
Application of Induced Pluripotent Stem Cell Technology to the Study of Hematological Diseases


Abstract
:1. Introduction
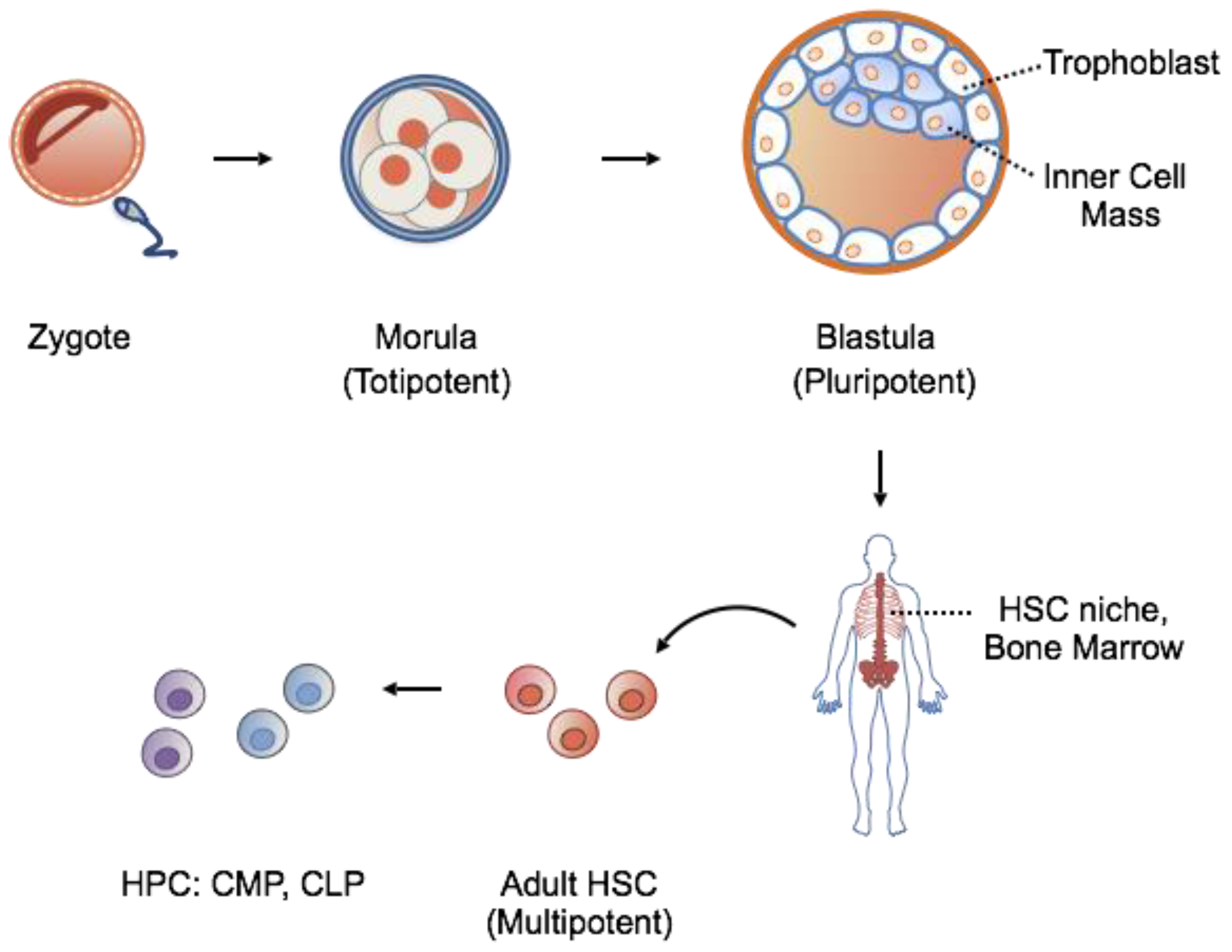
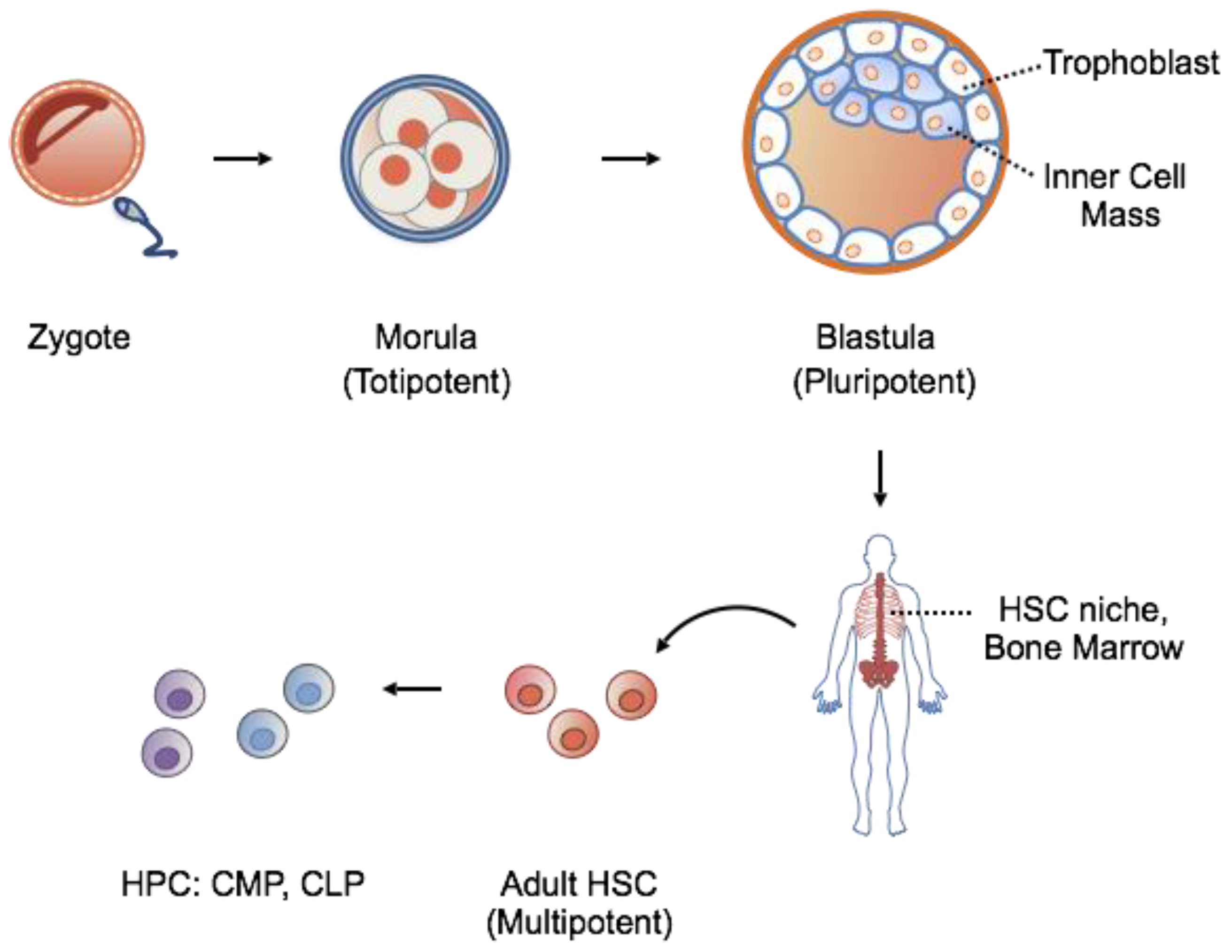
1.1. Stem Cells: Features
1.2. Stem Cells: Source for Potential Therapy
2. Induced Pluripotent Stem Cells: A Novel Solution to an Old Problem
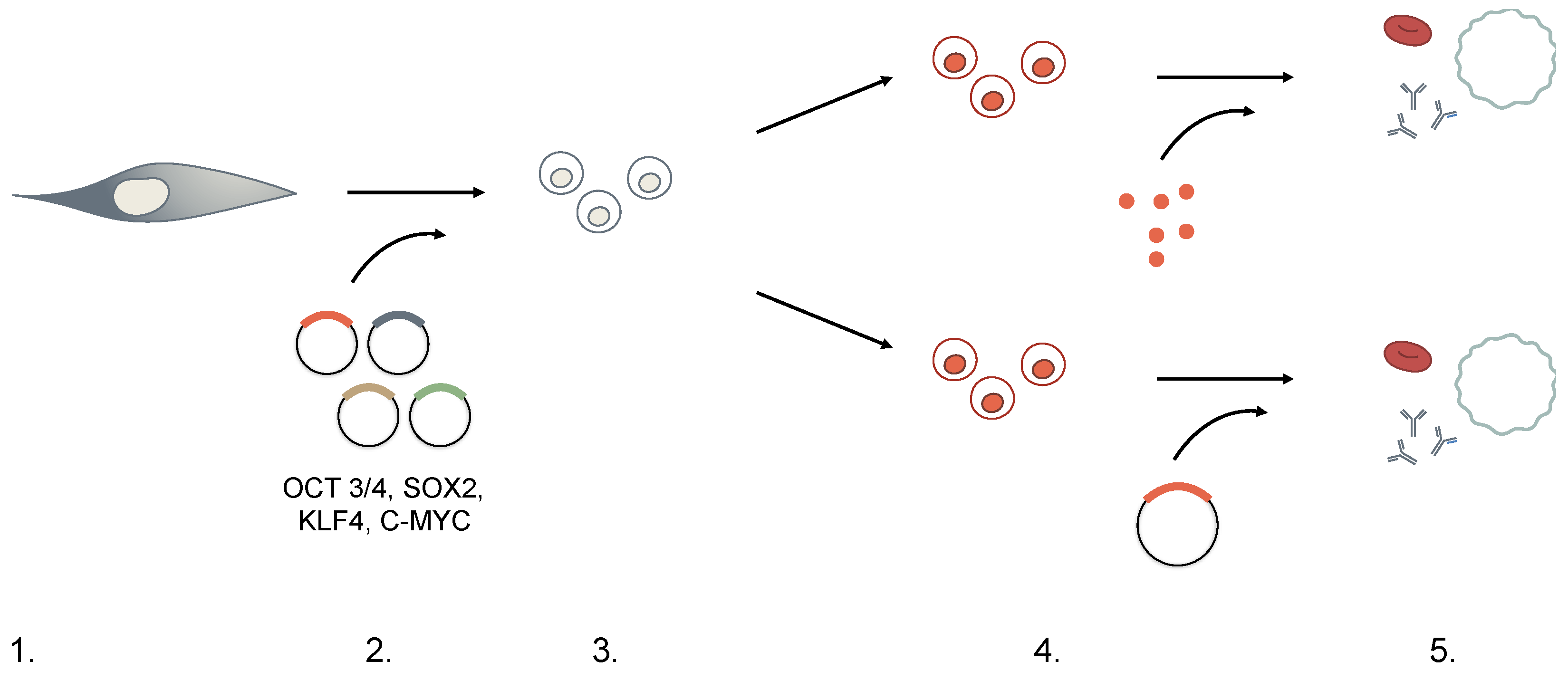
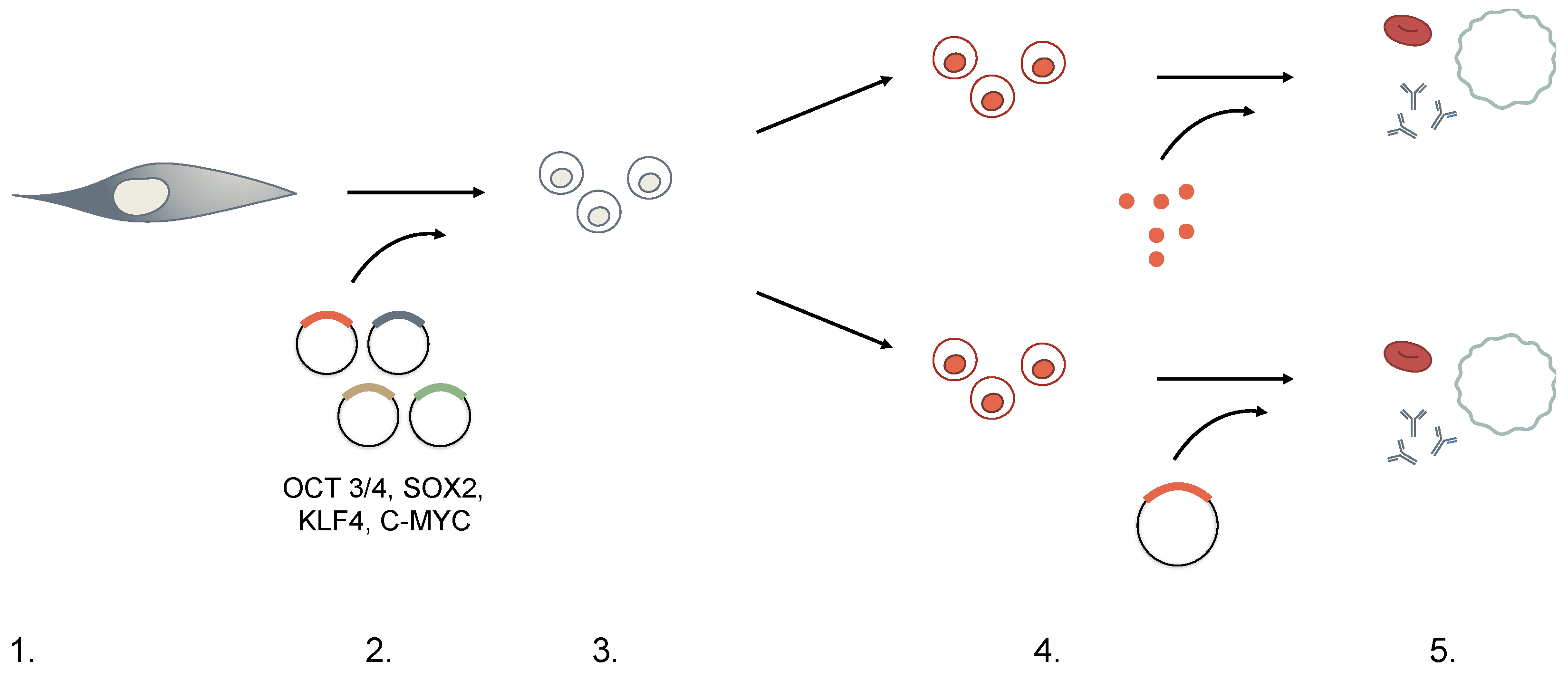
3. Generation of iPSCs
3.1. Moloney-Based Retrovirus
3.2. HIV-Based Lentivirus
3.3. Transient Transfection and Adenovirus
3.4. Small Molecules
3.5. Protein Transduction
3.6. Genome Editing
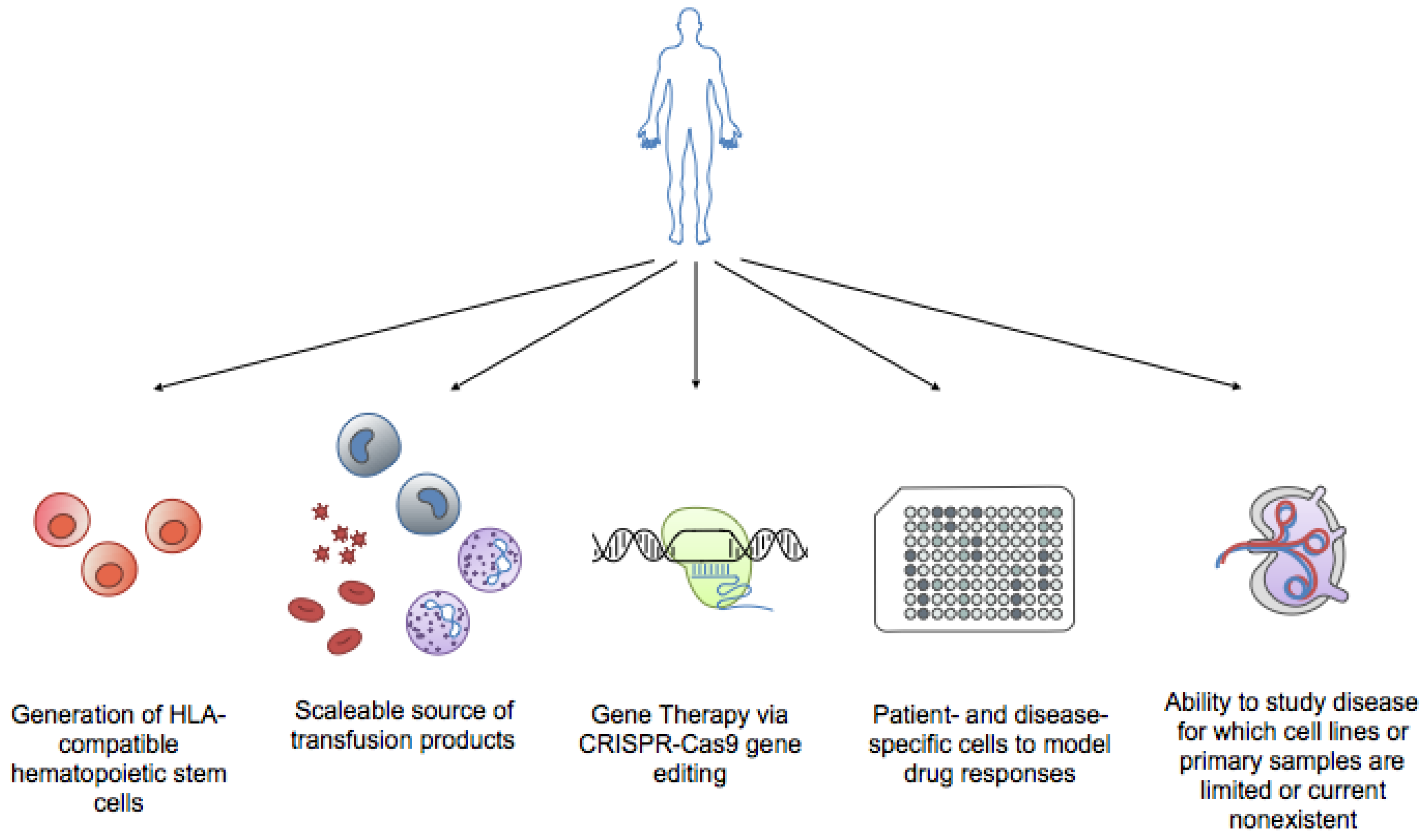
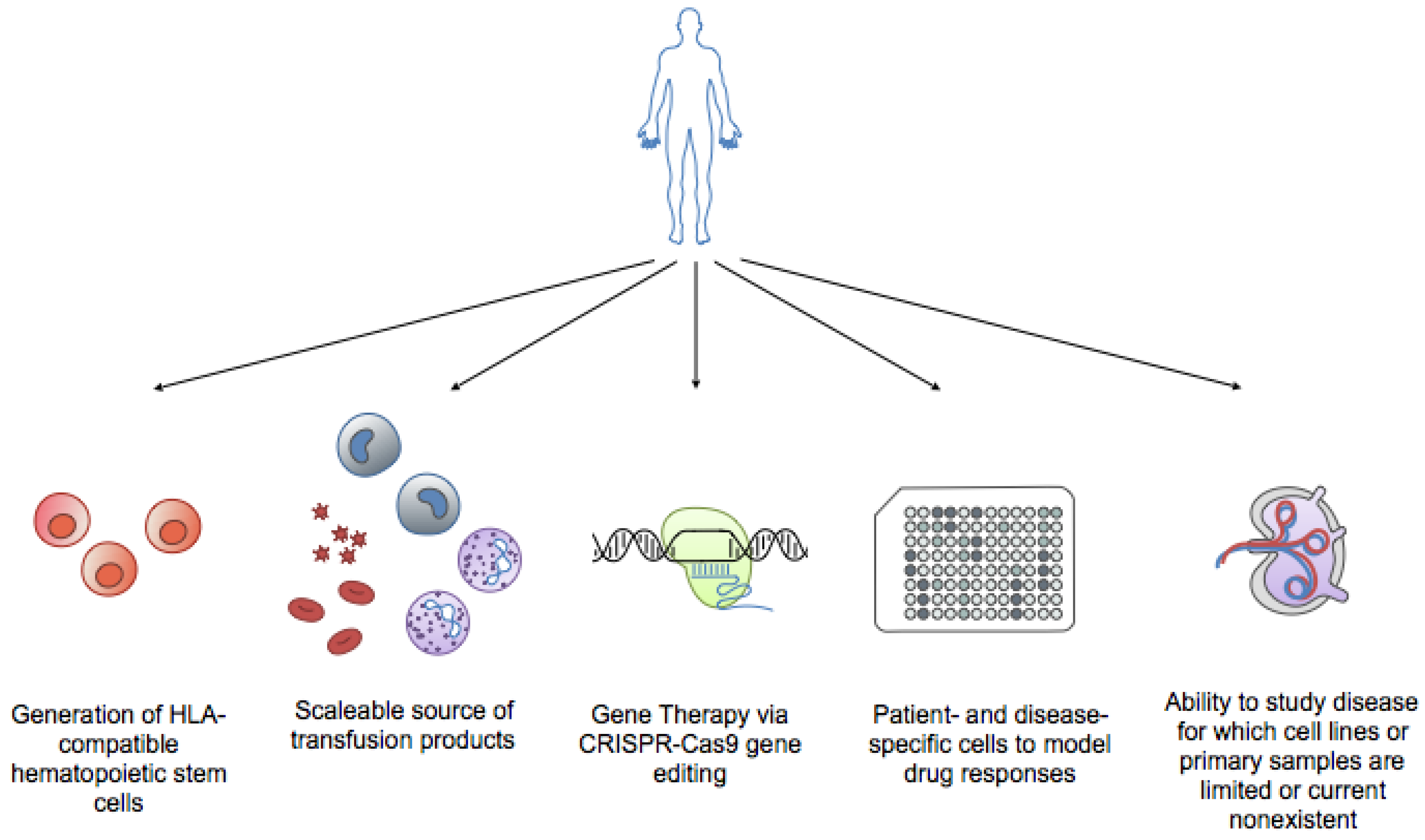
4. iPSCs and Hematopoiesis: Are We There Yet?
4.1. Red Blood Cells/Platelets
4.2. Neutrophils and Monocytes/Macrophages
4.3. Dendritic cells and Natural Killer (NK) Cells
4.4. T and B Lymphocytes
4.5. Hematopoietic Stem Cells
5. iPSCs Hematological Disorders
5.1. iPSCs in Congenital Hematopoietic Diseases
5.1.1. Fanconia Anemia
5.1.2. β-Thalassemia
5.2. Generation of iPSCs from Hematologic Malignancy
5.2.1. Myeloproliferative Neoplasms
5.2.2. Myelodysplastic Syndrome
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Gluckman, E.; Rocha, V.; Boyer-Chammard, A.; Locatelli, F.; Arcese, W.; Pasquini, R.; Ortega, J.; Souillet, G.; Ferreira, E.; Laporte, J.P.; et al. Outcome of cord-blood transplantation from related and unrelated donors. Eurocord Transplant Group and the European Blood and Marrow Transplantation Group. N. Engl. J. Med. 1997, 337, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, W.I.; Martin, P.J.; Storer, B.; Clift, R.; Forman, S.J.; Negrin, R.; Kashyap, A.; Flowers, M.E.; Lilleby, K.; Chauncey, T.R.; et al. Transplantation of bone marrow as compared with peripheral-blood cells from HLA-identical relatives in patients with hematologic cancers. N. Engl. J. Med. 2001, 344, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Gragert, L.; Eapen, M.; Williams, E.; Freeman, J.; Spellman, S.; Baitty, R.; Hartzman, R.; Rizzo, J.D.; Horowitz, M.; Confer, D.; et al. HLA match likelihoods for hematopoietic stem-cell grafts in the U.S. registry. N. Engl. J. Med. 2014, 371, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Confer, D.L. Unrelated marrow donor registries. Curr. Opin. Hematol. 