Modes of Interaction of KMT2 Histone H3 Lysine 4 Methyltransferase/COMPASS Complexes with Chromatin


Institute of Biochemistry and Molecular Biology, Medical School, RWTH Aachen University, Pauwelsstrasse 30, 52057 Aachen, Germany
*
Author to whom correspondence should be addressed.
Cells 2018, 7(3), 17; https://doi.org/10.3390/cells7030017
Submission received: 18 January 2018
/
Revised: 22 February 2018
/
Accepted: 27 February 2018
/
Published: 2 March 2018
(This article belongs to the Section Cell Nuclei: Function, Transport and Receptors)
Abstract
:Regulation of gene expression is achieved by sequence-specific transcriptional regulators, which convey the information that is contained in the sequence of DNA into RNA polymerase activity. This is achieved by the recruitment of transcriptional co-factors. One of the consequences of co-factor recruitment is the control of specific properties of nucleosomes, the basic units of chromatin, and their protein components, the core histones. The main principles are to regulate the position and the characteristics of nucleosomes. The latter includes modulating the composition of core histones and their variants that are integrated into nucleosomes, and the post-translational modification of these histones referred to as histone marks. One of these marks is the methylation of lysine 4 of the core histone H3 (H3K4). While mono-methylation of H3K4 (H3K4me1) is located preferentially at active enhancers, tri-methylation (H3K4me3) is a mark found at open and potentially active promoters. Thus, H3K4 methylation is typically associated with gene transcription. The class 2 lysine methyltransferases (KMTs) are the main enzymes that methylate H3K4. KMT2 enzymes function in complexes that contain a necessary core complex composed of WDR5, RBBP5, ASH2L, and DPY30, the so-called WRAD complex. Here we discuss recent findings that try to elucidate the important question of how KMT2 complexes are recruited to specific sites on chromatin. This is embedded into short overviews of the biological functions of KMT2 complexes and the consequences of H3K4 methylation.
1. Introduction
Gene expression is affected by many endogenous and exogenous signals and cues that help to shape the phenotypic traits of cells and distinguish between cell types as well as different metabolic and disease states. In eukaryotes, chromatin as a dynamic and complex three-dimensional structure, controls gene expression through the precise packaging of DNA combined with many layers of signaling and regulatory factors. These complex regulations support the development of over 200 different cell types in the mammalian organism [1]. The fundamental unit of chromatin is the nucleosome, which is composed of two copies of each of the four core histones (H2A, H2B, H3, H4 and several variants thereof) and a 147 base pair fragment of genomic DNA wrapped around the histone octamer [2]. Histones are small, alkaline proteins with a globular structure, while the N-terminal tails of core histones are unstructured, highly conserved regions of these proteins. Core histones and in particular their N-terminal tails are modified by many different post-translational modifications (PTMs). A large number of histone PTMs (aka histone marks) have been described, including methylation and several others such as acetylation, acylations, phosphorylation, ADP-ribosylation, β-N-acetylglucosaminylation, ubiquitinylation, and sumoylation (for review [3,4]). These PTMs control the interactions of histone and their N-terminal tails with chromatin, co-factors and transcriptional regulators, and affect the stability and positioning of nucleosomes [5]. Thus, PTMs of core histones can function as road signs for transcription factors and chromatin remodeling complexes, controlling all aspects of gene transcription. Moreover, histone modifications are involved in processes such as DNA replication [6], chromatin condensation during mitosis [7], and the response to DNA damage [8]. Deregulated modification of core histones by PTMs as well as mutations in core histones that affect their modification have been linked to diseases, including various types of cancers and neurological and autoimmune disorders [9,10,11]. While for some histone marks the molecular consequences are well characterized, for many the functional relevance is only poorly defined. Moreover, the cross-talk of individual histone marks is only slowly being unraveled, leaving open many opportunities to study distinct PTMs that all together are proposed to create a histone code [12,13].
One of the best-studied PTMs of core histones is methylation of lysine (K) residues. Various lysines have been demonstrated to be mono-(me1), di-(me2), and/or tri-methylated (me3), a modification that is catalyzed by different lysine methyltransferases (KMTs), referred to as writers. Although methylation of core histones was discovered in 1964 [14], the first enzyme, SUV39H1, which was demonstrated to transfer methyl groups onto lysine 9 of histone H3 (H3K9), was identified in 2000 [15]. Finding proteins able to erase methylation marks was challenging. Initially it was speculated that methylation can only be removed when the modified core histones are recycled. However, in 2004, the first histone demethylase was discovered [16], demonstrating that lysine methylation is a reversible PTM. Since then around 30 KMTs [17] and over 20 lysine demethylases (KDMs or erasers) [18] have been discovered and their different capabilities and specificities are being characterized.
The molecular analysis of some of the histone lysine methylations has allowed categorizing their functional relevance broadly either in gene activation or in gene repression. For example, tri-methylation of H3K9 and H3K27 are typically associated with gene repression [19,20], while methylation of H3K4 and H3K36 are strongly connected with open chromatin and gene expression [21,22]. Here we will address H3K4 methylation, which is catalyzed predominantly by MLL and SET1 enzymes (summarized as KMT2 for lysine-specific methyltransferases subclass 2 [23]). These enzymes function in complexes referred to as KMT2 complexes or COMPASS (complex of proteins associated with Set1), first identified in yeast [24,25]. While yeast has one, mammals possess 6 different KMT2 complexes that have been identified. In this review, we will discuss the different mammalian KMT2 complexes. First, we will briefly summarize the functional activities of the different complexes and refer to reviews that describe various issues in more detail. In view of the importance of H3K4 methylation both at promoters and at enhancers, a relevant question is how KMT2 complexes are recruited to DNA and chromatin and how locus-specific effects are achieved. We will focus on the various mechanisms that have been described to target these complexes to specific chromosomal sites.
