Classical Transient Receptor Potential 1 (TRPC1): Channel or Channel Regulator?



Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Basic Features of the TRPC1 Gene and Protein

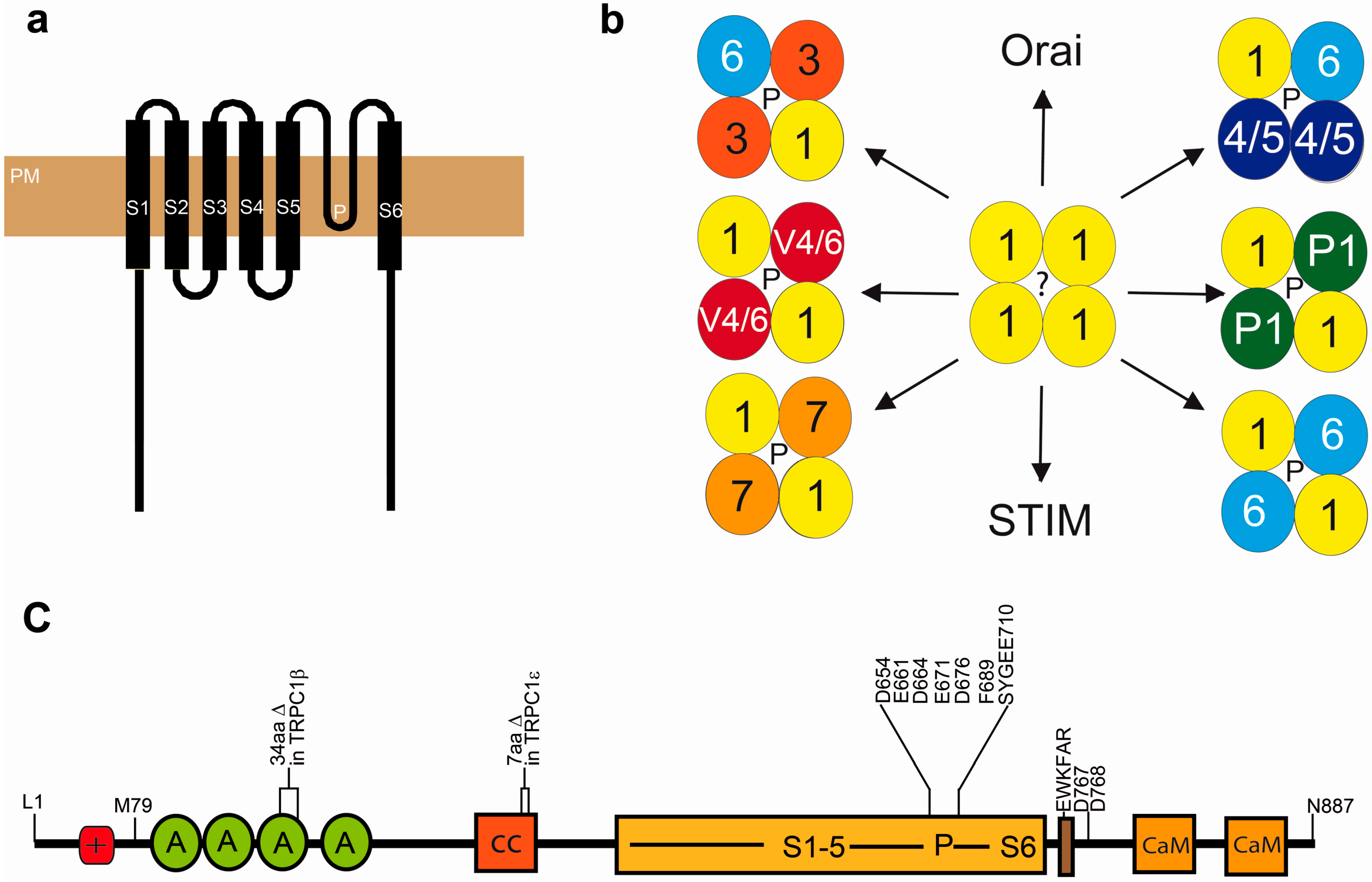
3. Molecular Make-Up and Regulation of Functional TRPC1 Channels
4. Contribution of TRPC1 to Store-Operated Ca2+ Entry (SOCE)
5. TRPC1 Channel Function in a Physiological Setting: Lessons from a TRPC1-Deficient Mouse Model
5.1. TRPC1 in the Kidney
5.2. TRPC1 in Acinar Cells of the Salivary Glands and the Pancreas
5.3. TRPC1 in Skeletal Muscle Function and Development
5.4. TRPC1 in Development and Bone Formation

5.5. TRPC1 in Platelets and Immune Cells
5.6. TRPC1 and Neuronal Function
5.7. TRPC1 in the Cardiopulmonary System
5.8. TRPC1 as a Tumor Marker
6. Conclusions
List of Abbreviations
| Aa | amino acids |
| Ang II | angiotensin II |
| AFM | atomic force microscopy |
| [Ca2+]i | intracellular Ca2+ concentration |
| CaM | calmodulin |
| CHO | Chinese hamster ovary cells |
| ClC-3 | voltage gated Cl− channel 3 |
| COS | African green monkey kidney cells |
| DMD | Duchenne muscular dystrophy |
| DN | diabetic nephropathy |
| DRG | dorsal root ganglions |
| EPSC | excitatory postsynaptic conductance |
| ER | endoplasmic reticulum |
| EST | expressed sequence tag |
| GFR | glomerular filtration rate |
| GST | glutathione-S-transferase |
| HEK | human embryonic kidney cells |
| HCG | cultured human submandibular glands |
| HPV | hypoxic pulmonary vasoconstriction |
| IL1β | interleukin 1β |
| I-mfa | inhibitor of MyoD family a |
| LPS | lipopolysaccharide |
| MARCKS | myristoylated alanine rich C-kinase substrate |
| M-CSF | macrophage colony stimulating factor |
| mGluR1 | metabotropic glutamate receptor 1 |
| NFAT | nuclear factor of activated T-Cells |
| PASMC | precapillary pulmonary arterial smooth muscle cells |
| PD | Parkinson’s disease |
| PDGFB | platelet-derived growth factor B |
| PKD | polycystic kidney disease |
| Sh-RNA | small hairpin RNA |
| Si-RNA | small interfering RNA |
| RANKL | Receptor Activator of NFκB-Ligand |
| ROCE | receptor-operated Ca2+ entry |
| SOCE | store-operated Ca2+ entry |
| TAC | thoracic aortic constriction |
| STIM | stromal interaction molecule |
| TIRF | total internal reflecting fluorescence |
| TRPC1 | Classical Transient Receptor Potential 1 |
| WT | wild-type |
| XTRPC1 | Xenopus homologue of TRPC1 |
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Nilius, B.; Szallasi, A. Transient receptor potential channels as drug targets: From the science of basic research to the art of medicine. Pharm. Rev. 2014, 66, 676–814. [Google Scholar] [PubMed]
- Wu, L.J.; Sweet, T.B.; Clapham, D.E. International union of basic and clinical pharmacology. Lxxvi. Current progress in the mammalian trp ion channel family. Pharm. Rev. 2010, 62, 381–404. [Google Scholar] [PubMed]
- Montell, C.; Rubin, G.M. Molecular characterization of the drosophila trp locus: A putative integral membrane protein required for phototransduction. Neuron 1989, 2, 1313–1323. [Google Scholar] [PubMed]
- Zhu, X.; Chu, P.B.; Peyton, M.; Birnbaumer, L. Molecular cloning of a widely expressed human homologue for the drosophila trp gene. FEBS Lett. 1995, 373, 193–198. [Google Scholar] [PubMed]
- Wes, P.D.; Chevesich, J.; Jeromin, A.; Rosenberg, C.; Stetten, G.; Montell, C. TRPC1, a human homolog of a drosophila store-operated channel. Proc. Natl. Acad. Sci. USA 1995, 92, 9652–9656. [Google Scholar]
- Zitt, C.; Zobel, A.; Obukhov, A.G.; Harteneck, C.; Kalkbrenner, F.; Luckhoff, A.; Schultz, G. Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion. Neuron 1996, 16, 1189–1196. [Google Scholar] [PubMed]
- Clapham, D.E.; Julius, D.; Montell, C.; Schultz, G. International union of pharmacology. Xlix. Nomenclature and structure-function relationships of transient receptor potential channels. Pharm. Rev. 2005, 57, 427–450. [Google Scholar] [PubMed]
- Dietrich, A.; Chubanov, V.; Kalwa, H.; Rost, B.R.; Gudermann, T. Cation channels of the transient receptor potential superfamily: Their role in physiological and pathophysiological processes of smooth muscle cells. Pharmacol. Ther. 2006, 112, 744–760. [Google Scholar] [PubMed]
