
Skeletal Muscle Laminopathies: A Review of Clinical and Molecular Features

Abstract
:1. Introduction
2. Genetics and Pathogenesis
3. Skeletal Muscle Involvement in Laminopathies
4. Cardiac Involvement
5. Atypical Cases in Laminopathies
6. Genotype-Phenotype Correlation
7. Treatment
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Bonne, G.; Yaou, R.B.; Béroud, C.; Boriani, G.; Brown, S.; de Visser, M.; Duboc, D.; Ellis, J.; Hausmanowa-Petrusewicz, I.; Lattanzi, G.; et al. 108th ENMC International Workshop, 3rd Workshop of the MYO-CLUSTER project: EUROMEN, 7th International Emery-Dreifuss Muscular Dystrophy (EDMD) Workshop, 13–15 September 2002, Naarden, The Netherlands. Neuromuscul. Disord. 2003, 13, 508–515. [Google Scholar] [CrossRef]
- Bertrand, A.T.; Chikhaoui, K.; Ben Yaou, R.; Bonne, G. Clinical and genetic heterogeneity in laminopathies. Biochem. Soc. Trans. 2011, 39, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J.; Bonne, G. “Laminopathies”: A wide spectrum of human diseases. Exp. Cell Res. 2007, 313, 2121–2133. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J. Nuclear lamins and laminopathies. J. Pathol. 2012, 226, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; D’Amico, A.; Pini, A.; Sivo, S.; Pane, M.; Ricci, G.; Vercelli, L.; D’Ambrosio, P.; Travaglini, L.; Sala, S.; et al. LMNA-associated myopathies: The Italian experience in a large cohort of patients. Neurology 2014, 83, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Carboni, N.; Politano, L.; Floris, M.; Mateddu, A.; Solla, E.; Olla, S.; Maggi, L.; Maioli, M.A.; Piras, R.; Cocco, E.; et al. Overlapping syndromes in laminopathies: A meta-analysis of the reported literature. Acta Myol. 2013, 32, 7–17. [Google Scholar] [PubMed]
- Bönnemann, C.G.; Wang, C.H.; Quijano-Roy, S.; Deconinck, N.; Bertini, E.; Ferreiro, A.; Muntoni, F.; Sewry, C.; Béroud, C.; Mathews, K.D.; et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul. Disord. 2014, 24, 289–311. [Google Scholar] [CrossRef] [PubMed]
- Gruenbaum, Y.; Margalit, A.; Goldman, R.D.; Shumaker, D.K.; Wilson, K.L. The nuclear lamina comes of age. Nat. Rev. Mol. Cell Biol. 2005, 6, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Gruenbaum, Y.; Foisner, R. Lamins: Nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu. Rev. Biochem. 2015, 84, 131–164. [Google Scholar] [CrossRef] [PubMed]
- Gruenbaum, Y.; Medalia, O. Lamins: The structure and protein complexes. Curr. Opin. Cell Biol. 2015, 32, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.N.; Wilson, K.L. Partners and post-translational modifications of nuclear lamins. Chromosoma 2013, 122, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Barton, R.M.; Worman, H.J. Prenylated prelamin A interacts with Narf, a novel nuclear protein. J. Biol. Chem. 1999, 274, 30008–30018. [Google Scholar] [CrossRef] [PubMed]
- Osmanagic-Myers, S.; Dechat, T.; Foisner, R. Lamins at the crossroads of mechanosignaling. Genes Dev. 2015, 29, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Mattioli, E.; Columbaro, M.; Capanni, C.; Maraldi, N.M.; Cenni, V.; Scotlandi, K.; Marino, M.T.; Merlini, L.; Squarzoni, S.; Lattanzi, G. Prelamin A-mediated recruitment of SUN1 to the nuclear envelope directs nuclear positioning in human muscle. Cell Death Differ. 2011, 18, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Shimi, T.; Pfleghaar, K.; Kojima, S.; Pack, C.G.; Solovei, I.; Goldman, A.E.; Adam, S.A.; Shumaker, D.K.; Kinjo, M.; Cremer, T.; et al. The A- and B-type nuclear lamin networks: Microdomains involved in chromatin organization and transcription. Genes Dev. 2008, 22, 3409–3421. [Google Scholar] [CrossRef] [PubMed]
- Camozzi, D.; Capanni, C.; Cenni, V.; Mattioli, E.; Columbaro, M.; Squarzoni, S.; Lattanzi, G. Diverse lamin-dependent mechanisms interact to control chromatin dynamics. Focus on laminopathies. Nucleus 2014, 5, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Cesarini, E.; Mozzetta, C.; Marullo, F.; Gregoretti, F.; Gargiulo, A.; Columbaro, M.; Cortesi, A.; Antonelli, L.; Di Pelino, S.; Squarzoni, S.; et al. Lamin A/C sustains PcG protein architecture, maintaining transcriptional repression at target genes. J. Cell Biol. 2015, 211, 533–551. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; Pavlidis, P.; Decostre, V.; Herron, A.J.; Arimura, T.; Bonne, G.; Worman, H.J. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Investig. 2007, 117, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Dechat, T.; Adam, S.A.; Taimen, P.; Shimi, T.; Goldman, R.D. Nuclear lamins. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 1993, 268, 16321–16326. [Google Scholar] [PubMed]
