
Antisense-Based Progerin Downregulation in HGPS-Like Patients’ Cells

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Delivery of Morpholino Oligonucleotides
2.3. Morpholino Sequences
2.4. RNA Isolation, Transcriptional Analysis and Real-Time Quantitative PCR
2.5. Western-Blot
2.6. Senescence Assay
2.7. Quantitation of Abnormal Nuclear Morphology
2.8. Statistical Analyses
3. Results
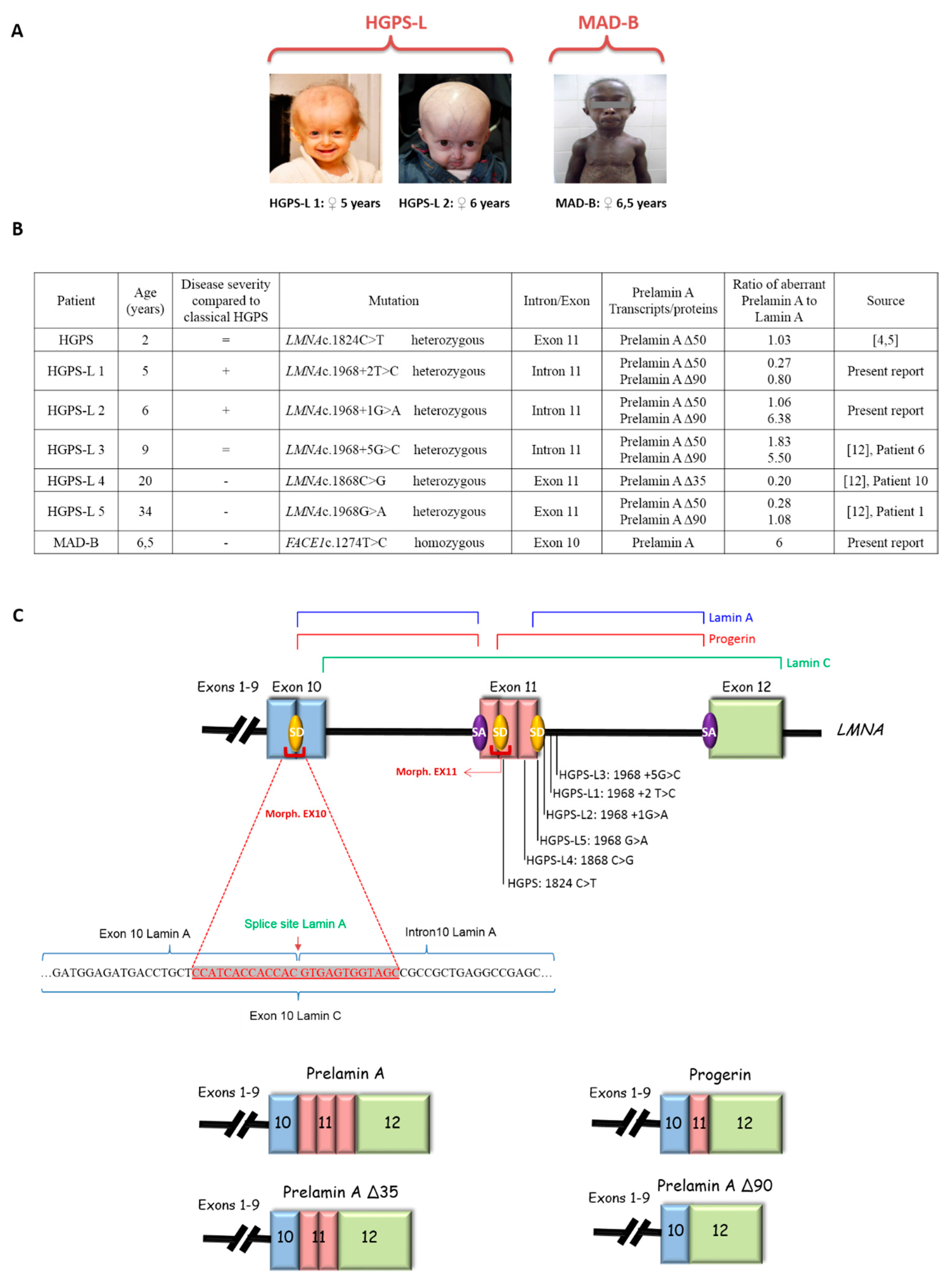
3.1. Patients’ Molecular and Clinical Features
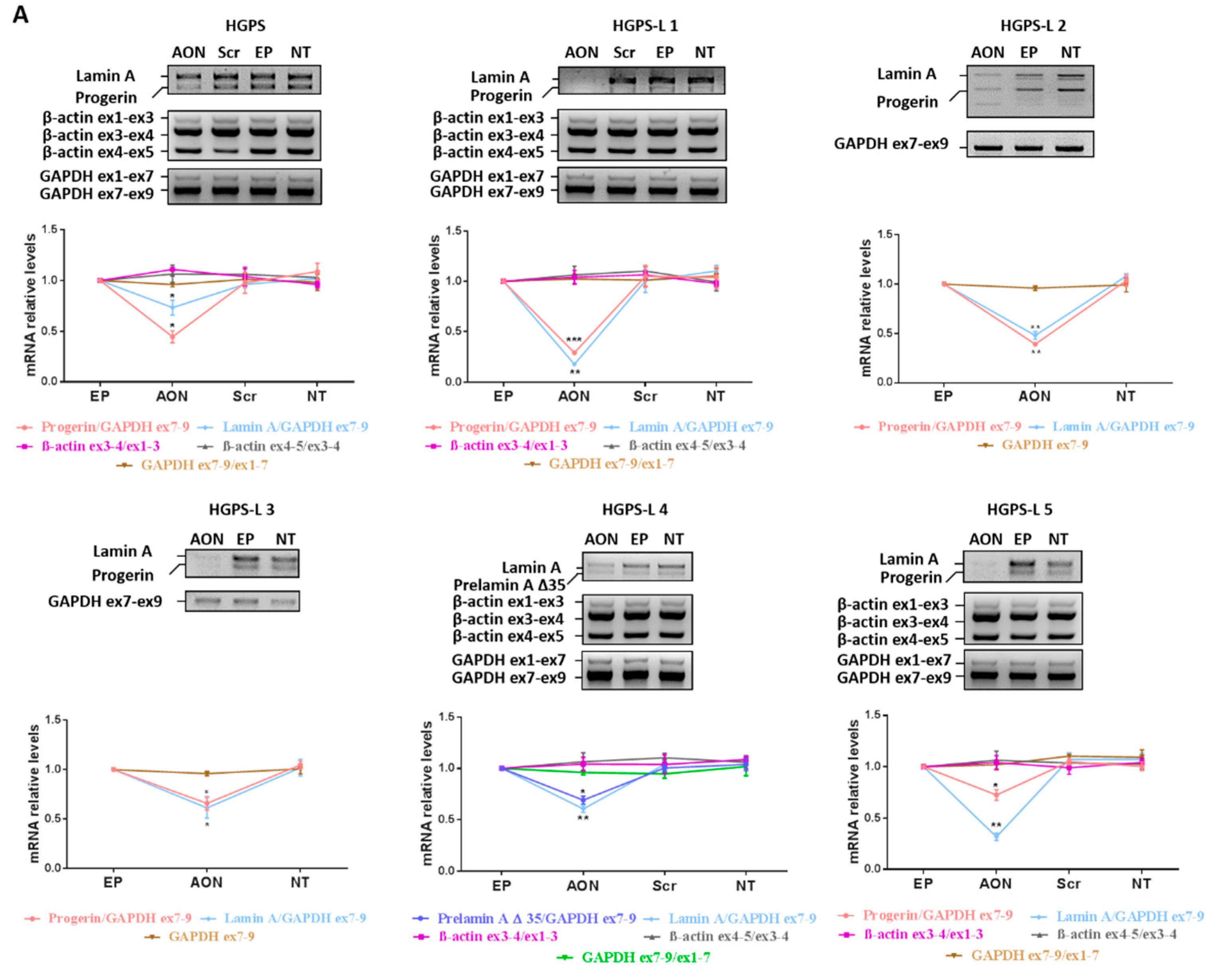
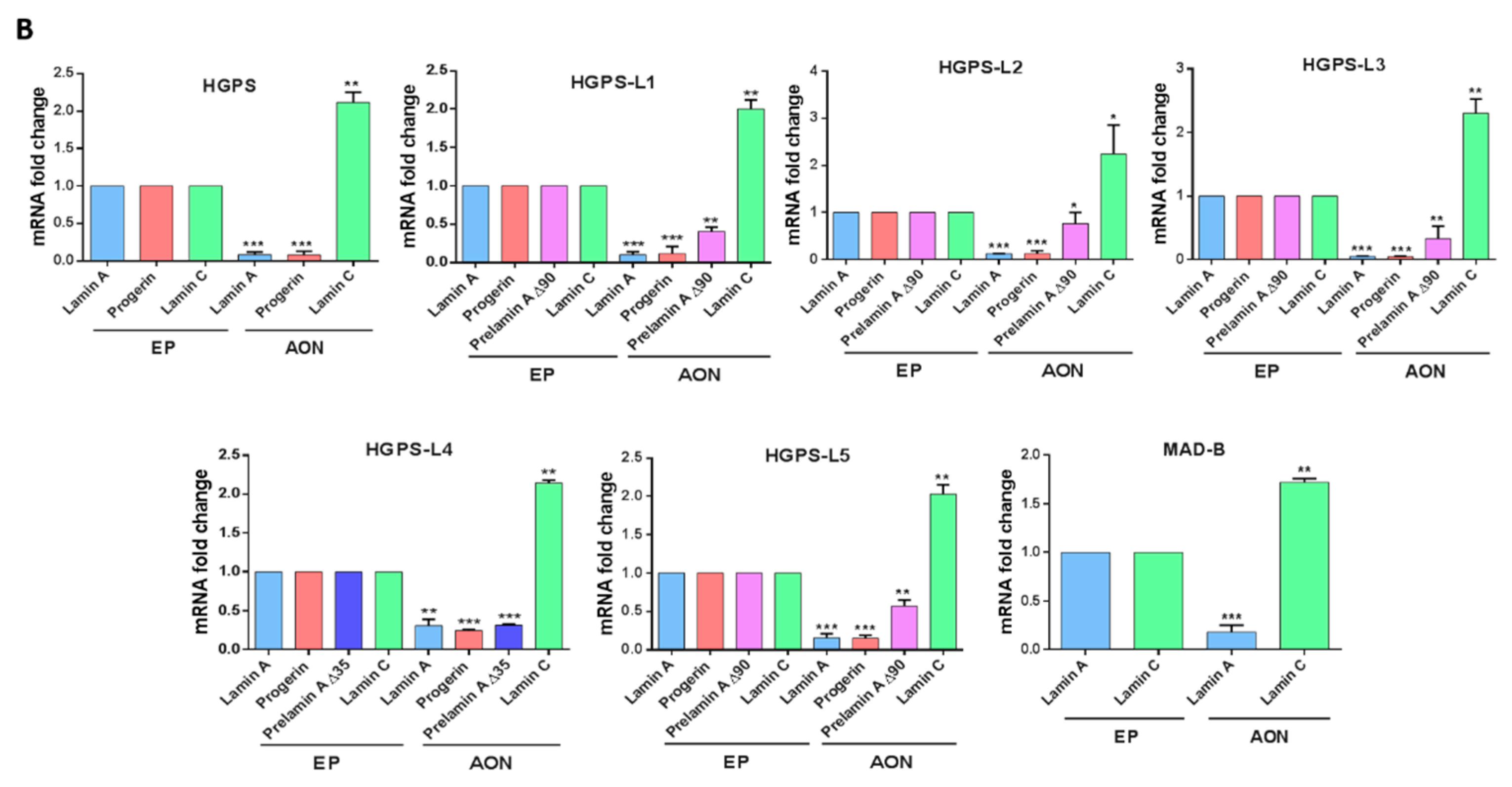
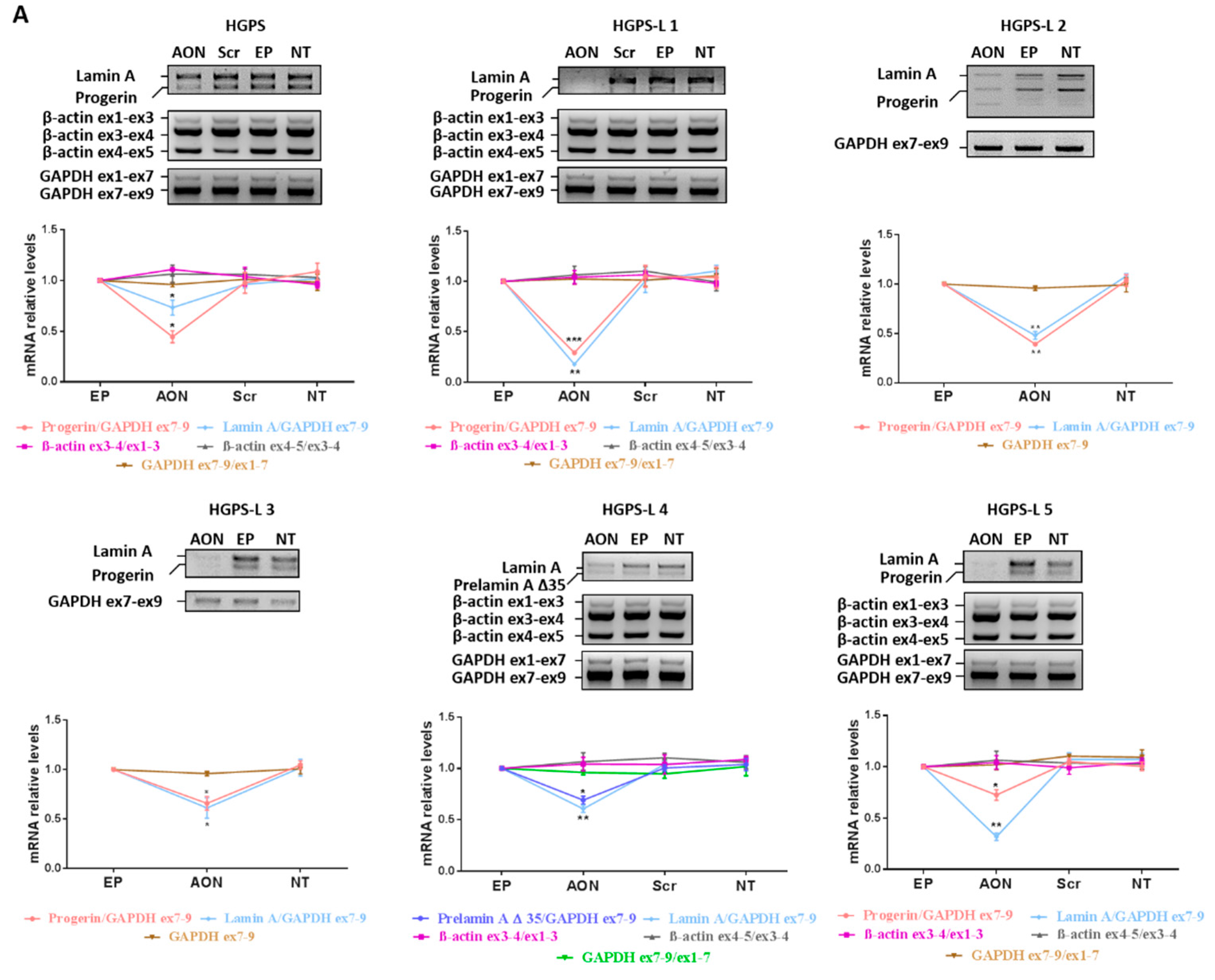
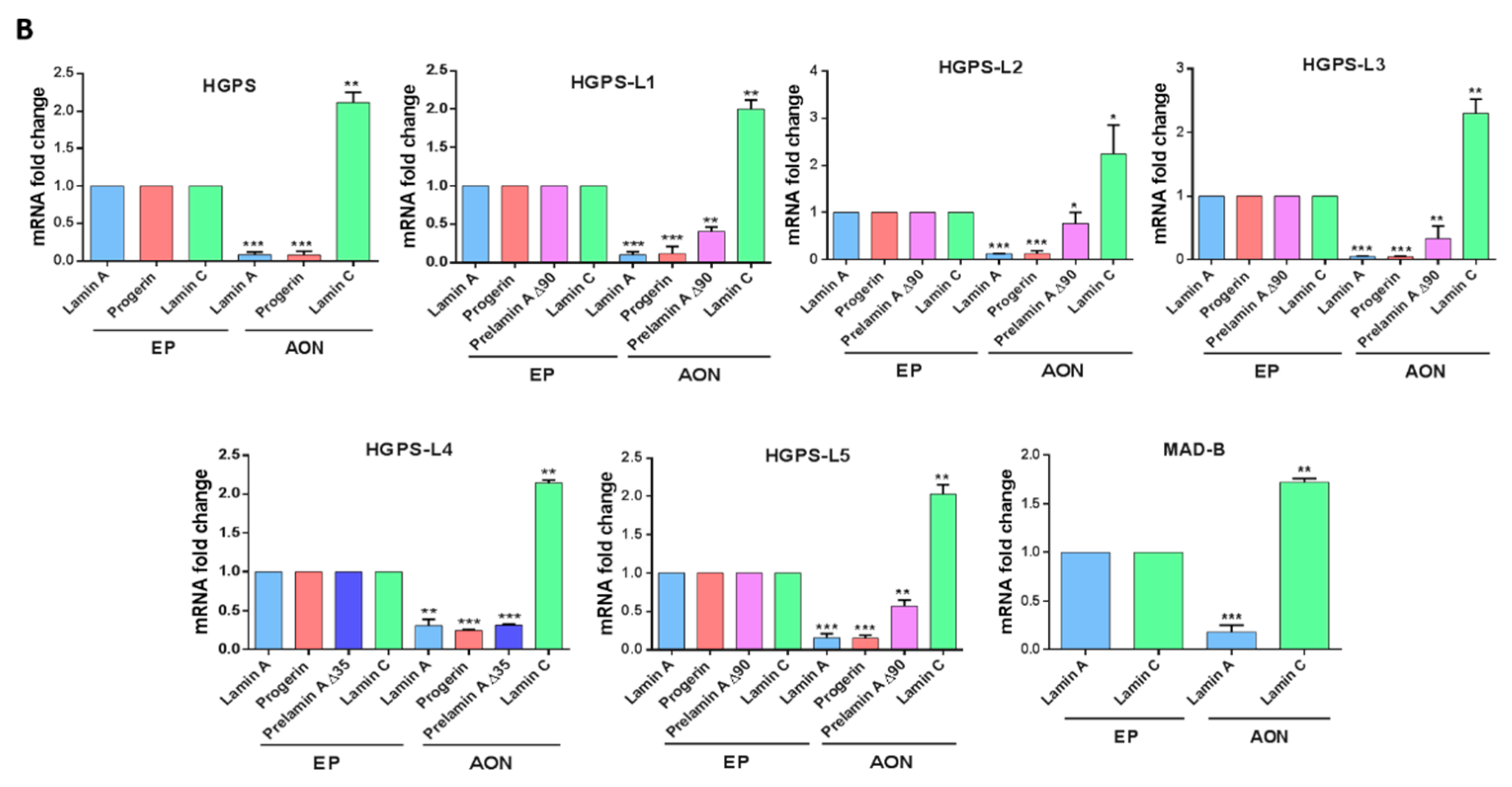
3.2. Efficient Reduction of Aberrant Pre-mRNA Splicing in HGPS-Like and MAB-B Patient Cells Using Antisense Morpholinos
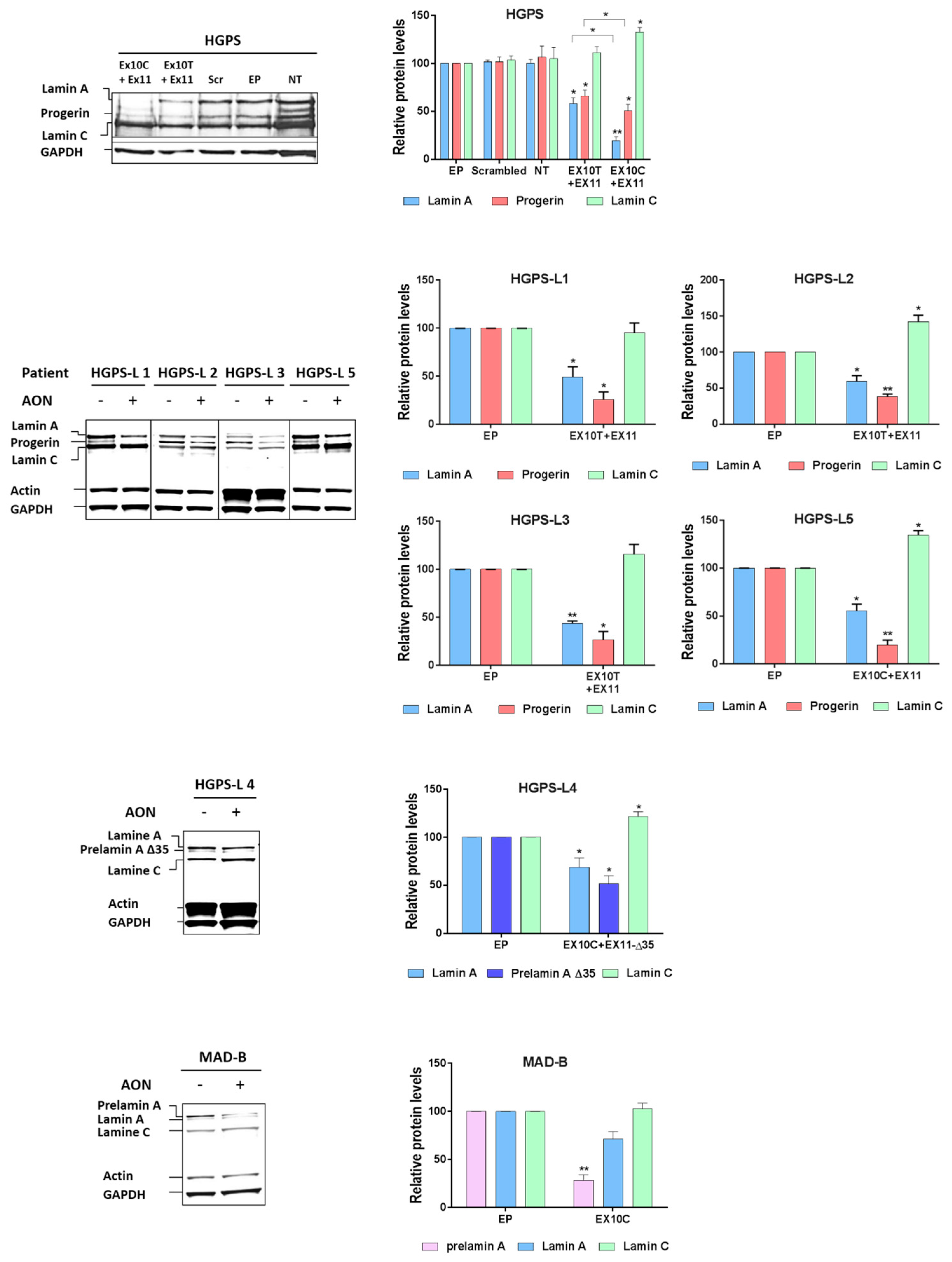
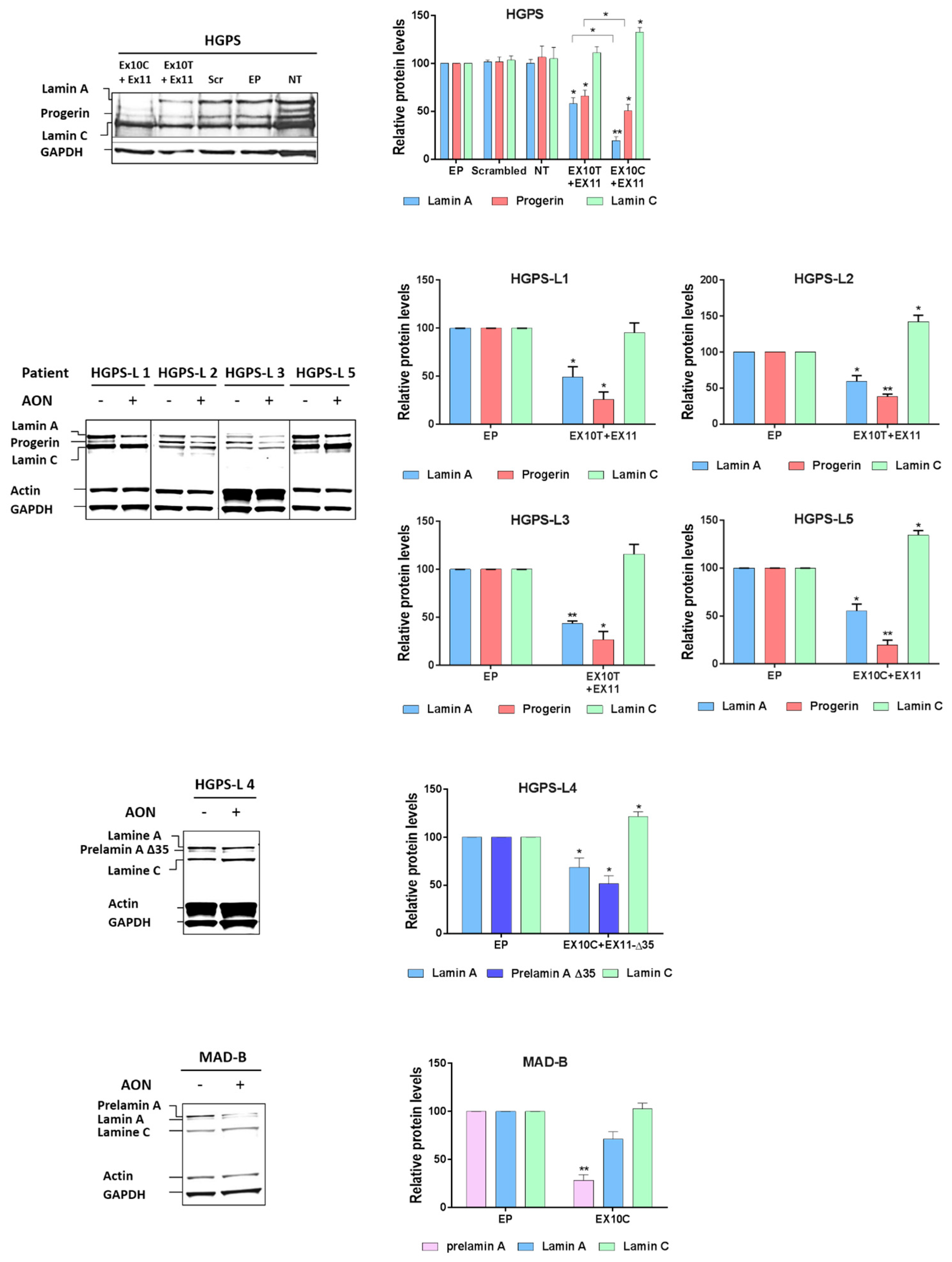
3.3. Antisense Morpholinos Reduce Aberrant Prelamin A Levels in HGPS-Like and MAB-B Patient Cells
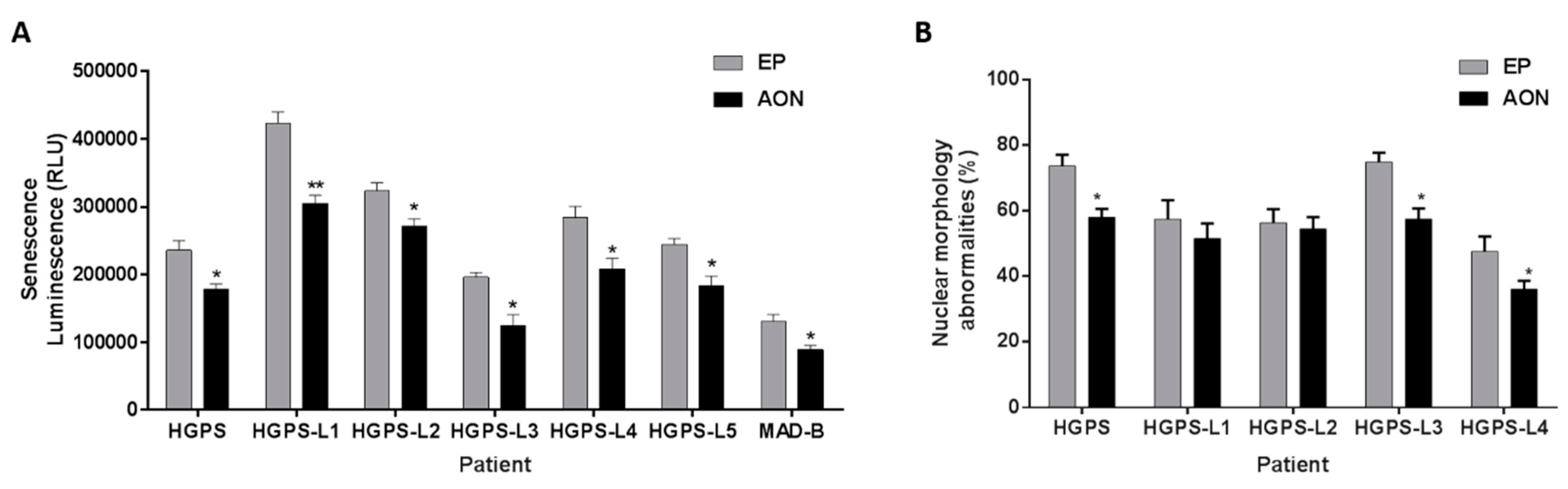
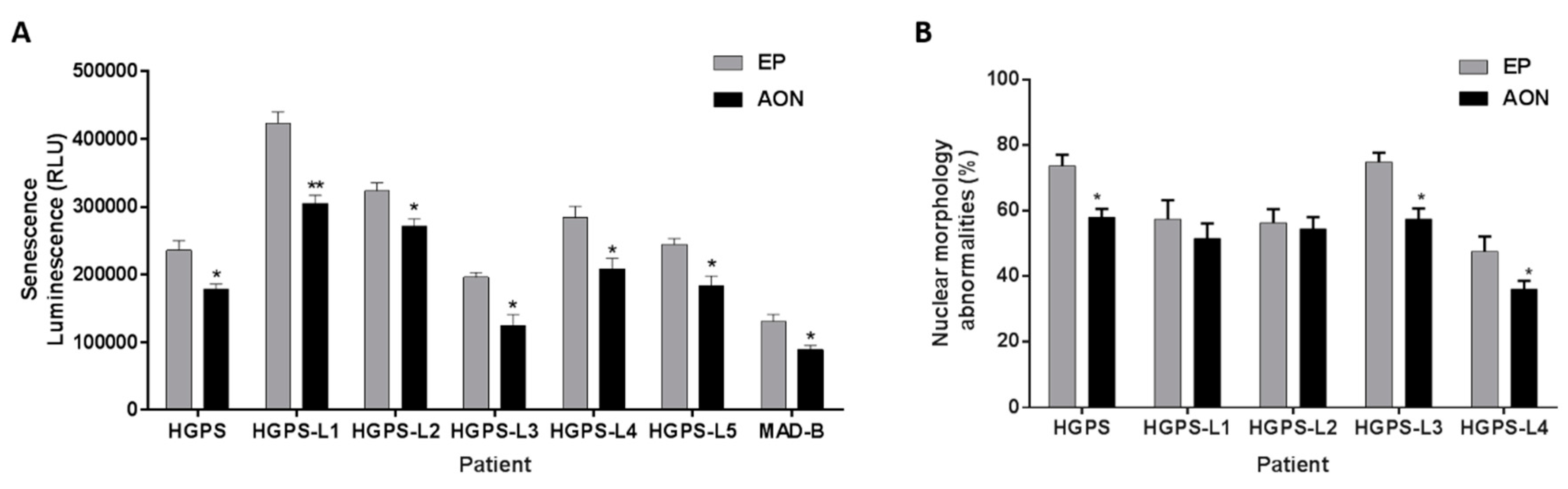
3.4. Antisense Morpholinos Reduce Senescence in HGPS-Like and MAB-B Patient Cells and Improve Nuclear Shape Abnormalities
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cau, P.; Navarro, C.; Harhouri, K.; Roll, P.; Sigaudy, S.; Kaspi, E.; Perrin, S.; De Sandre-Giovannoli, A.; Levy, N. Nuclear matrix, nuclear envelope and premature aging syndromes in a translational research perspective. Semin. Cell Dev. Biol. 2014, 19, 125–147. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.Z.; Chaudhary, N.; Blobel, G. Cdna sequencing of nuclear lamins a and c reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 6450–6454. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin a and nuclear lamin c. J. Biol. Chem. 1993, 268, 16321–16326. [Google Scholar] [PubMed]