- Sakura, H.; Ashcroft, F.M. Identification of four TRP1 gene variants murine pancreatic beta-cells. Diabetologia 1997, 40, 528–532. [Google Scholar] [PubMed]
- Ong, E.C.; Nesin, V.; Long, C.L.; Bai, C.X.; Guz, J.L.; Ivanov, I.P.; Abramowitz, J.; Birnbaumer, L.; Humphrey, M.B.; Tsiokas, L. A TRPC1 protein-dependent pathway regulates osteoclast formation and function. J. Biol. Chem. 2013, 288, 22219–22232. [Google Scholar] [PubMed]
- Rychkov, G.; Barritt, G.J. TRPC1 Ca2+-permeable channels in animal cells. Handb. Exp. Pharmacol. 2007, 23–52. [Google Scholar]
- Nesin, V.; Tsiokas, L. TRPC1. Handb. Exp. Pharmacol. 2014, 222, 15–51. [Google Scholar] [PubMed]
- Boulay, G.; Brown, D.M.; Qin, N.; Jiang, M.; Dietrich, A.; Zhu, M.X.; Chen, Z.; Birnbaumer, M.; Mikoshiba, K.; Birnbaumer, L. Modulation of Ca2+ entry by polypeptides of the inositol 1,4,5-trisphosphate receptor (IP3R) that bind transient receptor potential (TRP): Evidence for roles of TRP and IP3R in store depletion-activated Ca2+ entry. Proc. Natl. Acad. Sci. USA 1999, 96, 14955–14960. [Google Scholar] [PubMed]
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112. [Google Scholar] [PubMed]
- Maroto, R.; Raso, A.; Wood, T.G.; Kurosky, A.; Martinac, B.; Hamill, O.P. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat. Cell Biol. 2005, 7, 179–185. [Google Scholar] [PubMed]
- Gottlieb, P.; Folgering, J.; Maroto, R.; Raso, A.; Wood, T.G.; Kurosky, A.; Bowman, C.; Bichet, D.; Patel, A.; Sachs, F.; et al. Revisiting TRPC1 and TRPC6 mechanosensitivity. Pflugers Arch. 2008, 455, 1097–1103. [Google Scholar]
- Dietrich, A.; Kalwa, H.; Storch, U.; Mederos y Schnitzler, M.; Salanova, B.; Pinkenburg, O.; Dubrovska, G.; Essin, K.; Gollasch, M.; Birnbaumer, L.; et al. Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflugers Arch. 2007, 455, 465–477. [Google Scholar]
- Mederos y Schnitzler, M.; Storch, U.; Meibers, S.; Nurwakagari, P.; Breit, A.; Essin, K.; Gollasch, M.; Gudermann, T. Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J. 2008, 27, 3092–3103. [Google Scholar]
- Storch, U.; Mederos y Schnitzler, M.; Gudermann, T. G protein-mediated stretch reception. Am. J. Physiol Heart Circ. Physiol 2012, 302, H1241–H1249. [Google Scholar] [PubMed]
- Eijkelkamp, N.; Quick, K.; Wood, J.N. Transient receptor potential channels and mechanosensation. Annu Rev. Neurosci 2013, 36, 519–546. [Google Scholar] [PubMed]
- Hanlon, M.R.; Wallace, B.A. Structure and function of voltage-dependent ion channel regulatory beta subunits. Biochemistry 2002, 41, 2886–2894. [Google Scholar] [PubMed]
- Hofmann, T.; Schaefer, M.; Schultz, G.; Gudermann, T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc. Natl Acad Sci USA 2002, 99, 7461–7466. [Google Scholar] [PubMed]
- Alfonso, S.; Benito, O.; Alicia, S.; Angelica, Z.; Patricia, G.; Diana, K.; Vaca, L. Regulation of the cellular localization and function of human transient receptor potential channel 1 by other members of the TRPC family. Cell Calcium 2008, 43, 375–387. [Google Scholar] [PubMed]
- Cheng, K.T.; Liu, X.; Ong, H.L.; Swaim, W.; Ambudkar, I.S. Local Ca2+ entry via orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca2+ signals required for specific cell functions. PLoS Biol. 2011, 9, e1001025. [Google Scholar] [PubMed]
- Souza, L.B.; Ambudkar, I.S. Trafficking mechanisms and regulation of TRPC channels. Cell Calcium 2014, 56, 43–50. [Google Scholar] [PubMed]
- Barrera, N.P.; Shaifta, Y.; McFadzean, I.; Ward, J.P.T.; Henderson, R.M.; Edwardson, J.M. Afm imaging reveals the tetrameric structure of the TRPC1 channel. Biochem. Biophys. Res. Commun. 2007, 358, 1086–1090. [Google Scholar]
- Beech, D.J.; Muraki, K.; Flemming, R. Non-selective cationic channels of smooth muscle and the mammalian homologues of drosophila trp. J. Physiol. 2004, 559, 685–706. [Google Scholar] [PubMed]