- Lattanzi, G.; Columbaro, M.; Mattioli, E.; Cenni, V.; Camozzi, D.; Wehnert, M.; Santi, S.; Riccio, M.; Del Coco, R.; Maraldi, N.M.; et al. Pre-Lamin A processing is linked to heterochromatin organization. J. Cell Biochem. 2007, 102, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Chatzifrangkeskou, M.; Bonne, G.; Muchir, A. Nuclear envelope and striated muscle diseases. Curr. Opin. Cell Biol. 2015, 32, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Davidson, P.M.; Lammerding, J. Broken nuclei—Lamins, nuclear mechanics, and disease. Trends Cell Biol. 2014, 24, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Zwerger, M.; Jaalouk, D.E.; Lombardi, M.L.; Isermann, P.; Mauermann, M.; Dialynas, G.; Herrmann, H.; Wallrath, L.L.; Lammerding, J. Myopathic lamin mutations impair nuclear stability in cells and tissue and disrupt nucleo-cytoskeletal coupling. Hum. Mol. Genet. 2013, 22, 2335–2349. [Google Scholar] [CrossRef] [PubMed]
- Azibani, F.; Muchir, A.; Vignier, N.; Bonne, G.; Bertrand, A.T. Striated muscle laminopathies. Semin. Cell Dev. Biol. 2014, 29, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Ramos, F.J.; Chen, S.C.; Garelick, M.G.; Dai, D.F.; Liao, C.Y.; Schreiber, K.H.; MacKay, V.L.; An, E.H.; Strong, R.; Ladiges, W.C.; et al. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med. 2012, 4, 144ra103. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; Worman, H.J. Targeting mitogen-activated protein kinase signaling in mouse models of cardiomyopathy caused by lamin A/C gene mutations. Methods Enzymol. 2016, 568, 557–580. [Google Scholar] [PubMed]
- Muchir, A.; Kim, Y.J.; Reilly, S.A.; Wu, W.; Choi, J.C.; Worman, H.J. Inhibition of extracellular signal-regulated kinase 1/2 signaling has beneficial effects on skeletal muscle in a mouse model of Emery-Dreifuss muscular dystrophy caused by lamin A/C gene mutation. Skelet. Muscle 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Webster, B.M.; Lusk, C.P. Border safety: Quality control at the nuclear envelope. Trends Cell Biol. 2016, 26, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.C.; Woman, H.J. Reactivation of autophagy ameliorates LMNA cardiomyopathy. Autophagy 2013, 9, 110–111. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J.; Schirmer, E.C. Nuclear membrane diversity: Underlying tissue-specific pathologies in disease? Curr. Opin. Cell Biol. 2015, 34, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, G.L.; Muntoni, F.; Miocic, S.; Sinagra, G.; Sewry, C.; Mestroni, L. Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation 2000, 101, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Poppe, M.; Quinlivan, R.; Messina, S.; Kinali, M.; Demay, L.; Bourke, J.; Richard, P.; Sewry, C.; Pike, M.; et al. Extreme variability of phenotype in patients with an identical missense mutation in the lamin A/C gene: From congenital onset with severe phenotype to milder classic Emery-Dreifuss variant. Arch. Neurol. 2004, 61, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Menezes, M.P.; Waddell, L.B.; Evesson, F.J.; Cooper, S.; Webster, R.; Jones, K.; Mowat, D.; Kiernan, M.C.; Johnston, H.M.; Corbett, A.; et al. Importance and challenge of making an early diagnosis in LMNA-related muscular dystrophy. Neurology 2012, 78, 1258–1263. [Google Scholar] [CrossRef] [PubMed]
- Carboni, N.; Mura, M.; Marrosu, G.; Cocco, E.; Marini, S.; Solla, E.; Mateddu, A.; Maioli, M.A.; Piras, R.; Mallarini, G.; et al. Muscle imaging analogies in a cohort of patients with different phenotypes caused by LMNA gene mutations. Muscle Nerve 2010, 41, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Clements, E.; Offiah, A.; Pichiecchio, A.; Vasco, G.; Bianco, F.; Berardinelli, A.; Manzur, A.; Pane, M.; Messina, S.; et al. Muscle magnetic resonance imaging involvement in muscular dystrophies with rigidity of the spine. Ann. Neurol. 2010, 67, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Andrés, D.; Dabaj, I.; Mompoint, D.; Hankiewicz, K.; Azzi, V.; Ioos, C.; Romero, N.B.; Ben Yaou, R.; Bergounioux, J.; Bonne, G.; et al. Pediatric laminopathies: Whole-body MRI fingerprint and comparison with SEPN1-myopathy. Muscle Nerve 2015. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G.; Di Barletta, M.R.; Varnous, S.; Becane, H.; Hammouda, E.H.; Merlini, L.; Muntoni, F.; Greenberg, C.R.; Gary, F.; Urtizberea, J.A.; et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery–Dreifuss muscular dystrophy. Nat. Genet. 1999, 21, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E.H. Emery-Dreifuss muscular dystrophy: A 40-year retrospective. Neuromuscul. Disord. 2000, 10, 228–232. [Google Scholar] [CrossRef]
