1. Introduction
The clustered regularly interspaced short palindromic repeats (CRISPR)-associated sequence 9 (CRISPR/Cas9) system has become the tool of choice for targeted gene disruption and the analysis of gene and protein function in cultured mammalian cells [
1]. The CRISPR/Cas9 system requires the nuclease Cas9 and a guide RNA (gRNA) to introduce targeted double-strand breaks within DNA. In general, DNA double-strand breaks activate the cellular DNA non-homologous-end-joining (NHEJ) repair program, which frequently leads to the introduction of so-called indels (insertions/deletions), and hence to a frameshift mutation or an in-frame amino acid insertion or deletion. When located within the coding region of a gene, the presence of premature stop-codons can lead to the “knockout” of the gene of interest, especially when located within one of the first permanent exons.
For screening purposes, transfected cells are diluted to obtain single-cell clones whose changes within the DNA are identical and should exhibit the same phenotype, i.e., complete absence of expression of the protein of interest. It is considered as sufficient to validate the presence of a frameshift mutation in the putative knockout clone by sequencing of the genomic DNA, followed by assessment of protein expression and/or functional testing [
2]. However, not all indels necessarily lead to premature stop codons. They might also interfere with hnRNA splicing if sequences that regulate splicing are altered. Therefore, a single indel mutation might theoretically give rise to different mRNA products within a clonal cell population. This would, instead of a complete protein knockout, result in the production of altered proteins with unpredictable functional consequences, as these proteins might, for example, gain dominant negative functions.