1997, 4, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Ballen, K.K.; Gluckman, E.; Broxmeyer, H.E. Umbilical cord blood transplantation: The first 25 years and beyond. Blood 2013, 122, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Delaney, C.; Heimfeld, S.; Brashem-Stein, C.; Voorhies, H.; Manger, R.L.; Bernstein, I.D. Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat. Med. 2010, 16, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Laughlin, M.J.; Barker, J.; Bambach, B.; Koc, O.N.; Rizzieri, D.A.; Wagner, J.E.; Gerson, S.L.; Lazarus, H.M.; Cairo, M.; Stevens, C.E.; et al. Hematopoietic engraftment and survival in adult recipients of umbilical-cord blood from unrelated donors. N. Engl. J. Med. 2001, 344, 1815–1822. [Google Scholar] [CrossRef] [PubMed]
- Ledran, M.H.; Krassowska, A.; Armstrong, L.; Dimmick, I.; Renstrom, J.; Lang, R.; Yung, S.; Santibanez-Coref, M.; Dzierzak, E.; Stojkovic, M.; et al. Efficient hematopoietic differentiation of human embryonic stem cells on stromal cells derived from hematopoietic niches. Cell Stem Cell 2008, 3, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, J.; Wernig, M.; Markoulaki, S.; Sun, C.W.; Meissner, A.; Cassady, J.P.; Beard, C.; Brambrink, T.; Wu, L.C.; Townes, T.M.; et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science 2007, 318, 1920–1923. [Google Scholar] [CrossRef] [PubMed]
- Hochedlinger, K.; Jaenisch, R. Nuclear reprogramming and pluripotency. Nature 2006, 441, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhu, F.; Yong, J.; Zhang, P.; Hou, P.; Li, H.; Jiang, W.; Cai, J.; Liu, M.; Cui, K.; et al. Generation of induced pluripotent stem cells from adult rhesus monkey fibroblasts. Cell Stem Cell 2008, 3, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoi, T.; Yae, K.; Nakagawa, M.; Ichisaka, T.; Okita, K.; Takahashi, K.; Chiba, T.; Yamanaka, S. Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science 2008, 321, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Lowry, W.E.; Richter, L.; Yachechko, R.; Pyle, A.D.; Tchieu, J.; Sridharan, R.; Clark, A.T.; Plath, K. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc. Natl. Acad. Sci. USA 2008, 105, 2883–2888. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.G.; Adam, M.A.; Miller, A.D. Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol. Cell Biol. 1990, 10, 4239–4242. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Blomer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Lois, C.; Hong, E.J.; Pease, S.; Brown, E.J.; Baltimore, D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 2002, 295, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Blelloch, R.; Venere, M.; Yen, J.; Ramalho-Santos, M. Generation of induced pluripotent stem cells in the absence of drug selection. Cell Stem Cell 2007, 1, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of mouse induced pluripotent stem cells without viral vectors. Science 2008, 322, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Snitow, M.; Chen, A.E.; Melton, D.A. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol. 2008, 26, 795–797. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Do, J.T.; Desponts, C.; Hahm, H.S.; Scholer, H.R.; Ding, S. A combined chemical and genetic approach for the generation of induced pluripotent stem cells. Cell Stem Cell 2008, 2, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Gump, J.M.; Dowdy, S.F. TAT transduction: The molecular mechanism and therapeutic prospects. Trends Mol. Med. 2007, 13, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Urnov, F.D.; Miller, J.C.; Lee, Y.L.; Beausejour, C.M.; Rock, J.M.; Augustus, S.; Jamieson, A.C.; Porteus, M.H.; Gregory, P.D.; Holmes, M.C. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 2005, 435, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Marraffini, L.A. CRISPR-Cas immunity in prokaryotes. Nature 2015, 526, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Lapillonne, H.; Kobari, L.; Mazurier, C.; Tropel, P.; Giarratana, M.C.; Zanella-Cleon, I.; Kiger, L.; Wattenhofer-Donze, M.; Puccio, H.; Hebert, N. Red blood cell generation from human induced pluripotent stem cells: Perspectives for transfusion medicine. Haematologica 2010, 95, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Takayama, N.; Nishimura, S.; Nakamura, S.; Shimizu, T.; Ohnishi, R.; Endo, H.; Yamaguchi, T.; Otsu, M.; Nishimura, K.; Nakanishi, M.; et al. Transient activation of c-MYC expression is critical for efficient platelet generation from human induced pluripotent stem cells. J. Exp. Med. 2010, 207, 2817–2830. [Google Scholar] [CrossRef] [PubMed]