- Hetzer, M.W.; Wente, S.R. Border control at the nucleus: Biogenesis and organization of the nuclear membrane and pore complexes. Dev. Cell 2009, 17, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.; Plessmann, U.; Traub, P. Maturation of nuclear lamin a involves a specific carboxy-terminal trimming, which removes the polyisoprenylation site from the precursor; implications for the structure of the nuclear lamina. FEBS Lett. 1989, 257, 411–414. [Google Scholar] [CrossRef]
- Varela, I.; Pereira, S.; Ugalde, A.P.; Navarro, C.L.; Suarez, M.F.; Cau, P.; Cadinanos, J.; Osorio, F.G.; Foray, N.; Cobo, J.; et al. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat. Med. 2008, 14, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef] [PubMed]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin a truncation in hutchinson-gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin a cause hutchinson-gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Hennekam, R.C. Hutchinson-gilford progeria syndrome: Review of the phenotype. Am. J. Med. Genet. A 2006, 140, 2603–2624. [Google Scholar] [CrossRef] [PubMed]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin a causes progressive changes in nuclear architecture in hutchinson-gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [PubMed]
- Barthelemy, F.; Navarro, C.; Fayek, R.; Da Silva, N.; Roll, P.; Sigaudy, S.; Oshima, J.; Bonne, G.; Papadopoulou-Legbelou, K.; Evangeliou, A.E.; et al. Truncated prelamin A expression in HGPS-like patients: A transcriptional study. Eur. J. Hum. Genet. 