Flotillin-1 and -2 are ubiquitously expressed, highly conserved proteins that localize into specific cholesterol-rich microdomains (membrane rafts). Functionally, flotillins are associated with various signaling pathways and have been found to be overexpressed in many types of cancer (reviewed in [
3]). Flotillin-1 is an important regulator of MAP kinase (mitogen-activated protein kinase) signaling, and it interacts with several components of the MAPK pathway [
4]. Thus, flotillin-1 is required for a proper EGFR activation and downstream signaling [
5]. Interestingly, flotillins not only regulate MAP kinase signaling, but they themselves are target genes of EGFR/MAP kinase mediated transcriptional regulation [
6,
7]. The flotillin-1 gene is located on chromosome 6 and contains 13 exons, encoding a protein with 427 residues [
8].
Until recently, we have investigated the function of flotillins using siRNA or shRNA mediated knockdown. However, these methods have the disadvantage that the cells still show a varying degree of residual flotillin expression that may mask the functional consequences of loss of flotillin expression [
5,
9,
10]. In the present study, we have used the CRISPR/Cas9 method to knock out the
FLOT1 gene in HeLa cells to obtain full genetic ablation of flotillin-1 expression. Analysis of the obtained single-cell clones by Western Blot revealed a successful
FLOT1 knockout, and the genomic changes observed were indel mutations, as expected. However, we also analyzed the consequences of the genomic changes at the mRNA level. We here show that minor genomic indel mutations caused by gRNA-mediated knockout may result in radically altered or randomized exon splicing of the respective hnRNA. These mRNA products in turn may bear the risk of giving rise to functionally altered protein products. Thus, the results of our study stress the importance of analysis of the consequences also at the mRNA and translational level.
3. Results
Flotillin-1 knockout HeLa cells were produced by transfecting the cells with gRNA CRISPR/Cas9 plasmids targeting the human
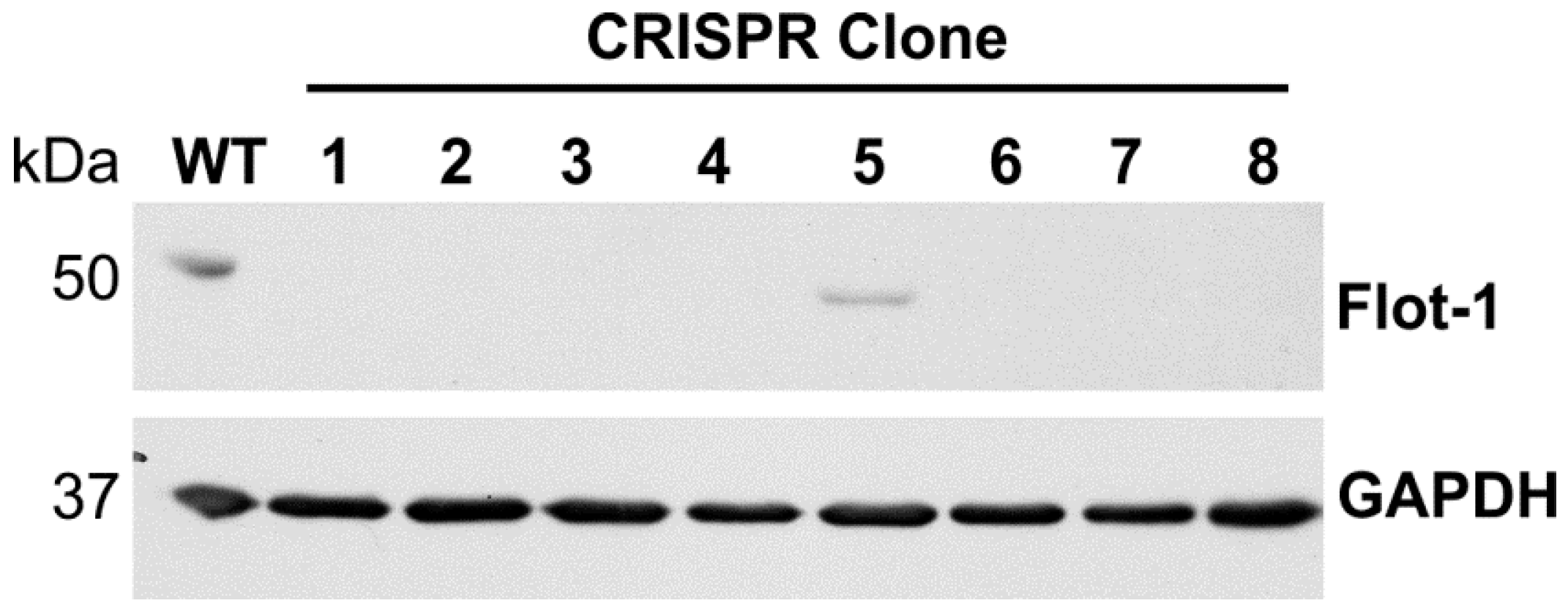
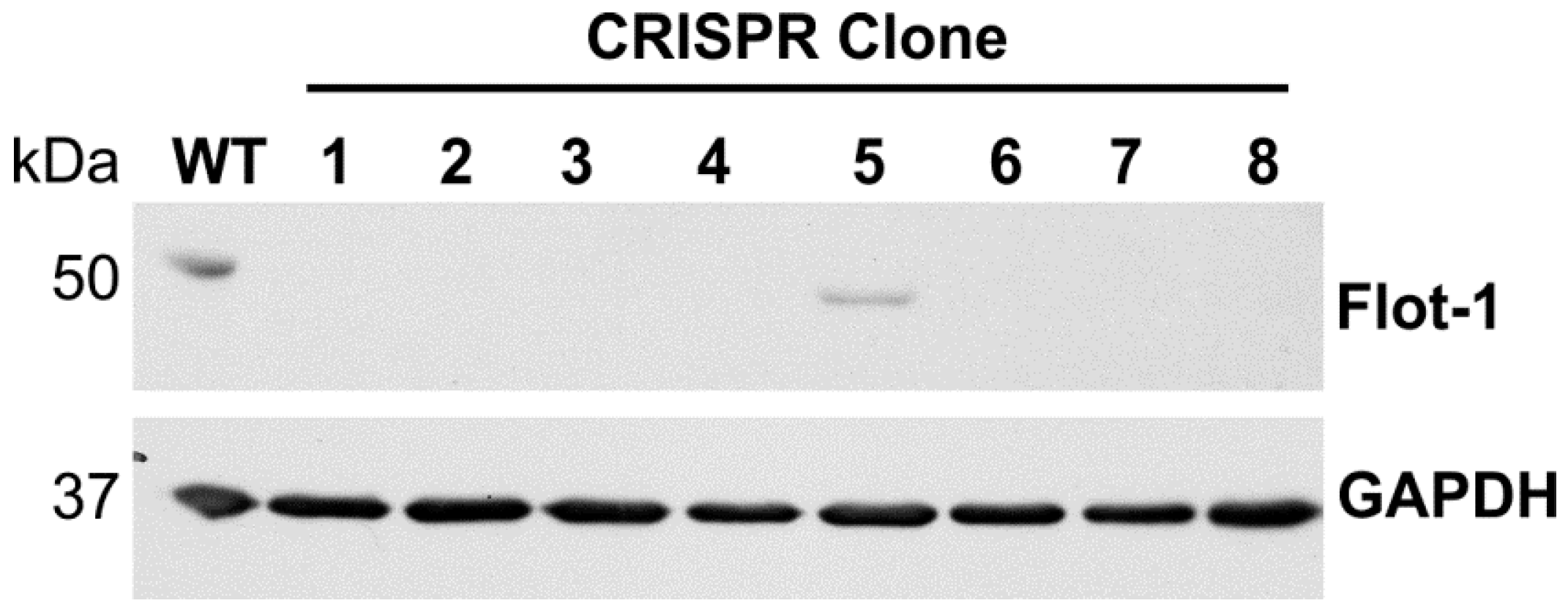
FLOT1 gene. Five gRNA sequences were tested (data not shown), of which gRNA 165723 showed superior knockout efficiency and was chosen for further analysis. HeLa cells were transfected with this gRNA, and single-cell clones originating from two separate experiments were selected by limiting dilution. The clones were tested by means of Western Blot (
Figure 1), and clones showing a complete lack of detectable full-length flotillin-1 protein were expanded and cultured further.
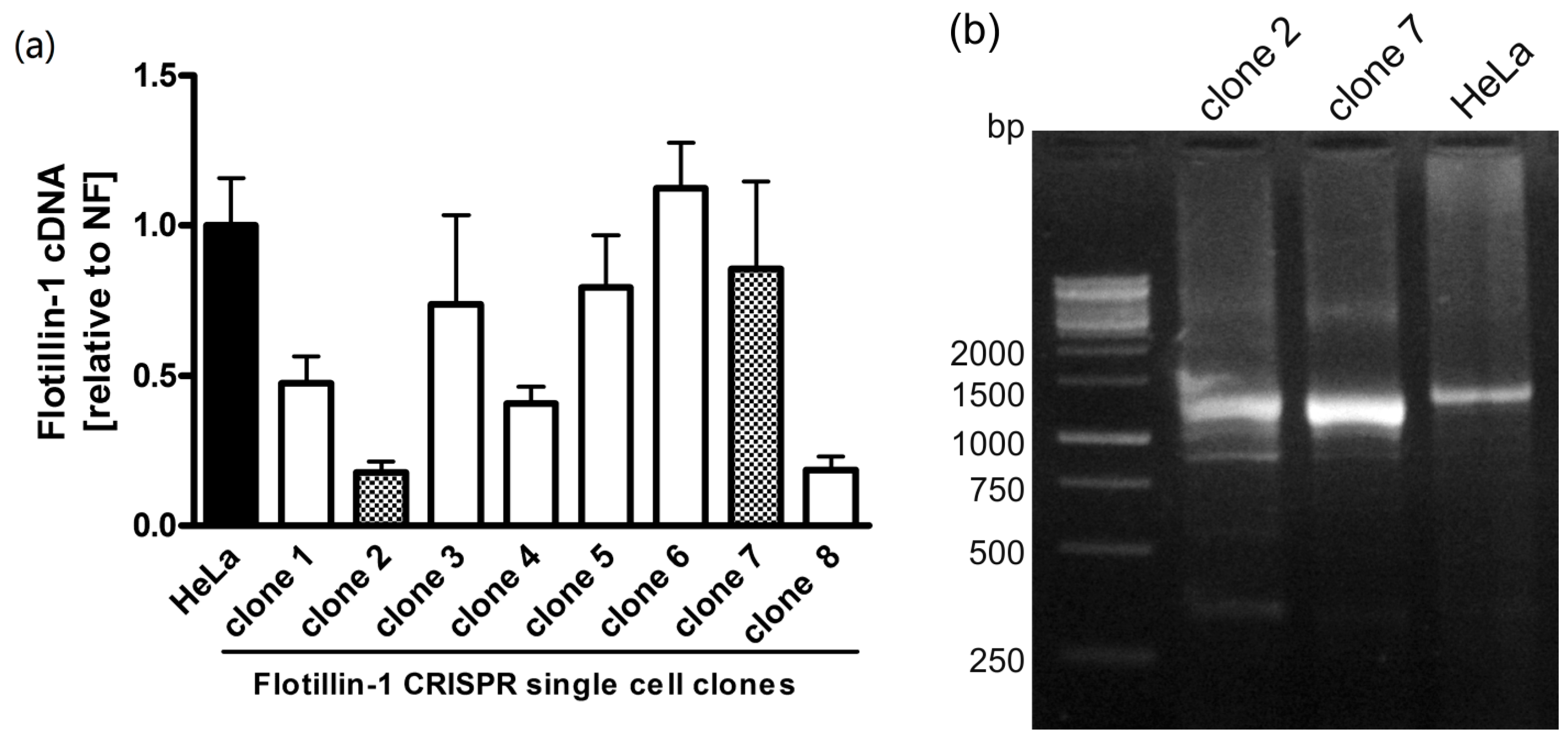
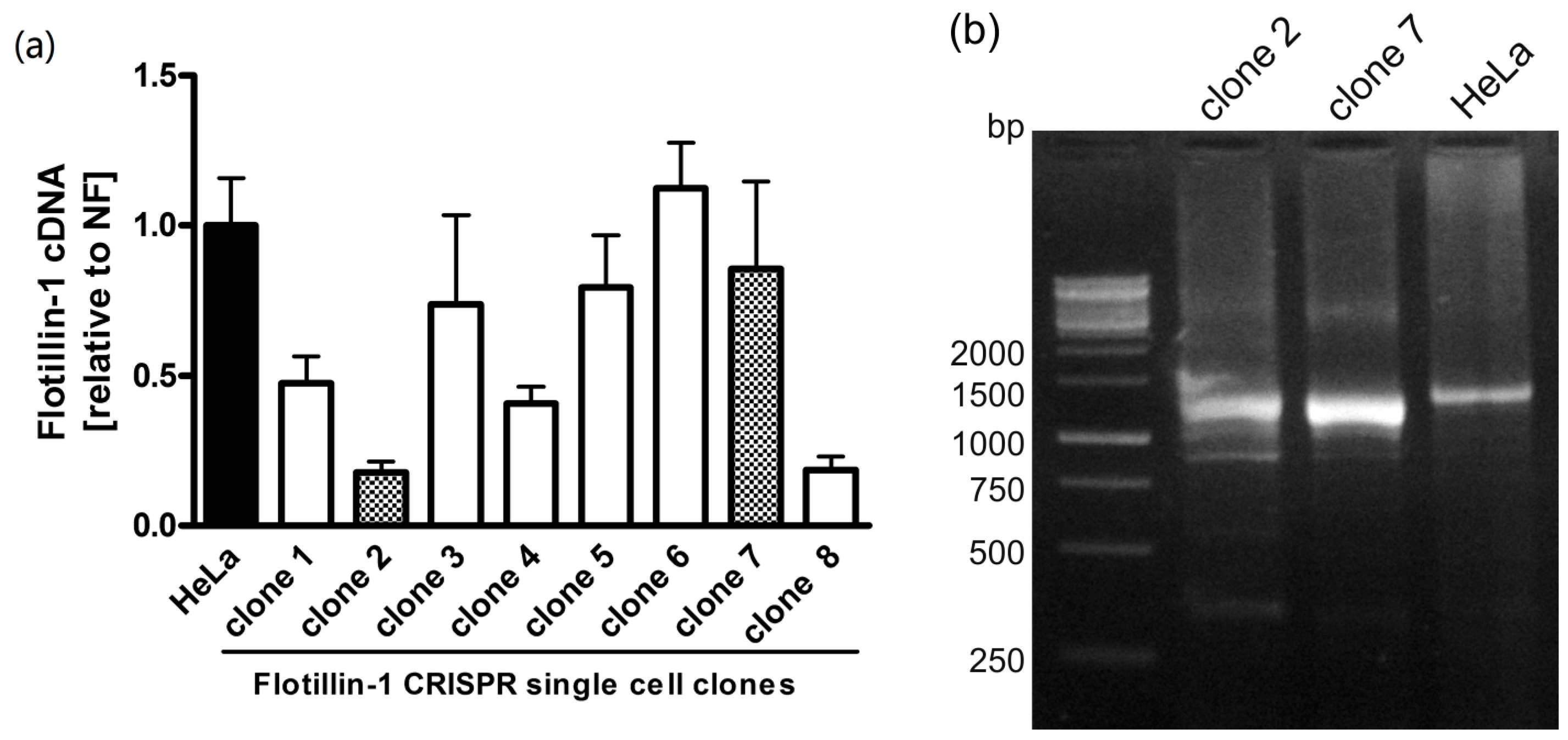
To check the effect of
FLOT1 gene knockout on the abundance of the respective mRNA, qPCR was performed. As shown in
Figure 2, the single-cell clones showed varying amounts of flotillin-1 mRNA, ranging from wild-type levels to about 20%.