- Lintschinger, B.; Balzer-Geldsetzer, M.; Baskaran, T.; Graier, W.F.; Romanin, C.; Zhu, M.X.; Groschner, K. Coassembly of TRP1 and TRP3 proteins generates diacylglycerol- and Ca2+-sensitive cation channels. J. Biol. Chem. 2000, 275, 27799–27805. [Google Scholar] [PubMed]
- Storch, U.; Forst, A.L.; Philipp, M.; Gudermann, T.; Mederos y Schnitzler, M. Transient receptor potential channel 1 (TRPC1) reduces calcium permeability in heteromeric channel complexes. J. Biol. Chem. 2012, 287, 3530–3540. [Google Scholar] [PubMed]
- Strubing, C.; Krapivinsky, G.; Krapivinsky, L.; Clapham, D.E. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron 2001, 29, 645–655. [Google Scholar] [PubMed]
- Strubing, C.; Krapivinsky, G.; Krapivinsky, L.; Clapham, D.E. Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J. Biol. Chem. 2003, 278, 39014–39019. [Google Scholar] [PubMed]
- Goel, M.; Sinkins, W.G.; Schilling, W.P. Selective association of TRPC channel subunits in rat brain synaptosomes. J. Biol. Chem. 2002, 277, 48303–48310. [Google Scholar] [PubMed]
- Zagranichnaya, T.K.; Wu, X.; Villereal, M.L. Endogenous TRPC1, TRPC3, and TRPC7 proteins combine to form native store-operated channels in hek-293 cells. J. Biol. Chem. 2005, 280, 29559–29569. [Google Scholar] [PubMed]
- Kim, J.; Kwak, M.; Jeon, J.P.; Myeong, J.; Wie, J.; Hong, C.; Kim, S.Y.; Jeon, J.H.; Kim, H.J.; So, I. Isoform- and receptor-specific channel property of canonical transient receptor potential (TRPC)1/4 channels. Pflugers Arch. 2014, 466, 491–504. [Google Scholar] [PubMed]
- Tsiokas, L.; Arnould, T.; Zhu, C.; Kim, E.; Walz, G.; Sukhatme, V.P. Specific association of the gene product of pkd2 with the TRPC1 channel. Proc. Natl. Acad. Sci. USA 1999, 96, 3934–3939. [Google Scholar] [PubMed]
- Bai, C.X.; Giamarchi, A.; Rodat-Despoix, L.; Padilla, F.; Downs, T.; Tsiokas, L.; Delmas, P. Formation of a new receptor-operated channel by heteromeric assembly of TRPP2 and TRPC1 subunits. EMBO Rep. 2008, 9, 472–479. [Google Scholar] [PubMed]
- Kobori, T.; Smith, G.D.; Sandford, R.; Edwardson, J.M. The transient receptor potential channels TRPP2 and TRPC1 form a heterotetramer with a 2:2 stoichiometry and an alternating subunit arrangement. J. Biol. Chem. 2009, 284, 35507–35513. [Google Scholar] [PubMed]
- Schindl, R.; Fritsch, R.; Jardin, I.; Frischauf, I.; Kahr, H.; Muik, M.; Riedl, M.C.; Groschner, K.; Romanin, C. Canonical transient receptor potential (TRPC) 1 acts as a negative regulator for vanilloid TRPV6-mediated Ca2+ influx. J. Biol. Chem. 2012, 287, 35612–35620. [Google Scholar] [PubMed]
- Courjaret, R.; Hubrack, S.; Daalis, A.; Dib, M.; Machaca, K. The xenopus TRPV6 homolog encodes a Mg2+ -permeant channel that is inhibited by interaction with TRPC1. J. Cell. Physiol 2013, 228, 2386–2398. [Google Scholar] [PubMed]
- Ma, X.; Cheng, K.T.; Wong, C.O.; O’Neil, R.G.; Birnbaumer, L.; Ambudkar, I.S.; Yao, X. Heteromeric TRPV4-c1 channels contribute to store-operated Ca2+ entry in vascular endothelial cells. Cell Calcium 2011, 50, 502–509. [Google Scholar] [PubMed]
- Du, J.; Ma, X.; Shen, B.; Huang, Y.; Birnbaumer, L.; Yao, X. TRPV4, TRPC1, and TRPP2 assemble to form a flow-sensitive heteromeric channel. FASEB J. 2014. [Google Scholar]
- Singh, B.B.; Liu, X.; Tang, J.; Zhu, M.X.; Ambudkar, I.S. Calmodulin regulates Ca2+-dependent feedback inhibition of store-operated Ca2+ influx by interaction with a site in the c terminus of TRPC1. Mol. Cell 2002, 9, 739–750. [Google Scholar] [PubMed]
- Yuan, J.P.; Kiselyov, K.; Shin, D.M.; Chen, J.; Shcheynikov, N.; Kang, S.H.; Dehoff, M.H.; Schwarz, M.K.; Seeburg, P.H.; Muallem, S.; et al. Homer binds TRPC family channels and is required for gating of TRPC1 by IP3 receptors. Cell 2003, 114, 777–789. [Google Scholar] [PubMed]
- Stiber, J.A.; Zhang, Z.S.; Burch, J.; Eu, J.P.; Zhang, S.; Truskey, G.A.; Seth, M.; Yamaguchi, N.; Meissner, G.; Shah, R.; et al. Mice lacking homer 1 exhibit a skeletal myopathy characterized by abnormal transient receptor potential channel activity. Mol. Cell Biol. 2008, 28, 2637–2647. [Google Scholar] [PubMed]