2015, 23, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Hisama, F.M.; Lessel, D.; Leistritz, D.; Friedrich, K.; McBride, K.L.; Pastore, M.T.; Gottesman, G.S.; Saha, B.; Martin, G.M.; Kubisch, C.; et al. Coronary artery disease in a werner syndrome-like form of progeria characterized by low levels of progerin, a splice variant of lamin A. Am. J. Med. Genet. A 2011, 155, 3002–3006. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, K.; Katsuya, T.; Sugimoto, K.; Kuremura, M.; Kim, H.D.; Li, L.; Ogihara, T. Lmna mutation in a 45 year old japanese subject with hutchinson-gilford progeria syndrome. J. Med. Genet. 2004, 41, e67. [Google Scholar] [CrossRef] [PubMed]
- Moulson, C.L.; Fong, L.G.; Gardner, J.M.; Farber, E.A.; Go, G.; Passariello, A.; Grange, D.K.; Young, S.G.; Miner, J.H. Increased progerin expression associated with unusual lmna mutations causes severe progeroid syndromes. Hum. Mutat. 2007, 28, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Shalev, S.A.; De Sandre-Giovannoli, A.; Shani, A.A.; Levy, N. An association of hutchinson-gilford progeria and malignancy. Am. J. Med. Genet. A 2007, 143, 1821–1826. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Fryns, J.P.; Auchus, R.J.; Garg, A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum. Mol. Genet. 2003, 12, 1995–2001. [Google Scholar] [CrossRef] [PubMed]
- Shackleton, S.; Smallwood, D.T.; Clayton, P.; Wilson, L.C.; Agarwal, A.K.; Garg, A.; Trembath, R.C. Compound heterozygous ZMPSTE24 mutations reduce prelamin a processing and result in a severe progeroid phenotype. J. Med. Genet. 2005, 42, e36. [Google Scholar] [CrossRef] [PubMed]
- Ben Yaou, R.; Navarro, C.; Quijano-Roy, S.; Bertrand, A.T.; Massart, C.; De Sandre-Giovannoli, A.; Cadinanos, J.; Mamchaoui, K.; Butler-Browne, G.; Estournet, B.; et al. Type B mandibuloacral dysplasia with congenital myopathy due to homozygous ZMPSTE24 missense mutation. Eur. J. Hum. Genet. 