To analyze the consequences of gRNA-mediated
FLOT1 knockout at genomic and mRNA level in detail, we chose clones 2 and 7, which originate from separate transfections, for further analysis. The
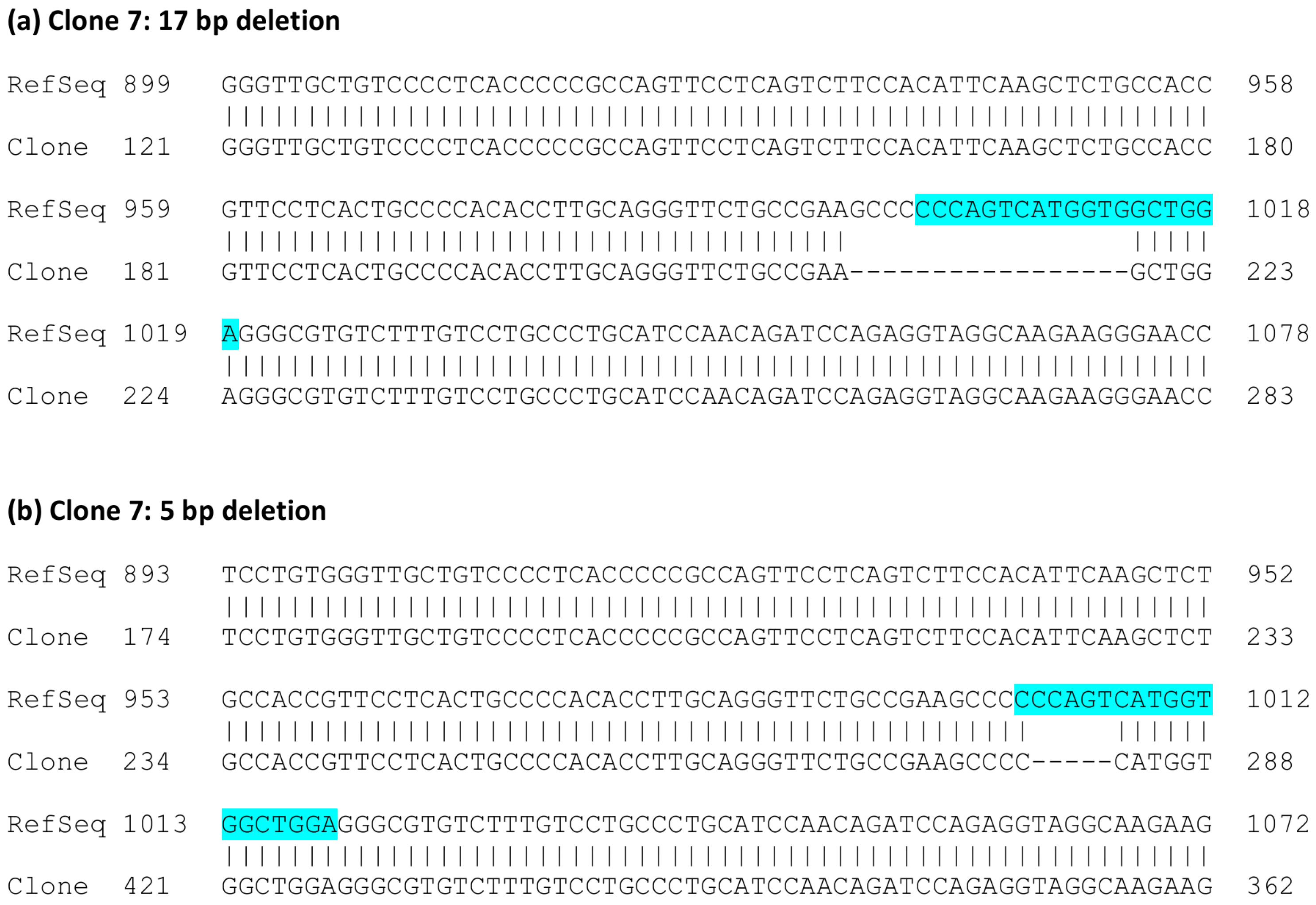
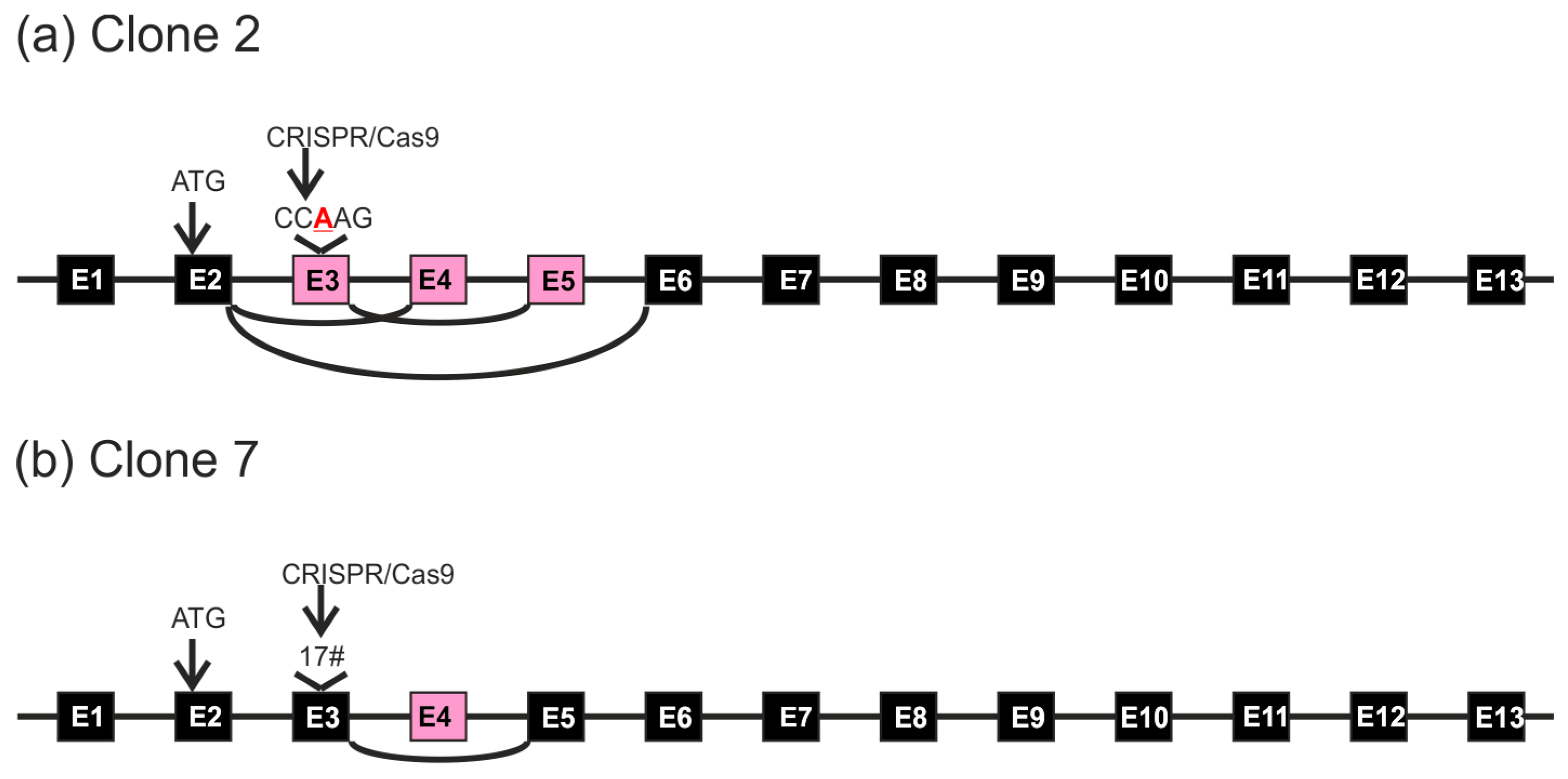
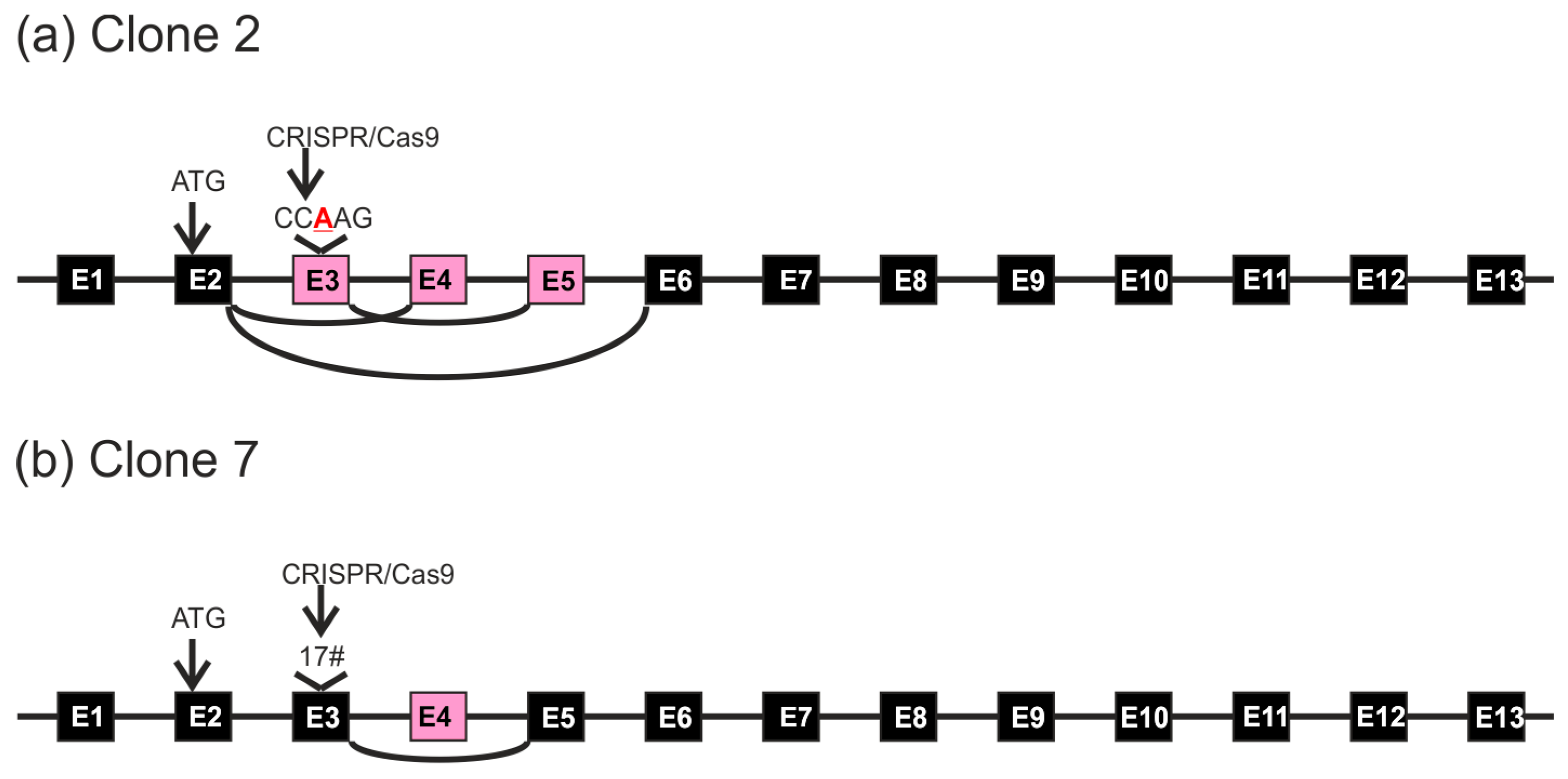
FLOT1 gene resides on human chromosome 6. Since HeLa cells are polyploid and exhibit three copies of chromosome 6, direct genomic sequencing was not feasible. Thus, the genomic region around the targeted exon 3 was PCR amplified with primers that reside within the flanking