
Microinjection of Antibodies Targeting the Lamin A/C Histone-Binding Site Blocks Mitotic Entry and Reveals Separate Chromatin Interactions with HP1, CenpB and PML


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Construction
2.2. Cell Culture and Transfections
2.3. Antibodies
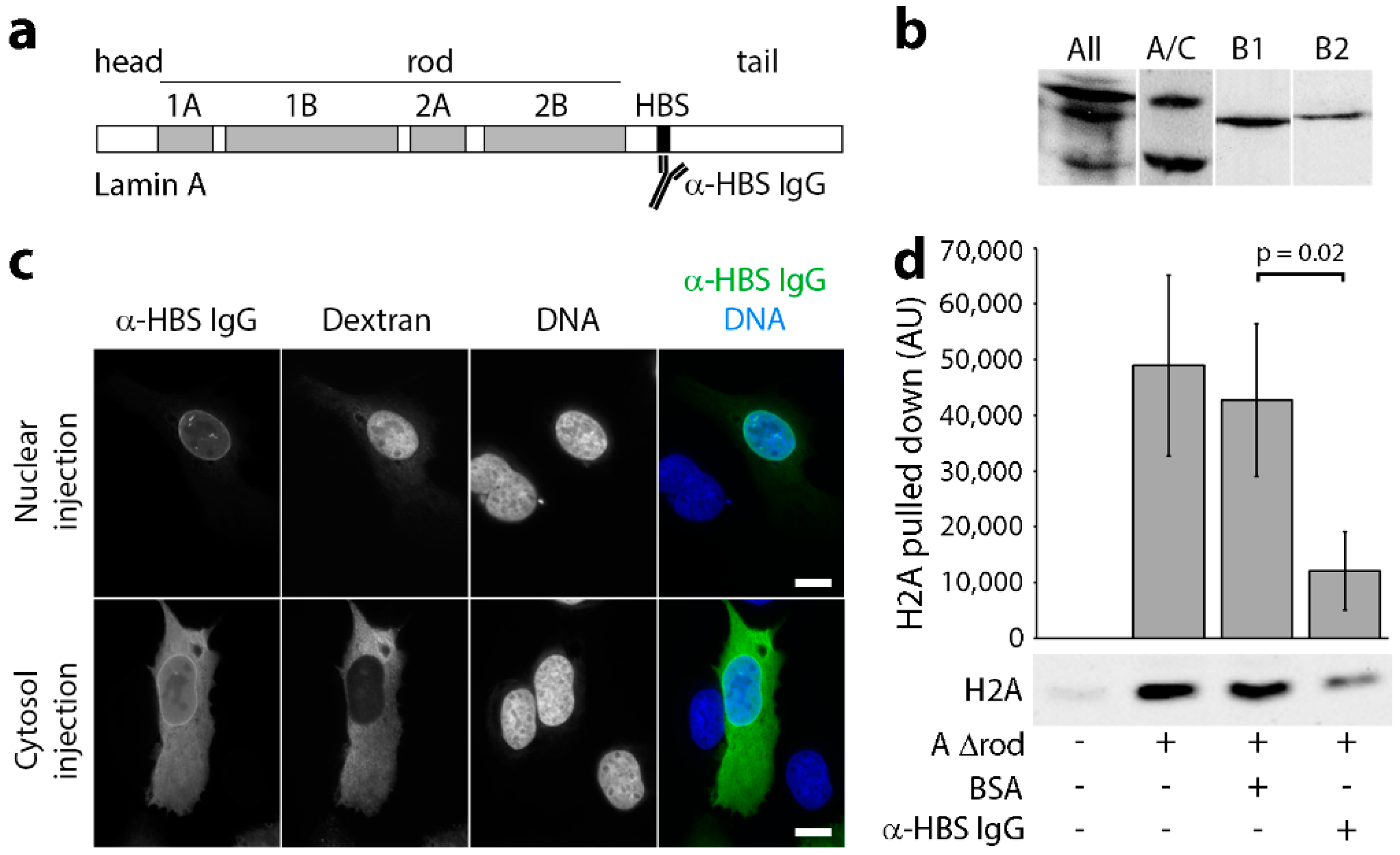
2.4. Testing Antibody Blocking
2.5. Microinjection
2.6. Microscopy
2.7. Image and Bioinformatics Analysis and Statistics
3. Results
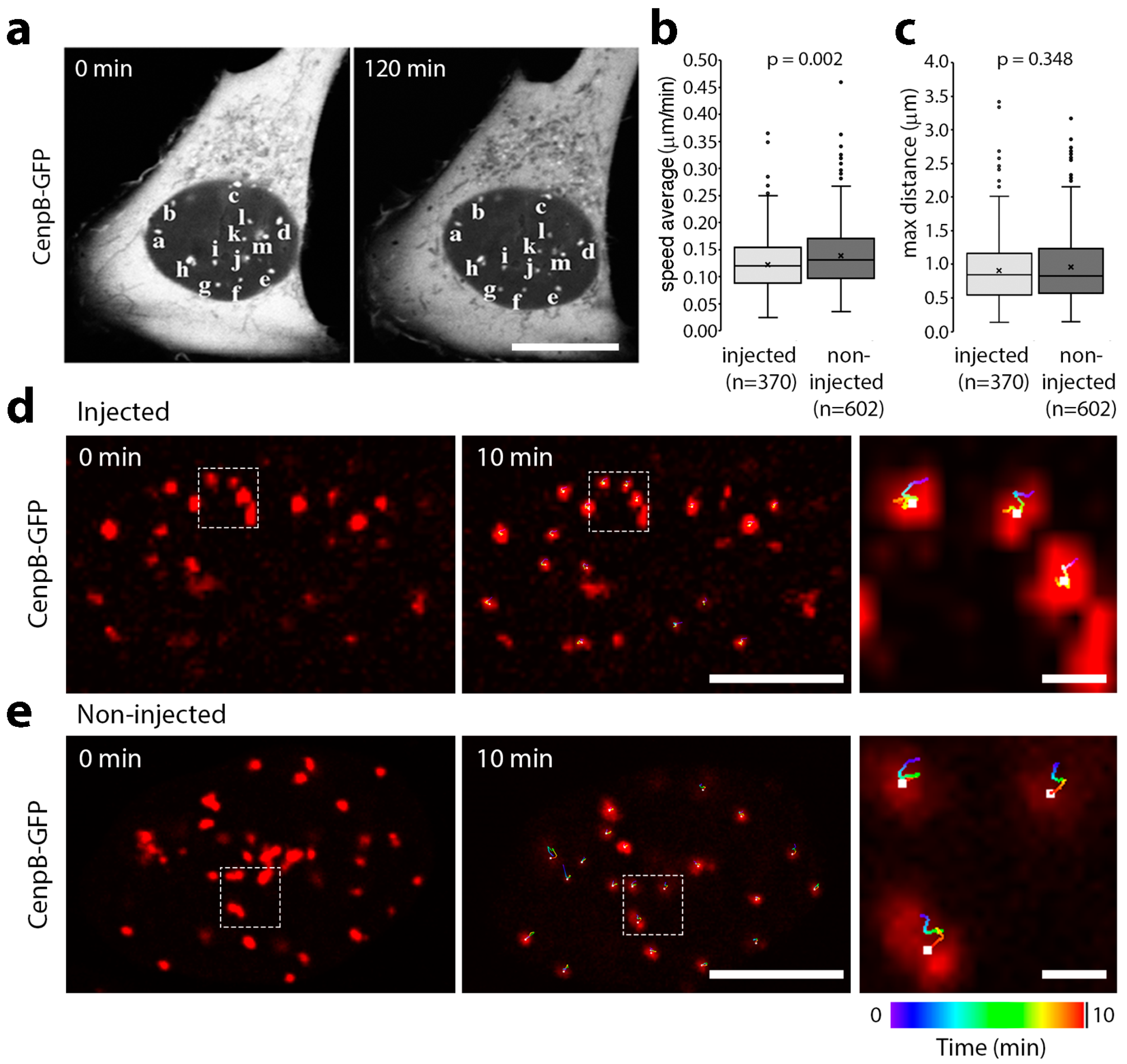
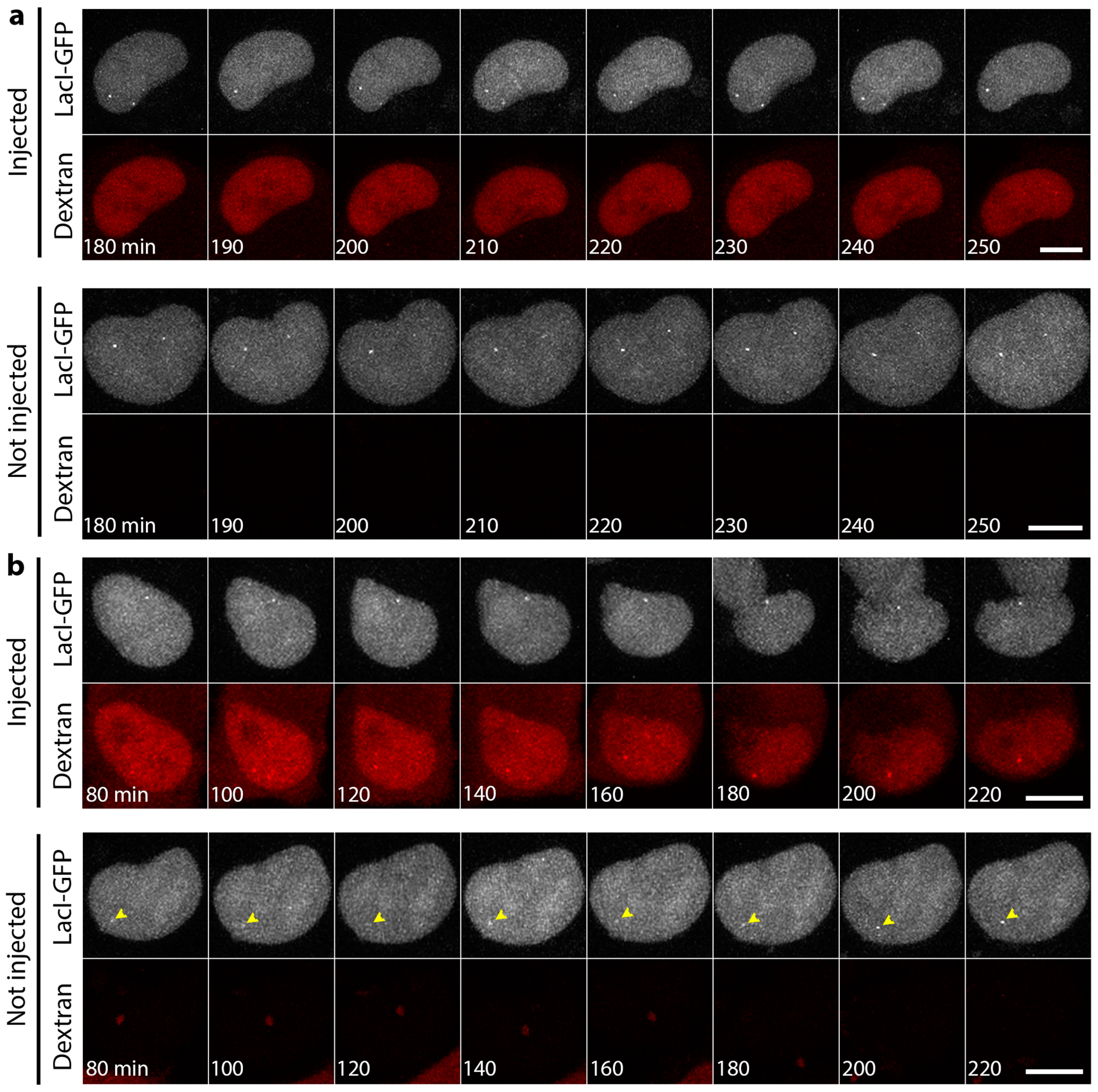
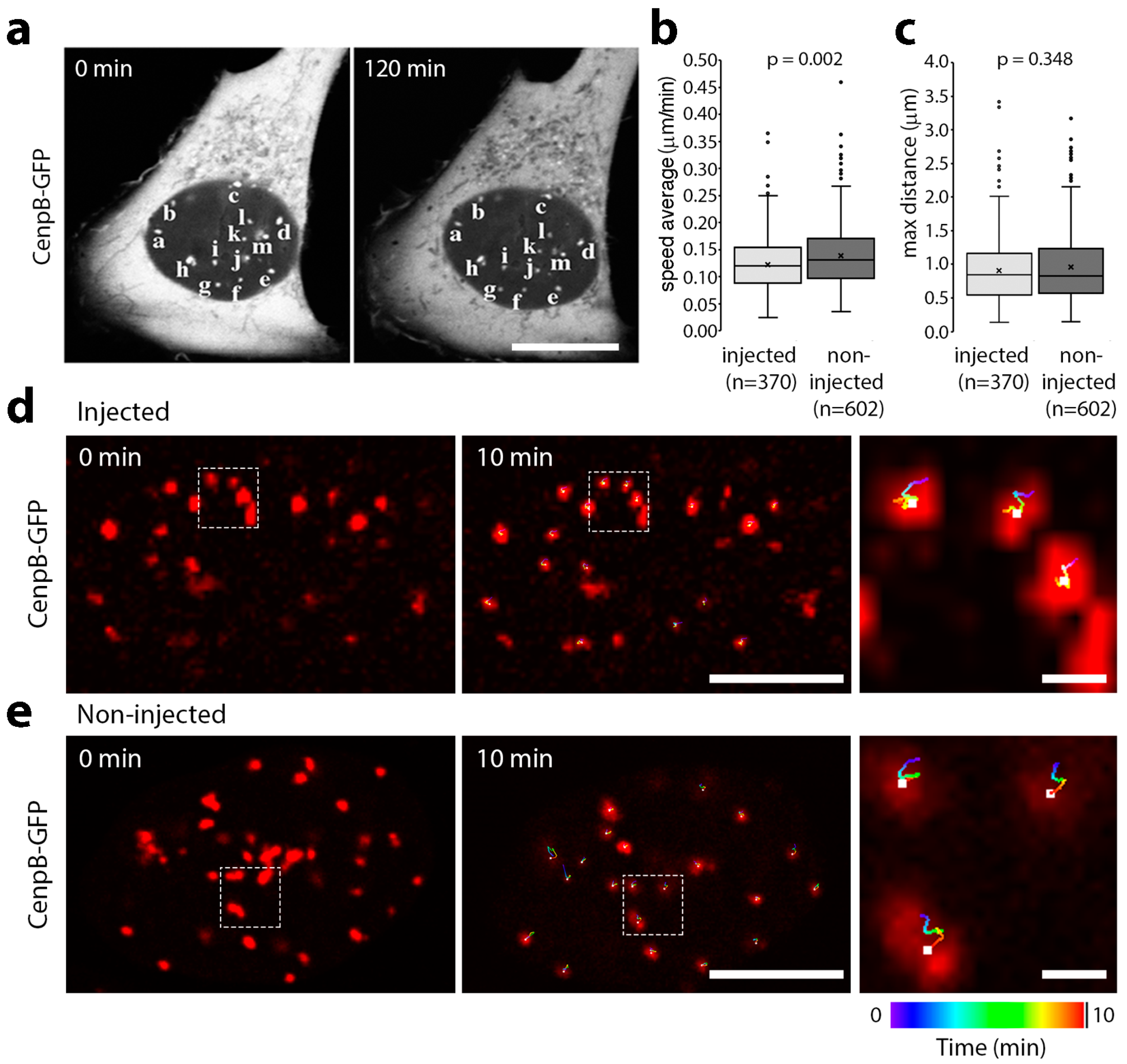
3.1. Microinjection of Lamin Histone-Binding Site Antibodies Has Little Effect on Overall Genome Mobility
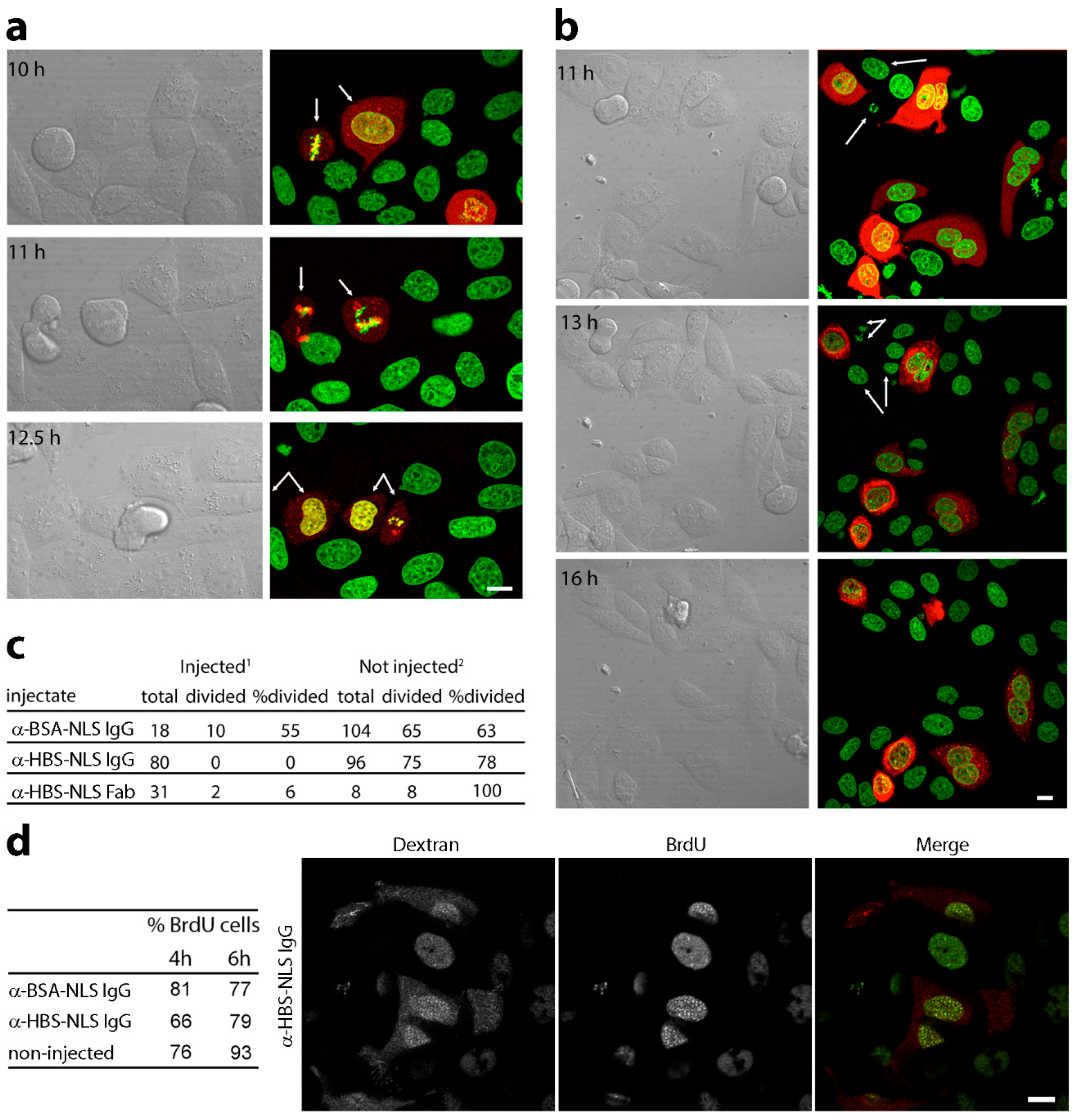
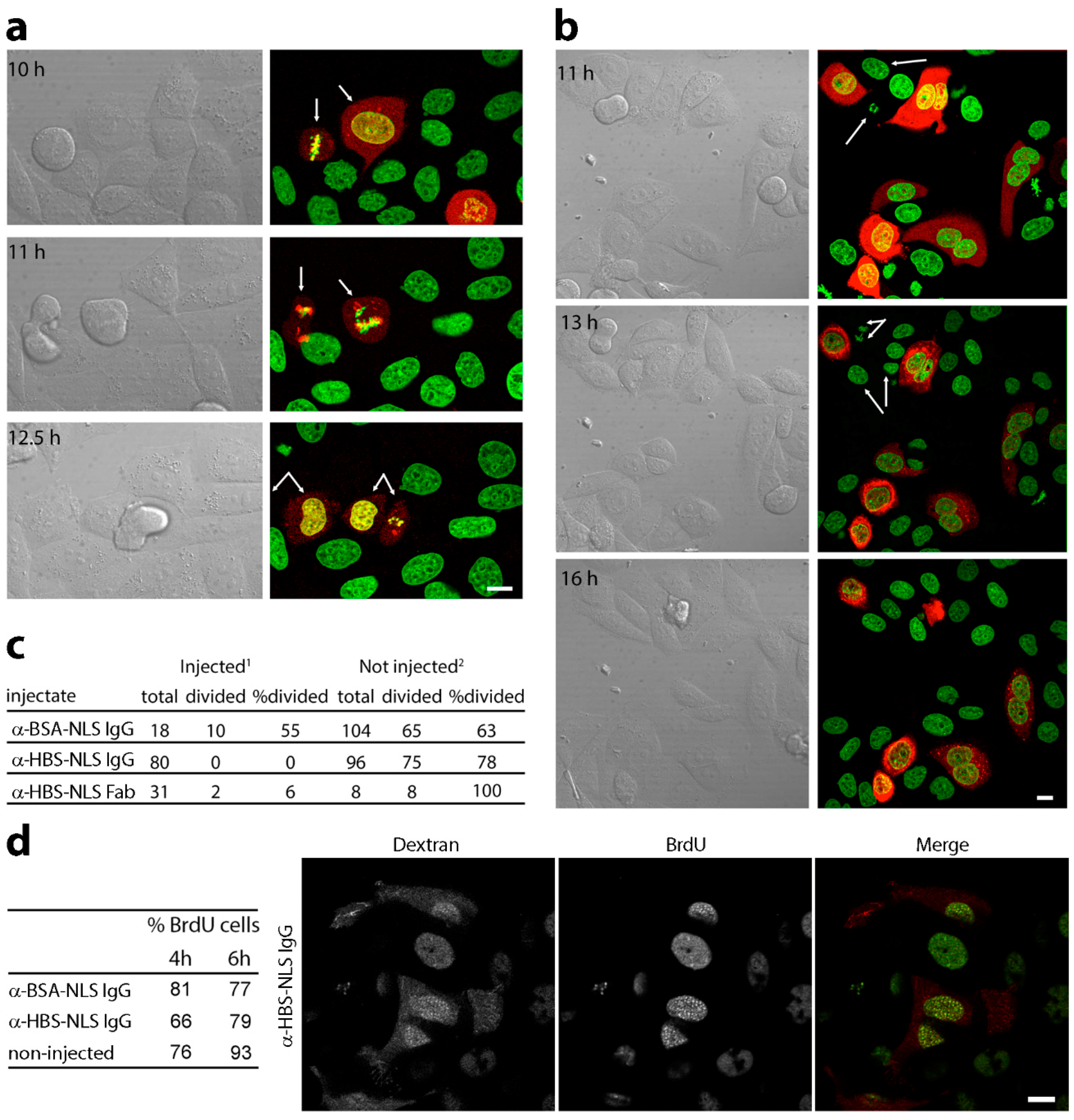
3.2. Microinjection of Lamin Histone-Binding Site Antibodies Blocks Mitotic Entry
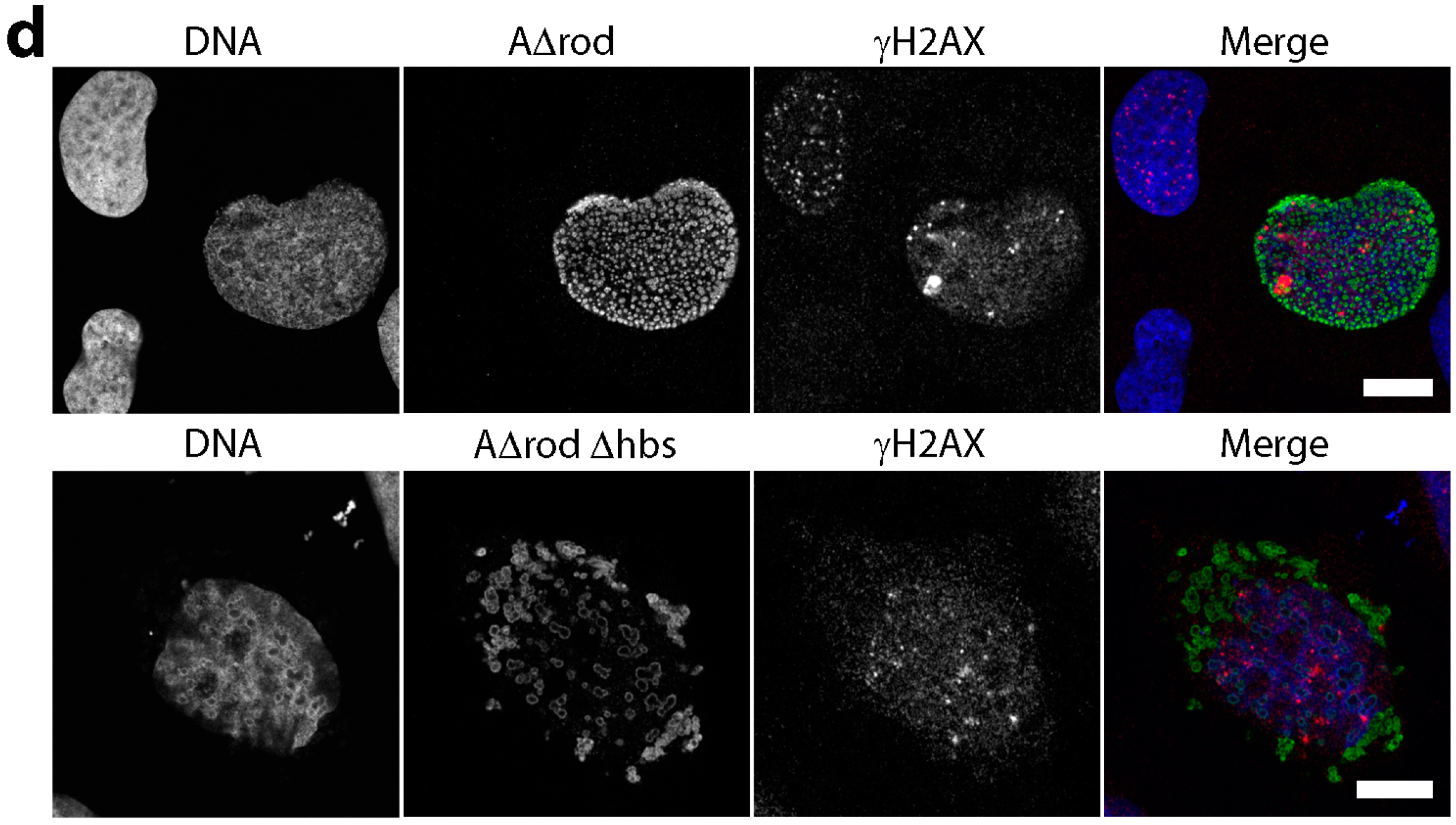
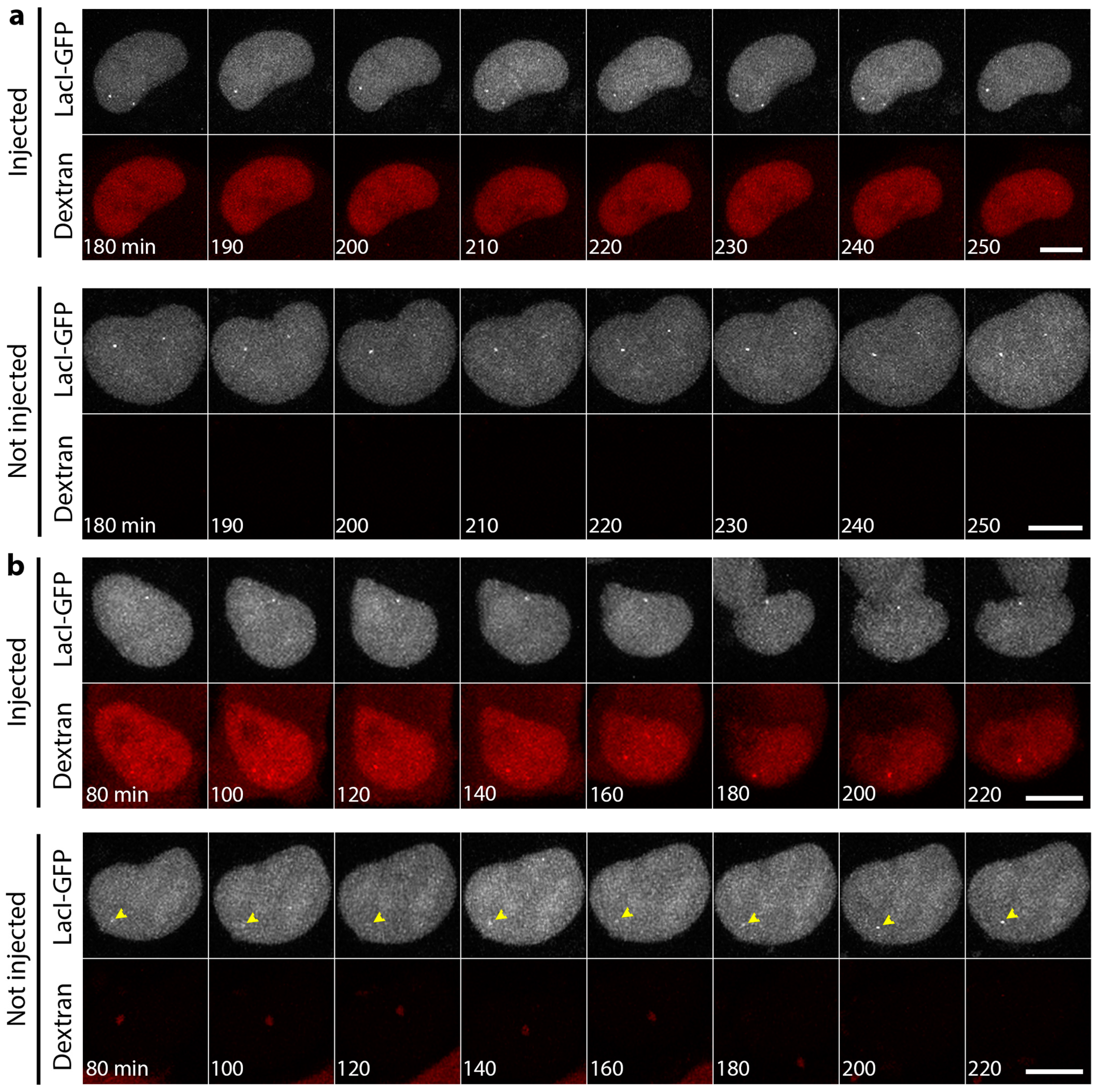
3.3. DNA Replication Is Partly Delayed in Cells Microinjected with Lamin Histone-Binding Site Antibodies
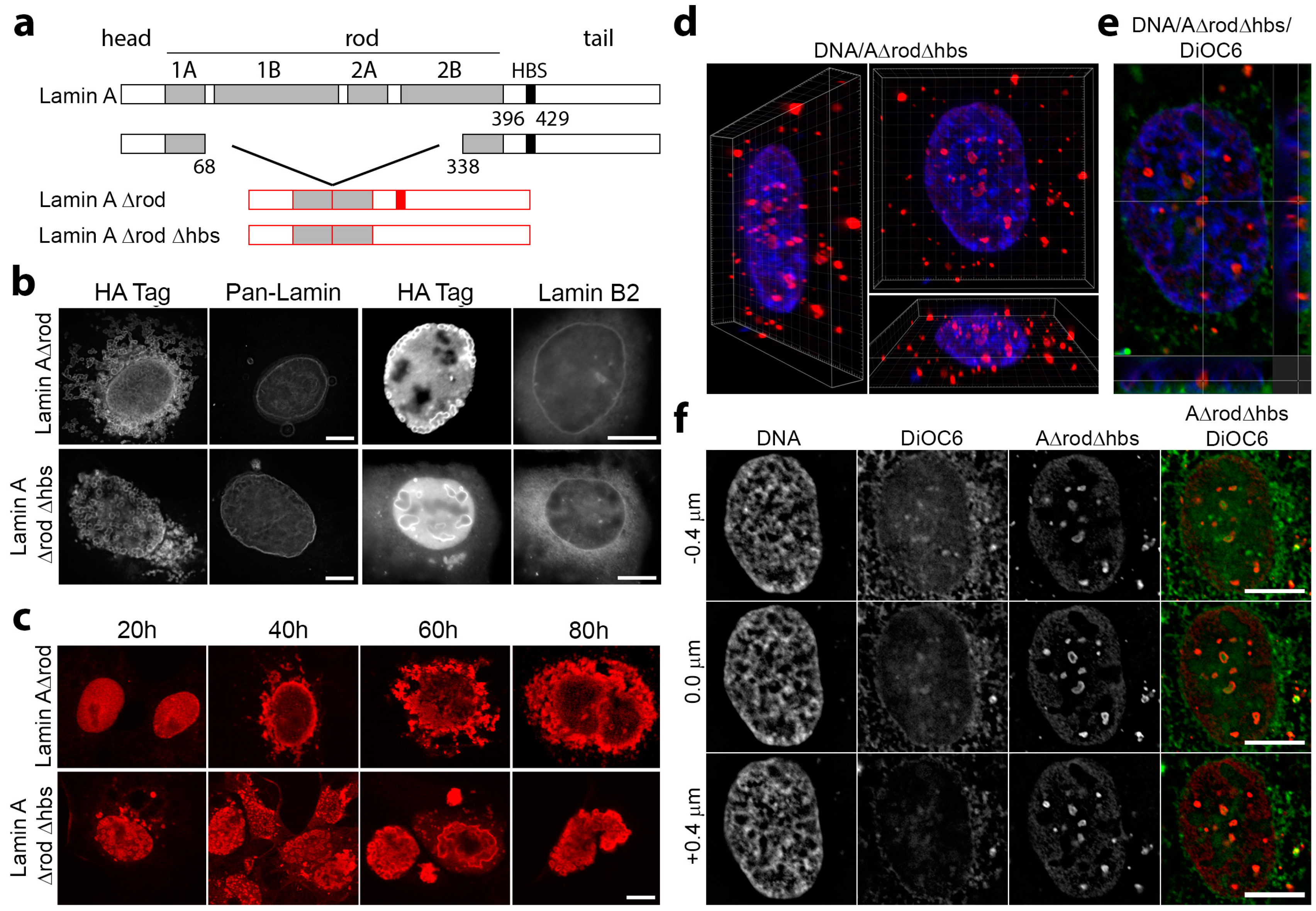
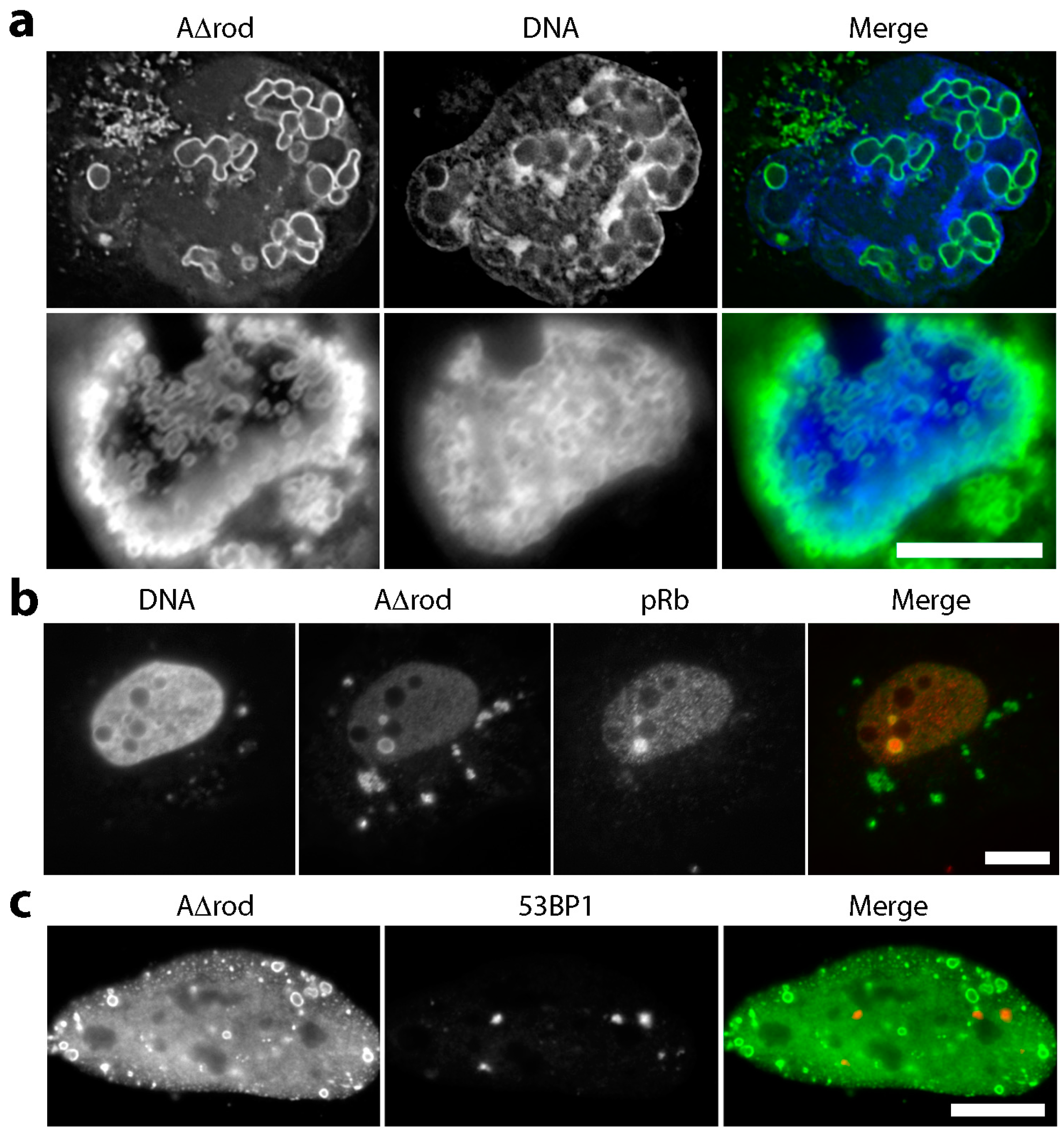
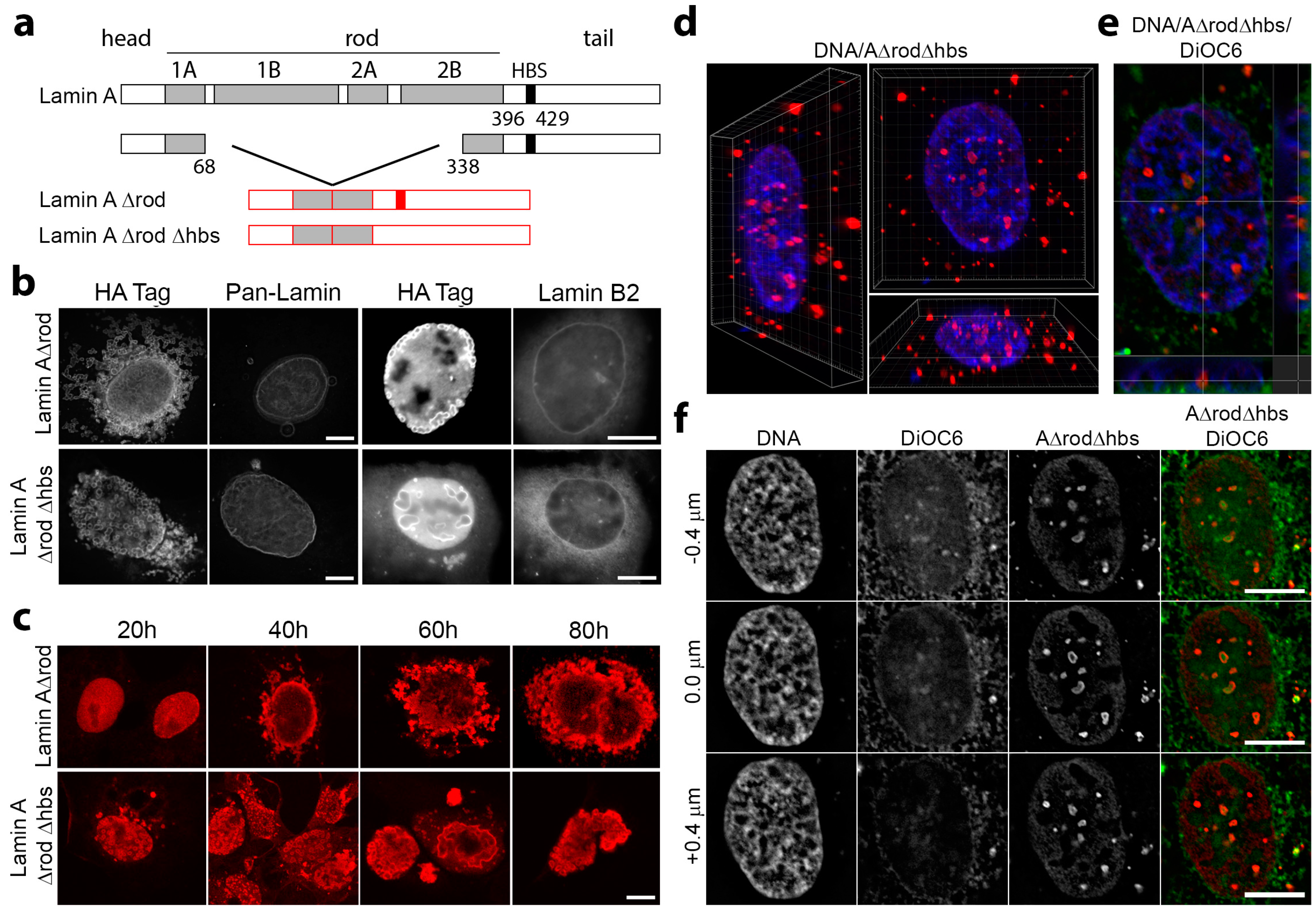
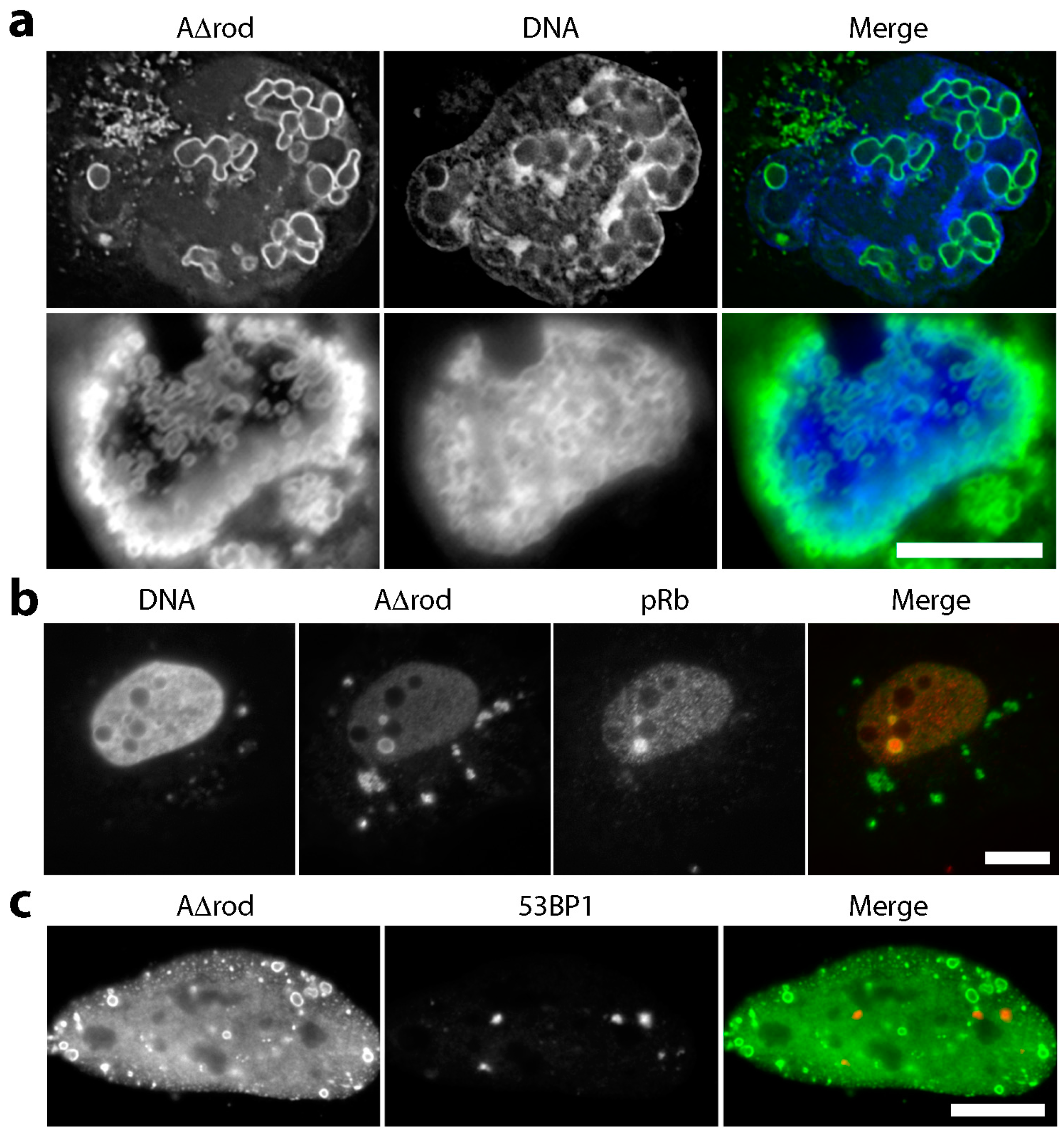
3.4. Mini-Lamin A Structures Accumulate DNA
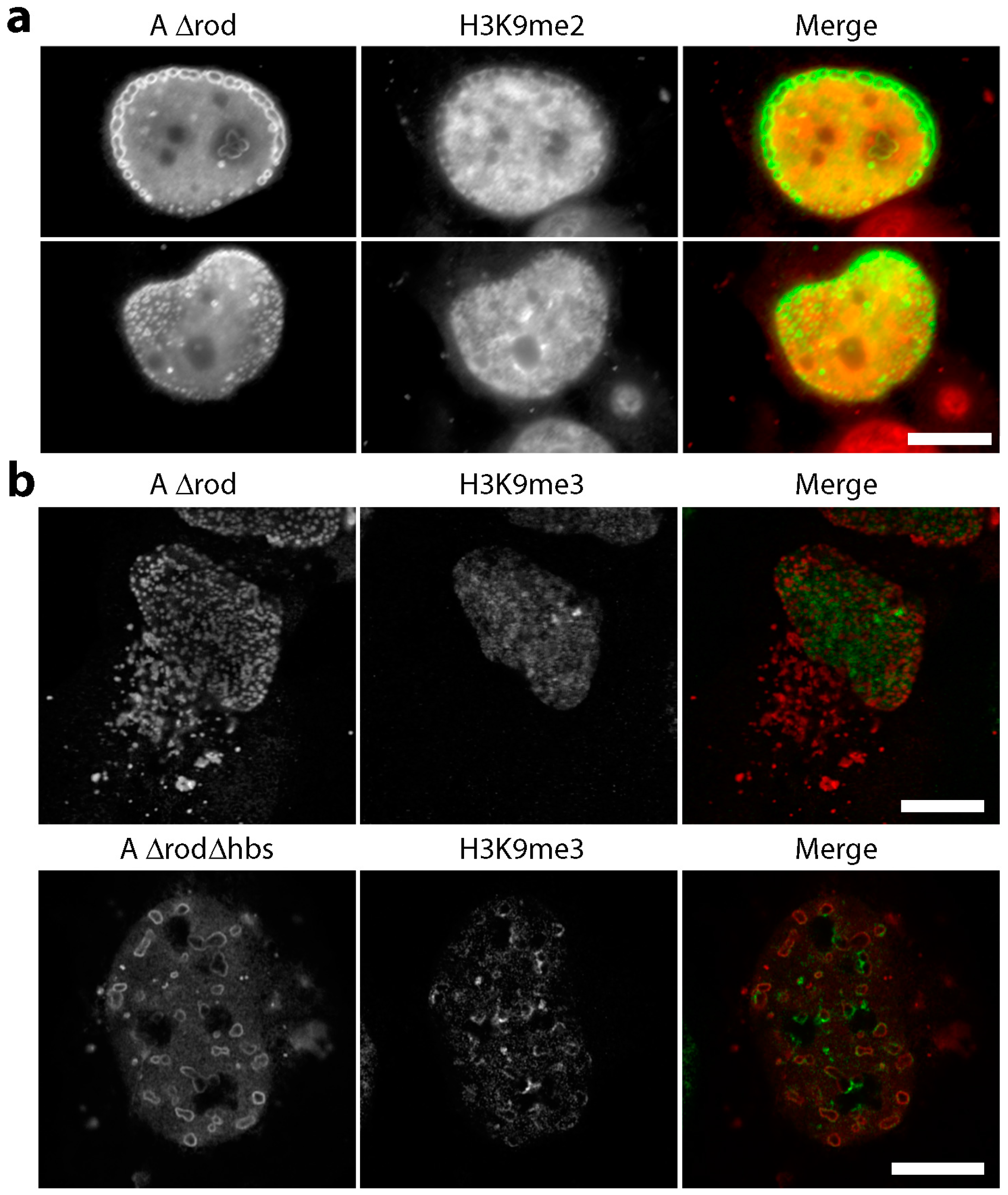
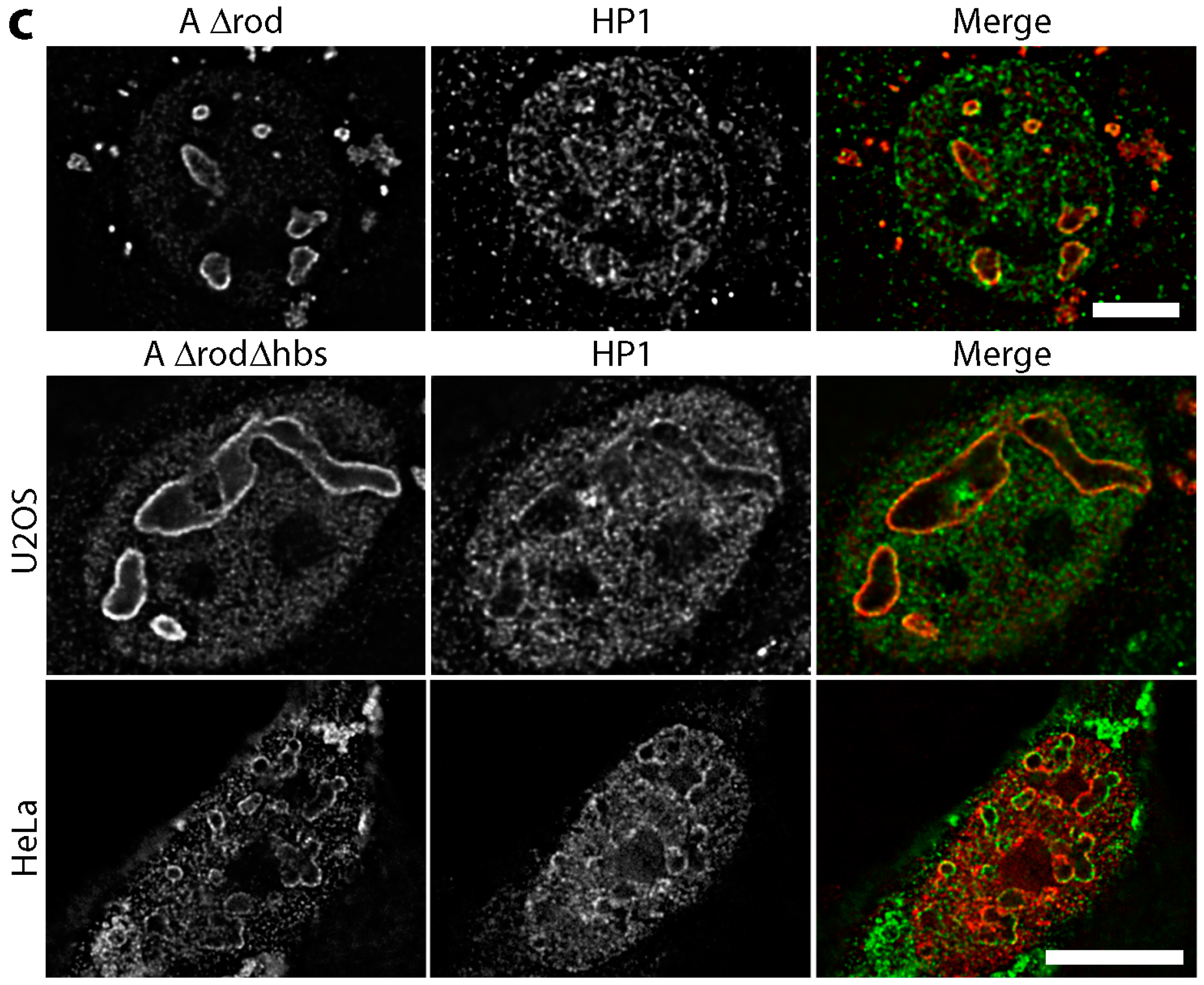
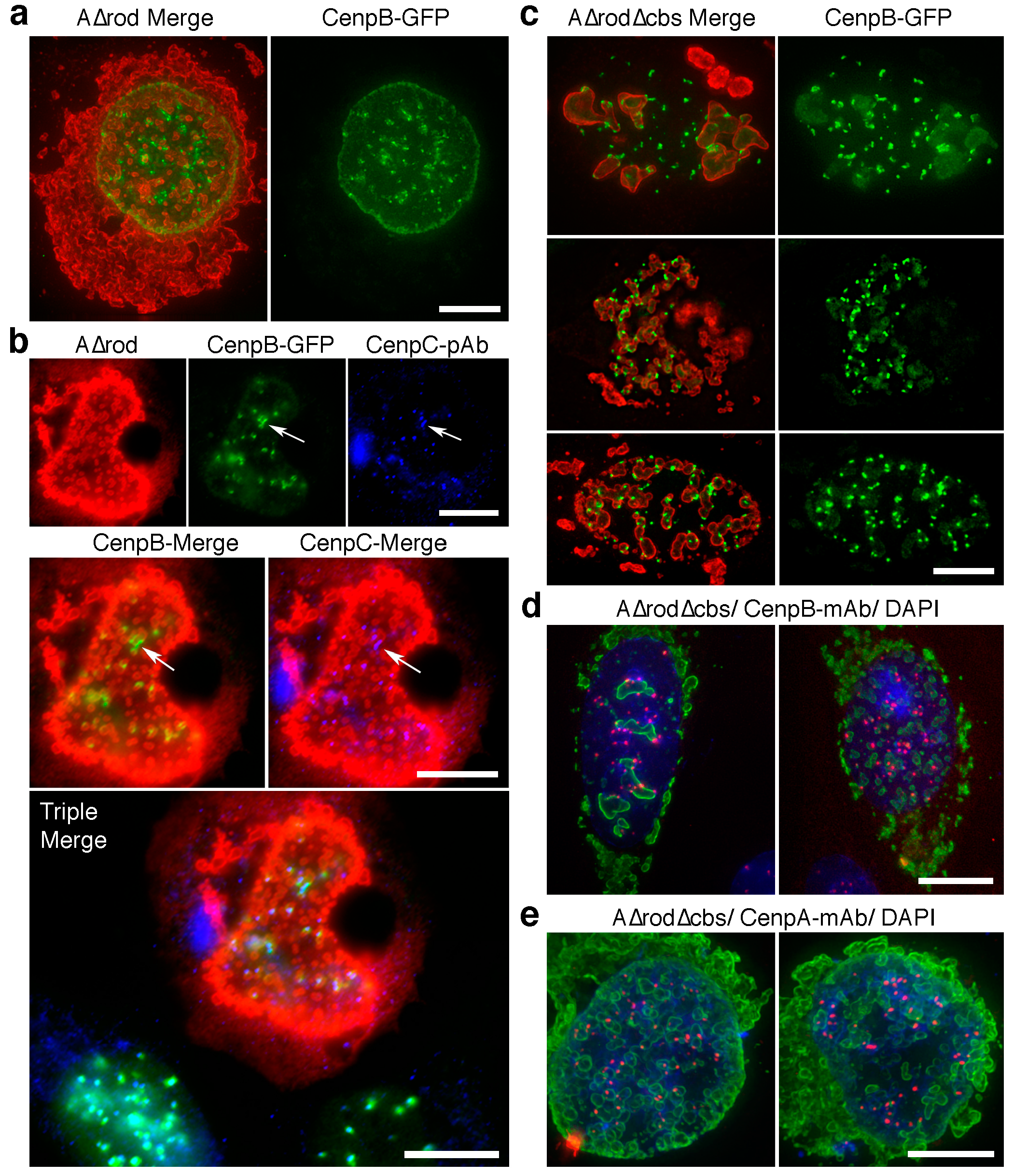
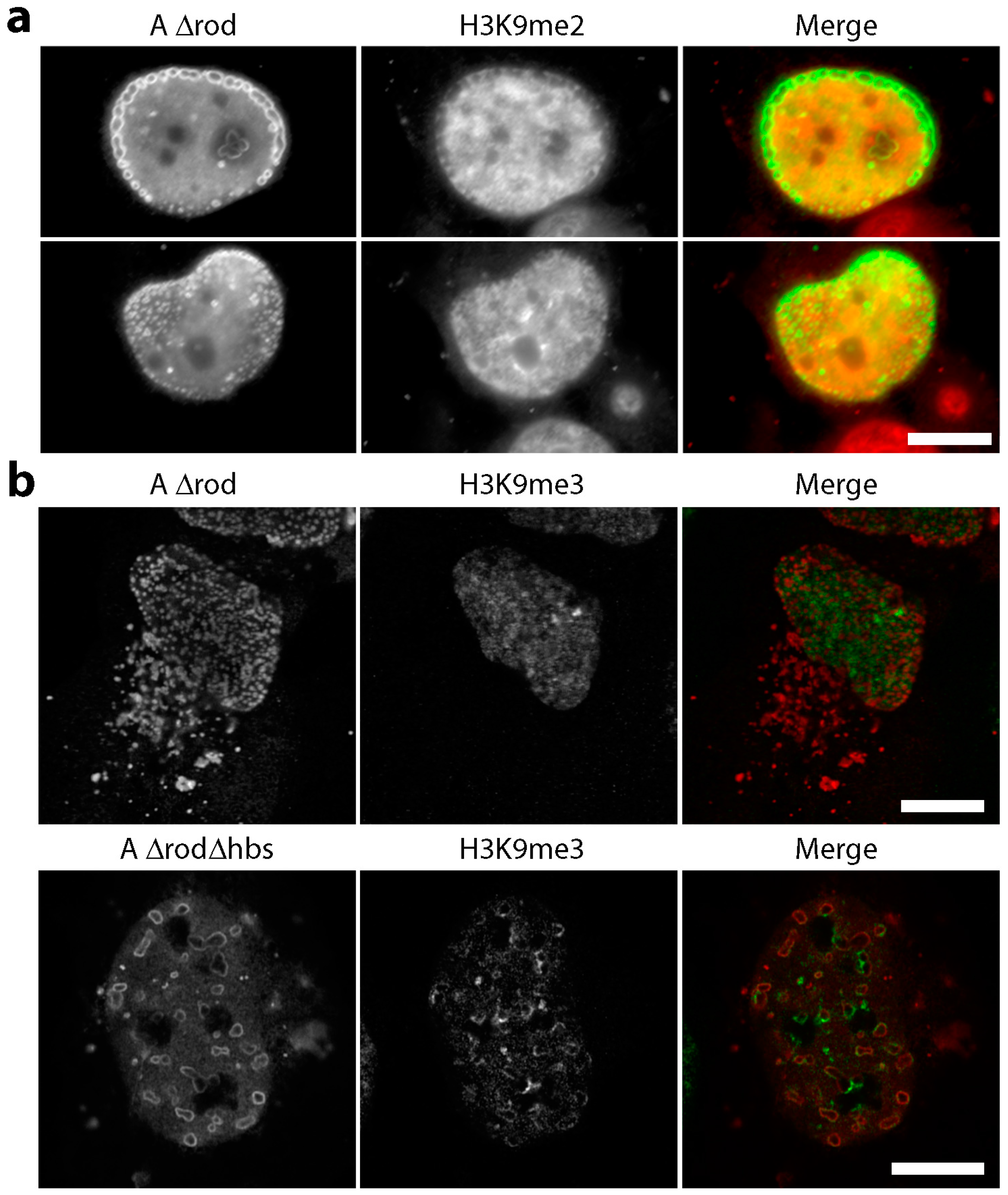
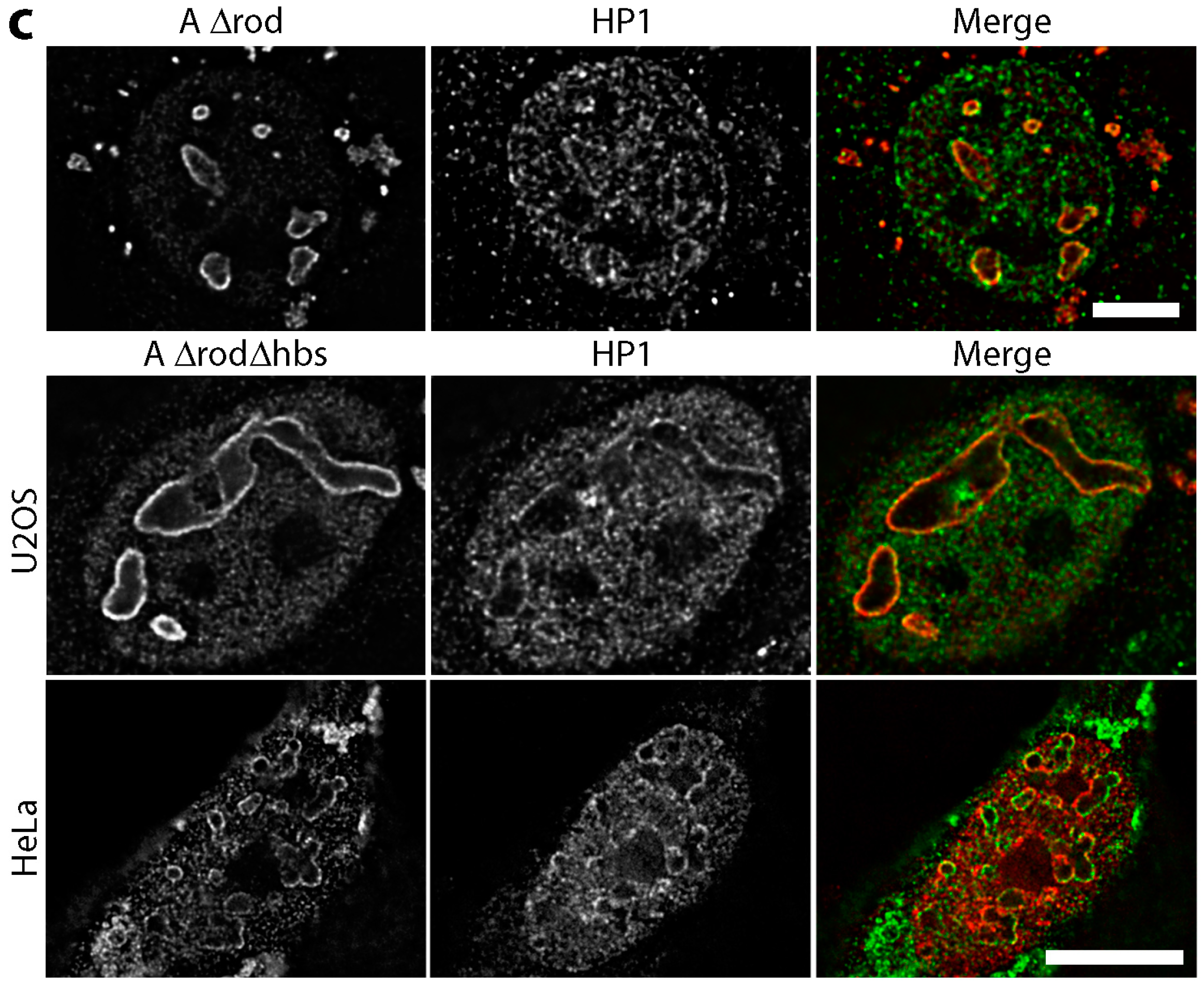
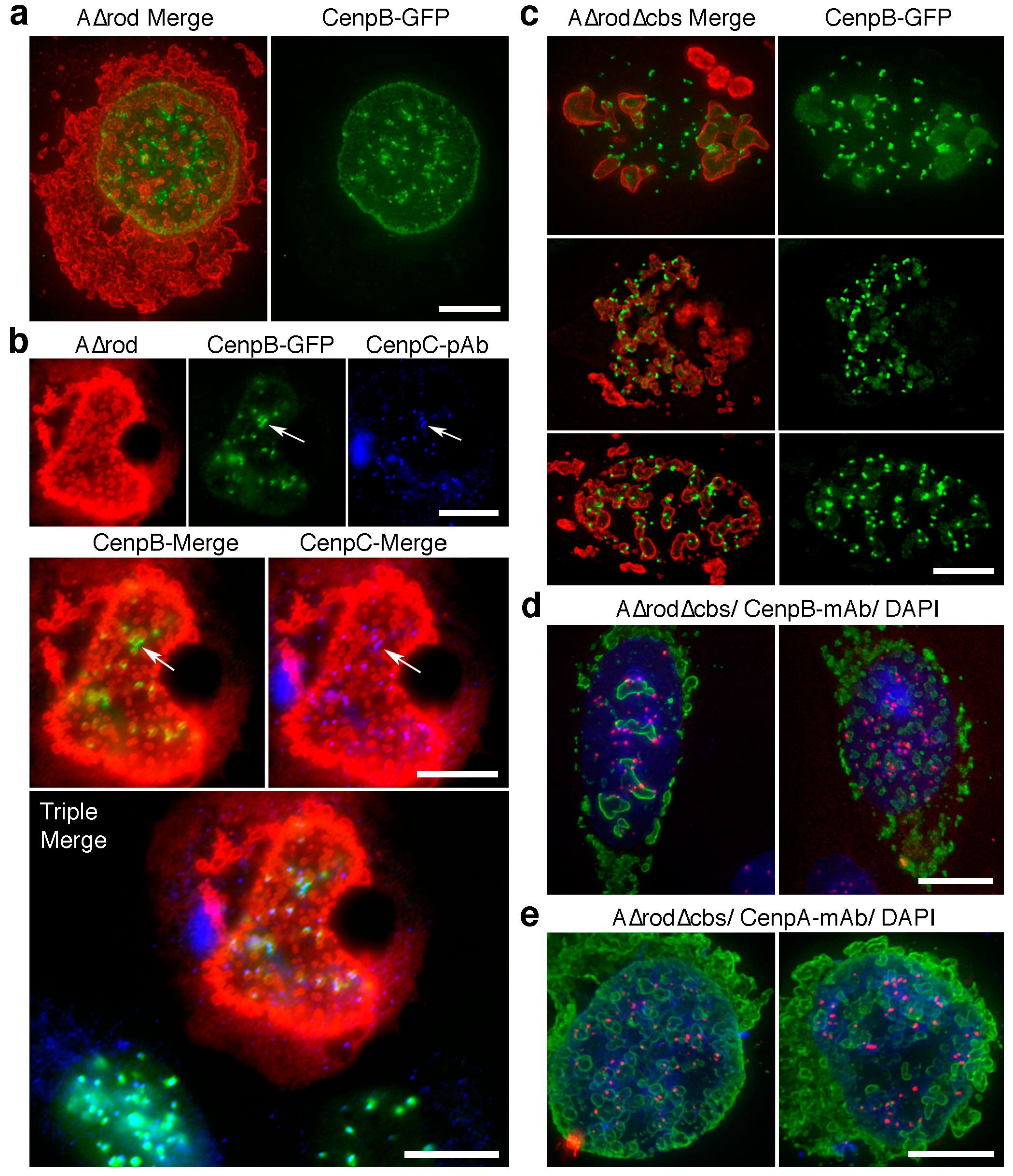
3.5. A∆rod Mutants Affect Specific Chromatin Regions Containing Centromere Proteins
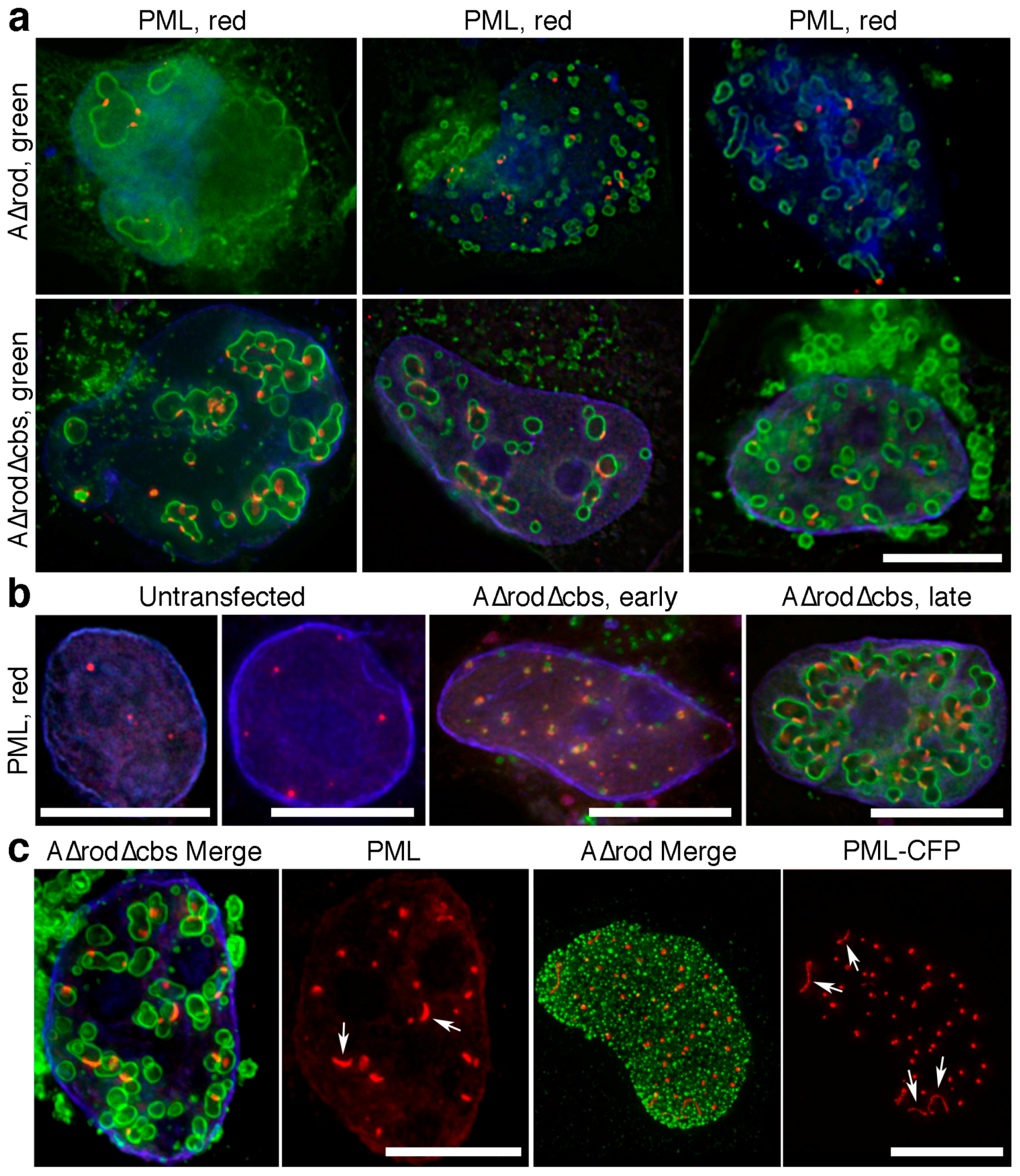
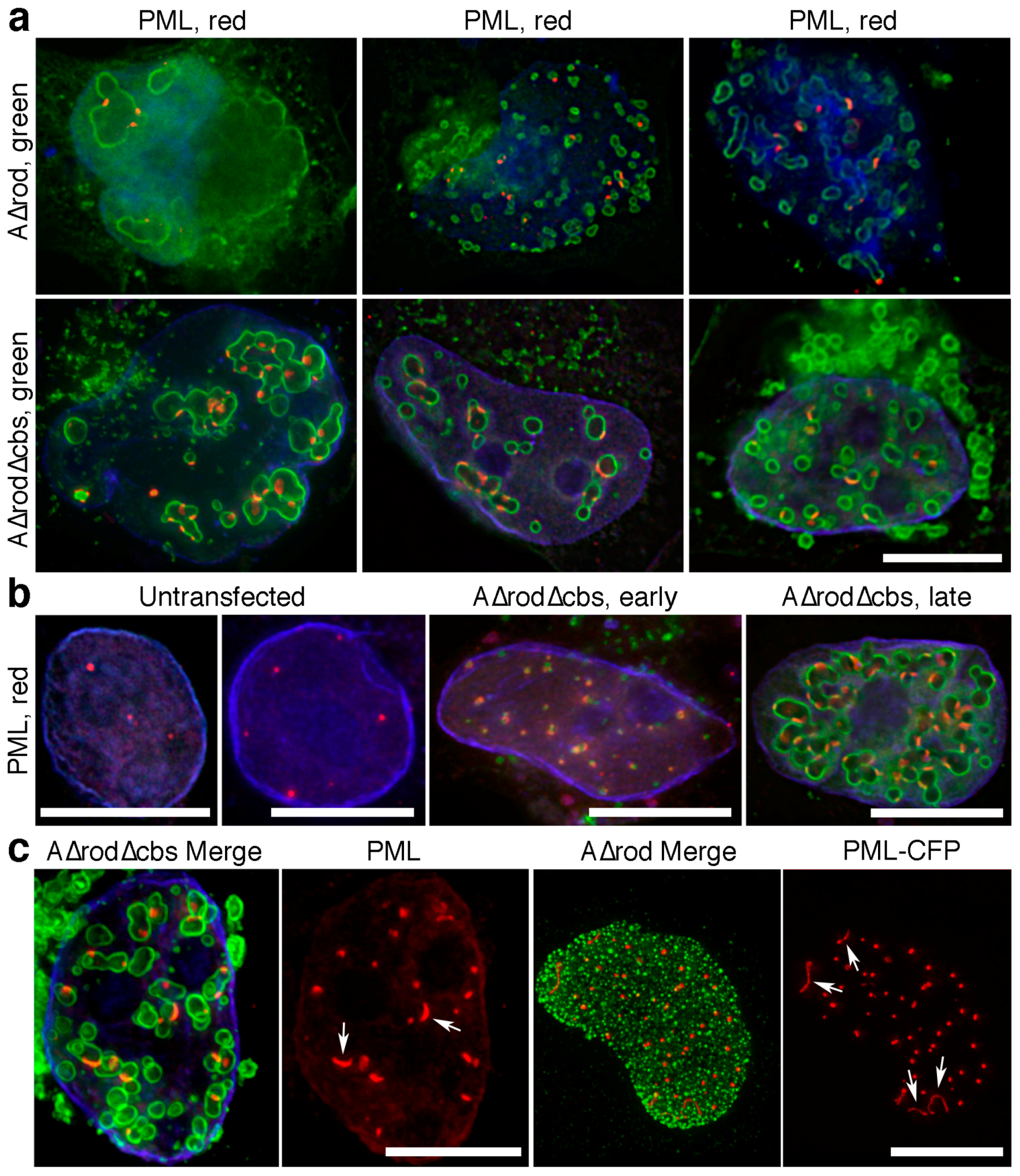
3.6. A∆rod Mutants also Affect Pro-Myelocytic Leukaemia Protein Distribution
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PML | promyelocytic leukaemia protein |
| HP1 | heterochromatin protein 1 |
References
- Meaburn, K.J.; Cabuy, E.; Bonne, G.; Levy, N.; Morris, G.E.; Novelli, G.; Kill, I.R.; Bridger, J.M. Primary laminopathy fibroblasts display altered genome organization and apoptosis. Aging Cell 2007, 6, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Mehta, I.S.; Eskiw, C.H.; Arican, H.D.; Kill, I.R.; Bridger, J.M. Farnesyltransferase inhibitor treatment restores chromosome territory positions and active chromosome dynamics in Hutchinson-Gilford progeria syndrome cells. Genome Biol. 2011, 12, R74. [Google Scholar] [CrossRef] [PubMed]