2011, 19, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Blondel, S.; Jaskowiak, A.L.; Egesipe, A.L.; Le Corf, A.; Navarro, C.; Cordette, V.; Martinat, C.; Laabi, Y.; Djabali, K.; de Sandre-Giovannoli, A.; et al. Induced pluripotent stem cells reveal functional differences between drugs currently investigated in patients with hutchinson-gilford progeria syndrome. Stem Cells Transl. Med. 2014, 3, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Cenni, V.; Capanni, C.; Columbaro, M.; Ortolani, M.; D’Apice, M.R.; Novelli, G.; Fini, M.; Marmiroli, S.; Scarano, E.; Maraldi, N.M.; et al. Autophagic degradation of farnesylated prelamin A as a therapeutic approach to lamin-linked progeria. Eur. J. Histochem. 2011, 55, e36. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Graziotto, J.J.; Blair, C.D.; Mazzulli, J.R.; Erdos, M.R.; Krainc, D.; Collins, F.S. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in hutchinson-gilford progeria syndrome cells. Sci. Transl. Med. 2011, 3, 89ra58. [Google Scholar] [CrossRef] [PubMed]
- Toth, J.I.; Yang, S.H.; Qiao, X.; Beigneux, A.P.; Gelb, M.H.; Moulson, C.L.; Miner, J.H.; Young, S.G.; Fong, L.G. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc. Natl. Acad. Sci. USA 2005, 102, 12873–12878. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Bergo, M.O.; Toth, J.I.; Qiao, X.; Hu, Y.; Sandoval, S.; Meta, M.; Bendale, P.; Gelb, M.H.; Young, S.G.; et al. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted hutchinson-gilford progeria syndrome mutation. Proc. Natl. Acad. Sci. USA 2005, 102, 10291–10296. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.G.; Navarro, C.L.; Cadinanos, J.; Lopez-Mejia, I.C.; Quiros, P.M.; Bartoli, C.; Rivera, J.; Tazi, J.; Guzman, G.; Varela, I.; et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci. Transl. Med. 2011, 3, 106ra107. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Reversal of the cellular phenotype in the premature aging disease hutchinson-gilford progeria syndrome. Nat. Med. 2005, 11, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Parra, M.K.; Gee, S.; Mohandas, N.; Conboy, J.G. Efficient in vivo manipulation of alternative pre-mrna splicing events using antisense morpholinos in mice. J. Biol. Chem. 2011, 286, 6033–6039. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.G.; Barcena, C.; Soria-Valles, C.; Ramsay, A.J.; de Carlos, F.; Cobo, J.; Fueyo, A.; Freije, J.M.; Lopez-Otin, C. Nuclear lamina defects cause atm-dependent nf-kappab activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012, 26, 2311–2324. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Ugalde, A.P.; Fernandez, A.F.; Osorio, F.G.; Fueyo, A.; Freije, J.M.; Lopez-Otin, C. Insulin-like growth factor 1 treatment extends longevity in a mouse model of human premature aging by restoring somatotroph axis function. Proc. Natl. Acad. Sci. USA 2010, 107, 16268–16273. [Google Scholar] [CrossRef] [PubMed]
- Kubben, N.; Zhang, W.; Wang, L.; Voss, T.C.; Yang, J.; Qu, J.; Liu, G.H.; Misteli, T. Repression of the antioxidant NRF2 pathway in premature aging. Cell 2016, 165, 1361–1374. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, D.; Roedl, D.; Gordon, L.B.; Djabali, K. Sulforaphane enhances progerin clearance in hutchinson-gilford progeria fibroblasts. Aging cell 2015, 14, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Morcos, P.A.; Li, Y.; Jiang, S. Vivo-morpholinos: A non-peptide transporter delivers morpholinos into a wide array of mouse tissues. Biotechniques 2008, 45, 613–614. [Google Scholar] [CrossRef] [PubMed]