introns and contain suitable restriction sites, and the resulting PCR products were cloned into a plasmid and sequenced. Sequence analysis of several genomic clones (
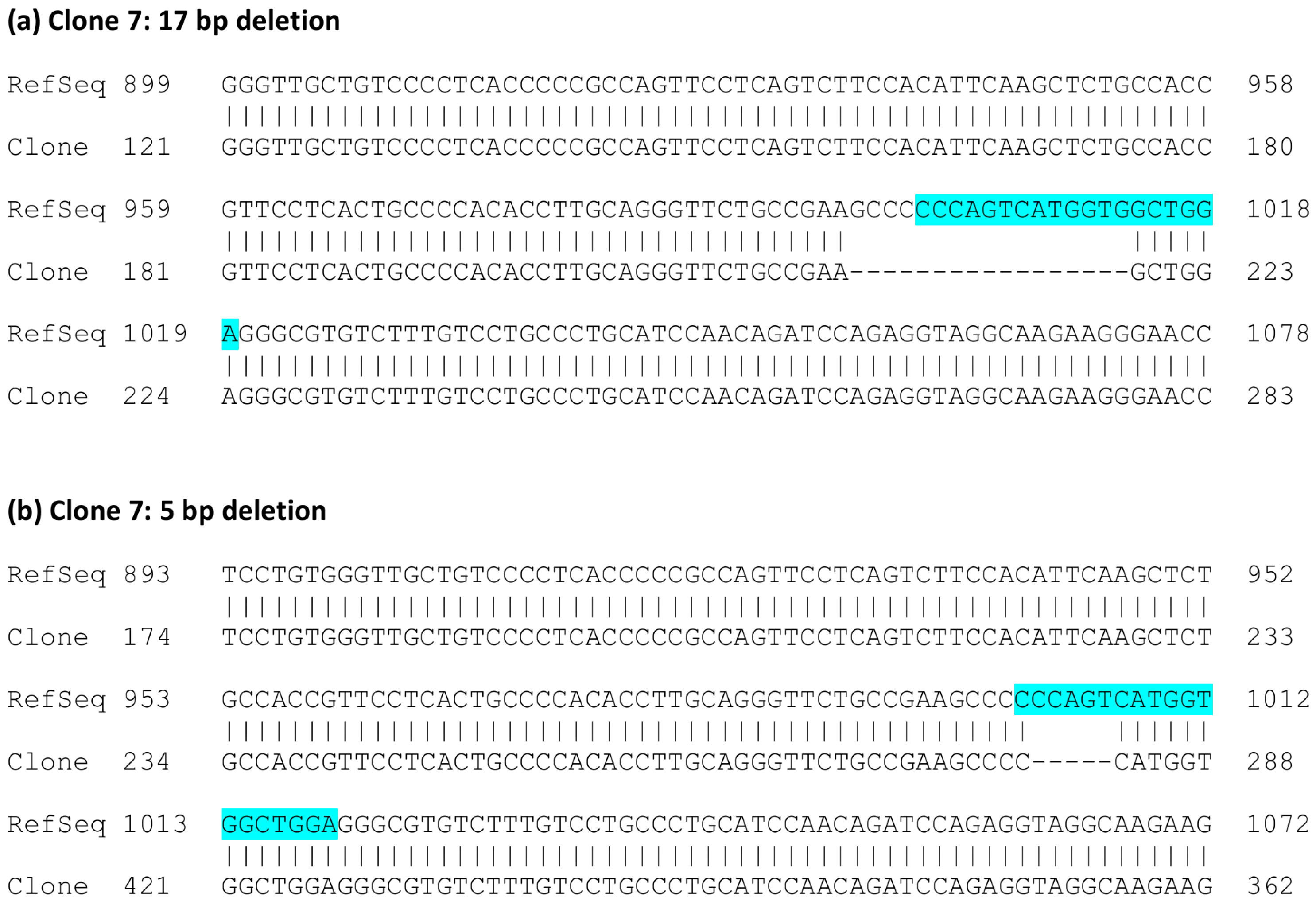
Figure 3) showed that clone 2 always exhibited the same alteration: an insertion of an extra base A in exon 3. The analyzed sequence also covers the flanking intron borders on both sides of exon 3. In the case of clone 7, deletions of 5 and 17 bp residing within the gRNA target sequence were observed (
Figure 4). No other sequence variants within the analyzed region were identified.