2. Methylation of Histone H3 at Lysine 4
Lysine methylation is catalyzed by KMTs, which contain an evolutionary conserved SET domain, which was named after the first three identified proteins harboring this domain in Drosophila, i.e., Su(var)3-9, Enhancer-of-zeste and Trithorax [26]. SET domains transfer methyl groups from S-adenosyl-l-methionine (SAM) onto the Nε group of a lysine in substrate proteins [27]. H3K4 can be mono-, di- or tri-methylated (H3K4me1, me2 or me3, respectively) by different enzymes that include mixed-lineage leukemia (MLL1-4 or KMT2A-D, respectively) and SET1A/B (KMT2F and G, respectively) methyltransferases (MTases) (Figure 1) [25,28,29]. KMT2 enzymes are considered the main H3K4 MTases [30]. Additional enzymes described to modify H3K4 are ASH1 (absent, small, or homeotic disks protein 1), SMYD1–3 (SET and MYND domain containing 1–3), SET7/9 (SET domain containing lysine methyltransferase 7), and PRDM9 (PR/SET domain 9) (Table 1) [29]. The ASH1L protein was suggested to modify H3K4 based on peptide work [31,32]. More detailed analysis using nucleosomes as substrates and functional studies suggest strongly that ASH1L is methylating predominantly H3K36, as with H3K4 methylation a positive histone mark [33]. PRDM9 (aka Meisetz) tri-methylates H3K4 in the context of recombination [34,35]. SET7/9 can mono-methylate H3K4 on isolated histones and peptides but shows only weak activity on nucleosomes. Its main function was suggested to be in methylating non-histone proteins ([36] and references therein). SMYD1–3 methylate H3K4, however, these activities appear weak compared to other sites in core histones and non-histone substrates. Moreover, the activity towards H3K4 appears locus-specific [37,38]. Together these findings support the notion that KMT2 complexes are the main cellular H3K4 MTases controlling gene transcription (Table 1).
The catalytic subunits of KMT2 complexes show little catalytic activity when analyzed in isolation [28,30]. All 6 MTases, MLL1-4 and SET1A/B, associate in cells with the four core subunits WDR5 (WD repeat domain 5), RbBP5 (Retinoblastoma binding protein 5), ASH2L (absent, small or homeotic 2-like), and DPY30 (Dumpy-30) (or for short WRAD) (Figure 1) [50]. The WRAD complex interacts with the SET domain of KMT2 enzymes and the core subunits are required for efficient catalytic activity in vitro and in cells (Figure 2). Biochemical reconstitution experiments have demonstrated that the WRAD complex stimulates the catalytic activity of the MTases up to several hundred-fold. Thus, the WRAD complex serves as a modulatory platform [51,52,53,54,55,56,57,58,59]. In addition to regulating activity, the components of the WRAD complex are contributing to targeting KMT2 complexes to chromatin through various mechanisms (see below). Of note is also that the WRAD complex can assemble in the absence of a KMT2 subunit [52,58,60]. Interestingly, WRAD complex proteins appear to be more abundant compared to all KMT2s taken together, which supports previous findings that the WRAD complex as well as the RAD sub-complex may exist separately and carry additional functions beyond their role within KMT2 complexes [50].
Further analyses revealed that additional subunits exist that are not shared among all KMT2 complexes [50]. The unique subunits of KMT2A and B are Menin and LEDGF (lens epithelium-derived growth factor, aka PSIP1/p75) [64,65,66]. For KMT2C and D, unique subunits are PTIP (pax transactivation domain-interacting protein) and NCOA6 (nuclear receptor coactivator 6) [67,68,69]. Finally, KMT2F and G are associated with CFP1 (CxxC finger protein 1) and WDR82 (WD repeat domain 82) [50,70]. Together, these observations suggest that KMT2 complexes are divers and may elicit binding characteristics for specific regions of the chromatin and/or have additional functions beyond H3K4 methylation that presently are only poorly understood [28,71].
The above summarized unique subunits of the different KMT2 complexes suggest three subgroups, with KMT2A and B, KMT2C and D, and KMT2F and G. This is consistent with domain organization and the methylation specificity of these three subgroups (Figure 1 and Table 1). The recombinant A/B complexes are predominantly mono- and di-methylating H3K4 with low tri-methylation activity [72]. In cells, however, these complexes are important for H3K4me3 [73]. The C/D complexes are preferentially mono-methylating H3K4 [74,75,76,77,78,79]. Finally, the F/G complexes show tri-methylation activity [80]. These findings agree with the localization of KMT2 complexes in chromatin in comparison to the distribution of distinct H3K4 methylations. The F/G complexes are found at promoters as is the H3K4me3 mark [81,82]. In contrast, the C/D complexes are localized to enhancer regions, which are enriched for H3K4me1 [74,83]. Moreover, these findings are consistent with methylation of H3K4 being associated with open chromatin, i.e., as pointed out above, H3K4me1 and H3K4me2/3 as marks for accessible enhancer regions and promoters, respectively [25,84,85]. In contrast, H3K4me1 located in promoter regions is thought to have repressive functions. Whether this is due to the recruitment of KMT2 complexes that specifically mono-methylate, i.e., KMT2C and D, has not been clarified. Rather it is thought that H3K4me1 is the remaining methylation in response to the activity of demethylases that removed one or two methyl-groups from H3K4me2/3 [86].