- Mewborn, S.K.; Puckelwartz, M.J.; Abuisneineh, F.; Fahrenbach, J.P.; Zhang, Y.; MacLeod, H.; Dellefave, L.; Pytel, P.; Selig, S.; Labno, C.M.; et al. Altered chromosomal positioning, compaction, and gene expression with a lamin A/C gene mutation. PLoS ONE 2010, 5, e14342. [Google Scholar] [CrossRef] [PubMed]
- Malhas, A.; Lee, C.F.; Sanders, R.; Saunders, N.J.; Vaux, D.J. Defects in lamin B1 expression or processing affect interphase chromosome position and gene expression. J. Cell Biol. 2007, 176, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [PubMed]
- Shumaker, D.K.; Dechat, T.; Kohlmaier, A.; Adam, S.A.; Bozovsky, M.R.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Khuon, S.; Collins, F.S.; et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. USA 2006, 103, 8703–8708. [Google Scholar] [CrossRef] [PubMed]
- Solovei, I.; Kreysing, M.; Lanctot, C.; Kosem, S.; Peichl, L.; Cremer, T.; Guck, J.; Joffe, B. Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell 2009, 137, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Solovei, I.; Wang, A.S.; Thanisch, K.; Schmidt, C.S.; Krebs, S.; Zwerger, M.; Cohen, T.V.; Devys, D.; Foisner, R.; Peichl, L.; et al. LBR and Lamin A/C Sequentially Tether Peripheral Heterochromatin and Inversely Regulate Differentiation. Cell 2013, 152, 584–598. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, R.I.; Muralikrishna, B.; Parnaik, V.K. Lamin A/C speckles mediate spatial organization of splicing factor compartments and RNA polymerase II transcription. J. Cell Biol. 2002, 159, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Ellis, D.J.; Jenkins, H.; Whitfield, W.G.; Hutchison, C.J. GST-lamin fusion proteins act as dominant negative mutants in Xenopus egg extract and reveal the function of the lamina in DNA replication. J. Cell Sci. 1997, 110, 2507–2518. [Google Scholar] [PubMed]
- Kennedy, B.K.; Barbie, D.A.; Classon, M.; Dyson, N.; Harlow, E. Nuclear organization of DNA replication in primary mammalian cells. Genes Dev. 2000, 14, 2855–2868. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.; Campbell, K.H.; Ford, C.C.; Stick, R.; Hutchison, C.J. The role of lamin LIII in nuclear assembly and DNA replication, in cell- free extracts of Xenopus eggs. J. Cell Sci. 1991, 98, 271–279. [Google Scholar] [PubMed]