- Takayama, N.; Nishikii, H.; Usui, J.; Tsukui, H.; Sawaguchi, A.; Hiroyama, T.; Eto, K.; Nakauchi, H. Generation of functional platelets from human embryonic stem cells in vitro via ES-sacs, VEGF-promoted structures that concentrate hematopoietic progenitors. Blood 2008, 111, 5298–5306. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, L.; Malladi, R.; Yang, C.T.; French, A.; Pilkington, K.J.; Forsey, R.W.; Sloane-Stanley, J.; Silk, K.M.; Davies, T.J.; Fairchild, P.J.; et al. Human induced pluripotent stem cells are capable of B-cell lymphopoiesis. Blood 2011, 117, 4008–4011. [Google Scholar] [CrossRef] [PubMed]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.D.; Vodyanik, M.A.; Slukvin, I.I. Generation of mature human myelomonocytic cells through expansion and differentiation of pluripotent stem cell-derived lin-CD34+CD43+CD45+ progenitors. J. Clin. Investig. 2009, 119, 2818–2829. [Google Scholar] [CrossRef] [PubMed]
- Woll, P.S.; Grzywacz, B.; Tian, X.; Marcus, R.K.; Knorr, D.A.; Verneris, M.R.; Kaufman, D.S. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood 2009, 113, 6094–6101. [Google Scholar] [CrossRef] [PubMed]
- Lengerke, C.; Daley, G.Q. Autologous blood cell therapies from pluripotent stem cells. Blood Rev. 2010, 24, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Daley, G.Q. From embryos to embryoid bodies: Generating blood from embryonic stem cells. Ann. N. Y. Acad. Sci. 2003, 996, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Mitra, K.; Koya, M.; Velho, M.; Desprat, R.; Lenz, J.; Bouhassira, E.E. Production of embryonic and fetal-like red blood cells from human induced pluripotent stem cells. PLoS ONE 2011, 6, e25761. [Google Scholar] [CrossRef] [PubMed]
- Giani, F.C.; Fiorini, C.; Wakabayashi, A.; Ludwig, L.S.; Salem, R.M.; Jobaliya, C.D.; Regan, S.N.; Ulirsch, J.C.; Liang, G.; Steinberg-Shemer, O.; et al. Targeted Application of Human Genetic Variation Can Improve Red Blood Cell Production from Stem Cells. Cell Stem Cell 2016, 18, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Y.; Gao, Y.; Forbes, J.A.; Qayyum, R.; Becker, L.; Cheng, L.; Wang, Z.Z. Efficient generation of megakaryocytes from human induced pluripotent stem cells using food and drug administration-approved pharmacological reagents. Stem Cells Transl. Med. 2015, 4, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Wahlster, L.; Daley, G.Q. Progress towards generation of human haematopoietic stem cells. Nat. Cell Biol. 2016, 18, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.L.; Yeap, W.H.; Tai, J.J.; Ong, S.M.; Dang, T.M.; Wong, S.C. The three human monocyte subsets: Implications for health and disease. Immunol. Res. 2012, 53, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Yanagimachi, M.D.; Niwa, A.; Tanaka, T.; Honda-Ozaki, F.; Nishimoto, S.; Murata, Y.; Yasumi, T.; Ito, J.; Tomida, S.; Oshima, K.; et al. Robust and highly-efficient differentiation of functional monocytic cells from human pluripotent stem cells under serum- and feeder cell-free conditions. PLoS ONE 2013, 8, e59243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Z.; Knorr, D.A.; Clouser, C.L.; Hexum, M.K.; Southern, P.; Mansky, L.M.; Park, I.H.; Kaufman, D.S. Human pluripotent stem cells produce natural killer cells that mediate anti-HIV-1 activity by utilizing diverse cellular mechanisms. J. Virol. 2011, 85, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Knorr, D.A.; Ni, Z.; Hermanson, D.; Hexum, M.K.; Bendzick, L.; Cooper, L.J.; Lee, D.A.; Kaufman, D.S. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl. Med. 2013, 2, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Vizcardo, R.; Masuda, K.; Yamada, D.; Ikawa, T.; Shimizu, K.; Fujii, S.; Koseki, H.; Kawamoto, H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell 2013, 12, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Kaneko, S.; Kawana-Tachikawa, A.; Tajima, Y.; Goto, H.; Zhu, D.; Nakayama-Hosoya, K.; Iriguchi, S.; Uemura, Y.; Shimizu, T.; et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013, 12, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Amabile, G.; Welner, R.S.; Nombela-Arrieta, C.; D'Alise, A.M.; Di Ruscio, A.; Ebralidze, A.K.; Kraytsberg, Y.; Ye, M.; Kocher, O.; Neuberg, D.S.; et al. In vivo generation of transplantable human hematopoietic cells from induced pluripotent stem cells. Blood 2013, 121, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Zhan, H.; Mali, P.; Dowey, S.; Williams, D.M.; Jang, Y.Y.; Dang, C.V.; Spivak, J.L.; Moliterno, A.R.; Cheng, L. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood 2009, 114, 5473–5480. [Google Scholar] [CrossRef] [PubMed]
- Carette, J.E.; Pruszak, J.; Varadarajan, M.; Blomen, V.A.; Gokhale, S.; Camargo, F.D.; Wernig, M.; Jaenisch, R.; Brummelkamp, T.R. Generation of iPSCs from cultured human malignant cells. Blood 2010, 115, 4039–4042. [Google Scholar] [CrossRef] [PubMed]