Methylation of H3K4 is reversed by several demethylases, including LSD1 (KDM1) and four different JARID1 proteins (KDM5A-D) (Table 1) [87,88,89,90,91,92,93]. The number of enzymes capable to methylate/demethylate H3K4 supports the notion that this amino acid and its modification are important to control gene transcription.
3. KMT2 Complexes Are Essential for Development and Are Affected in Diseases
Knockout studies of different KMT2 family members have demonstrated the importance of these enzymes for mouse development and their functionality in different tissues. Well studied is MLL1 (KMT2A). Its homozygous deletion is embryonically lethal, whereas the Mll1+/− heterozygotes show retarded growth and several additional abnormalities [94,95]. Together with its well-documented role in specific forms of leukemia, it has been suggested that MLL1 is particularly relevant in the hematopoietic system [96]. Indeed, several studies have demonstrated a key role for MLL1 in hematopoietic stem and progenitor cells [97,98,99,100]. Mll2 (KMT2B), the closest homolog of Mll1, is essential for early embryonic development in the mouse [101]. However, the phenotypes are different as for example different Hox genes are deregulated in Mll1 and Mll2 knockout cells. Moreover, MLL2 is required in oocytes and during spermatogenesis but not late in embryogenesis and during homeostasis of somatic cells [42,102]. Recently, SET1A and B were also shown to be essential, again with different phenotypes [45]. This indicates that despite the homology between the different KMT2 enzymes, in particular also the high homology between SET1A and B, and between MLL1 and MLL2 (Figure 1), these MTases are functionally different. Aspects that are potentially relevant are their expression pattern, their substrate specificity also beyond H3K4, and differences in the components of the protein complexes: The latter may be particularly relevant to position KMT2 and associated factors to specific sites in the chromatin, an aspect that has been hampered at least in part by the lack of high-quality antibodies specific for the different KMT2 enzymes.
Some subunits of the WRAD complex have also been analyzed by knockout strategies in mice. Ash2l is essential for mouse embryonal development [103]. The embryos die very early in development at a pre-implantation stage indicating that this protein fulfills an essential function. Of note are that heterozygous animals were normal ([103] and our unpublished findings). The inducible knockout of Ash2l in mouse hepatocytes provokes a disintegration of the liver and subsequent death of the animals [104]. A recent study indicates that Dpy30 is necessary in hematopoiesis [105]. Loss of Dpy30 results in the accumulation of hematopoietic stem and progenitor cells, while peripheral cells are depleted. This is consistent with the analysis of Mll1, which indicated that this MTase is required for proper development of the hematopoietic system [73]. Thus, the components of the WRAD complex, which have been analyzed thus far by knockout studies, are essential for mouse and/or organ development. It is tempting to speculate that these effects are due to the activating functions of Ash2l and Dpy30 in KMT2 complexes. However, other activities might be relevant as well, including targeting to chromatin sites and functions independent of KMT2 complexes (see below).
Many of the enzymes that control H3K4 methylation are involved in diseases, most prominently cancer [28,106]. The association of KMT2 complexes with cancer is best studied with regards to MLL1 as translocations of the MLL1 gene are associated with around 10% of human leukemias [107]. MLL1 translocation may occur as a failure in DNA double strand repair and have been shown to provoke MLL1 fusion to over 80 different partners. Frequently the fusion protein partners are involved, directly or indirectly, in the recruitment of DOT1L (disruptor of telomeric silencing 1-like), a H3K79 dimethyltransferase. H3K79me2 has multiple functions, both in activation and repression of gene transcription, but its role in transformation is not fully understood [108]. Some fusion partner regulate transcriptional elongation of RNA polymerase II (RNAP II) through interaction with the so-called super elongation complex (SEC) [109]. The MLL-SEC interaction stabilizes abnormally the localization of the whole complex on MLL target genes thus misregulating the transcription elongation checkpoint control (TECC) step and the release of RNAP II. The cooperation with SEC might partially explain at least some MLL1 translocations, which have comparable phenotypes. Nevertheless, the transforming activity of some MLL fusion proteins is still elusive [110,111]. MLL1 mutations are also linked to the rare Wiedemann-Steiner syndrome [112,113,114]. Moreover, other KMT2 methyltransferase genes have also been connected to tumor development (e.g., [115,116,117]) and mutations in MLL2 contribute to the Kabuki syndrome (e.g., [118]). In addition to the methyltransferases, other components of the KMT2 complexes appear to contribute to cancer or cancer related processes, well documented for Menin [119,120]. ASH2L cooperates with an activated form of RAS in transforming rat embryo fibroblasts [121]. In support of a potential role of ASH2L in tumor formation, ASH2L protein but not mRNA is overexpressed in the large majority of human tumors of different origin [121,122]. Consistent with these findings, low ASH2L expression correlates with a favorable prognosis in patients with acute myeloid leukemia [123]. Similarly, high expression of WDR5, particularly when combined with high expression of MLL1, is correlated with high-risk acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML) [124].