- Wegner, L.; Anthonsen, S.; Bork-Jensen, J.; Dalgaard, L.; Hansen, T.; Pedersen, O.; Poulsen, P.; Vaag, A. LMNA rs4641 and the muscle lamin a and c isoforms in twins—Metabolic implications and transcriptional regulation. J. Clin. Endocrinol. Metab. 2010, 95, 3884–3892. [Google Scholar] [CrossRef] [PubMed]
- Mesa, J.L.; Loos, R.J.; Franks, P.W.; Ong, K.K.; Luan, J.; O’Rahilly, S.; Wareham, N.J.; Barroso, I. Lamin A/C polymorphisms, type 2 diabetes, and the metabolic syndrome: Case-control and quantitative trait studies. Diabetes 2007, 56, 884–889. [Google Scholar] [CrossRef] [PubMed]
- Wegner, L.; Andersen, G.; Sparso, T.; Grarup, N.; Glumer, C.; Borch-Johnsen, K.; Jorgensen, T.; Hansen, T.; Pedersen, O. Common variation in LMNA increases susceptibility to type 2 diabetes and associates with elevated fasting glycemia and estimates of body fat and height in the general population: Studies of 7495 danish whites. Diabetes 2007, 56, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of hutchinson-gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Nobumori, C.; Tu, Y.; Choi, C.; Yang, S.H.; Jung, H.J.; Vickers, T.A.; Rigo, F.; Bennett, C.F.; Young, S.G.; et al. Modulation of lmna splicing as a strategy to treat prelamin a diseases. J. Clin. Investig. 2016, 126, 1592–1602. [Google Scholar] [CrossRef] [PubMed]
- Nissan, X.; Blondel, S.; Navarro, C.; Maury, Y.; Denis, C.; Girard, M.; Martinat, C.; De Sandre-Giovannoli, A.; Levy, N.; Peschanski, M. Unique preservation of neural cells in hutchinson-Gilford progeria syndrome is due to the expression of the neural-specific miR-9 microRNA. Cell Rep. 2012, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Coffinier, C.; Choe, Y.; Beigneux, A.P.; Davies, B.S.; Yang, S.H.; Barnes, R.H., 2nd; Hong, J.; Sun, T.; Pleasure, S.J.; et al. Regulation of prelamin A but not lamin C by mir-9, a brain-specific microRNA. Proc. Natl. Acad. Sci. USA 2012, 109, E423–E431. [Google Scholar] [CrossRef] [PubMed]