- Meune, C.; Van Berlo, J.H.; Anselme, F.; Bonne, G.; Pinto, Y.M.; Duboc, D. Primary prevention of sudden death in patients with lamin A/C gene mutations. N. Engl. J. Med. 2006, 354, 209–210. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G.; Mercuri, E.; Muchir, A.; Urtizberea, A.; Bécane, H.M.; Recan, D.; Merlini, L.; Wehnert, M.; Boor, R.; Reuner, U.; et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol. 2000, 48, 170–180. [Google Scholar] [CrossRef]
- Bécane, H.M.; Bonne, G.; Varnous, S.; Muchir, A.; Ortega, V.; Hammouda, E.H.; Urtizberea, J.A.; Lavergne, T.; Fardeau, M.; Eymard, B.; et al. High incidence of sudden death with conduction system and myocardial disease due to lamins A and C gene mutation. Pacing Clin. Electrophysiol. 2000, 23, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Boriani, G.; Gallina, M.; Merlini, L.; Bonne, G.; Toniolo, D.; Amati, S.; Biffi, M.; Martignani, C.; Frabetti, L.; Bonvicini, M.; et al. Clinical relevance of atrial fibrillation/flutter, stroke, pacemaker implant, and heart failure in Emery-Dreifuss muscular dystrophy: A long-term longitudinal study. Stroke 2003, 34, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Pasotti, M.; Klersy, C.; Pilotto, A.; Marziliano, N.; Rapezzi, C.; Serio, A.; Mannarino, S.; Gambarin, F.; Favalli, V.; Grasso, M.; et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J. Am. Coll. Cardiol. 2008, 52, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Manera, J.; Alejaldre, A.; González, L.; Olivé, M.; Gómez-Andrés, D.; Muelas, N.; Vílchez, J.J.; Llauger, J.; Carbonell, P.; Márquez-Infante, C.; et al. Muscle imaging in muscle dystrophies produced by mutations in the EMD and LMNA genes. Neuromuscul. Disord. 2016, 26, 33–40. [Google Scholar]
- Gueneau, L.; Bertrand, A.T.; Jais, J.P.; Salih, M.A.; Stojkovic, T.; Wehnert, M.; Hoeltzenbein, M.; Spuler, S.; Saitoh, S.; Verschueren, A.; et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am. J. Hum. Genet. 2009, 85, 338–353. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; Bonne, G.; van der Kooi, A.J.; van Meegen, M.; Baas, F.; Bolhuis, P.A.; de Visser, M.; Schwartz, K. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum. Mol. Genet. 2000, 9, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Quijano-Roy, S.; Mbieleu, B.; Bönnemann, C.G.; Jeannet, P.Y.; Colomer, J.; Clarke, N.F.; Cuisset, J.M.; Roper, H.; De Meirleir, L.; D’Amico, A.; et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann. Neurol. 2008, 64, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Komaki, H.; Hayashi, Y.K.; Tsuburaya, R.; Sugie, K.; Kato, M.; Nagai, T.; Imataka, G.; Suzuki, S.; Saitoh, S.; Asahina, N.; et al. Inflammatory changes in infantile-onset LMNA-associated myopathy. Neuromuscul. Disord. 2011, 21, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Hattori, A.; Komaki, H.; Kawatani, M.; Sakuma, H.; Saito, Y.; Nakagawa, E.; Sugai, K.; Sasaki, M.; Hayashi, Y.K.; Nonaka, I.; et al. A novel mutation in the LMNA gene causes congenital muscular dystrophy with dropped head and brain involvement. Neuromuscul. Disord. 2012, 22, 149–151. [Google Scholar] [CrossRef] [PubMed]
- De Sandre-Giovannoli, A.; Chaouch, M.; Kozlov, S.; Vallat, J.M.; Tazir, M.; Kassouri, N.; Szepetowski, P.; Hammadouche, T.; Vandenberghe, A.; Stewart, C.L.; et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am. J. Hum. Genet. 2002, 70, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Chaouch, M.; Allal, Y.; De Sandre-Giovannoli, A.; Vallat, J.M.; Amer-el-Khedoud, A.; Kassouri, N.; Chaouch, A.; Sindou, P.; Hammadouche, T.; Tazir, M.; et al. The phenotypic manifestations of autosomal recessive axonal Charcot-Marie-Tooth due to a mutation in Lamin A/C gene. Neuromuscul. Disord. 2003, 13, 60–67. [Google Scholar] [CrossRef]
- Tazir, M.; Azzedine, H.; Assami, S.; Sindou, P.; Nouioua, S.; Zemmouri, R.; Hamadouche, T.; Chaouch, M.; Feingold, J.; Vallat, J.M.; et al. Phenotypic variability in autosomal recessive axonal Charcot-Marie-Tooth disease due to the R298C mutation in lamin A/C. Brain 2004, 127, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Goizet, C.; Yaou, R.B.; Demay, L.; Richard, P.; Bouillot, S.; Rouanet, M.; Hermosilla, E.; Le Masson, G.; Lagueny, A.; Bonne, G.; et al. A new mutation of the lamin A/C gene leading to autosomal dominant axonal neuropathy, muscular dystrophy, cardiac disease, and leuconychia. J. Med. Genet. 2004, 41, e29. [Google Scholar] [CrossRef] [PubMed]
- Leiden Muscular Dystrophy. Available online: http://www.dmd.nl (accessed on 13 June 2016).