A variety of AML and ALL treatment strategies have been developed, which include the inhibition of KMT2 complexes through their interactors or fusion partners [125]. Targeting can be achieved by inhibiting the interaction with the WRAD complex, i.e., the binding to WDR5 [40,126,127], with other KMT2 complex partner including Menin [128,129] or MYC [130], as well as with DOT1L, the only known H3K79 methyltransferase [131,132]. Moreover, inhibition of the KMT2 antagonist LSD1 (lysine-specific demethylase 1) [133] and of chromatin modifiers belonging to the BET (bromodomain and extra terminal) family [134] and others [135,136] have also been suggested to be of therapeutic relevance. Through these various treatments, the proliferation of leukemic cells can be modulated, and/or the cells are sensitized to chemotherapy.
Together, the findings summarized above document the importance of KMT2 enzymes and associated factors in embryogenesis and in normal organ development and maintenance. This is further supported by the many genetic alterations associated with the genes encoding these factors that are linked to diseases.
4. Recruitment of KMT2 Complexes to Genes
Considering the close correlation of H3K4 methylation with open chromatin and gene expression, and of the essential functions of the KMT2 methyltransferase complexes, an important question is how specificity is achieved. In other words, how are KMT2 complexes selectively recruited to distinct sites on chromatin to catalyze gene-specific effects. Moreover, how are individual H3K4 methylation marks, i.e., mono-, di- and tri-methylation, generated at these distinct chromatin sites. H3K4me2/3 marks are positioned near transcription start site (TSS) of genes that are either actively transcribed or are in a state that allows their rapid activation [78,137]. In contrast, the distribution of H3K4me1 is broader, often at distant, active regulatory elements such as enhancers [79], while H3K4me1 appears to be a sign of gene silencing when localized in the proximity of TSSs (see also above) [86]. These locations of the different H3K4 methylation marks also relate to the question regarding their functional consequences, which we will briefly discuss below.
Several possibilities can be envisaged regarding how large multi-subunit KMT2 complexes are recruited to specific loci, i.e., in particular to promoters and enhancers. These possibilities include the recruitment by sequence-specific transcription factors (sTFs), specific forms of chromatin that are e.g., defined by certain patterns of histone marks, transcriptional co-factors, basal transcription factors (bTFs), and long non-coding RNAs (lncRNAs) (Figure 3). Considering the common and the unique subunits of the 6 different KMT2 complexes, site-specific recruitments are likely to occur through the latter, while the common subunits, i.e., the WRAD core complex, might contribute to stabilizing the interactions. In the following we discuss the different possibilities.
4.1. Recruitment of KMT2 Complexes by Sequence-Specific Transcription Factors
Arguably the most obvious mechanism is the recruitment of KMT2 complexes by sTFs, similar to many other co-factors that affect chromatin and ultimately control gene expression [138]. This would suggest that once an activating sTF has recognized its response element, e.g., as an enhancer or promoter element, binding of KMT2 complexes would represent part of the assembly of different transcriptional co-factors that modify the surrounding chromatin and contribute to the activity of a given locus (Figure 3). sTFs can bind to promoter proximal and distal response elements. It is possible that at proximal and distal sites distinct KMT2 complexes are recruited, i.e., complexes that result in H3K4me2/3 and H3K4me1, respectively. At present, little information is available whether sTFs can interact with distinct KMT2 complexes dependent on whether the factors bind near core promoters or at distal enhancers. Most studies refer to sTFs that interact at proximal sites and only few have been described to recruit KMT2 complexes to enhancer. Nevertheless, in a few cases site-specific recruitment of distinct KMT2 complexes have been noted and will be discussed below.
Considering the high complexity of the protein composition and the crowded environment in the nucleus, in which the many interactions must be controlled in a time and location-specific manner, locating multi-subunit complexes to specific sites is challenging. The use of different co-immunoprecipitation (co-IP) and chromatin immunoprecipitation (ChIP) protocols to study the interaction and distribution of factors and histone marks combined with in vitro interaction assays, and gene expression measurements are required to define causality of functionally relevant assemblies of proteins at selective sites in chromatin. Most of the studies that have reported KMT2 recruitments to chromatin do not allow to specify how the enzymes interact with DNA and chromatin, and thus some uncertainty about direct or indirect interaction remains. Also, indirect interactions might involve more than one component bridging KMT2 enzymes with DNA, an aspect that is difficult to analyze. It is worth remembering that protein-protein crosslinks with formaldehyde are less likely than protein-DNA crosslinks because of the proximity of the DNA binding domain of a sTF and the multiple sites on DNA that can be modified by formaldehyde. In contrast, stabilizing protein-protein interactions are limited by the typically limited number of sites that are in sufficient proximity for being crosslinked, which can make it difficult to detect proteins that are indirectly bound to DNA by ChIP [139].
As discussed above, the WRAD core complex appears to be associated with all KMT2 family proteins, which is necessary to assemble functional MTase complexes [50]. In that study, using label-free quantitative mass spectrometry, it was also noted that the WRAD complex components were more abundant than the KMT2 family members. Moreover, also the WRAD complex subunits appeared to have distinct cellular concentrations, suggesting that the different components are present in cells in part independent of KMT2 complexes. For example, WDR5, the most abundant WRAD component in HeLa cells, is in a more than 50-fold molar excess over all KMT2 enzymes [50]. Indeed, WRAD complex subunits associate with several additional factors indicating that these subunits participate in other protein-protein interactions and complexes [50]. Thus, these studies suggest that WRAD subunits need to be considered with care as they most certainly have additional functions, which might be relevant for the regulation of chromatin and gene expression independent of the activities of KMT2 MTases.