- Fong, L.G.; Ng, J.K.; Lammerding, J.; Vickers, T.A.; Meta, M.; Cote, N.; Gavino, B.; Qiao, X.; Chang, S.Y.; Young, S.R.; et al. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J. Clin. Investig. 2006, 116, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Mejia, I.C.; Vautrot, V.; De Toledo, M.; Behm-Ansmant, I.; Bourgeois, C.F.; Navarro, C.L.; Osorio, F.G.; Freije, J.M.; Stevenin, J.; De Sandre-Giovannoli, A.; et al. A conserved splicing mechanism of the lmna gene controls premature aging. Hum. Mol. Genet. 2011, 20, 4540–4555. [Google Scholar] [CrossRef] [PubMed]
- Strandgren, C.; Nasser, H.A.; McKenna, T.; Koskela, A.; Tuukkanen, J.; Ohlsson, C.; Rozell, B.; Eriksson, M. Transgene silencing of the hutchinson-gilford progeria syndrome mutation results in a reversible bone phenotype, whereas resveratrol treatment does not show overall beneficial effects. FASEB J. 2015, 29, 3193–3205. [Google Scholar] [CrossRef] [PubMed]
- Fong, L.G.; Ng, J.K.; Meta, M.; Cote, N.; Yang, S.H.; Stewart, C.L.; Sullivan, T.; Burghardt, A.; Majumdar, S.; Reue, K.; et al. Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice. Proc. Natl. Acad. Sci. USA 2004, 101, 18111–18116. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Griffith, G.; Babbs, A.; El Andaloussi, S.; Ezzat, K.; Avril, A.; Dugovic, B.; Chaussenot, R.; Ferry, A.; Voit, T.; et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat. Med. 2015, 21, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.L.; Cirak, S.; Partridge, T. What can we learn from clinical trials of exon skipping for DMD? Mol. Ther. Nucleic Acids 2014, 3, e152. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | rs4641 Allele (Verified on the Aberrant Prelamin A cDNA) | Morpholino Name | Morpholino Sequence |