- The UMD–LMNA Mutations Database. Available online: http://www.umd.be/LMNA/ (accessed on 13 June 2016).
- Nigro, G.; Russo, V.; Rago, A.; Papa, A.A.; Carbone, N.; Marchel, M.; Palladino, A.; Hausmanowa-Petrusewicz, I.; Russo, M.G.; Politano, L. Regional and transmural dispersion of repolarisation in patients with Emery-Dreifuss muscular dystrophy. Kardiol. Pol. 2012, 70, 1154–1159. [Google Scholar] [PubMed]
- Fatkin, D.; MacRae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J., Jr.; Spudich, S.; De Girolami, U.; et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Van Berlo, J.H.; de Voogt, W.G.; van der Kooi, A.J.; van Tintelen, J.P.; Bonne, G.; Yaou, R.B.; Duboc, D.; Rossenbacker, T.; Heidbüchel, H.; de Visser, M.; et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: Do lamin A/C mutations portend a high risk of sudden death? J. Mol. Med. 2005, 83, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Carboni, N.; Mateddu, A.; Marrosu, G.; Cocco, E.; Marrosu, M.G. Genetic and clinical characteristics of skeletal and cardiac muscle in patients with lamin A/C gene mutations. Muscle Nerve 2013, 48, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, R.; Mason, P.K. Lamin A/C deficiency as a cause of familial dilated cardiomyopathy. Curr. Opin. Cardiol. 2009, 24, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Van Rijsingen, I.A.; Nannenberg, E.A.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Hermans-van Ast, J.F.; van der Kooi, A.J.; van Tintelen, J.P.; van den Berg, M.P.; Grasso, M.; et al. Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur. J. Heart Fail. 2013, 15, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Maioli, M.A.; Marrosu, G.; Mateddu, A.; Solla, E.; Carboni, N.; Tacconi, P.; Lai, C.; Marrosu, M.G. A novel mutation in the central rod domain of lamin A/C producing a phenotype resembling