- Strotmann, R.; Semtner, M.; Kepura, F.; Plant, T.D.; Schoneberg, T. Interdomain interactions control Ca2+-dependent potentiation in the cation channel TRPV4. PLoS One 2010, 5, e10580. [Google Scholar] [PubMed]
- TRPC1/interactors. Available online: http://trpchannel.org/summaries/TRPC1 (accessed on 23 September 2014).
- Hoth, M.; Penner, R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 1992, 355, 353–356. [Google Scholar] [PubMed]
- Dietrich, A.; Kalwa, H.; Rost, B.R.; Gudermann, T. The diacylgylcerol-sensitive TRPC3/6/7 subfamily of cation channels: Functional characterization and physiological relevance. Pflugers Arch. 2005, 451, 72–80. [Google Scholar] [PubMed]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in orai1 causes immune deficiency by abrogating crac channel function. Nature 2006, 441, 179–185. [Google Scholar] [PubMed]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. Cracm1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006, 312, 1220–1223. [Google Scholar] [PubMed]
- Cahalan, M.D. Stimulating store-operated Ca2+ entry. Nat. Cell Biol 2009, 11, 669–677. [Google Scholar] [PubMed]
- Liao, Y.; Plummer, N.W.; George, M.D.; Abramowitz, J.; Zhu, M.X.; Birnbaumer, L. A role for orai in TRPC-mediated Ca2+ entry suggests that a TRPC:Orai complex may mediate store and receptor operated Ca2+ entry. Proc. Natl Acad Sci USA 2009, 106, 3202–3206. [Google Scholar] [PubMed]
- Lee, K.P.; Yuan, J.P.; So, I.; Worley, P.F.; Muallem, S. Stim1-dependent and stim1-independent function of Transient Receptor Potential Canonical (TRPC) channels tunes their store-operated mode. J. Biol. Chem. 2010, 285, 38666–38673. [Google Scholar] [PubMed]
- Cheng, K.T.; Ong, H.L.; Liu, X.B.; Ambudkar, I.S. Contribution and regulation of TRPC channels in store-operated Ca2+ entry. Curr. Top. Membr 2013, 71, 149–179. [Google Scholar] [PubMed]
- DeHaven, W.I.; Jones, B.F.; Petranka, J.G.; Smyth, J.T.; Tomita, T.; Bird, G.S.; Putney, J.W. TRPC channels function independently of stim1 and orai1. J. Physiol-London 2009, 587, 2275–2298. [Google Scholar] [PubMed]
- Ong, H.L.; Chen, J.; Chataway, T.; Brereton, H.; Zhang, L.; Downs, T.; Tsiokas, L.; Barritt, G. Specific detection of the endogenous transient receptor potential (trp)-1 protein in liver and airway smooth muscle cells using immunoprecipitation and western-blot analysis. Biochem. J. 2002, 364, 641–648. [Google Scholar] [PubMed]
- Liu, X.; Cheng, K.T.; Bandyopadhyay, B.C.; Pani, B.; Dietrich, A.; Paria, B.C.; Swaim, W.D.; Beech, D.; Yildrim, E.; Singh, B.B.; et al. Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1(-/-) mice. Proc. Natl. Acad. Sci. USA 2007, 104, 17542–17547. [Google Scholar] [PubMed]
- Seth, M.; Zhang, Z.S.; Mao, L.; Graham, V.; Burch, J.; Stiber, J.; Tsiokas, L.; Winn, M.; Abramowitz, J.; Rockman, H.A.; et al. TRPC1 channels are critical for hypertrophic signaling in the heart. Circ. Res. 2009, 105, 1023–1030. [Google Scholar] [PubMed]
- Malczyk, M.; Veith, C.; Fuchs, B.; Hofmann, K.; Storch, U.; Schermuly, R.T.; Witzenrath, M.; Ahlbrecht, K.; Fecher-Trost, C.; Flockerzi, V.; et al. Classical transient receptor potential channel 1 in hypoxia-induced pulmonary hypertension. Am. J. Resp. Crit. Care Med. 2013, 188, 1451–1459. [Google Scholar] [PubMed]
- Varga-Szabo, D.; Authi, K.S.; Braun, A.; Bender, M.; Ambily, A.; Hassock, S.R.; Gudermann, T.; Dietrich, A.; Nieswandt, B. Store-operated Ca2+ entry in platelets occurs independently of Transient Receptor Potential (TRP) c1. Pflugers Arch. 2008, 457, 377–387. [Google Scholar] [PubMed]
- Berbey, C.; Weiss, N.; Legrand, C.; Allard, B. Transient receptor potential canonical type 1 (TRPC1) operates as a sarcoplasmic reticulum calcium leak channel in skeletal muscle. J. Biol. Chem. 2009, 284, 36387–36394. [Google Scholar] [PubMed]
- Tajeddine, N.; Zanou, N.; Van Schoor, M.; Lebacq, J.; Gailly, P. TRPC1: Subcellular localization? J. Biol Chem 2010, 285, le1. [Google Scholar] [CrossRef]
- DeCaen, P.G.; Delling, M.; Vien, T.N.; Clapham, D.E. Direct recording and molecular identification of the calcium channel of primary cilia. Nature 2013, 504, 315–318. [Google Scholar] [PubMed]
