubiquitin-Proteasome System by Pathogenic Bacteria

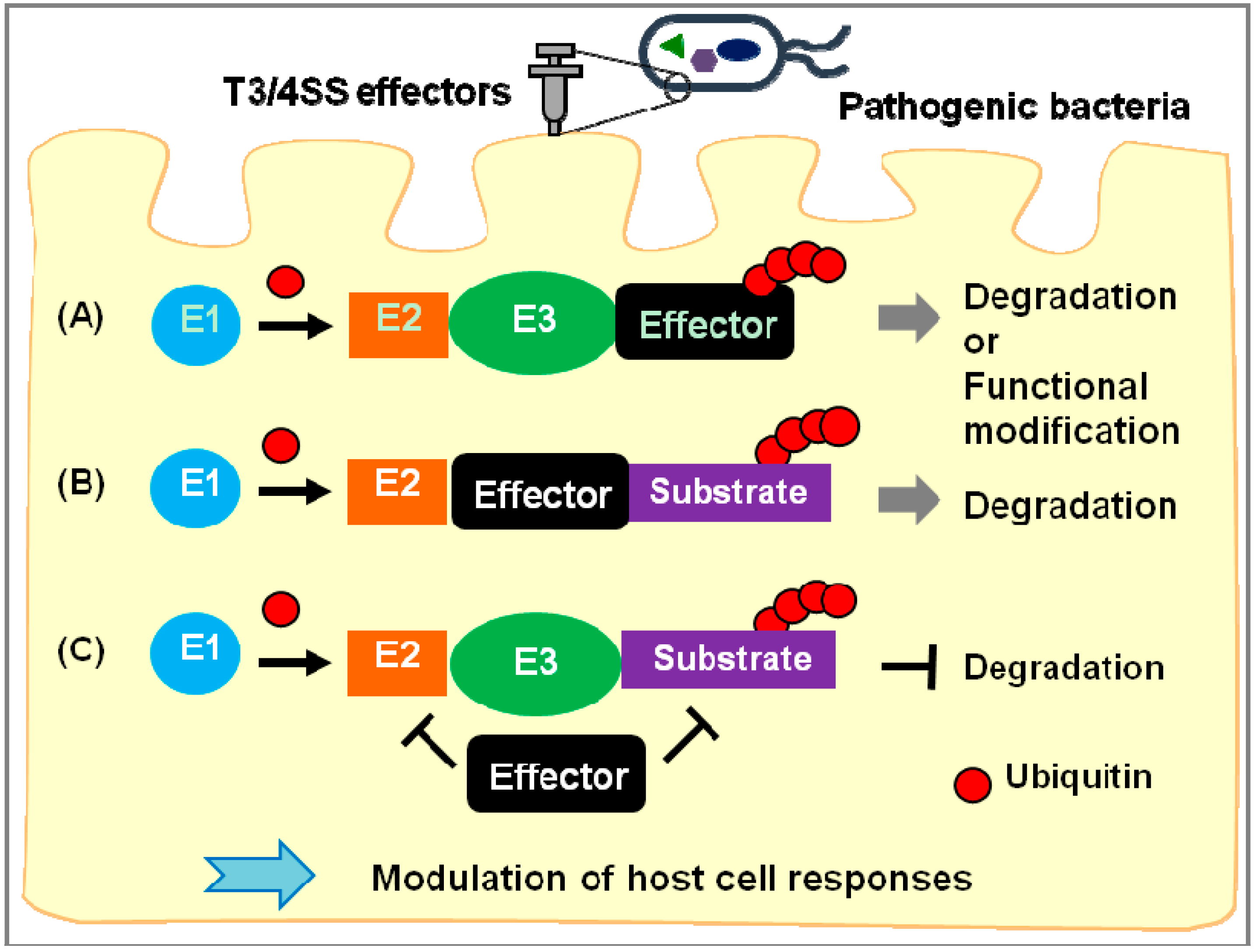{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Function | Pathogens | Effector | Targets | Mechanism of Action | Ref | |
|---|---|---|---|---|---|---|
| ubiquitin ligase (E3) | HECT-like | enterohemorrhagic E.coli (EHEC) | NleL | Unknown | Pedestal formation | [22,23] |
| S. Typhimurium | SopA | Unknown | Regulation of inflammation | [24,25] | ||
| RING/U-box | EHEC | NleG | Unknown | Unknown | [26] | |
| L. pneumophila | LubX | Clk1, SidH | Unknown | [27,28] | ||
| P. syringae | AvrPtoB | Fen, CERK1, FLS2, BAK1 | Suppression of plant immunity | [29,30,31,32] | ||
| Novel E3 ligase (NEL) | Rhizobium | NopM | Unknown | Unknown | [33] | |
| S. Typhimurium | Slrp | Trx, ERdj3 | Induction of cell death Interference with unfolded protein response | [34,35] | ||
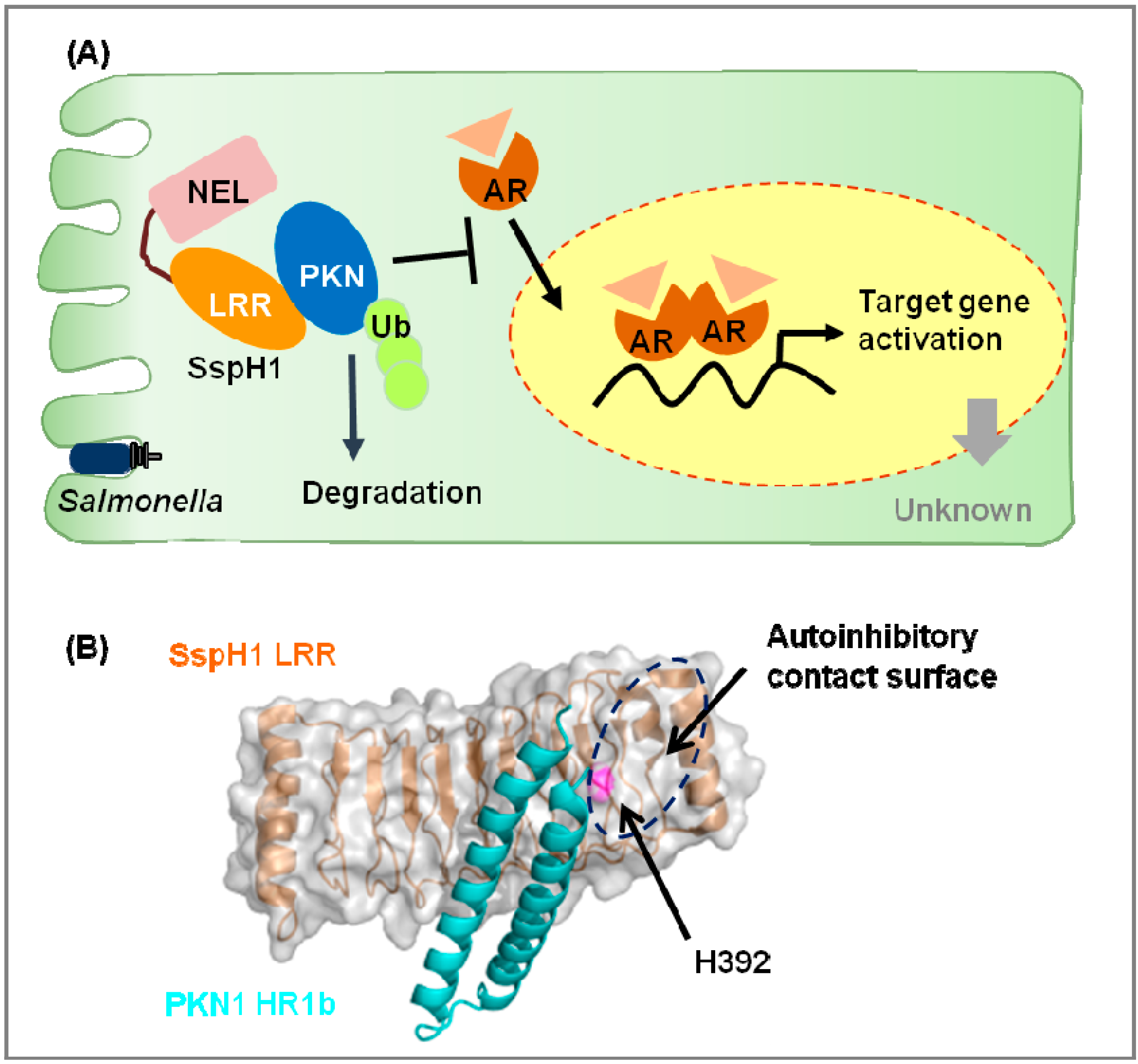
| SspH1 | PKN1 | Inhibition of androgen receptor signal | [36,37] | |||
| SspH2 | NOD1, SGT1 | Up-regulation of inflammation | [38] | |||
| Shigella | IpaH1.4 | Unknown | Unknown | [39] | ||
| IpaH3 | Unknown | Unknown | [40] | |||
| IpaH4.5 | p65 | Suppression of inflammation | [41] | |||
| IpaH9.8 | Ste7, U2AF 35, NEMO | Suppression of inflammation/splicing | [42,43,44] | |||
| IpaH0772 | TRAF2 | Suppression of inflammation | [45] | |||
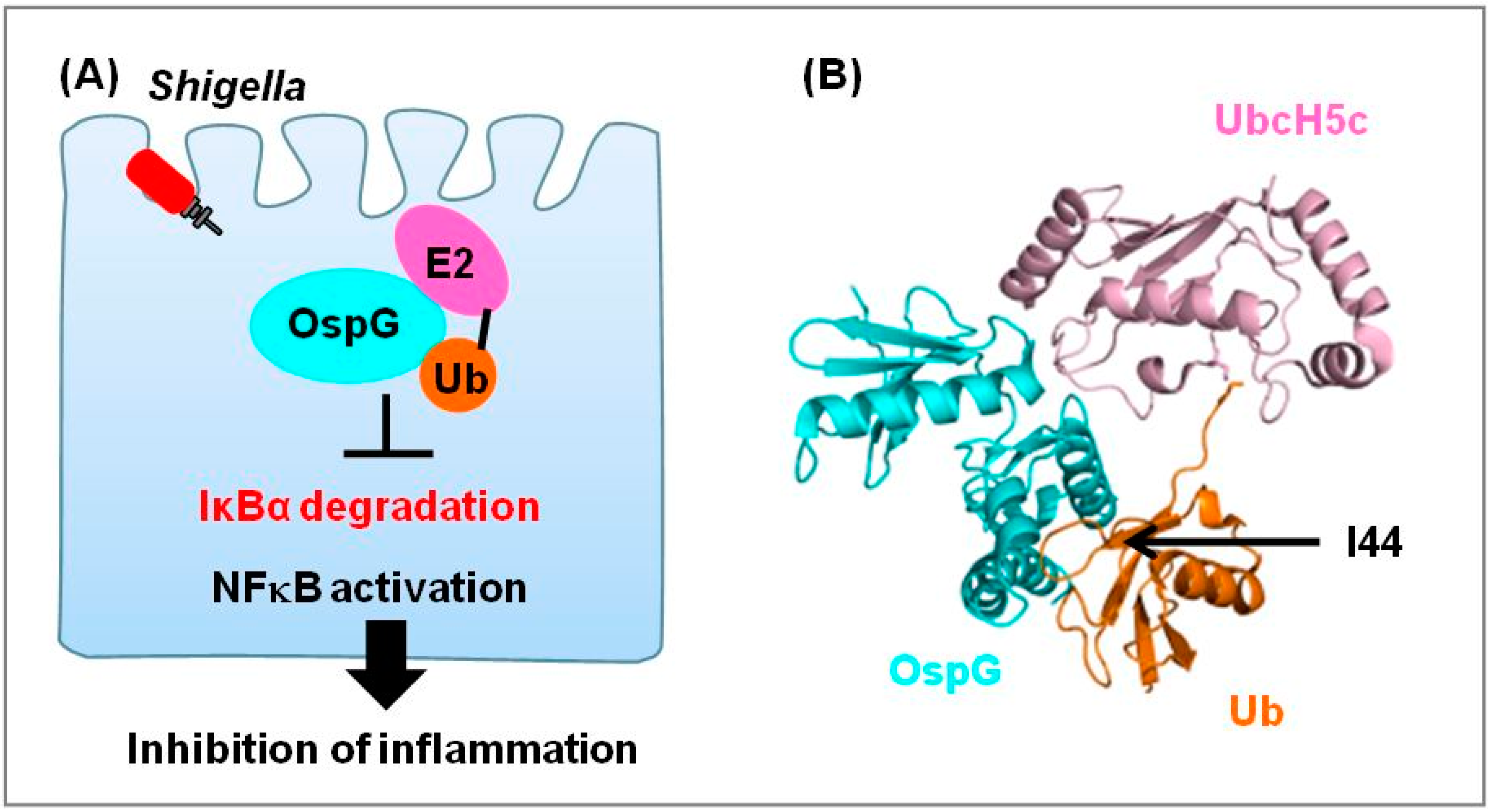
| Kinase | Shigella | OspG | UbcH5 | Suppression of inflammation | [46,47] | |
| EHEC | OspG | Unknown | Unknown | [48] | ||
| Y. enterocolitica | OspG | Unknown | Unknown | [48] | ||
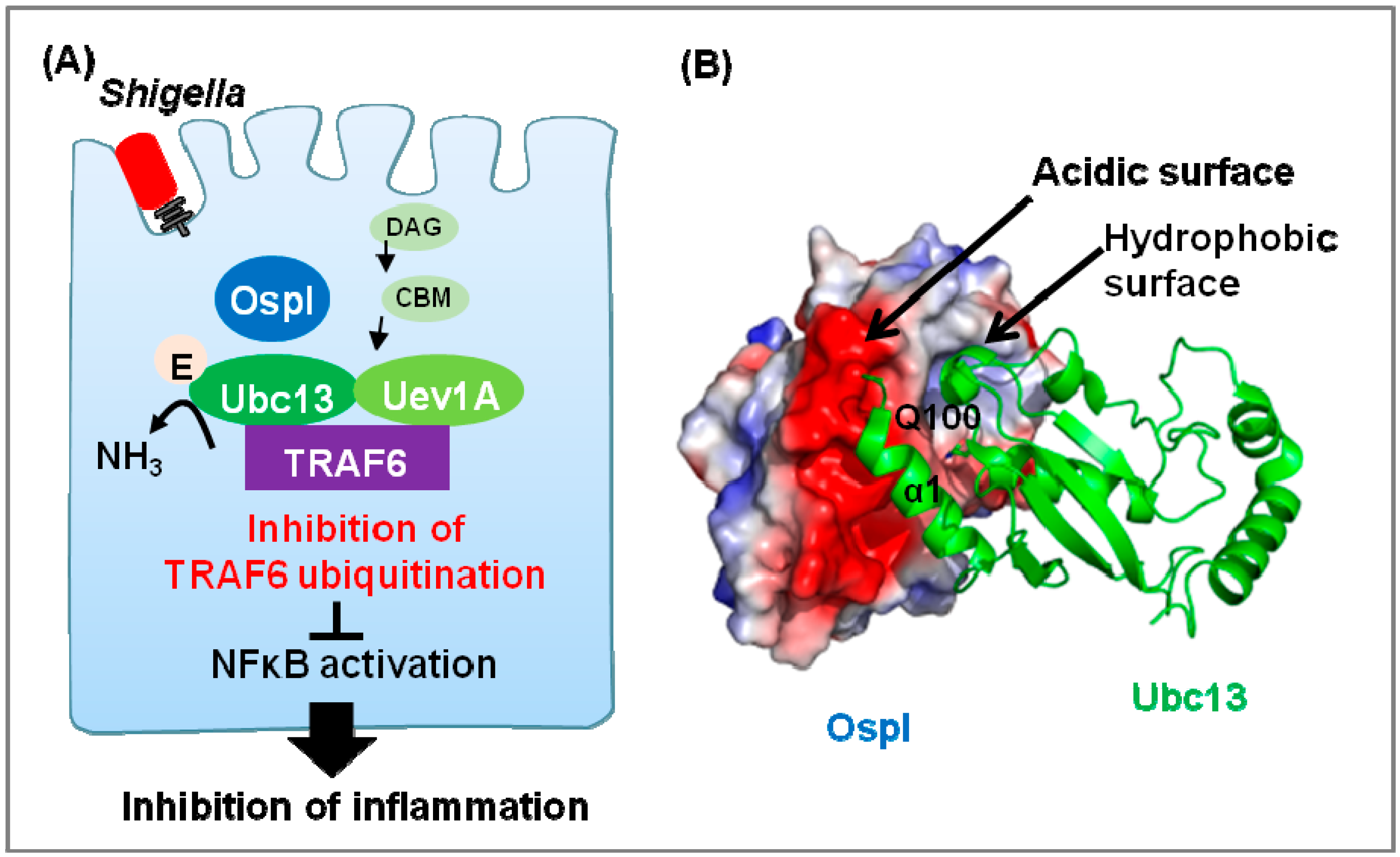
| Deamidase | Shigella | OspI | Ubc13 | Suppression of inflammation | [49,50,51] | |
| enteropathogenic Escherichia coli (EPEC)/EHEC | Cif | NEDD8, Ub | Inhibition of Cullin-RING ligases (CRL) activation | [52,53,54,55,56] | ||
| B. pseudomallei | CHBP | NEDD8, Ub | Inhibition of CRL activation | [52,57,58,59] | ||
| P. luminescens | CifPl | NEDD8 | Inhibition of CRL activation | [57] | ||
| Y. pseudotuberculosis | CifYp | NEDD8, Ub | Inhibition of CRL activation | [60] | ||

2.1. Novel E3 Ligases and Their Substrates

2.2 The Deamidase OspI Targets Ubc13

2.3. The Deamidase Cif Targets Ubiquitin or NEDD8

2.4. Kinase OspG Regulates E2

3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Niyogi, S.K. Shigellosis. J. Microbiol. 2005, 43, 133–143. [Google Scholar]
- Ochoa, T.J.; Contreras, C.A. Enteropathogenic escherichia coli infection in children. Curr. Opin. Infect. Dis. 2011, 24, 478–483. [Google Scholar] [CrossRef]
- Bassetti, M.; Merelli, M.; Temperoni, C.; Astilean, A. New antibiotics for bad bugs: where are we? Ann. Clin. Microbiol. Antimicrob. 2013, 12, 22. [Google Scholar] [CrossRef]