- Moir, R.D.; Montag-Lowy, M.; Goldman, R.D. Dynamic properties of nuclear lamins: Lamin B is associated with sites of DNA replication. J. Cell Biol. 1994, 125, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Spann, T.P.; Moir, R.D.; Goldman, A.E.; Stick, R.; Goldman, R.D. Disruption of nuclear lamin organization alters the distribution of replication factors and inhibits DNA synthesis. J. Cell Biol. 1997, 136, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Ivorra, C.; Kubicek, M.; Gonzalez, J.M.; Sanz-Gonzalez, S.M.; Alvarez-Barrientos, A.; O’Connor, J.E.; Burke, B.; Andres, V. A mechanism of AP-1 suppression through interaction of c-Fos with lamin A/C. Genes Dev. 2006, 20, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, E.; Dechat, T.; Foisner, R.; Quinlan, R.; Hutchison, C. Lamin A/C binding protein LAP2alpha is required for nuclear anchorage of retinoblastoma protein. Mol. Biol. Cell 2002, 13, 4401–4413. [Google Scholar] [CrossRef] [PubMed]
- Pekovic, V.; Harborth, J.; Broers, J.L.; Ramaekers, F.C.; van Engelen, B.; Lammens, M.; von Zglinicki, T.; Foisner, R.; Hutchison, C.; Markiewicz, E. Nucleoplasmic LAP2alpha-lamin A complexes are required to maintain a proliferative state in human fibroblasts. J. Cell Biol. 2007, 176, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Hamanaka, R.B.; Goldman, A.E.; Lucas, C.A.; Shumaker, D.K.; Kosak, S.T.; Chandel, N.S.; Goldman, R.D. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 2011, 25, 2579–2593. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Jaalouk, D.E.; Vartiainen, M.K.; Lammerding, J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 2013, 497, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [PubMed]
- Broers, J.L.; Peeters, E.A.; Kuijpers, H.J.; Endert, J.; Bouten, C.V.; Oomens, C.W.; Baaijens, F.P.; Ramaekers, F.C. Decreased mechanical stiffness in LMNA−/− cells is caused by defective nucleo-cytoskeletal integrity: Implications for the development of laminopathies. Hum. Mol. Genet. 2004, 13, 2567–2580. [Google Scholar] [CrossRef] [PubMed]
- Lammerding, J.; Fong, L.G.; Ji, J.Y.; Reue, K.; Stewart, C.L.; Young, S.G.; Lee, R.T. Lamins A and C but not lamin B1 regulate nuclear mechanics. J. Biol. Chem. 2006, 281, 25768–25780. [Google Scholar] [CrossRef] [PubMed]
- Lammerding, J.; Schulze, P.; Takahashi, T.; Kozlov, S.; Sullivan, T.; Kamm, R.; Stewart, C.; Lee, R. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Invest. 2004, 113, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Hale, C.M.; Panorchan, P.; Khatau, S.B.; George, J.P.; Tseng, Y.; Stewart, C.L.; Hodzic, D.; Wirtz, D. Nuclear lamin A/C deficiency induces defects in cell mechanics, polarization, and migration. Biophys. J. 2007, 93, 2542–2552. [Google Scholar] [CrossRef] [PubMed]