- Delling, M.; DeCaen, P.G.; Doerner, J.F.; Febvay, S.; Clapham, D.E. Primary cilia are specialized calcium signalling organelles. Nature 2013, 504, 311–314. [Google Scholar] [PubMed]
- Sours, S.; Du, J.; Chu, S.; Ding, M.; Zhou, X.J.; Ma, R. Expression of canonical transient receptor potential (TRPC) proteins in human glomerular mesangial cells. Am. J. Physiol. Renal. Physiol. 2006, 290, F1507–F1515. [Google Scholar] [PubMed]
- Du, J.; Sours-Brothers, S.; Coleman, R.; Ding, M.; Graham, S.; Kong, D.H.; Ma, R. Canonical transient receptor potential 1 channel is involved in contractile function of glomerular mesangial cells. J. Am. Soc. Nephrol. 2007, 18, 1437–1445. [Google Scholar] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [PubMed]
- Chen, K.; Jin, X.; Li, Q.; Wang, W.; Wang, Y.; Zhang, J. Association of TRPC1 gene polymorphisms with type 2 diabetes and diabetic nephropathy in han chinese population. Endocr. Res. 2013, 38, 59–68. [Google Scholar] [PubMed]
- Niehof, M.; Borlak, J. Hnf4 alpha and the ca-channel TRPC1 are novel disease candidate genes in diabetic nephropathy. Diabetes 2008, 57, 1069–1077. [Google Scholar] [PubMed]
- Zhang, D.; Freedman, B.I.; Flekac, M.; Santos, E.; Hicks, P.J.; Bowden, D.W.; Efendic, S.; Brismar, K.; Gu, H.F. Evaluation of genetic association and expression reduction of TRPC1 in the development of diabetic nephropathy. Am. J. Nephrol. 2009, 29, 244–251. [Google Scholar] [PubMed]
- Hong, J.H.; Li, Q.; Kim, M.S.; Shin, D.M.; Feske, S.; Birnbaumer, L.; Cheng, K.T.; Ambudkar, I.S.; Muallem, S. Polarized but differential localization and recruitment of stim1, orai1 and TRPC channels in secretory cells. Traffic 2011, 12, 232–245. [Google Scholar] [PubMed]
- Vandebrouck, C.; Martin, D.; Colson-Van Schoor, M.; Debaix, H.; Gailly, P. Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J. Cell Biol. 2002, 158, 1089–1096. [Google Scholar] [PubMed]
- Sabourin, J.; Cognard, C.; Constantin, B. Regulation by scaffolding proteins of canonical transient receptor potential channels in striated muscle. J. Muscle Res. Cell Motil. 2009, 30, 289–297. [Google Scholar]
- Sabourin, J.; Lamiche, C.; Vandebrouck, A.; Magaud, C.; Rivet, J.; Cognard, C.; Bourmeyster, N.; Constantin, B. Regulation of TRPC1 and TRPC4 cation channels requires an alpha1-syntrophin-dependent complex in skeletal mouse myotubes. J. Biol. Chem. 2009, 284, 36248–36261. [Google Scholar] [PubMed]
- Louis, M.; Zanou, N.; Van Schoor, M.; Gailly, P. TRPC1 regulates skeletal myoblast migration and differentiation. J. Cell Sci. 2008, 121, 3951–3959. [Google Scholar] [PubMed]
- Zanou, N.; Shapovalov, G.; Louis, M.; Tajeddine, N.; Gallo, C.; van Schoor, M.; Anguish, I.; Cao, M.L.; Schakman, O.; Dietrich, A.; et al. Role of TRPC1 channel in skeletal muscle function. Am. J. Physiol. Cell Physiol. 2010, 298, C149–C162. [Google Scholar] [PubMed]
- Zhang, B.T.; Whitehead, N.P.; Gervasio, O.L.; Reardon, T.F.; Vale, M.; Fatkin, D.; Dietrich, A.; Yeung, E.W.; Allen, D.G. Pathways of Ca2+ entry and cytoskeletal damage following eccentric contractions in mouse skeletal muscle. J. Appl. Physiol. 2012, 112, 2077–2086. [Google Scholar] [PubMed]
- Zanou, N.; Schakman, O.; Louis, P.; Ruegg, U.T.; Dietrich, A.; Birnbaumer, L.; Gailly, P. TRPC1 ion channel modulates phosphatidylinositol 3-kinase/akt pathway during myoblast differentiation and muscle regeneration. J. Biol. Chem. 2012, 287, 14524–14534. [Google Scholar] [PubMed]
- Vandebrouck, A.; Ducret, T.; Basset, O.; Sebille, S.; Raymond, G.; Ruegg, U.; Gailly, P.; Cognard, C.; Constantin, B. Regulation of store-operated calcium entries and mitochondrial uptake by minidystrophin expression in cultured myotubes. FASEB J. 2006, 20, 136–138. [Google Scholar] [PubMed]
- Fahlbusch, M. Analyse der Funktion des TRPC1-Proteins durch Charakterisierung eines TRPC1-defizienten Mausmodells. Doctoral Thesis, Philipps-University Marburg, Marburg, Germany, 15 October 2008. [Google Scholar]