- Montassier, E.; Batard, E.; Gastinne, T.; Potel, G.; de La Cochetiere, M.F. Recent changes in bacteremia in patients with cancer: A systematic review of epidemiology and antibiotic resistance. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 841–850. [Google Scholar] [CrossRef]
- Kim, M.; Ashida, H.; Ogawa, M.; Yoshikawa, Y.; Mimuro, H.; Sasakawa, C. Bacterial interactions with the host epithelium. Cell. Host Microbe. 2010, 8, 20–35. [Google Scholar] [CrossRef]
- Buttner, D. Protein export according to schedule: Architecture, assembly, and regulation of type III secretion systems from plant- and animal-pathogenic bacteria. Microbiol. Mol. Biol. Rev. 2012, 76, 262–310. [Google Scholar] [CrossRef]
- Bhavsar, A.P.; Guttman, J.A.; Finlay, B.B. Manipulation of host-cell pathways by bacterial pathogens. Nature 2007, 449, 827–834. [Google Scholar] [CrossRef]
- Carayol, N.; Tran Van Nhieu, G. The inside story of Shigella invasion of intestinal epithelial cells. Cold Spring Harb Perspect. Med. 2013, 3, a016717. [Google Scholar]
- Hunter, T. The age of crosstalk: Phosphorylation, ubiquitination, and beyond. Mol. Cell. 2007, 28, 730–738. [Google Scholar] [CrossRef]
- Ribet, D.; Cossart, P. Pathogen-mediated posttranslational modifications: A re-emerging field. Cell 2010, 143, 694–702. [Google Scholar] [CrossRef]
- Weissman, A.M.; Shabek, N.; Ciechanover, A. The predator becomes the prey: Regulating the ubiquitin system by ubiquitylation and degradation. Nat. Rev. Mol. Cell. Biol. 2011, 12, 605–620. [Google Scholar] [CrossRef]
- Ikeda, F.; Dikic, I. Atypical ubiquitin chains: New molecular signals. 'Protein Modifications: Beyond the Usual Suspects' review series. EMBO Rep. 2008, 9, 536–542. [Google Scholar] [CrossRef]
- Iwai, K. Diverse ubiquitin signaling in NF-kappaB activation. Trends Cell. Biol. 2012, 22, 355–364. [Google Scholar] [CrossRef]
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef]
- Bhoj, V.G.; Chen, Z.J. Ubiquitylation in innate and adaptive immunity. Nature 2009, 458, 430–437. [Google Scholar] [CrossRef]
- Hoeller, D.; Hecker, C.M.; Dikic, I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat. Rev. Cancer 2006, 6, 776–788. [Google Scholar] [CrossRef]
- Isaacson, M.K.; Ploegh, H.L. Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell. Host Microbe. 2009, 5, 559–570. [Google Scholar] [CrossRef]
- Mukhopadhyay, D.; Riezman, H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 2007, 315, 201–205. [Google Scholar] [CrossRef]
- Nakayama, K.I.; Nakayama, K. Ubiquitin ligases:Cell-cycle control and cancer. Nat. Rev. Cancer 2006, 6, 369–381. [Google Scholar] [CrossRef]