|---|---|---|---|
| HGPS | C | Ex10-allele C | 5′-GCTACCACTCACGTGGTGGTGATGG-3′ |
| Ex11-HGPSmut | 5′-GGGTCCACCCACCTGGGCTCCTGAG-3′ | ||
| HGPS-L1 | T | Ex10-allele T Ex11 | 5′-GCTACCACTCACATGGTGGTGATGG-3′ 5′-GGGTCCGCCCACCTGGGCTCCTGAG-3′ |
| HGPS-L2 | T | ||
| HGPS-L3 | T | ||
| HGPS-L4 | C | Ex10-allele C | 5′-GCTACCACTCACGTGGTGGTGATGG-3′ |
| Ex11-Δ35 | 5′-ACTGACCGTGACACTGGAGGCAGAA-3′ | ||
| HGPS-L5 | C | Ex10-allele C | 5′-GCTACCACTCACGTGGTGGTGATGG-3′ |
| Ex11 | 5′-GGGTCCGCCCACCTGGGCTCCTGAG-3′ | ||
| MAD-B | Homozygous C | Ex10-allele C | 5′-GCTACCACTCACGTGGTGGTGATGG-3′ |
| Negative controls | - | Ex10-scrambled | 5′-ATCGGCTTGTCGCGTGAGCGATCGA-3′ |
| Ex11-scrambled | 5′-ACCAGTGGCGTCGCCTCGCAGGTCC-3′ |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harhouri, K.; Navarro, C.; Baquerre, C.; Da Silva, N.; Bartoli, C.; Casey, F.; Mawuse, G.K.; Doubaj, Y.; Lévy, N.; De Sandre-Giovannoli, A. Antisense-Based Progerin Downregulation in HGPS-Like Patients’ Cells. Cells 2016, 5, 31. https://doi.org/10.3390/cells5030031
Harhouri K, Navarro C, Baquerre C, Da Silva N, Bartoli C, Casey F, Mawuse GK, Doubaj Y, Lévy N, De Sandre-Giovannoli A. Antisense-Based Progerin Downregulation in HGPS-Like Patients’ Cells. Cells. 2016; 5(3):31. https://doi.org/10.3390/cells5030031
Chicago/Turabian StyleHarhouri, Karim, Claire Navarro, Camille Baquerre, Nathalie Da Silva, Catherine Bartoli, Frank Casey, Guedenon Koffi Mawuse, Yassamine Doubaj, Nicolas Lévy, and Annachiara De Sandre-Giovannoli. 2016. "Antisense-Based Progerin Downregulation in HGPS-Like Patients’ Cells" Cells 5, no. 3: 31. https://doi.org/10.3390/cells5030031