WDR5 interacts with multiple sTFs. During the self-renewal of embryonic stem (ES) cells, WDR5 interacts with OCT4 and possibly also with SOX2 and NANOG, suggesting that WDR5 binds to multiple transcription factors [140]. Upon differentiation of ES cells, WDR5 is substantially down-regulated correlating with a decrease in H3K4me3, suggesting that these sTFs, which are characteristic for ES cell self-renewal and pluripotency, recruit KMT2 complexes through WDR5. Of note, global H3K4me1 and 2 were increased upon WDR5 knockdown [140]. Because WDR5 is present in excess over KMT2 enzymes and other WRAD subunits [50], this may indicate a switch from preferentially tri-methylating complexes such as those containing SET1A/KMT2F and SET1B/KMT2G to other KMT2 complexes that catalyze mono- and di-methylation or possibly to other complexes without MTase activity. Moreover, OCT4 interacts through SOX2 with ASH2L, providing an additional mechanism to recruit KMT2 complexes and to regulate H3K4 methylation [141]. In ES cells, the recruitment of KMT2 complexes can also occur via SMAD2/3 TF complexes, with DPY30 playing an important role [142]. SMAD2/3 cooperates with NANOG in controlling H3K4me3. OCT4 also interacts with the C-terminal region of SET1A/KMT2F, which is necessary for H3K4 methylation [82]. This is independent of WDR5, suggesting that OCT4 has at least two modes of interacting with KMT2 complexes. These two modes might mediate separate functions. Moreover, OCT4 may bind to WDR5 or WDR5-containing complexes distinct from KMT2 complexes and thus cause unrelated activities.
The MYC/MAX TF complex also interacts with KMT2 complexes [143]. This correlates with activation of gene expression. MYC binds directly to ASH2L and to WDR5. The interaction with KMT2 complexes is mediated primarily by ASH2L, while WDR5 appears to promote DNA binding of MYC/MAX complexes to response elements independent of other WRAD subunits [143,144]. In the latter it will be interesting to determine how WDR5 interacts with DNA and what its specificity is. Because ASH2L binds to the C-terminal one-third and WDR5 to a central region of MYC, it is possible that both interactions occur simultaneously. Of note is also that both studies demonstrate that WDR5 and MYC co-localize at many promoters and that WDR5 is bound prior to MYC, indicating that WDR5 plays a role in recruiting MYC/MAX complexes to response elements, which might then affect local chromatin organization by modulating KMT2 complexes [143,144].
Mef2d is a transcription factor that is involved in murine muscle cell differentiation. Upon p38 MAPK-dependent signaling, the phosphorylation of Mef2d promotes the binding of KMT2 complexes through Ash2l [145]. This stimulates the expression of muscle-specific genes, which contribute to the differentiation of myoblasts into multi-nucleated myotubes. PRMT4 (protein arginine N-methyltransferase-4, aka CARM1) is an arginine-specific MTase that has been demonstrated to regulate myogenic differentiation. It was observed that PRMT4 binds to the sTFs Myogenin and Mef2c and promotes methylation of H3R17 [146,147]. A more recent finding suggests that Pax7 (paired box transcription factor) regulates Myf5 transcription to stimulate myogenic differentiation when methylated by PRMT4. This modification of the N-terminal region of Pax7 results in binding to the C-terminal fragment of KMT2A/B (MLL1/2) [148]. This is associated with KMT2 complex recruitment and stimulation of H3K4me3 at the Myf5 promoter.
Additional DNA binding proteins that recruit KMT2 complexes through ASH2L and control H3K4 methylation include NF-Y (nuclear transcription factor Y) [149], NF-E2 (nuclear transcription factor erythroid 2) [150,151], and USF1 (upstream transcription factor 1) [81]. Moreover, in mouse neuroblastoma cells Ash2l interacts with Ap2delta, which is expressed specifically in the developing retina and the central nervous system [152,153]. This interaction stimulates the expression of some of the canonical KMT2 target genes, including Hoxc8 as well as genes such as Fgfr3, whose product is important in transcriptional regulation and signal transduction [154].
Beyond the WRAD subunits, additional proteins identified in KMT2 complexes have been described to associate with sTFs. For example, KMT2C/D (MLL3/4) interact through PTIP with Pax2 and Pax5 [50,68,69,155,156]. Similarly, the nuclear receptor coactivator NcoA6, which was found in sub-stoichiometric amounts in KMT2C/D (MLL3/4) complexes [50], targets H3K4 MTase complexes to sTFs [67,157]. These include nuclear receptors such as the farnesoid x receptor (FXR) [158] and the MAFA and B transcription factors responsible for cell-specific expression of the insulin gene in islet β-cells [159]. Similarly, the tumor suppressor and transcriptional regulator p53 may recruit KTM2 complexes through NcoA6 [160]. Other studies suggested that the histone acetyltransferase p300 is necessary for p53-KMT2 complex interaction [161,162]. Moreover, ASH2L is involved in p53-dependent gene transcription. Depletion of ASH2L interferes with target gene expression without affecting the recruitment of RNAP II, yet phosphorylation of the C-terminal domain at serine 5 was reduced, suggesting that the activation of RNAP II is dependent on ASH2L [163]. Because other subunits of WRAD are also recruited to p53 controlled promoters and H3K4me3 is enhanced, it is likely that KMT2 complexes are associating with p53. Together, these findings indicate that p53 possesses several different options to communicate with H3K4 selective MTases.