the Emery-Dreifuss muscular dystrophy phenotype. Muscle Nerve 2007, 36, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Van Engelen, B.G.; Muchir, A.; Hutchison, C.J.; van der Kooi, A.J.; Bonne, G.; Lammens, M. The lethal phenotype of a homozygous nonsense mutation in the lamin A/C gene. Neurology 2005, 64, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Sinkovec, M.; Petrovic, D.; Volk, M.; Peterlin, B. Familial progressive sinoatrial and atrioventricular conduction disease of adult onset with sudden death, dilated cardiomyopathy, and brachydactyly. A new type of heart-hand syndrome? Clin. Genet. 2005, 68, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Renou, L.; Stora, S.; Yaou, R.B.; Volk, M.; Sinkovec, M.; Demay, L.; Richard, P.; Peterlin, B.; Bonne, G. Heart-hand syndrome of Slovenian type: A new kind of laminopathy. J. Med. Genet. 2008, 45, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Speckman, R.A.; Bowcock, A.M. Multisystem dystrophy syndrome due to novel missense mutations in the amino-terminal head and alpha-helical rod domains of the Lamin A/C gene. Am. J. Med. 2002, 112, 549–555. [Google Scholar] [CrossRef]
- Di Barletta, M.R.; Ricci, E.; Galluzzi, G.; Tonali, P.; Mora, M.; Morandi, L.; Romorini, A.; Voit, T.; Orstavik, K.H.; Merlini, L.; et al. Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am. J. Hum. Genet. 2000, 66, 1407–1412. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Escrig, A.; Gobernado, I.; Garcia-Villanueva, M.; Sanchez-Herranz, A. Autosomal recessive Emery-Dreifuss muscular dystrophy caused by a novel mutation (R225Q) in the lamin A/C gene identified by exome sequencing. Muscle Nerve 2012, 45, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, S.; Menditto, I.; Degano, M.; Rodolico, C.; Merlini, L.; D’Amico, A.; Palmucci, L.; Berardinelli, A.; Pegoraro, E.; Trevisan, C.P.; et al. Phenotypic clustering of lamin A/C mutations in neuromuscular patients. Neurology 2007, 69, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Cattin, M.E.; Muchir, A.; Bonne, G. “State-of-the-heart” of cardiac laminopathies. Curr. Opin. Cardiol. 