- Cardozo, T.; Pagano, M. The SCF ubiquitin ligase: Insights into a molecular machine. Nat. Rev. Mol. Cell. Biol. 2004, 5, 739–751. [Google Scholar] [CrossRef]
- Rytkonen, A.; Holden, D.W. Bacterial interference of ubiquitination and deubiquitination. Cell. Host Microbe. 2007, 1, 13–22. [Google Scholar] [CrossRef]
- Lin, D.Y.; Diao, J.; Zhou, D.; Chen, J. Biochemical and structural studies of a HECT-like ubiquitin ligase from Escherichia coli O157:H7. J. Biol. Chem. 2011, 286, 441–449. [Google Scholar]
- Piscatelli, H.; Kotkar, S.A.; McBee, M.E.; Muthupalani, S.; Schauer, D.B.; Mandrell, R.E.; Leong, J.M.; Zhou, D. The EHEC type III effector NleL is an E3 ubiquitin ligase that modulates pedestal formation. PLoS One 2011, 6, e19331. [Google Scholar] [CrossRef] [Green Version]
- Diao, J.; Zhang, Y.; Huibregtse, J.M.; Zhou, D.; Chen, J. Crystal structure of SopA, a Salmonella effector protein mimicking a eukaryotic ubiquitin ligase. Nat. Struct. Mol. Biol. 2008, 15, 65–70. [Google Scholar] [CrossRef]
- Zhang, Y.; Higashide, W.M.; McCormick, B.A.; Chen, J.; Zhou, D. The inflammation-associated Salmonella SopA is a HECT-like E3 ubiquitin ligase. Mol. Microbiol. 2006, 62, 786–793. [Google Scholar] [CrossRef]
- Wu, B.; Skarina, T.; Yee, A.; Jobin, M.C.; Dileo, R.; Semesi, A.; Fares, C.; Lemak, A.; Coombes, B.K.; Arrowsmith, C.H.; Singer, A.U.; et al. NleG Type 3 effectors from enterohaemorrhagic Escherichia coli are U-Box E3 ubiquitin ligases. PLoS Pathog. 2010, 6, e1000960. [Google Scholar] [CrossRef]
- Kubori, T.; Hyakutake, A.; Nagai, H. Legionella translocates an E3 ubiquitin ligase that has multiple U-boxes with distinct functions. Mol. Microbiol. 2008, 67, 1307–1319. [Google Scholar] [CrossRef]
- Kubori, T.; Shinzawa, N.; Kanuka, H.; Nagai, H. Legionella metaeffector exploits host proteasome to temporally regulate cognate effector. PLoS Pathog. 2010, 6, e1001216. [Google Scholar] [CrossRef]
- Cheng, W.; Munkvold, K.R.; Gao, H.; Mathieu, J.; Schwizer, S.; Wang, S.; Yan, Y.B.; Wang, J.; Martin, G.B.; Chai, J. Structural analysis of Pseudomonas syringae AvrPtoB bound to host BAK1 reveals two similar kinase-interacting domains in a type III Effector. Cell. Host Microbe. 2011, 10, 616–626. [Google Scholar] [CrossRef]
- Gimenez-Ibanez, S.; Hann, D.R.; Ntoukakis, V.; Petutschnig, E.; Lipka, V.; Rathjen, J.P. AvrPtoB targets the LysM receptor kinase CERK1 to promote bacterial virulence on plants. Curr. Biol. 2009, 19, 423–429. [Google Scholar] [CrossRef]
- Gohre, V.; Spallek, T.; Haweker, H.; Mersmann, S.; Mentzel, T.; Boller, T.; de Torres, M.; Mansfield, J.W.; Robatzek, S. Plant pattern-recognition receptor FLS2 is directed for degradation by the bacterial ubiquitin ligase AvrPtoB. Curr. Biol. 2008, 18, 1824–1832. [Google Scholar] [CrossRef]