The estrogen receptor alpha (ERα) can directly interact with KMT2D/MLL4. The relevant region in KMT2D is the LXXLL motif, which is also present in KMT2C, resulting in H3K4 MTase recruitment [164]. In support of the functional relevance of this interaction, depletion of KMT2C/D but not KMT2A/B decreased estrogen-mediated induction of HOXC6 gene expression and, complementary, depletion of ERα abolished KMT2C/D binding to the HOXC6 promoter [165]. Yet, other observations indicate KMT2A/D or KMT2A/C as the relevant MTases in the estrogen response at SR-B1 or HOXB9 promoters, respectively [166,167]. Together, these analyses suggest that the interaction of ERα with different KMT2 complexes is promoter selective, possibly due to additional transcriptional regulators that determine specificity. Menin, which is only found in KMT2A/B complexes [50], can also associate with nuclear receptors such as the ER [168,169], as well as other sTFs, including c-MYB [170], thus providing an additional mechanism to recruit KMT2 complexes and control gene expression. Interestingly, amplification of the ASH2L locus in some breast cancers appears to result in ASH2L overexpression, which is positively correlated with increased expression of ERα. This is mediated by GATA3, which binds to an enhancer of the gene encoding ERα and recruits ASH2L and a KMT2 complex [171]. The forkhead box transcription factor FOXA1 has been described to facilitate binding of ERα to response elements in breast cancer cells [172]. Recent findings suggest that FOXA1 recruits a KMT2C complex to enhancers, controlling H3K4me1 [173]. It is postulated that this affects estrogen-dependent gene expression and cell proliferation. Together, the findings summarized here indicate that multiple interactions exist between KMT2 complexes and ERα, both direct and indirect, which control the expression, DNA binding, and function of ERα.
The E2F family of sTFs is closely associated with cell cycle-specific regulation of gene expression, for example by interacting with pocket proteins such as the tumor suppressor RB (retinoblastoma protein) [174]. Through HCF-1 (herpes simplex virus host cell factor-1) KMT2A and B, and KMT2F and G are recruited by the E2F family of transcription factors in a cell-cycle selective manner [175,176]. It has been suggested that the ANCCA/ATAD2, a AAA+ ATPase coactivator complex [177], facilitates the recruitment of KMT2 complexes [175,178,179].
Above we pointed out the transcription factors capable of interacting with KMT2 complexes, which are summarized in Table 2. Some of these factors belong to the group of pioneering TFs, including Pax7 and FOXA1. These proteins can sample genomic sites within closed chromatin. Thus, pioneering sTFs result, in the presence of additional non-pioneering factors, in the activation of promoters or enhancers that are embedded in poorly accessible chromatin. This has been well described as an important regulatory mechanism during embryogenesis and the subsequent differentiation into more specialized cell types [180]. The interaction of KMT2 complexes with pioneering factors is in support of regulatory mechanisms, in which methylation at H3K4 is important in the early stages of the activation of promoters and enhancers to allow cell type-specific chromatin opening and gene expression.
RNAP II and its interaction partners are basal transcription factors. Its positioning at the core promoter makes it an important platform to interact with transcriptional co-factors that control local chromatin as well as the chromatin along the transcribed region. WDR82 was found to interact with the C-terminal domain (CTD) of RNAP II and to mediate the recruitment of SET1A/KMT2F complexes [181]. WDR82 is one of the additional subunits, besides WRAD, that associates with some but not all KMT2s, in particular KMT2F and G complexes [50]. In yeast, but so far not analyzed in mammalian cells, the Set1 complex interacts with the CTD dependent on serine 5 phosphorylation but not when un-phosphorylated or serine 2 phosphorylated [182]. KMT2A has also been demonstrated to interact indirectly with RNAP II through hPaf1/PD2, a RNAP II-associated factor, by binding to a sequence flanking the CXXC domain of the MTase (Figure 1) [183,184]. These findings indicate that RNAP II contributes to the H3K4 methylation pattern. Indeed, it has been suggested recently that the length of binding of the Set1 complex to promoters in yeast correlates with H3K4 methylation [185]. Thus, H3K4me3 at promoters, in response to e.g., sTFs that recruit KMT2 complexes, is contributing to efficient RNAP II loading [186], but also to the efficiency of transcription in response to RNAP II-mediated methylation.
4.2. Recruitment of KMT2 Complexes by lncRNAs
With the advent of deep sequencing, large numbers of lncRNAs have been identified. The number of genes encoding lncRNAs appears to exceed those encoding proteins. Although typically less abundant than mRNAs, lncRNAs are frequently capped, spliced and poly-adenylated, similar to mRNAs [187,188]. lncRNAs have diverse functions including the regulation of chromatin, gene expression and signal transduction, and are associated with disease [189,190,191,192]. Interactions of lncRNAs with other macromolecules, including proteins, RNA and DNA, have been described. This offers several possibilities how lncRNAs may contribute to regulating transcription. lncRNAs can function as scaffold molecules in the organization of transcription-associated complexes and contribute to targeting of interacting proteins such as co-factors to distinct loci. This may occur by binding to sTFs, by interacting with DNA to form a triple helix or by forming R-loops [193,194,195].