- Robinson, L.J.; Mancarella, S.; Songsawad, D.; Tourkova, I.L.; Barnett, J.B.; Gill, D.L.; Soboloff, J.; Blair, H.C. Gene disruption of the calcium channel orai1 results in inhibition of osteoclast and osteoblast differentiation and impairs skeletal development. Lab. Invest. 2012, 92, 1071–1083. [Google Scholar] [PubMed]
- Hwang, S.Y.; Putney, J.W. Orai1-mediated calcium entry plays a critical role in osteoclast differentiation and function by regulating activation of the transcription factor nfatc1. FASEB J. 2012, 26, 1484–1492. [Google Scholar] [PubMed]
- Zhou, Y.; Lewis, T.L.; Robinson, L.J.; Brundage, K.M.; Schafer, R.; Martin, K.H.; Blair, H.C.; Soboloff, J.; Barnett, J.B. The role of calcium release activated calcium channels in osteoclast differentiation. J. Cell Physiol. 2011, 226, 1082–1089. [Google Scholar] [PubMed]
- Abed, E.; Labelle, D.; Martineau, C.; Loghin, A.; Moreau, R. Expression of transient receptor potential (TRP) channels in human and murine osteoblast-like cells. Mol. Membrane Biol. 2009, 26, 146–158. [Google Scholar]
- Den Dekker, E.; Molin, D.G.; Breikers, G.; van Oerle, R.; Akkerman, J.W.; van Eys, G.J.; Heemskerk, J.W. Expression of transient receptor potential mrna isoforms and Ca2+ influx in differentiating human stem cells and platelets. Biochim. Biophys. Acta 2001, 1539, 243–255. [Google Scholar]
- Grosse, J.; Braun, A.; Varga-Szabo, D.; Beyersdorf, N.; Schneider, B.; Zeitlmann, L.; Hanke, P.; Schropp, P.; Muhlstedt, S.; Zorn, C.; et al. An ef hand mutation in stim1 causes premature platelet activation and bleeding in mice. J. Clin. Invest. 2007, 117, 3540–3550. [Google Scholar] [PubMed]
- Rosado, J.A.; Brownlow, S.L.; Sage, S.O. Endogenously expressed TRP1 is involved in store-mediated Ca2+ entry by conformational coupling in human platelets. J. Biol. Chem. 2002, 277, 42157–42163. [Google Scholar] [PubMed]
- Galan, C.; Dionisio, N.; Smani, T.; Salido, G.M.; Rosado, J.A. The cytoskeleton plays a modulatory role in the association between stim1 and the Ca2+ channel subunits orai1 and TRPC1. Biochem. Pharmacol. 2011, 82, 400–410. [Google Scholar] [PubMed]
- Varga-Szabo, D.; Braun, A.; Kleinschnitz, C.; Bender, M.; Pleines, I.; Pham, M.; Renne, T.; Stoll, G.; Nieswandt, B. The calcium sensor stim1 is an essential mediator of arterial thrombosis and ischemic brain infarction. J. Exp. Med. 2008, 205, 1583–1591. [Google Scholar] [PubMed]
- Braun, A.; Varga-Szabo, D.; Kleinschnitz, C.; Pleines, I.; Bender, M.; Austinat, M.; Bosl, M.; Stoll, G.; Nieswandt, B. Orai1 (cracm1) is the platelet soc channel and essential for pathological thrombus formation. Blood 2009, 113, 2056–2063. [Google Scholar] [PubMed]
- Yildirim, E.; Carey, M.A.; Card, J.W.; Dietrich, A.; Flake, G.P.; Zhang, Y.; Bradbury, J.A.; Rebolloso, Y.; Germolec, D.R.; Morgan, D.L.; et al. Severely blunted allergen-induced pulmonary th2 cell response and lung hyperresponsiveness in type 1 transient receptor potential channel-deficient mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L539–L549. [Google Scholar] [PubMed]
- Sel, S.; Rost, B.R.; Yildirim, A.O.; Sel, B.; Kalwa, H.; Fehrenbach, H.; Renz, H.; Gudermann, T.; Dietrich, A. Loss of classical transient receptor potential 6 channel reduces allergic airway response. Clin. Exp. Allergy 2008, 38, 1548–1558. [Google Scholar] [PubMed]
- Py, B.F.; Jin, M.; Desai, B.N.; Penumaka, A.; Zhu, H.; Kober, M.; Dietrich, A.; Lipinski, M.M.; Henry, T.; Clapham, D.E.; et al. Caspase-11 controls interleukin-1beta release through degradation of TRPC1. Cell Rep. 2014, 6, 1122–1128. [Google Scholar]
- Wang, G.X.; Poo, M.M. Requirement of TRPC channels in netrin-1-induced chemotropic turning of nerve growth cones. Nature 2005, 434, 898–904. [Google Scholar] [PubMed]
- Shim, S.; Yuan, J.P.; Kim, J.Y.; Zeng, W.; Huang, G.; Milshteyn, A.; Kern, D.; Muallem, S.; Ming, G.L.; Worley, P.F. Peptidyl-prolyl isomerase fkbp52 controls chemotropic guidance of neuronal growth cones via regulation of TRPC1 channel opening. Neuron 2009, 64, 471–483. [Google Scholar] [PubMed]
- Dietrich, A.; Fahlbusch, M.; Gudermann, T.; LM-University of Munich, Munich, Germany. Unpublished work. 2014.