To study the effects at the mRNA level, cDNA was prepared from total RNA of WT and knockout HeLa cells and PCR amplified using primers specific for the coding region of flotillin-1 (
Figure 2b). The resulting fragments were cloned into a plasmid. The mRNA sequences obtained from the WT HeLa cells are identical to the
FLOT1 reference sequence (
Figure 5a). In the case of HeLa clone 2, we were expecting on the basis of the genomic alteration (insertion of an A) to detect mRNA clones that show the insertion and thus result in an early frame-shift and non-existent flotillin-1 protein. However, clones corresponding to such an mRNA species were never observed. Instead, all sequenced clones corresponded to mRNA species that had undergone differential splicing of exons 3–5 (
Figure 5).
Figure 5a shows the wild type flotillin-1 cDNA sequence with color coding of the exons. In several clones, exon 3 contained the expected A insertion, corresponding to the genomic defect, together with a deletion of exon 4 (
Figure 5b). In addition, clones deleted for exons 3, 4 and 5 (
Figure 5c) or exon 3 (
Figure 5d) were observed. These data suggest that the single nucleotide insertion within exon 3 profoundly alters the splicing of several exons in
FLOT1 mRNA. However, the splicing events appear to take place at the correct exon-intron borders.
The HeLa knockout clone 7, which contained genomic deletions within the gRNA target sequence, exhibited cDNA clones corresponding to mRNA species with the observed 17 bp deletion combined with a deletion of exon 4 (
Figure 5e). However, cDNA clones exhibiting the 5 bp deletion were never observed, implicating that such mRNA species may be unstable.
Since it is possible that such aberrant splicing might be specific for flotillin-1 or HeLa cells, we analyzed an independent gene, human
AGA (aspartylglucosaminidase) which was knocked out in HEK 293T cells. Similarly to CRISPR/Cas9 mediated flotillin-1 knockout, a gRNA residing in
AGA exon 6 resulted in altered splicing patterns of exons 2–6 (
Supplementary Materials Figure S1). These data show that the aberrant splicing induced by gRNAs is neither flotillin nor cell line specific.
To check the possible consequences of the observed splicing errors at the protein level, the cDNA sequences were translated
in silico.
Figure 6a shows the full-length human flotillin-1 protein sequence. Deletions of exons 3–5 or exon 3 result in an early stop codon after less than 20 residues from the N-terminus (
Figure 6b,c). These changes thus do not produce any functional protein product and can be expected to be true knockouts.
In contrast to the above mentioned errors, the insertion of one base (A) within exon 3 combined with exon 4 removal is predicted to produce a protein product that contains 21 amino terminal residues of flotillin-1, followed by 21 random amino acids and then again flotillin-1 protein sequence starting from residue 71 (
Figure 7a). This is due to the fact that although the insertion of a single A in exon 3 produces a frame shift, this is again compensated by the removal of exon 4, so that the reading frame from exon 5 onwards is corrected. Interestingly, also the 17 base deletion combined with exon 4 removal should produce a similarly altered protein product: 19 amino terminal residues of flotillin-1, 15 random ones, followed by the flotillin-1 sequence starting with residue 71 (
Figure 7b). These data suggest that these mRNA species could give rise to truncated proteins that may even act in a dominant negative fashion.
We performed a similar analysis for the mRNA species detected in
AGA gene knockout cells (
Supplementary Materials Figure S2). Also in this case, the splicing defect removing exons 2–6 resulted in an early STOP codon and a severely truncated protein. However, removal of exons 3–6, which does not result in a frameshift, could produce a truncated protein product in which residues 95–232 are missing. This change would be predicted to result in a truncated protein that is most likely highly unstable, in accordance with the effects described for
AGA gene mutations that result in aspartylglucosaminuria [
12].