- Rosebrock, T.R.; Zeng, L.; Brady, J.J.; Abramovitch, R.B.; Xiao, F.; Martin, G.B. A bacterial E3 ubiquitin ligase targets a host protein kinase to disrupt plant immunity. Nature 2007, 448, 370–374. [Google Scholar] [CrossRef]
- Xin, D.W.; Liao, S.; Xie, Z.P.; Hann, D.R.; Steinle, L.; Boller, T.; Staehelin, C. Functional analysis of NopM, a novel E3 ubiquitin ligase (NEL) domain effector of Rhizobium sp. strain NGR234. PLoS Pathog. 2012, 8, e1002707. [Google Scholar]
- Bernal-Bayard, J.; Cardenal-Munoz, E.; Ramos-Morales, F. The Salmonella type III secretion effector, salmonella leucine-rich repeat protein (SlrP), targets the human chaperone ERdj3. J. Biol. Chem. 2010, 285, 16360–16368. [Google Scholar]
- Bernal-Bayard, J.; Ramos-Morales, F. Salmonella type III secretion effector SlrP is an E3 ubiquitin ligase for mammalian thioredoxin. J. Biol. Chem. 2009, 284, 27587–27595. [Google Scholar] [CrossRef]
- Haraga, A.; Miller, S.I. A Salmonella type III secretion effector interacts with the mammalian serine/threonine protein kinase PKN1. Cell. Microbiol. 2006, 8, 837–846. [Google Scholar] [CrossRef]
- Keszei, A.F.; Tang, X.; McCormick, C.; Zeqiraj, E.; Rohde, J.R.; Tyers, M.; Sicheri, F. Structure of an SspH1-PKN1 complex reveals the basis for host substrate recognition and mechanism of activation for a bacterial E3 ubiquitin ligase. Mol. Cell. Biol. 2014, 34, 362–373. [Google Scholar] [CrossRef]
- Bhavsar, A.P.; Brown, N.F.; Stoepel, J.; Wiermer, M.; Martin, D.D.; Hsu, K.J.; Imami, K.; Ross, C.J.; Hayden, M.R.; Foster, L.J.; et al. The Salmonella type III effector SspH2 specifically exploits the NLR co-chaperone activity of SGT1 to subvert immunity. PLoS Pathog. 2013, 9, e1003518. [Google Scholar] [CrossRef]
- Singer, A.U.; Rohde, J.R.; Lam, R.; Skarina, T.; Kagan, O.; Dileo, R.; Chirgadze, N.Y.; Cuff, M.E.; Joachimiak, A.; Tyers, M.; Sansonetti, P.J.; Parsot, C.; Savchenko, A. Structure of the Shigella T3SS effector IpaH defines a new class of E3 ubiquitin ligases. Nat. Struct. Mol. Biol. 2008, 15, 1293–1301. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, H.; Hu, L.; Wang, J.; Zhou, Y.; Pang, Z.; Liu, L.; Shao, F. Structure of a Shigella effector reveals a new class of ubiquitin ligases. Nat. Struct. Mol. Biol. 2008, 15, 1302–1308. [Google Scholar] [CrossRef]
- Wang, F.; Jiang, Z.; Li, Y.; He, X.; Zhao, J.; Yang, X.; Zhu, L.; Yin, Z.; Li, X.; Wang, X.; et al. Shigella flexneri T3SS effector IpaH4.5 modulates the host inflammatory response via interaction with NF-kappaB p65 protein. Cell. Microbiol. 2013, 15, 474–485. [Google Scholar] [CrossRef]