- Willis, N.D.; Cox, T.R.; Rahman-Casans, S.F.; Smits, K.; Przyborski, S.A.; van den Brandt, P.; van Engeland, M.; Weijenberg, M.; Wilson, R.G.; de Bruine, A.; et al. Lamin A/C is a risk biomarker in colorectal cancer. PLoS ONE 2008, 3, e2988. [Google Scholar] [CrossRef] [PubMed]
- Luderus, M.E.; de Graaf, A.; Mattia, E.; den Blaauwen, J.L.; Grande, M.A.; de Jong, L.; van Driel, R. Binding of matrix attachment regions to lamin B1. Cell 1992, 70, 949–959. [Google Scholar] [CrossRef]
- Luderus, M.E.; den Blaauwen, J.L.; de Smit, O.J.; Compton, D.A.; van Driel, R. Binding of matrix attachment regions to lamin polymers involves single-stranded regions and the minor groove. Mol. Cell. Biol. 1994, 14, 6297–6305. [Google Scholar] [CrossRef] [PubMed]
- Shoeman, R.L.; Traub, P. The in vitro DNA-binding properties of purified nuclear lamin proteins and vimentin. J. Biol. Chem. 1990, 265, 9055–9061. [Google Scholar] [PubMed]
- Goldberg, M.; Harel, A.; Brandeis, M.; Rechsteiner, T.; Richmond, T.J.; Weiss, A.M.; Gruenbaum, Y. The tail domain of lamin Dm0 binds histones H2A and H2B. Proc. Natl. Acad. Sci. USA 1999, 96, 2852–2857. [Google Scholar] [CrossRef] [PubMed]
- Hoger, T.H.; Krohne, G.; Kleinschmidt, J.A. Interaction of Xenopus lamins A and LII with chromatin in vitro mediated by a sequence element in the carboxyterminal domain. Exp. Cell Res. 1991, 197, 280–289. [Google Scholar] [CrossRef]
- Taniura, H.; Glass, C.; Gerace, L. A chromatin binding site in the tail domain of nuclear lamins that interacts with core histones. J. Cell Biol. 1995, 131, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Baricheva, E.A.; Berrios, M.; Bogachev, S.S.; Borisevich, I.V.; Lapik, E.R.; Sharakhov, I.V.; Stuurman, N.; Fisher, P.A. DNA from Drosophila melanogaster beta-heterochromatin binds specifically to nuclear lamins in vitro and the nuclear envelope in situ. Gene 1996, 171, 171–176. [Google Scholar] [CrossRef]
- Dittmer, T.A.; Sahni, N.; Kubben, N.; Hill, D.E.; Vidal, M.; Burgess, R.C.; Roukos, V.; Misteli, T. Systematic identification of pathological lamin A interactors. Mol. Biol. Cell 2014, 25, 1493–1510. [Google Scholar] [CrossRef] [PubMed]
- Zastrow, M.S.; Vlcek, S.; Wilson, K.L. Proteins that bind A-type lamins: Integrating isolated clues. J. Cell Sci. 2004, 117, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.A.; Glass, J.R.; Taniura, H.; Hasel, K.W.; Blevitt, J.M.; Gerace, L. The alpha-helical rod domain of human lamins A and C contains a chromatin binding site. EMBO J. 1993, 12, 4413–4424. [Google Scholar] [PubMed]