2013, 28, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Yaou, R.B.; Toutain, A.; Arimura, T.; Demay, L.; Massart, C.; Peccate, C.; Muchir, A.; Llense, S.; Deburgrave, N.; Leturcq, F.; et al. Multitissular involvement in a family with LMNA and EMD mutations: Role of digenic mechanism? Neurology 2007, 68, 1883–1894. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Bonne, G.; Goldfarb, L.G.; Mercuri, E.; Piercy, R.J.; Burke, M.; Yaou, R.B.; Richard, P.; Récan, D.; Shatunov, A.; et al. Disease severity in dominant Emery Dreifuss is increased by mutations in both emerin and desmin proteins. Brain 2006, 129, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Granger, B.; Gueneau, L.; Drouin-Garraud, V.; Pedergnana, V.; Gagnon, F.; Ben Yaou, R.; Tezenas du Montcel, S.; Bonne, G. Modifier locus of the skeletal muscle involvement in Emery-Dreifuss muscular dystrophy. Hum. Genet. 2011, 129, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, É.; Labib, S.; Bertrand, A.T.; Decostre, V.; Bolongo, P.M.; Sylvius, N.; Bonne, G.; Tesson, F. Lamin A/C mutants disturb sumo1 localization and sumoylation in vitro and in vivo. PLoS ONE 2012, 7, e45918. [Google Scholar] [CrossRef] [PubMed]
- Arimura, T.; Onoue, K.; Takahashi-Tanaka, Y. Nuclear accumulation of androgen receptor in gender difference of dilated cardiomyopathy due to lamin A/C mutations. Cardiovasc. Res. 2013, 99, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Roncarati, R.; Anselmi, C.V.; Krawitz, P.; Lattanzi, G.; von Kodolitsch, Y.; Perrot, A.; di Pasquale, E.; Papa, L.; Portararo, P.; Columbaro, M.; et al. Doubly heterozygous LMNA and TTN mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy. Eur. J. Hum. Genet. 