- Ariano, P.; Dalmazzo, S.; Owsianik, G.; Nilius, B.; Lovisolo, D. TRPC channels are involved in calcium-dependent migration and proliferation in immortalized gnrh neurons. Cell Calcium 2011, 49, 387–394. [Google Scholar] [PubMed]
- Boudes, M.; Uvin, P.; Pinto, S.; Freichel, M.; Birnbaumer, L.; Voets, T.; de Ridder, D.; Vennekens, R. Crucial role of TRPC1 and TRPC4 in cystitis-induced neuronal sprouting and bladder overactivity. PLoS One 2013, 8, e69550. [Google Scholar] [PubMed]
- Kim, S.J.; Kim, Y.S.; Yuan, J.P.; Petralia, R.S.; Worley, P.F.; Linden, D.J. Activation of the TRPC1 cation channel by metabotropic glutamate receptor mglur1. Nature 2003, 426, 285–291. [Google Scholar] [PubMed]
- Hartmann, J.; Dragicevic, E.; Adelsberger, H.; Henning, H.A.; Sumser, M.; Abramowitz, J.; Blum, R.; Dietrich, A.; Freichel, M.; Flockerzi, V.; et al. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron 2008, 59, 392–398. [Google Scholar] [PubMed]
- Staaf, S.; Maxvall, I.; Lind, U.; Husmark, J.; Mattsson, J.P.; Ernfors, P.; Pierrou, S. Down regulation of TRPC1 by shrna reduces mechanosensitivity in mouse dorsal root ganglion neurons in vitro. Neurosci. Lett. 2009, 457, 3–7. [Google Scholar] [PubMed]
- Alessandri-Haber, N.; Dina, O.A.; Chen, X.; Levine, J.D. TRPC1 and TRPC6 channels cooperate with TRPV4 to mediate mechanical hyperalgesia and nociceptor sensitization. J. Neurosci. 2009, 29, 6217–6228. [Google Scholar] [PubMed]
- Garrison, S.R.; Dietrich, A.; Stucky, C.L. TRPC1 contributes to light-touch sensation and mechanical responses in low-threshold cutaneous sensory neurons. J. Neurophysiol. 2012, 107, 913–922. [Google Scholar] [PubMed]
- Phelan, K.D.; Mock, M.M.; Kretz, O.; Shwe, U.T.; Kozhemyakin, M.; Greenfield, L.J.; Dietrich, A.; Birnbaumer, L.; Freichel, M.; Flockerzi, V.; et al. Heteromeric canonical transient receptor potential 1 and 4 channels play a critical role in epileptiform burst firing and seizure-induced neurodegeneration. Mol. Pharmacol. 2012, 81, 384–392. [Google Scholar] [PubMed]
- Phelan, K.D.; Shwe, U.T.; Abramowitz, J.; Wu, H.; Rhee, S.W.; Howell, M.D.; Gottschall, P.E.; Freichel, M.; Flockerzi, V.; Birnbaumer, L.; et al. Canonical transient receptor channel 5 (TRPC5) and TRPC1/4 contribute to seizure and excitotoxicity by distinct cellular mechanisms. Mol. Pharmacol. 2013, 83, 429–438. [Google Scholar] [PubMed]
- Selvaraj, S.; Sun, Y.; Watt, J.A.; Wang, S.; Lei, S.; Birnbaumer, L.; Singh, B.B. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of akt/mtor signaling. J. Clin. Invest. 2012, 122, 1354–1367. [Google Scholar] [PubMed]
- Dietrich, A.; Kalwa, H.; Fuchs, B.; Grimminger, F.; Weissmann, N.; Gudermann, T. In vivo TRPC functions in the cardiopulmonary vasculature. Cell Calcium 2007, 42, 233–244. [Google Scholar] [PubMed]
- Dietrich, A.; Gudermann, T. Trp channels in the cardiopulmonary vasculature. Adv. Exp. Med. Biol. 2011, 704, 781–810. [Google Scholar] [PubMed]
- Ohba, T.; Watanabe, H.; Murakami, M.; Takahashi, Y.; Iino, K.; Kuromitsu, S.; Mori, Y.; Ono, K.; Iijima, T.; Ito, H. Upregulation of TRPC1 in the development of cardiac hypertrophy. J. Mol. Cell. Cardiol. 2007, 42, 498–507. [Google Scholar] [PubMed]
- Molkentin, J.D.; Dorn, G.W., Jr. Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu Rev. Physiol. 2001, 63, 391–426. [Google Scholar] [PubMed]
- Wu, X.; Eder, P.; Chang, B.; Molkentin, J.D. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 7000–7005. [Google Scholar] [PubMed]
- Paria, B.C.; Malik, A.B.; Kwiatek, A.M.; Rahman, A.; May, M.J.; Ghosh, S.; Tiruppathi, C. Tumor necrosis factor-alpha induces nuclear factor-kappab-dependent TRPC1 expression in endothelial cells. J. Biol. Chem. 2003, 278, 37195–37203. [Google Scholar] [PubMed]