- Zhao, K.; Harel, A.; Stuurman, N.; Guedalia, D.; Gruenbaum, Y. Binding of matrix attachment regions to nuclear lamin is mediated by the rod domain and depends on the lamin polymerization state. FEBS Lett. 1996, 380, 161–164. [Google Scholar] [CrossRef]
- Schirmer, E.C.; Guan, T.; Gerace, L. Involvement of the lamin rod domain in heterotypic lamin interactions important for nuclear organization. J. Cell Biol. 2001, 153, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Östlund, C.; Bonne, G.; Schwartz, K.; Worman, H. Properties of lamin A mutants found in Emery-Dreifuss muscular dystrophy, cardiomyopathy and Dunnigan-type partial lipodystrophy. J. Cell Sci. 2001, 114, 4435–4445. [Google Scholar] [PubMed]
- Raharjo, W.; Enarson, P.; Sullivan, T.; Stewart, C.; Burke, B. Nuclear envelope defects associated with LMNA mutations cause dilated cardiomyopathy and Emery-Dreifuss muscular dystrophy. J. Cell Sci. 2001, 114, 4447–4457. [Google Scholar] [PubMed]
- Vigouroux, C.; Auclair, M.; Dubosclard, E.; Pouchelet, M.; Capeau, J.; Courvalin, J.-C.; Buendia, B. Nuclear envelope disorganization in fibroblasts from lipodystrophic patients with heterozygous R482Q/W mutations in the lamin A/C gene. J. Cell Sci. 2001, 114, 4459–4468. [Google Scholar] [PubMed]
- Shelby, R.D.; Hahn, K.M.; Sullivan, K.F. Dynamic elastic behavior of alpha-satellite DNA domains visualized in situ in living human cells. J. Cell Biol. 1996, 135, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Sullivan, K.F.; Wahl, G.M. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol. 1998, 8, 377–385. [Google Scholar] [CrossRef]
- Chubb, J.R.; Boyle, S.; Perry, P.; Bickmore, W.A. Chromatin motion is constrained by association with nuclear compartments in human cells. Curr. Biol. 2002, 12, 439–445. [Google Scholar] [CrossRef]
- Allan, C.; Burel, J.M.; Moore, J.; Blackburn, C.; Linkert, M.; Loynton, S.; Macdonald, D.; Moore, W.J.; Neves, C.; Patterson, A.; et al. OMERO: Flexible, model-driven data management for experimental biology. Nat. Methods 2012, 9, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Zuleger, N.; Boyle, S.; Kelly, D.A.; de Las Heras, J.I.; Lazou, V.; Korfali, N.; Batrakou, D.G.; Randles, K.N.; Morris, G.E.; Harrison, D.J.; et al. Specific nuclear envelope transmembrane proteins can promote the location of chromosomes to and from the nuclear periphery. Genome Biol. 2013, 14, R14. [Google Scholar] [CrossRef] [PubMed]