2013, 21, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Zastrow, M.S.; Vlcek, S.; Wilson, K.L. Proteins that bind A-type lamins: Integrating isolated clues. J. Cell Sci. 2004, 117, 979–987. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov (accessed on 13 June 2016).
- Catelain, C.; Riveron, S.; Papadopoulos, A.; Mougenot, N.; Jacquet, A.; Vauchez, K.; Yada, E.; Pucéat, M.; Fiszman, M.; Butler-Browne, G.; et al. Myoblasts and embryonic stem cells differentially engraft in a mouse model of genetic dilated cardiomyopathy. Mol. Ther. 2013, 21, 1064–1075. [Google Scholar] [CrossRef] [PubMed]
- Scharner, J.; Figeac, N.; Ellis, J.A.; Zammit, P.S. Ameliorating pathogenesis by removing an exon containing a missense mutation: A potential exon-skipping therapy for laminopathies. Gene Ther. 2015, 22, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chen, L.; Libina, N.; Janes, J.; Martin, G.M.; Campisi, J.; Oshima, J. Correction of cellular phenotypes of Hutchinson-Gilford Progeria cells by RNA interference. Hum. Genet. 2005, 118, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 2005, 11, 440–445. [Google Scholar] [CrossRef] [PubMed]
- McClintock, D.; Gordon, L.B.; Djabali, K. Hutchinson–Gilford progeria mutant lamin A primarily targets human vascular cells as detected by an anti-Lamin A G608G antibody. Proc. Natl. Acad. Sci. USA 2006, 103, 2154–2159. [Google Scholar] [CrossRef] [PubMed]
- Wehnert, M.S.; Bonne, G. The nuclear muscular dystrophies. Semin. Pediatr. Neurol. 2002, 9, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.C.; Muchir, A.; Wu, W.; Iwata, S.; Homma, S.; Morrow, J.P.; Worman, H.J. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med. 2012, 4, 144ra102. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Muchir, A.; Shan, J.; Bonne, G.; Worman, H.J. Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin A/C gene. Circulation 2011, 123, 53–61. [Google Scholar] [CrossRef] [PubMed]