- Paria, B.C.; Vogel, S.M.; Ahmmed, G.U.; Alamgir, S.; Shroff, J.; Malik, A.B.; Tiruppathi, C. Tumor necrosis factor-alpha-induced TRPC1 expression amplifies store-operated Ca2+ influx and endothelial permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L1303–L1313. [Google Scholar] [PubMed]
- Paria, B.C.; Bair, A.M.; Xue, J.; Yu, Y.; Malik, A.B.; Tiruppathi, C. Ca2+ influx induced by protease-activated receptor-1 activates a feed-forward mechanism of TRPC1 expression via nuclear factor-kappab activation in endothelial cells. J. Biol. Chem. 2006, 281, 20715–20727. [Google Scholar] [PubMed]
- Tiruppathi, C.; Freichel, M.; Vogel, S.M.; Paria, B.C.; Mehta, D.; Flockerzi, V.; Malik, A.B. Impairment of store-operated Ca2+ entry in TRPC4(-/-) mice interferes with increase in lung microvascular permeability. Circ. Res. 2002, 91, 70–76. [Google Scholar] [PubMed]
- Singh, I.; Knezevic, N.; Ahmmed, G.U.; Kini, V.; Malik, A.B.; Mehta, D. Galphaq-TRPC6-mediated Ca2+ entry induces rhoa activation and resultant endothelial cell shape change in response to thrombin. J. Biol. Chem. 2007, 282, 7833–7843. [Google Scholar] [PubMed]
- Weissmann, N.; Sydykov, A.; Kalwa, H.; Storch, U.; Fuchs, B.; Mederos y Schnitzler, M.; Brandes, R.P.; Grimminger, F.; Meissner, M.; Freichel, M.; et al. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nature commun. 2012, 3, 649. [Google Scholar]
- Kunichika, N.; Yu, Y.; Remillard, C.V.; Platoshyn, O.; Zhang, S.; Yuan, J.X. Overexpression of TRPC1 enhances pulmonary vasoconstriction induced by capacitative Ca2+ entry. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L962–L969. [Google Scholar] [PubMed]
- Kumar, B.; Dreja, K.; Shah, S.S.; Cheong, A.; Xu, S.Z.; Sukumar, P.; Naylor, J.; Forte, A.; Cipollaro, M.; McHugh, D.; et al. Upregulated TRPC1 channel in vascular injury in vivo and its role in human neointimal hyperplasia. Circ. Res. 2006, 98, 557–563. [Google Scholar]
- Weissmann, N.; Dietrich, A.; Fuchs, B.; Kalwa, H.; Ay, M.; Dumitrascu, R.; Olschewski, A.; Storch, U.; Mederos y Schnitzler, M.; Ghofrani, H.A.; et al. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc. Natl. Acad. Sci. USA 2006, 103, 19093–19098. [Google Scholar] [PubMed]
- Xia, Y.; Yang, X.R.; Fu, Z.; Paudel, O.; Abramowitz, J.; Birnbaumer, L.; Sham, J.S. Classical transient receptor potential 1 and 6 contribute to hypoxic pulmonary hypertension through differential regulation of pulmonary vascular functions. Hypertension 2014, 63, 173–180. [Google Scholar] [PubMed]
- Dhennin-Duthille, I.; Gautier, M.; Faouzi, M.; Guilbert, A.; Brevet, M.; Vaudry, D.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. High expression of transient receptor potential channels in human breast cancer epithelial cells and tissues: Correlation with pathological parameters. Cell. Physiol. Biochem. 2011, 28, 813–822. [Google Scholar] [PubMed]
- Bomben, V.C.; Turner, K.L.; Barclay, T.T.; Sontheimer, H. Transient receptor potential canonical channels are essential for chemotactic migration of human malignant gliomas. J. Cell. Physiol. 2011, 226, 1879–1888. [Google Scholar] [PubMed]
- Cuddapah, V.A.; Turner, K.L.; Sontheimer, H. Calcium entry via TRPC1 channels activates chloride currents in human glioma cells. Cell Calcium 2013, 53, 187–194. [Google Scholar] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dietrich, A.; Fahlbusch, M.; Gudermann, T. Classical Transient Receptor Potential 1 (TRPC1): Channel or Channel Regulator? Cells 2014, 3, 939-962. https://doi.org/10.3390/cells3040939
Dietrich A, Fahlbusch M, Gudermann T. Classical Transient Receptor Potential 1 (TRPC1): Channel or Channel Regulator? Cells. 2014; 3(4):939-962. https://doi.org/10.3390/cells3040939
Chicago/Turabian StyleDietrich, Alexander, Meike Fahlbusch, and Thomas Gudermann. 2014. "Classical Transient Receptor Potential 1 (TRPC1): Channel or Channel Regulator?" Cells 3, no. 4: 939-962. https://doi.org/10.3390/cells3040939