- Ashida, H.; Kim, M.; Schmidt-Supprian, M.; Ma, A.; Ogawa, M.; Sasakawa, C. A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKgamma to dampen the host NF-kappaB-mediated inflammatory response. Nat. Cell. Biol. 2010, 12, 66–73. [Google Scholar] [CrossRef]
- Okuda, J.; Toyotome, T.; Kataoka, N.; Ohno, M.; Abe, H.; Shimura, Y.; Seyedarabi, A.; Pickersgill, R.; Sasakawa, C. Shigella effector IpaH9.8 binds to a splicing factor U2AF(35) to modulate host immune responses. Biochem. Biophys. Res. Commun. 2005, 333, 531–539. [Google Scholar] [CrossRef]
- Rohde, J.R.; Breitkreutz, A.; Chenal, A.; Sansonetti, P.J.; Parsot, C. Type III secretion effectors of the IpaH family are E3 ubiquitin ligases. Cell. Host Microbe. 2007, 1, 77–83. [Google Scholar] [CrossRef]
- Ashida, H.; Nakano, H.; Sasakawa, C. Shigella IpaH0722 E3 ubiquitin ligase effector targets TRAF2 to inhibit PKC-NF-kappaB activity in invaded epithelial cells. PLoS Pathog. 2013, 9, e1003409. [Google Scholar] [CrossRef]
- Kim, D.W.; Lenzen, G.; Page, A.L.; Legrain, P.; Sansonetti, P.J.; Parsot, C. The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. USA 2005, 102, 14046–14051. [Google Scholar] [CrossRef]
- Pruneda, J.N.; Smith, F.D.; Daurie, A.; Swaney, D.L.; Villen, J.; Scott, J.D.; Stadnyk, A.W.; Le Trong, I.; Stenkamp, R.E.; Klevit, R.E.; et al. E2~Ub conjugates regulate the kinase activity of Shigella effector OspG during pathogenesis. EMBO J. 2014, 33, 437–449. [Google Scholar]
- Nobe, R.; Nougayrede, J.P.; Taieb, F.; Bardiau, M.; Cassart, D.; Navarro-Garcia, F.; Mainil, J.; Hayashi, T.; Oswald, E. Enterohaemorrhagic Escherichia coli serogroup O111 inhibits NF-(kappa)B-dependent innate responses in a manner independent of a type III secreted OspG orthologue. Microbiology 2009, 155, 3214–3225. [Google Scholar] [CrossRef]
- Fu, P.; Zhang, X.; Jin, M.; Xu, L.; Wang, C.; Xia, Z.; Zhu, Y. Complex structure of OspI and Ubc13: The molecular basis of Ubc13 deamidation and convergence of bacterial and host E2 recognition. PLoS Pathog. 2013, 9, e1003322. [Google Scholar] [CrossRef]
- Nishide, A.; Kim, M.; Takagi, K.; Himeno, A.; Sanada, T.; Sasakawa, C.; Mizushima, T. Structural basis for the recognition of Ubc13 by the Shigella flexneri effector OspI. J. Mol. Biol. 2013, 425, 2623–2631. [Google Scholar] [CrossRef]
- Sanada, T.; Kim, M.; Mimuro, H.; Suzuki, M.; Ogawa, M.; Oyama, A.; Ashida, H.; Kobayashi, T.; Koyama, T.; Nagai, S.; et al. The Shigella flexneri effector OspI deamidates UBC13 to dampen the inflammatory response. Nature 2012, 483, 623–626. [Google Scholar] [CrossRef]
- Cui, J.; Yao, Q.; Li, S.; Ding, X.; Lu, Q.; Mao, H.; Liu, L.; Zheng, N.; Chen, S.; Shao, F. Glutamine deamidation and dysfunction of ubiquitin/NEDD8 induced by a bacterial effector family. Science 2010, 329, 1215–1218. [Google Scholar] [CrossRef]