- Amabile, G.; Di Ruscio, A.; Muller, F.; Welner, R.S.; Yang, H.; Ebralidze, A.K.; Zhang, H.; Levantini, E.; Qi, L.; Martinelli, G.; et al. Dissecting the role of aberrant DNA methylation in human leukaemia. Nat. Commun. 2015, 6, 7091. [Google Scholar] [CrossRef] [PubMed]
- Khincha, P.P.; Savage, S.A. Genomic characterization of the inherited bone marrow failure syndromes. Semin Hematol. 2013, 50, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Joenje, H.; Patel, K.J. The emerging genetic and molecular basis of Fanconi anaemia. Nat. Rev. Genet. 2001, 2, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Grompe, M.; D’Andrea, A. Fanconi anemia and DNA repair. Hum. Mol. Genet. 2001, 10, 2253–2259. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.M.; Niwa, A.; Yabe, M.; Hira, A.; Okada, C.; Amano, N.; Watanabe, A.; Watanabe, K.; Heike, T.; Takata, M.; et al. Pluripotent cell models of fanconi anemia identify the early pathological defect in human hemoangiogenic progenitors. Stem Cells Transl. Med. 2015, 4, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Fan, Y.; He, W.; Zhu, D.; Niu, X.; Wang, D.; Ou, Z.; Luo, M.; Sun, X. Improved hematopoietic differentiation efficiency of gene-corrected beta-thalassemia induced pluripotent stem cells by CRISPR/Cas9 system. Stem Cells Dev. 2015, 24, 1053–1065. [Google Scholar] [CrossRef] [PubMed]
- Kumano, K.; Arai, S.; Hosoi, M.; Taoka, K.; Takayama, N.; Otsu, M.; Nagae, G.; Ueda, K.; Nakazaki, K.; Kamikubo, Y.; et al. Generation of induced pluripotent stem cells from primary chronic myelogenous leukemia patient samples. Blood 2012, 119, 6234–6242. [Google Scholar] [CrossRef] [PubMed]
- Cordoba, I.; Gonzalez-Porras, J.R.; Nomdedeu, B.; Luno, E.; de Paz, R.; Such, E.; Tormo, M.; Vallespi, T.; Collado, R.; Xicoy, B.; et al. Better prognosis for patients with del(7q) than for patients with monosomy 7 in myelodysplastic syndrome. Cancer 2012, 118, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Kotini, A.G.; Chang, C.J.; Boussaad, I.; Delrow, J.J.; Dolezal, E.K.; Nagulapally, A.B.; Perna, F.; Fishbein, G.A.; Klimek, V.M.; Hawkins, R.D.; et al. Functional analysis of a chromosomal deletion associated with myelodysplastic syndromes using isogenic human induced pluripotent stem cells. Nat. Biotechnol. 2015, 33, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, N.J.; Ny, L.; Nilsson, J.A.; Larsson, E. Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat. Genet. 2014, 46, 1258–1263. [Google Scholar] [CrossRef] [PubMed]