- Makatsori, D.; Kourmouli, N.; Polioudaki, H.; Shultz, L.D.; McLean, K.; Theodoropoulos, P.A.; Singh, P.B.; Georgatos, S.D. The inner nuclear membrane protein lamin B receptor forms distinct microdomains and links epigenetically marked chromatin to the nuclear envelope. J. Biol. Chem. 2004, 279, 25567–25573. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Worman, H.J. Interaction between an integral protein of the nuclear envelope inner membrane and human chromodomain proteins homologous to Drosophila HP1. J. Biol. Chem. 1996, 271, 14653–14656. [Google Scholar] [PubMed]
- Maul, G.G.; Negorev, D.; Bell, P.; Ishov, A.M. Review: Properties and assembly mechanisms of ND10, PML bodies, or PODs. J. Struct. Biol. 2000, 129, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Maraldi, N.M.; Squarzoni, S.; Sabatelli, P.; Lattanzi, G.; Ognibene, A.; Manzoli, F.A. Emery-Dreifuss muscular dystrophy, nuclear cell signaling and chromatin remodeling. Adv. Enzyme Regul. 2002, 42, 1–18. [Google Scholar] [CrossRef]
- Rabl, C. Über Zelltheilung. Morphol. Jahrb. 1885, 10, 214–330. [Google Scholar]
- Hadlaczky, G.; Went, M.; Ringertz, N.R. Direct evidence for the non-random localization of mammalian chromosomes in the interphase nucleus. Exp. Cell Res. 1986, 167, 1–15. [Google Scholar] [CrossRef]
- Scherthan, H.; Weich, S.; Schwegler, H.; Heyting, C.; Harle, M.; Cremer, T. Centromere and telomere movements during early meiotic prophase of mouse and man are associated with the onset of chromosome pairing. J. Cell Biol. 1996, 134, 1109–1125. [Google Scholar] [CrossRef] [PubMed]
- Weimer, R.; Haaf, T.; Kruger, J.; Poot, M.; Schmid, M. Characterization of centromere arrangements and test for random distribution in G0, G1, S, G2, G1, and early S’ phase in human lymphocytes. Hum. Genet. 1992, 88, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.; Ward, D.C. Cell cycle dependent chromosomal movement in pre-mitotic human T-lymphocyte nuclei. Chromosoma 1992, 101, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Olins, A.; Herrmann, H.; Lichter, P.; Kratzmeier, M.; Doenecke, D.; Olins, D. Nuclear envelope and chromatin compositional differences comparing undifferentiated and retinoic acid- and phorbol ester-treated HL-60 cells. Exp. Cell Res. 2001, 268, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Aquiles Sanchez, J.; Karni, R.J.; Wangh, L.J. Fluorescent in situ hybridization (FISH) analysis of the relationship