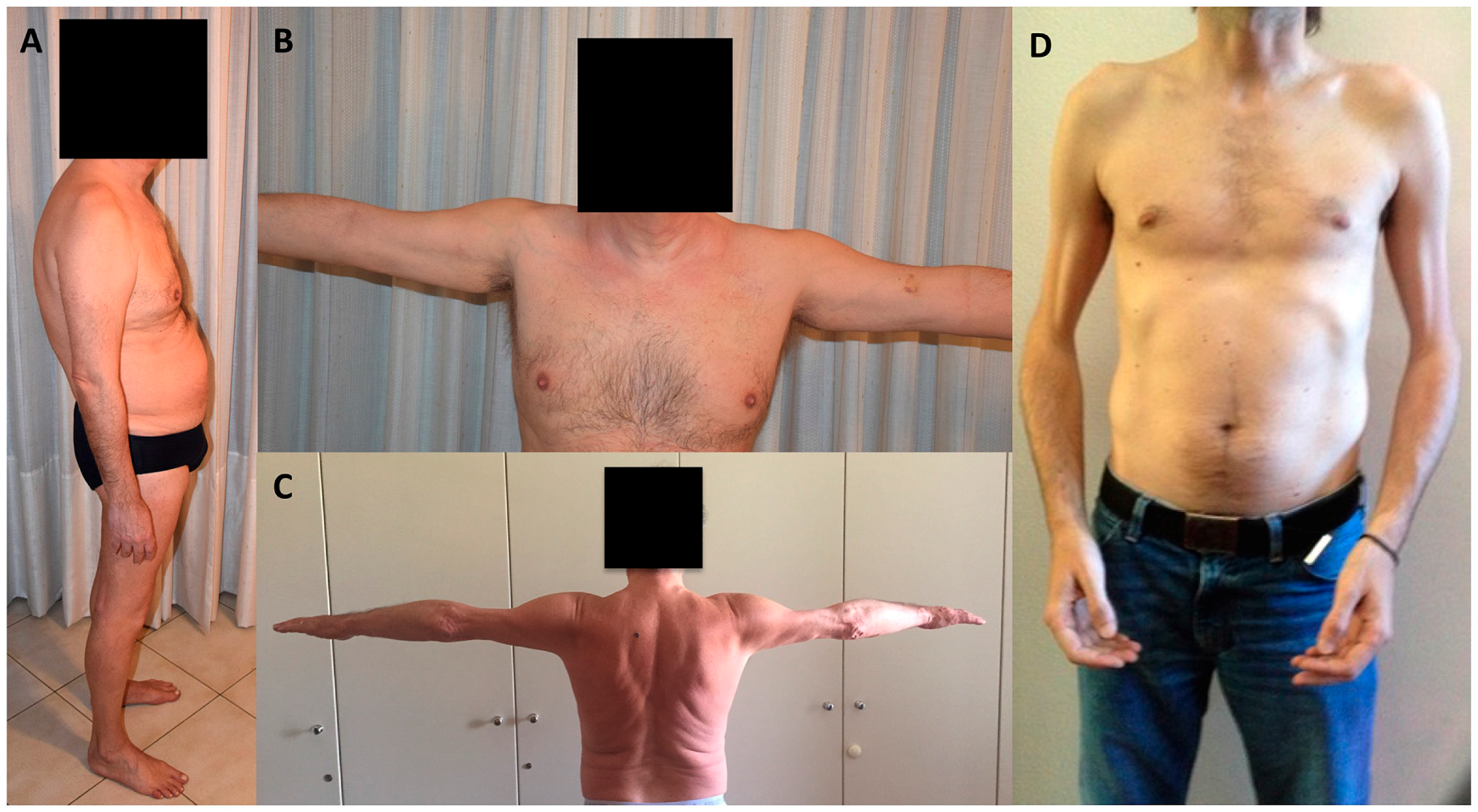

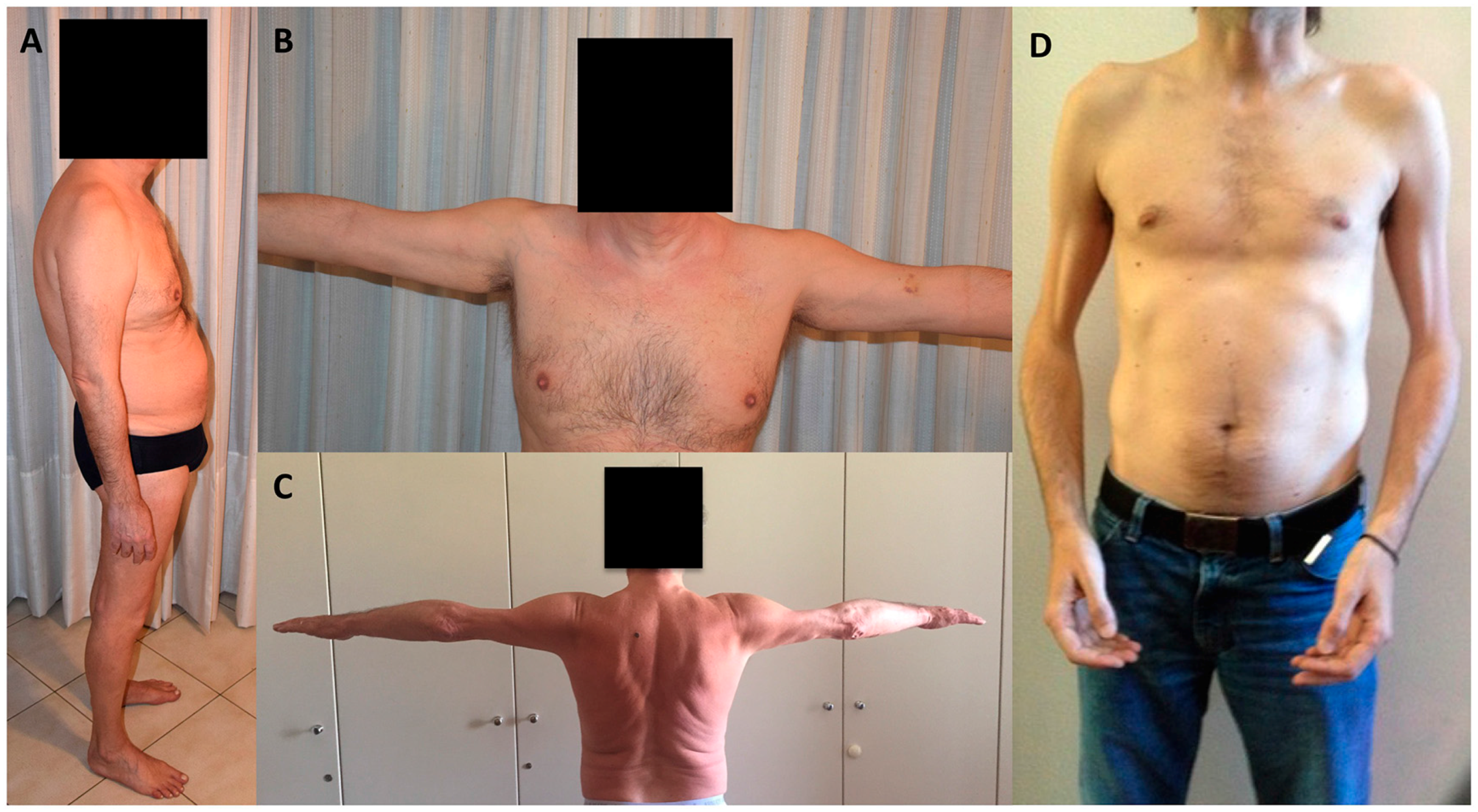{kind=link}
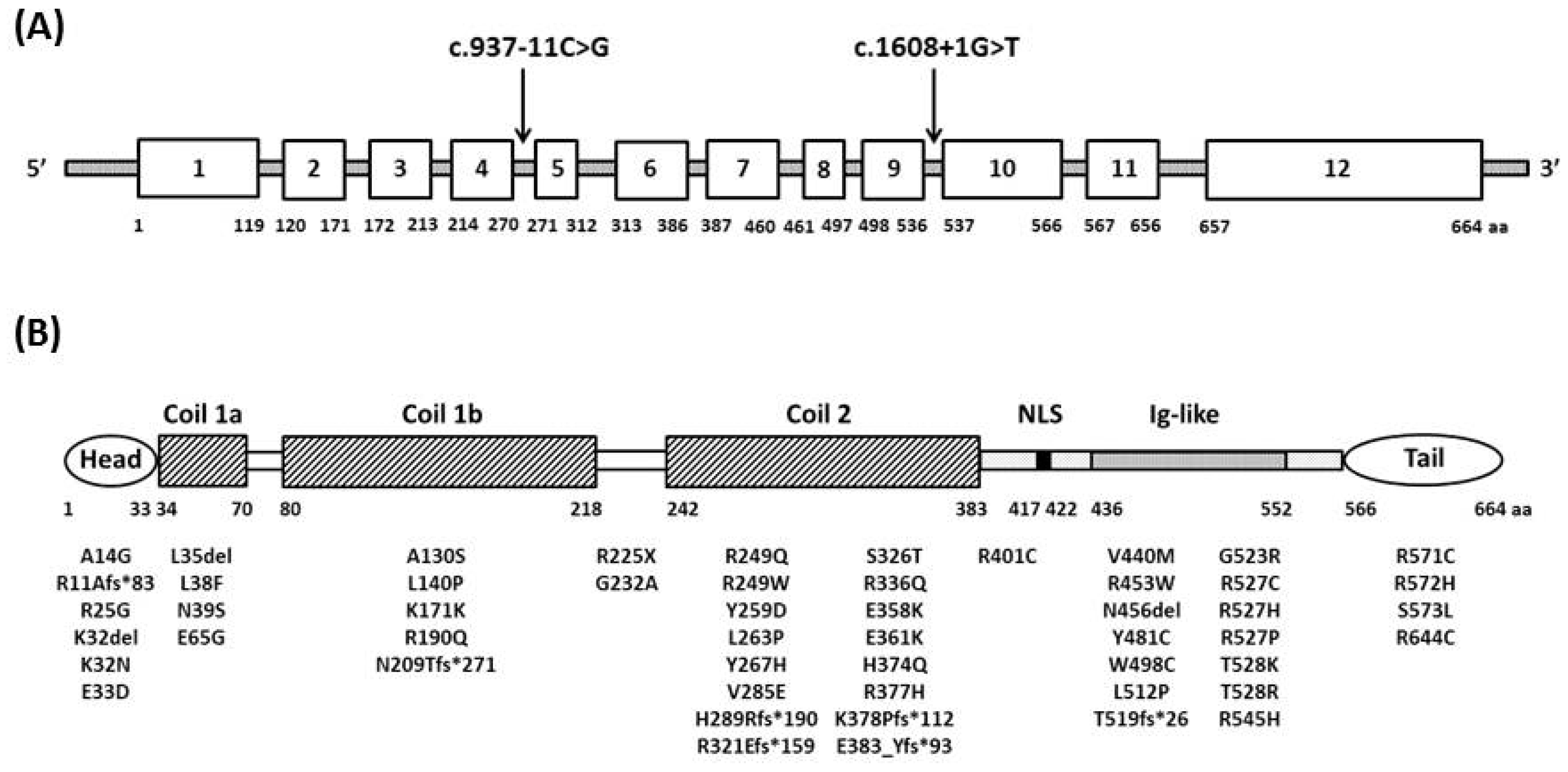
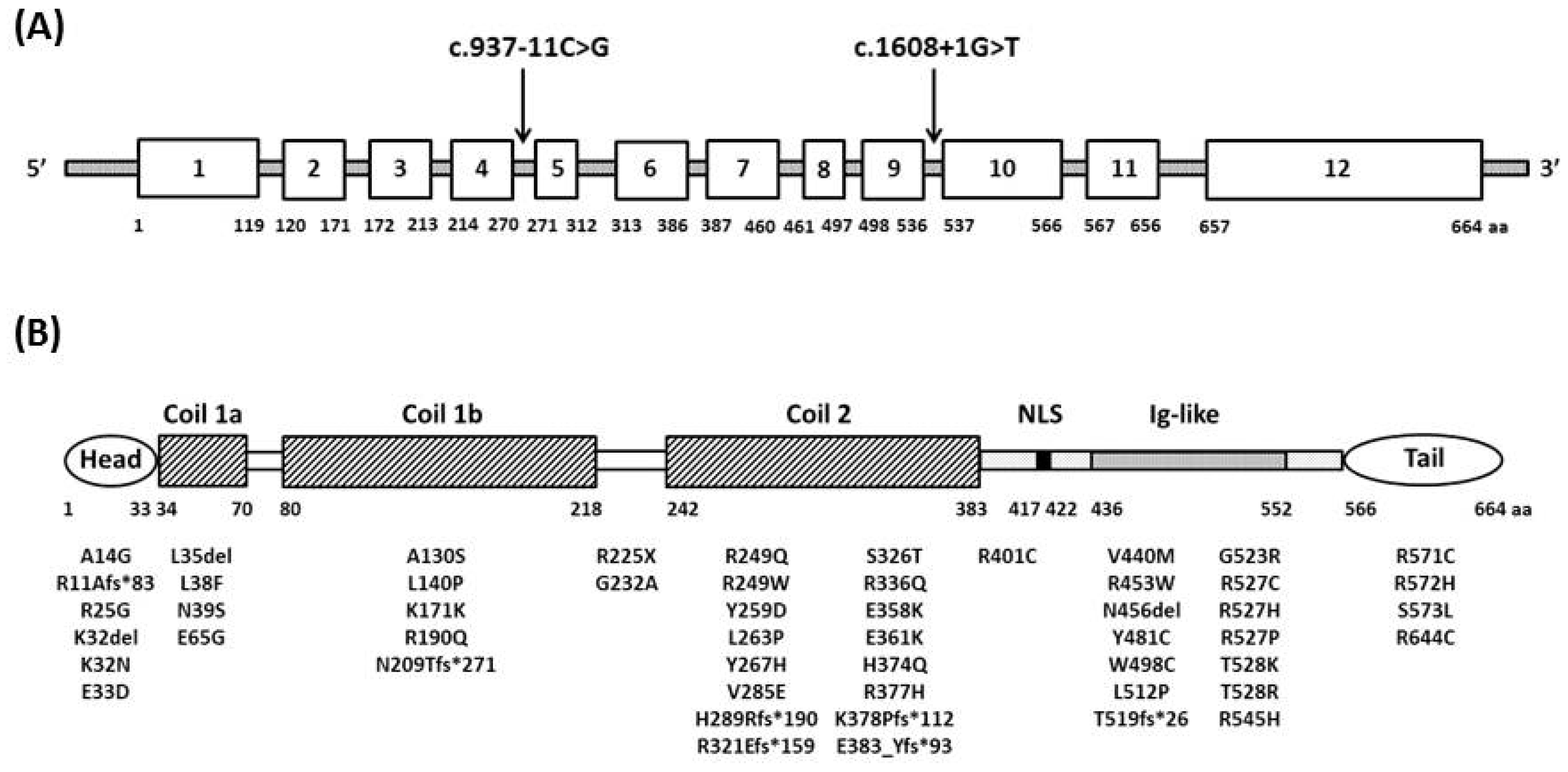{kind=link}
| Onset | EDMD2 2nd–3rd Decade | LGMD1B 3rd–4th Decade | LMNA-CMD <2 Years |
|---|---|---|---|
| Weakness distribution | scapulo/humero/peroneal | pelvic/scapular | diffuse or DHS |
| Tendon contractures | frequent, elbow quite specific | relatively frequent | frequent |
| Axial involvement | frequent | rare | frequent |
| Scoliosis | frequent | rare | frequent |
| Rigid spine | frequent | rare | frequent |
| Dysphagia | very rare | very rare | rare |
| Facial weakness | rare | very rare | rare |
| Non-autonomous ambulation | rare and late | rare and late | frequent |
| Heart involvement | almost invariably with age | almost invariably with age | relatively frequent |
| Respiratory involvement | rare | rare | relatively frequent |
| Muscle biopsy | Unspecific myopathic | unspecific myopathic | myopathic or dystrophic |
| Type of mutations | missense | frameshift | missense |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maggi, L.; Carboni, N.; Bernasconi, P. Skeletal Muscle Laminopathies: A Review of Clinical and Molecular Features. Cells 2016, 5, 33. https://doi.org/10.3390/cells5030033
Maggi L, Carboni N, Bernasconi P. Skeletal Muscle Laminopathies: A Review of Clinical and Molecular Features. Cells. 2016; 5(3):33. https://doi.org/10.3390/cells5030033
Chicago/Turabian StyleMaggi, Lorenzo, Nicola Carboni, and Pia Bernasconi. 2016. "Skeletal Muscle Laminopathies: A Review of Clinical and Molecular Features" Cells 5, no. 3: 33. https://doi.org/10.3390/cells5030033