- Scudellari, M. How iPS cells changed the world. Nature 2016, 534, 310–312. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Embryonic Stem Cells (ESCs) | Hematopoietic Stem Cells (HSCs) | Induced Pluripotent Stem Cells (iPSCs) | |
|---|---|---|---|
| Source | Inner mass cells of blastocyst | Bone marrow donations; umbilical cord blood | Any somatic cell |
| Application | Basic science research; limited clinical application currently | Hematopoietic stem cell transplantation | Basic science research |
| Markers | SOX2, NANOG, Oct-4, SSEA-1, SSEA-3, SSEA-4 TRA-1-60, TRA-1-81 Frizzled5 | CD34+, c-Kit−/low, Lin-, CD38-,Flt-3/Flk-2 | Reactivation of embryonic stem cell markers, e.g., SOX2, NANOG, OCT-4, KLF4, SSEA-4, TRA-1-60 |
| Derivation | Isolation from in vitro fertilized embryos | Purification fromdonations | Ectopic expression of ESC transcription factors: OCT3/4, SOX2, KLF4, c-MYC |
| Pros | Able to generate all three germ layers; Amenable to cell culture expansion while maintaining pluripotency | Not controversial; Can be harvested from bone marrow blood, can be mobilized to peripheral blood upon granulocyte-colony stimulating factor (G-CSF) induction, or obtained from umbilical cord blood donations; Rise in biobanking of umbilical cord blood increases amount of source material | Non-invasive isolation; Avoids Human Leukocyte Antigen Loci (HLA)-compatibility issues Can be genetically altered before transfusion; Expands available research areas; Recapitulates patient genome; Theoretically unlimited source material |
| Cons | Ethical concerns of using embryonic-derived cells; Limited source material | Restricted lineage differentiation; Dependent on HLA compatibility; Transplantation-associated risks: immune suppression, graft rejection, graft vs. host disease | Low efficiency of reprogramming;Incomplete programming, or “epigenetic memory”; No standardized protocol for production; Genetically unstable; Safety concerns; Maintenance of germline mutations; Insertional mutagenesis for integrating vectors |
| Induced Pluripotent Stem Cells (iPSCs) Protocol | Consideration | |
|---|---|---|
| 1. Choice of Cell Type | Adult mouse and human fibroblasts were used in first iPSCs experiments. | While iPSCs can in principle be generated from any somatic cell, in practice, there seems to be an inverse relationship between degree of differentiation and ease of reprogramming. Additionally, there is expanding concern for “memory” of the original cell, hindering the re-differentiation process downstream. |