Some recent reports have revealed that KMT2 complexes are linked to lncRNAs, although the information available is still sketchy (Figure 3). A first connection between KMT2 complexes and lncRNA was established when the lncRNA HOTTIP was identified as a regulator of the expression of the HOXA gene cluster [196]. The HOTTIP gene is located at the 5′ end of the HOXA cluster and interacts with the genes that are in the 5′ half of the cluster as determined by chromosome conformation capture-carbon copy (5C) analysis, indicating looping and thus close association with the promoters and regulatory sequences of these genes. The knockdown of HOTTIP reduces the expression of these HOXA genes, suggesting that the lncRNA itself is necessary for gene expression. The knockdown also correlates with reduced H3K4me3 in the 5′ half of the HOXA cluster. From these findings it was argued that HOTTIP controls histone modifications. Indeed, HOTTIP lncRNA interacts with the WRAD subunit WDR5 and affects H3K4 methylation [196,197]. This is consistent with a key role of KMT2 complexes in the regulation of HOX genes [198]. Interestingly, HOTTIP has been found associated with cancer (e.g., [197,199,200,201] for review [202]), possibly by affecting H3K4 methylation, which would be in support of a role of KMT2 complexes in tumors (as discussed above).
In addition to genes of the HOXA cluster, also HOXB genes are regulated by a lncRNA and KMT2 complexes. HoxBlinc is encoded by a gene in the HOXB cluster and, similar to HOTTIP, the knockdown or knockout of HoxBlinc results in reduced expression of HOXB genes, accompanied by a decrease in KMT2 complex recruitment and in H3K4me3 [203]. Through this mechanism, HoxBlinc controls hematopoietic lineage commitment, but beyond this, little is known about the biological function of this lncRNA.
Fendrr is another lncRNA that interacts with KMT2 complexes [204]. Fendrr was identified as an RNA that is essential for the development of tissues that are derived from lateral mesoderm [204]. More recent observations suggest a role of Fendrr in tumorigenesis [205,206,207]. Mechanistic studies indicate that the loss of Fendrr promotes H3K4me3 and/or represses H3K27me3 at some genes. These two histone marks are important to define bivalent chromatin, which is specified by the co-occurrence of the two marks. This is characteristic for poised promoters, which can easily switch from a repressed state to either open or closed chromatin and thus allows quick adjustment to altered gene expression needs, for example in stem and cancer cells [71]. Consistent with this is the observation that Fendrr interact with KMT2 complexes and with polycomb repressive complex 2 (PRC2) in cells [208]. Thus, the effects in response to a loss of Fendrr are complex, which is reflected in the antagonistic consequences on PRC2 and KMT2 complex recruitment. As the latter was evaluated using the core component WDR5 as a surrogate of KMT2 complexes, one word of caution is indicated. Both HOTTIP and Fendrr lncRNAs have been shown to interact with WDR5 [196,204]. As discussed above, WDR5 has been reported to bind to several other proteins beyond KMT2 complexes. In particular, WDR5 interacts with many different lncRNAs, which has been suggested to occur independently of KMT2 MTases [196,209]. Mutation of the binding site for RNA in WDR5 interferes with its cellular functions without apparent effects on KMT2 complexes [209]. Thus, additional experimentation will be necessary to further evaluate the concept.
How precisely HOTTIP, HoxBlinc and Fendrr are controlling the activity and the targeting of KMT2 complexes to specific loci is not fully understood. It is possible that these lncRNAs are involved in the local organization of chromatin such as looping [210,211]. By positioning the respective lncRNA, one or several KMT2 complexes could be recruited to and brought into the vicinity of the regulated promoters. Whether the positioning of the lncRNAs are due to interaction with DNA, locally synthesized RNA or protein has not been resolved [193]. The lncRNAs may have scaffolding function and through this contribute to stabilizing different transcriptional co-factors, including KMT2 complexes, at enhancers and promoters [212].
While HOTTIP and Fendrr bind to WDR5, HoxBlinc interacts with the SET domain of some KMT2 enzymes [203]. Yeast Set1 shares with KMT2F and G an RNA recognition motive [204]. The deletion of the Set1 RNA recognition motif (RRM) causes impaired H3K4 methylation, suggesting that RNA binding is also important for recruitment and/or stabilization of the complex on chromatin in yeast [213]. Moreover, the N-SET domain of Set1 was identified as an additional RNA binding motive, which is also present in both KMT2F and G. Thus, the interaction with RNA appears to be a conserved mechanism to position KMT2 complexes on chromatin, which is potentially relevant for the site-specific targeting of these MTases.
4.3. Direct DNA Binding
In the two sections above, we summarized how sTFs and lncRNAs contribute to the recruitment and positioning of KMT2 complexes. An additional mode can be the direct interaction of these complexes with DNA (Figure 3). It is of interest to note that KMT2 MTases possess DNA binding domains. These include the CXXC zinc finger domain, the AT-hook region, and the high mobility group I (HMG-I) binding motif (Figure 1). The CXXC domain is present in a number of proteins and has been found to interact with non-methylated CpG-rich DNA [214]. CpG islands function frequently as promoters and in their non-methylated state correlate with H3K4me3 and histone acetylation, marks of open, transcriptionally active genes [215]. In contrast, methylation of CpG islands results typically in repression of promoter activity and gene transcription [216]. Indeed, the CXXC domains of KMT2A and B bind to non-methylated CpG sequences [217,218,219,220]. This is also consistent with KMT2A protecting CpG clusters from becoming methylated [221]. Moreover, the binding of KMT2B complexes with CpG-rich regions promotes their H3K4me3 and this correlates with increased expression at least on some genes [220]. In addition to KMT2A and B, CFP1 is another protein that contains a CXXC domain [214]. This is worth noting because CFP1 is part of KMT2G and F complexes [50]. These two MTases do not possess any recognizable DNA binding domains and thus it is possible that they use CFP1 to directly interact with CpG-rich DNA. Therefore, these findings suggest that the CXXC zinc finger domain is sufficient for KMT2 complexes to interact with non-methylated CpG-rich DNA and thus can potentially target most human promoters. An obvious question is how selectivity is achieved, in addition to methylating CpG-rich DNA [215]. Are additional factors, for example sTFs or additional chromatin marks, required for efficient KMT2 complex recruitment? Another possibility is that once a KMT2 complex is bound its activity is controlled in a promoter-specific fashion. It will be interesting to evaluate the different options.