between chromosome location and nuclear morphology in human neutrophils. Chromosoma 1997, 106, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Chaly, N.; Munro, S.B. Centromeres reposition to the nuclear periphery during L6E9 myogenesis in vitro. Exp. Cell Res. 1996, 223, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Olszewska, M.; Wiland, E.; Kurpisz, M. Positioning of chromosome 15, 18, X and Y centromeres in sperm cells of fertile individuals and infertile patients with increased level of aneuploidy. Chromosome Res. 2008, 16, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Zhou, Z.; Wang, Y.; Wang, J.; Kallgren, S.P.; Kurchuk, T.; Miller, E.A.; Chang, F.; Jia, S. Csi1 links centromeres to the nuclear envelope for centromere clustering. J. Cell Biol. 2012, 199, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Taimen, P.; Pfleghaar, K.; Shimi, T.; Moller, D.; Ben-Harush, K.; Erdos, M.R.; Adam, S.A.; Herrmann, H.; Medalia, O.; Collins, F.S.; et al. A progeria mutation reveals functions for lamin A in nuclear assembly, architecture, and chromosome organization. Proc. Natl. Acad. Sci. USA 2009, 106, 20788–20793. [Google Scholar] [CrossRef] [PubMed]
- Wiblin, A.E.; Cui, W.; Clark, A.J.; Bickmore, W.A. Distinctive nuclear organisation of centromeres and regions involved in pluripotency in human embryonic stem cells. J. Cell Sci. 2005, 118, 3861–3868. [Google Scholar] [CrossRef] [PubMed]
- De Vos, W.H.; Houben, F.; Kamps, M.; Malhas, A.; Verheyen, F.; Cox, J.; Manders, E.M.; Verstraeten, V.L.; van Steensel, M.A.; Marcelis, C.L.; et al. Repetitive disruptions of the nuclear envelope invoke temporary loss of cellular compartmentalization in laminopathies. Hum. Mol. Genet. 2011, 20, 4175–4186. [Google Scholar] [CrossRef] [PubMed]
- Pluta, A.F.; Earnshaw, W.C.; Goldberg, I.G. Interphase-specific association of intrinsic centromere protein CENP-C with HDaxx, a death domain-binding protein implicated in Fas-mediated cell death. J. Cell Sci. 1998, 111, 2029–2041. [Google Scholar] [PubMed]
- Ishov, A.M.; Sotnikov, A.G.; Negorev, D.; Vladimirova, O.V.; Neff, N.; Kamitani, T.; Yeh, E.T.; Strauss, J.F., 3rd; Maul, G.G. PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol. 1999, 147, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Earnshaw, W.C.; Pluta, A.F.; Sternsdorf, T.; Ainsztein, A.M.; Carmena, M.; Ruchaud, S.; Hsu, W.L.; Orr, A. A dynamic connection between centromeres and ND10 proteins. J. Cell Sci. 1999, 112, 3443–3454. [Google Scholar] [PubMed]