| 2. Dedifferentiation | Retroviral- or lentiviral-mediated expression of four pluripotent-specific genes: OCT3/4, SOX2, KLF4, and c-MYC (OSKM). | Concern for the transforming potential of c-MYC led to the identification of other factor substitutes. c-MYC was later deemed dispensible, and other factor combinations (Nanog, Lin28) have successfully generated iPSCs.Methods of delivery must also consider the effects of insertional mutagenesis when using integrating vectors. Non-integrating viruses, small-molecules, RNA- and transposon-based technologies are also currently being explored. |
| 3. Selection | Transduced cells are cultured in embryonic stem cell (ESC) medium + antibiotics for 2–4 weeks with an ESC-specific marker, Fbx15, driving antibiotic resistance. Only reprogrammed cells can survive the selection process. | Although Fbx15 is expressed only in ESCs, it is not essential to ESC development and explains the partial reprogramming observed initially. Currently, Nanog-driven selection is favored instead. |
| 4. Differentiation | Cultured with feeder cells and cytokines directing lineage-specific differentiation. | iPSCs can differentiate through (direct) addition of lineage-specific transcription factors or (indirect) culture in lineage-specific cytokines and growth factors. Protocols vary among laboratories. |
| 5. Functional Testing | Expression of lineage-specific markers measured through PCR or immunofluorescence. | Functional tests are not standardized. Definition of lineage-specific characteristics vary among laboratories. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Cascino, P.; Ummarino, S.; Di Ruscio, A. Application of Induced Pluripotent Stem Cell Technology to the Study of Hematological Diseases. Cells 2017, 6, 7. https://doi.org/10.3390/cells6010007
Li M, Cascino P, Ummarino S, Di Ruscio A. Application of Induced Pluripotent Stem Cell Technology to the Study of Hematological Diseases. Cells. 2017; 6(1):7. https://doi.org/10.3390/cells6010007
Chicago/Turabian StyleLi, Mailin, Pasquale Cascino, Simone Ummarino, and Annalisa Di Ruscio. 2017. "Application of Induced Pluripotent Stem Cell Technology to the Study of Hematological Diseases" Cells 6, no. 1: 7. https://doi.org/10.3390/cells6010007