- Hsu, Y.; Jubelin, G.; Taieb, F.; Nougayrede, J.P.; Oswald, E.; Stebbins, C.E. Structure of the cyclomodulin Cif from pathogenic Escherichia coli. J. Mol. Biol. 2008, 384, 465–477. [Google Scholar] [CrossRef]
- Jubelin, G.; Taieb, F.; Duda, D.M.; Hsu, Y.; Samba-Louaka, A.; Nobe, R.; Penary, M.; Watrin, C.; Nougayrede, J.P.; Schulman, B.A.; et al. Pathogenic bacteria target NEDD8-conjugated cullins to hijack host-cell signaling pathways. PLoS Pathog. 2010, 6, e1001128. [Google Scholar] [CrossRef]
- Morikawa, H.; Kim, M.; Mimuro, H.; Punginelli, C.; Koyama, T.; Nagai, S.; Miyawaki, A.; Iwai, K.; Sasakawa, C. The bacterial effector Cif interferes with SCF ubiquitin ligase function by inhibiting deneddylation of Cullin1. Biochem. Biophys. Res. Commun. 2010, 401, 268–274. [Google Scholar] [CrossRef]
- Samba-Louaka, A.; Nougayrede, J.P.; Watrin, C.; Oswald, E.; Taieb, F. The enteropathogenic Escherichia coli effector Cif induces delayed apoptosis in epithelial cells. Infect. Immun. 2009, 77, 5471–5477. [Google Scholar] [CrossRef]
- Crow, A.; Race, P.R.; Jubelin, G.; Varela Chavez, C.; Escoubas, J.M.; Oswald, E.; Banfield, M.J. Crystal structures of Cif from bacterial pathogens Photorhabdus luminescens and Burkholderia pseudomallei. PLoS One 2009, 4, e5582. [Google Scholar]
- Yao, Q.; Cui, J.; Wang, J.; Li, T.; Wan, X.; Luo, T.; Gong, Y.N.; Xu, Y.; Huang, N.; Shao, F. Structural mechanism of ubiquitin and NEDD8 deamidation catalyzed by bacterial effectors that induce macrophage-specific apoptosis. Proc. Natl. Acad. Sci. USA 2012, 109, 20395–20400. [Google Scholar]
- Yao, Q.; Cui, J.; Zhu, Y.; Wang, G.; Hu, L.; Long, C.; Cao, R.; Liu, X.; Huang, N.; Chen, S.; et al. A bacterial type III effector family uses the papain-like hydrolytic activity to arrest the host cell cycle. Proc. Natl. Acad. Sci. USA 2009, 106, 3716–3721. [Google Scholar]
- Crow, A.; Hughes, R.K.; Taieb, F.; Oswald, E.; Banfield, M.J. The molecular basis of ubiquitin-like protein NEDD8 deamidation by the bacterial effector protein Cif. Proc. Natl. Acad. Sci. USA 2012, 109, E1830–E1838. [Google Scholar]
- Kubori, T.; Galan, J.E. Temporal regulation of salmonella virulence effector function by proteasome-dependent protein degradation. Cell 2003, 115, 333–342. [Google Scholar]
- Gaus, K.; Hentschke, M.; Czymmeck, N.; Novikova, L.; Trulzsch, K.; Valentin-Weigand, P.; Aepfelbacher, M.; Ruckdeschel, K. Destabilization of YopE by the ubiquitin-proteasome pathway fine-tunes Yop delivery into host cells and facilitates systemic spread of Yersinia enterocolitica in host lymphoid tissue. Infect. Immun. 2011, 79, 1166–1175. [Google Scholar] [CrossRef]