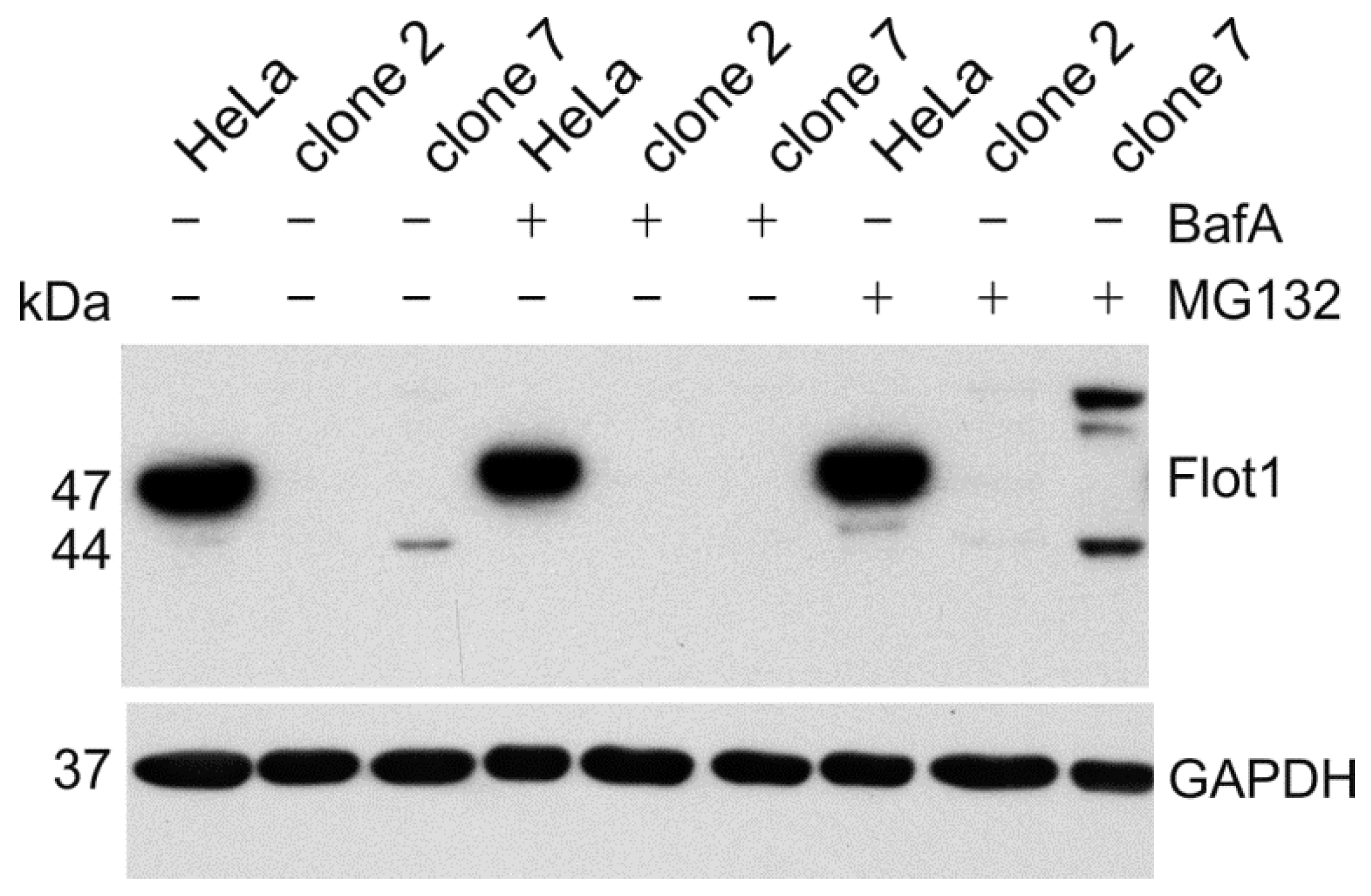
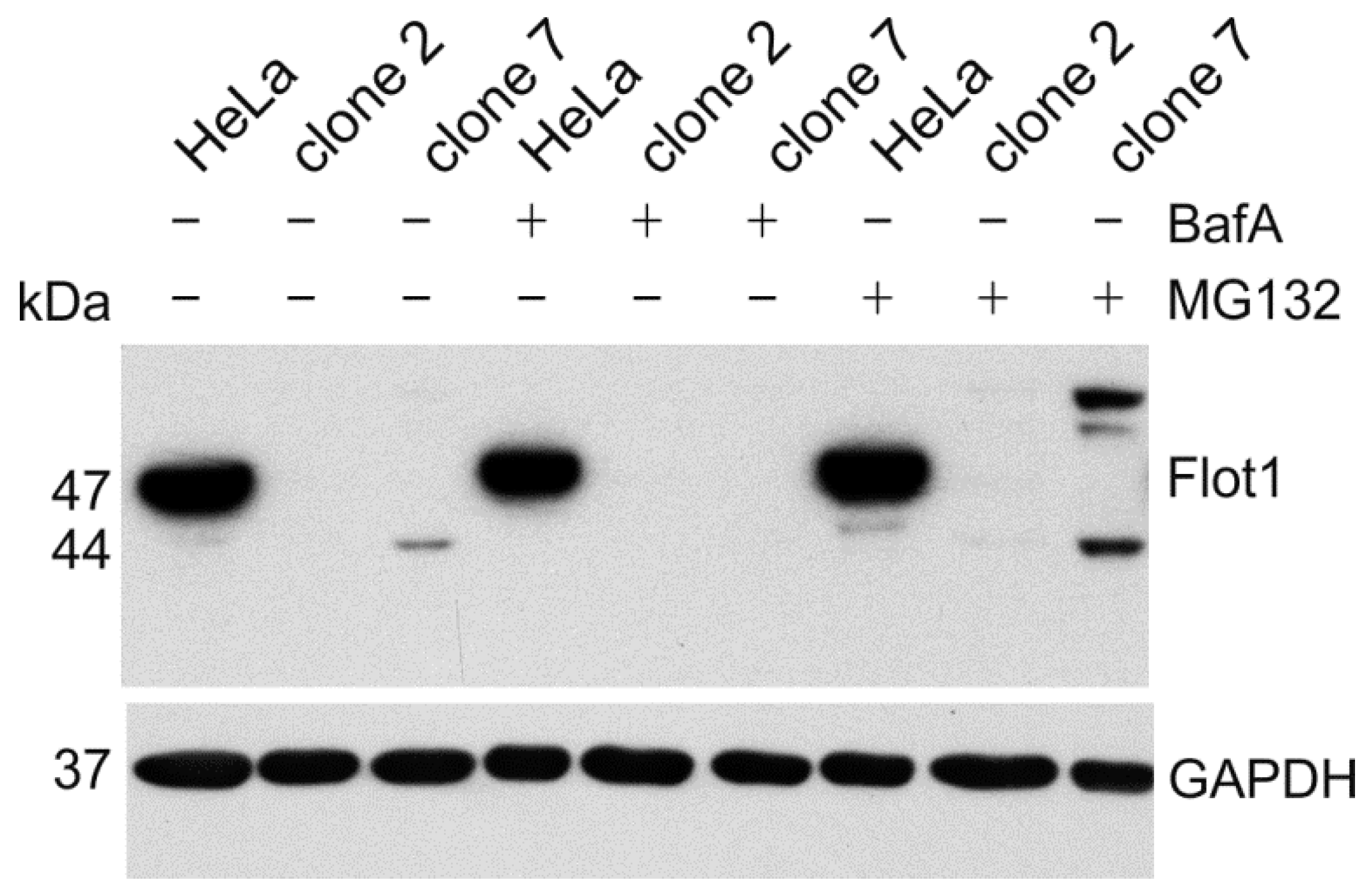
To demonstrate that the aberrant flotillin-1 protein products are expressed, we performed Western Blot analysis of the cell lysates (
Figure 8). Since the aberrant proteins may be unstable, we used inhibitors of lysosomal acidification (bafilomycin A) or the proteasome (MG132) to inhibit protein degradation. In clone 2, we were not able to detect any protein fragments for flotillin-1. However, in clone 7, we observed after a prolonged exposure of the blot a truncated flotillin-1 protein with a molecular mass of about 44 kDa, which is well in accordance with the calculated 43.5 kDa for the aberrant protein depicted in
Figure 7b. In addition, a protein product with a higher molecular mass than the WT flotillin-1 protein was observed. This signal probably originates from the ubiquitinated protein that remains undegraded upon inhibition of the proteasome. Unfortunately, a similar analysis could not be performed for the
AGA knockouts, as none of the available antibodies is sensitive enough to detect endogenous AGA protein fragments if they are poorly expressed.