- Hollenbach, A.D.; McPherson, C.J.; Mientjes, E.J.; Iyengar, R.; Grosveld, G. Daxx and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. J. Cell Sci. 2002, 115, 3319–3330. [Google Scholar] [PubMed]
- Zink, D.; Bornfleth, H.; Visser, A.; Cremer, C.; Cremer, T. Organization of early and late replicating DNA in human chromosome territories. Exp. Cell Res. 1999, 247, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Pope, B.D.; Ryba, T.; Dileep, V.; Yue, F.; Wu, W.; Denas, O.; Vera, D.L.; Wang, Y.; Hansen, R.S.; Canfield, T.K.; et al. Topologically associating domains are stable units of replication-timing regulation. Nature 2014, 515, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, Y.; Minden, J.S.; Swedlow, J.R.; Sedat, J.W.; Agard, D.A. Focal points for chromosome condensation and decondensation revealed by three-dimensional in vivo time-lapse microscopy. Nature 1989, 342, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Heald, R.; McKeon, F. Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell 1990, 61, 579–589. [Google Scholar] [CrossRef]
- Furukawa, K. LAP2 binding protein 1 (L2BP1/BAF) is a candidate mediator of LAP2- chromatin interaction. J. Cell Sci. 1999, 112, 2485–2492. [Google Scholar] [PubMed]
- Robson, M.I.; de Las Heras, J.I.; Czapiewski, R.; Le Thanh, P.; Booth, D.G.; Kelly, D.A.; Webb, S.; Kerr, A.R.; Schirmer, E.C. Tissue-Specific Gene Repositioning by Muscle Nuclear Membrane Proteins Enhances Repression of Critical Developmental Genes during Myogenesis. Mol. Cell 2016, 62, 834–847. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dixon, C.R.; Platani, M.; Makarov, A.A.; Schirmer, E.C. Microinjection of Antibodies Targeting the Lamin A/C Histone-Binding Site Blocks Mitotic Entry and Reveals Separate Chromatin Interactions with HP1, CenpB and PML. Cells 2017, 6, 9. https://doi.org/10.3390/cells6020009
Dixon CR, Platani M, Makarov AA, Schirmer EC. Microinjection of Antibodies Targeting the Lamin A/C Histone-Binding Site Blocks Mitotic Entry and Reveals Separate Chromatin Interactions with HP1, CenpB and PML. Cells. 2017; 6(2):9. https://doi.org/10.3390/cells6020009
Chicago/Turabian StyleDixon, Charles R., Melpomeni Platani, Alexandr A. Makarov, and Eric C. Schirmer. 2017. "Microinjection of Antibodies Targeting the Lamin A/C Histone-Binding Site Blocks Mitotic Entry and Reveals Separate Chromatin Interactions with HP1, CenpB and PML" Cells 6, no. 2: 9. https://doi.org/10.3390/cells6020009