- Hentschke, M.; Trulzsch, K.; Heesemann, J.; Aepfelbacher, M.; Ruckdeschel, K. Serogroup-related escape of Yersinia enterocolitica YopE from degradation by the ubiquitin-proteasome pathway. Infect. Immun. 2007, 75, 4423–4431. [Google Scholar] [CrossRef]
- Knodler, L.A.; Winfree, S.; Drecktrah, D.; Ireland, R.; Steele-Mortimer, O. Ubiquitination of the bacterial inositol phosphatase, SopB, regulates its biological activity at the plasma membran. Cell. Microbiol. 2009, 11, 1652–1670. [Google Scholar] [CrossRef]
- Patel, J.C.; Hueffer, K.; Lam, T.T.; Galan, J.E. Diversification of a Salmonella virulence protein function by ubiquitin-dependent differential localization. Cell 2009, 137, 283–294. [Google Scholar] [CrossRef]
- Rytkonen, A.; Poh, J.; Garmendia, J.; Boyle, C.; Thompson, A.; Liu, M.; Freemont, P.; Hinton, J.C.; Holden, D.W. SseL, a Salmonella deubiquitinase required for macrophage killing and virulence. Proc. Natl. Acad. Sci. USA 2007, 104, 3502–3507. [Google Scholar] [CrossRef]
- Janjusevic, R.; Abramovitch, R.B.; Martin, G.B.; Stebbins, C.E. A bacterial inhibitor of host programmed cell death defenses is an E3 ubiquitin ligase. Science 2006, 311, 222–226. [Google Scholar] [CrossRef]
- Munkvold, K.R.; Martin, G.B. Advances in experimental methods for the elucidation of Pseudomonas syringae effector function with a focus on AvrPtoB. Mol. Plant. Pathol. 2009, 10, 777–793. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Zhang, L.; Ding, X.; Cui, J.; Xu, H.; Chen, J.; Gong, Y.N.; Hu, L.; Zhou, Y.; Ge, J.; Lu, Q.; Liu, L.; Chen, S.; Shao, F. Cysteine methylation disrupts ubiquitin-chain sensing in NF-kappaB activation. Nature 2012, 481, 204–208. [Google Scholar]
- Iwai, H.; Kim, M.; Yoshikawa, Y.; Ashida, H.; Ogawa, M.; Fujita, Y.; Muller, D.; Kirikae, T.; Jackson, P.K.; Kotani, S.; et al. A bacterial effector targets Mad2L2, an APC inhibitor, to modulate host cell cycling. Cell 2007, 130, 611–623. [Google Scholar] [CrossRef]
- Hicks, S.W.; Galan, J.E. Hijacking the host ubiquitin pathway: structural strategies of bacterial E3 ubiquitin ligases. Curr. Opin. Microbiol. 2010, 13, 41–46. [Google Scholar] [CrossRef]
- Hicks, S.W.; Charron, G.; Hang, H.C.; Galan, J.E. Subcellular targeting of Salmonella virulence proteins by host-mediated S-palmitoylation. Cell. Host Microbe. 2011, 10, 9–20. [Google Scholar] [CrossRef]
- Metzger, E.; Muller, J.M.; Ferrari, S.; Buettner, R.; Schule, R. A novel inducible transactivation domain in the androgen receptor: implications for PRK in prostate cancer. EMBO J. 2003, 22, 270–280. [Google Scholar] [CrossRef]
- Mukai, H. The structure and function of PKN, a protein kinase having a catalytic domain homologous to that of PKC. J. Biochem. 2003, 133, 17–27. [Google Scholar] [CrossRef]