4. Discussion
The CRISPR/Cas9 method and its variations have in the past years developed into a widely-used tool that facilitates targeted genome editing in mammalian cells. Not only gene knockout but also targeted correction of, for example, disease mutations can be accomplished [
13,
14,
15,
16,
17], and high hopes have been raised concerning the therapeutic use of these methods. In most cases, researches control the success of the targeted gene ablation by sequencing the genomic region around the gRNA target site and by demonstrating the lack of protein expression by means of Western Blot. However, only very rarely are the consequences of the gRNA-mediated approach studied at the mRNA level. We here show, using the human
FLOT1 and
AGA genes as examples, that very benign genomic changes may have heterogeneous consequences for the splicing of the respective hnRNA product, giving rise to alternative splicing products that may produce dysfunctional proteins instead of resulting in a complete gene knockout.
In this study, we intended to knock out the expression of flotillin-1 in HeLa cells. Our analysis at the genomic level revealed expected indel alterations in the targeted exon number 3 of the
FLOT1 gene. Western Blot analysis revealed the absence of a band at the right molecular weight position in several single-cell clones originating from different transfections. However, qPCR demonstrated that a varying percentage of flotillin-1 mRNA was still detectable in the cell clones. This prompted us to carry out an analysis of the flotillin-1 mRNA species in these cell clones. A summary of the genomic changes vs. mRNA alterations is provided for
FLOT1 gene in
Figure 9. Although the respective genomic indel mutations were present in the mRNA species, they were frequently coupled with altered splicing of the nearby exons. In the mRNA species containing a single nucleotide insertion in exon 3, splicing appeared to have occurred at the normal intron-exon junctions, consistent with the unaltered consensus splice sites. However, the inclusion or exclusion of exons 3–5 apparently took place in a random manner, and we cannot exclude that yet further mRNA species may arise due to this genomic change. On the other hand, a 17 bp deletion in exon 3 was consistently associated with the lack of exon 4, implicating that different genomic alterations produced by the same gRNA can alter the splicing in various ways. Similar aberrant splicing events were detected upon knockout of the human
AGA gene in HEK cells, demonstrating that the aberrant splicing may be a general consequence of CRISPR/Cas9-mediated genomic alterations.
There is no evidence from our own studies or in the literature that alternative splicing would be a typical feature of the
FLOT1 gene, and only a single protein product is detected in all cells we have studied so far. The only genomic alterations we detected after gRNA-mediated knockout of
FLOT1 gene in HeLa cells were associated with the target exon 3. In general, splicing of hnRNA and inclusion or exclusion of exons is frequently regulated by short, conserved sequences within the exons, known as ESE (exonic splicing enhancers) and ESS (exonic splicing silencers) that provide binding sites for splice regulator proteins [
18]. Mutations that alter such ESE or ESS sites are also known to cause various diseases, such as Becker muscular dystrophy or X-linked Parkinsonism with spasticity, by altering the splicing of the respective hnRNA [
19,
20,
21]. Therefore, it is possible that an ESE or ESS was altered by the insertion of an A in the exon 3 in
FLOT1 gene, resulting in a seemingly random inclusion of exon 3. However, why the splicing of exons 4 and 5 is also altered is not clear, since these exons exhibited no sequence alterations. It is possible that especially exon 5 may be a cassette exon that is removed upon changes in splicing of the previous exons.
Prediction of the protein products that could arise from the altered mRNA species revealed that in many cases, a frame shift due to altered splicing resulted in an early stop codon and absence of any protein product. These findings in clone 2 were also consistent with the observed reduction of the mRNA level (to about 20%), as premature stop codons frequently result in mRNA decay [
22]. Thus, these altered splicing products are expected to produce a true knockout of flotillin-1 expression. However, we also observed mRNA species that were predicted to give rise to an altered protein product that would contain most of the flotillin-1 protein sequence, with an aberrant sequence included in its near-N-terminal region. However, since the mRNA levels in this clone are highly reduced, we were not able to detect such protein products by Western Blot. In clone 7, the observed mRNA species would be expected to produce an altered protein with a molecular mass of about 44 kDa, as compared to the 47 kDa WT protein. Since the mRNA level was only slightly reduced (to about 80%), we were indeed able to detect the respective aberrant protein product by Western Blot. However, this aberrant protein appears to become rapidly degraded, since proteasome inhibitors resulted in an increase in the amount of this product. Furthermore, a protein product with a higher molecular mass than the WT flotillin-1 was also detected, probably representing the ubiquitinated form. The rapid degradation of the aberrant product may be due to the fact that it is missing the palmitoylation sequence in Cys34 that is important for the membrane association of flotillin-1 [
23,
24]. Nevertheless, such an aberrant protein, even if it is short-lived, might exhibit dominant negative effects that obscure the interpretation of the functional consequences of the gene ablation.
Although most indel mutations are likely to produce a true knockout, we have here shown that at least in some cases, they may result in altered splicing and even expression of an aberrant protein. Therefore, the results of our study should encourage researchers who are using the CRISPR-mediated genome editing to study the consequences also at the mRNA level. Such an analysis will help to select cell clones that are true knockouts, which will give more reliable data when analyzing the functional consequences of the targeted gene ablation.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}