- PyMOL. Available online: http://www.pymol.org/ (accessed on 1 January 2014).
- Chou, Y.C.; Keszei, A.F.; Rohde, J.R.; Tyers, M.; Sicheri, F. Conserved structural mechanisms for autoinhibition in IpaH ubiquitin ligases. J. Biol. Chem. 2012, 287, 268–275. [Google Scholar]
- Quezada, C.M.; Hicks, S.W.; Galan, J.E.; Stebbins, C.E. A family of Salmonella virulence factors functions as a distinct class of autoregulated E3 ubiquitin ligases. Proc. Natl. Acad. Sci. USA 2009, 106, 4864–4869. [Google Scholar] [CrossRef]
- Sansonetti, P.J. War and peace at mucosal surfaces. Nat. Rev. Immunol. 2004, 4, 953–964. [Google Scholar]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Sanada, T.; Kim, M.; Mimuro, H.; Ashida, H.; Ogawa, M.; Mizushima, T.; Sasakawa, C. A bacterial effector targets the TRAF6-NFkappaB pathway to modulate the acute inflammatory response to bacterial invasion of epithelial cells. Virulence 2012, 3, 518–521. [Google Scholar] [CrossRef]
- Chen, Z.J. Ubiquitin signalling in the NF-kappaB pathway. Nat. Cell. Biol. 2005, 7, 758–765. [Google Scholar] [CrossRef]
- Taieb, F.; Nougayrede, J.P.; Oswald, E. Cycle inhibiting factors (cifs): Cyclomodulins that usurp the ubiquitin-dependent degradation pathway of host cells. Toxins (Basel) 2011, 3, 356–368. [Google Scholar] [CrossRef]
- Petroski, M.D.; Deshaies, R.J. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol. Cell. Biol. 2005, 6, 9–20. [Google Scholar] [CrossRef]
- Canova, M.J.; Molle, V. Bacterial Serine/Threonine Protein Kinases in Host-Pathogen Interactions. J. Biol. Chem. 2014. [Google Scholar]
- Cohen, P.; Tcherpakov, M. Will the ubiquitin system furnish as many drug targets as protein kinases? Cell 2010, 143, 686–693. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kim, M.; Otsubo, R.; Morikawa, H.; Nishide, A.; Takagi, K.; Sasakawa, C.; Mizushima, T. Bacterial Effectors and Their Functions in the Ubiquitin-Proteasome System: Insight from the Modes of Substrate Recognition. Cells 2014, 3, 848-864. https://doi.org/10.3390/cells3030848
Kim M, Otsubo R, Morikawa H, Nishide A, Takagi K, Sasakawa C, Mizushima T. Bacterial Effectors and Their Functions in the Ubiquitin-Proteasome System: Insight from the Modes of Substrate Recognition. Cells. 2014; 3(3):848-864. https://doi.org/10.3390/cells3030848
Chicago/Turabian StyleKim, Minsoo, Ryota Otsubo, Hanako Morikawa, Akira Nishide, Kenji Takagi, Chihiro Sasakawa, and Tsunehiro Mizushima. 2014. "Bacterial Effectors and Their Functions in the Ubiquitin-Proteasome System: Insight from the Modes of Substrate Recognition" Cells 3, no. 3: 848-864. https://doi.org/10.3390/cells3030848