AT-hooks and HMG domains were originally discovered in high-mobility group proteins, which are non-histone chromatin proteins. High-mobility group proteins have multiple functions in chromatin organization, in part by competing with histone H1 and by interacting with sTFs, but also by binding to RNA components [222,223]. HMG domains come in different flavors, with AT-hooks as in KMT2A and HMG-I domains as in KMT2C and D being highly related [224]. AT-hooks and HMG-I domains are short motifs that interact with the minor groove of AT-rich DNA sequences [222,225,226]. Neither individual HMG domains nor AT-hooks are usually sufficient for selectively locating proteins to DNA, but support promoter recognition and stabilize DNA binding. The HMG domain as well as AT-hooks of KMT2s can also recognize so called cruciform DNA, four way junctions that can develop during multiple processes including replication and transcription [227]. Cruciform DNA can occur when longer than 6 bp inverted repeats in DNA sequences are extruded from the super-helical DNA. Such inverted repeats have been found in many promoter regions and near replication start sites potentially explaining the localization of KMT2 complexes to these sites [227]. Furthermore, AT-rich sequences may be important for regulation as they disfavor nucleosome formation, and thus allow easier access of KMT2 complexes as well as other TFs to DNA. Human promoters and enhancers are enriched in AT base pairs in comparison to the surrounding sequences [228]. Interestingly, a feature of some HGM domains is the induction of DNA bending and thus potentially juxtaposing nonadjacent sites thereby supporting DNA looping [229]. Together, through these DNA binding modes, KMT2 complexes are proposed to influence chromatin such that other transcription factors can bind more easily, a function that might even be independent of catalytic activity.
In addition to the catalytic subunits of KMT2 complexes, the WRAD subunit ASH2L has also been shown to directly interact with DNA. The crystal structure of an N-terminal fragment of ASH2L revealed a forkhead-like helix-wing-helix (HWH) domain that binds DNA [230,231]. Mutants in the DNA binding domain of ASH2L were analyzed in cells and were demonstrated to affect both H3K4 methylation and gene expression [230,231]. Similar effects were noted after knockdown or loss of ASH2L, correlating with reduced cell proliferation [104,121,232]. The DNA binding activity of ASH2L was also used to define consensus sequences. Both in vitro and in cells G-rich elements were identified [230,232]. The ASH2L-DNA binding affinity is rather weak [230,231], suggesting that this interaction may assist the recruitment of KMT2 complexes to DNA rather than being responsible for selective binding. A word of caution is that the analyzed mutants were not tested whether they support KMT2 complex formation and/or activity. The loss of H3K4 methylation and gene expression may be a consequence of KMT2 complex destabilization due to mutant ASH2L rather than effects on DNA binding, an aspect that needs to be resolved.
The findings summarized in this section suggest that KMT2 complexes can interact directly with DNA. This has implications for co-factor functioning. While most histone modifying enzymes require sTFs for site-selective recruitment, KMT2 complexes may bind directly to DNA, for example CpG islands, and thus affect chromatin at promoters and/or enhancers independent of sTFs. This offers an additional mode for H3K4 methylation at certain loci.
4.4. Interaction with Chromatin
The subunits of KMT2 complexes possess domains that suggest binding to chromatin, independent of the above summarized mechanisms that allow sequence-specific binding to certain loci (Figure 3). It is conceivable that some combinations of histone and DNA marks, including H3K4 methylation, support or antagonize the interaction with KMT2 complexes. This might be relevant for example for spreading H3K4 methylation as has been proposed for H3K27 methylation by the PRC2 and for DNA methylation [233,234,235]. H3K4 methylation is prevalent at promoters and enhancers, suggesting that these histone marks are functionally important and spreading might enhance the activity of these DNA elements. Epigenetic readers such as the plant homeodomain (PHD) finger, the double tudor domain (DTD), the MBT (malignant brain tumor) domain, the PWWP (Pro-Trp-Trp-Pro) domain, and the double chromodomain (DCD) have been described to recognize and bind to methylated H3K4 by enclosing the extended methylated side chain in a hydrophobic cage [236,237,238]. Through these different domains, it is conceivable that reading H3K4 methylation has multiple functions, including propagating a chromatin state, connecting to the basal transcription machinery and to serve as a “bookmark”.
It is interesting to note that some KMT2 MTases contain PHD fingers (Figure 1). PHD fingers are structurally conserved, represented by the canonical C4HC2C/H sequence coordinating two zinc ions. For some PHD fingers high-affinity binding was observed to certain tri-methylated lysine residues of histone H3. Therefore, KMT2s might potentially recognize the histone marks they generate via their PHD fingers and propagate H3K4 methylation further on chromatin. This could serve as part of a positive feedback loop that is independent of the underlying DNA sequence. Of the many PHD fingers only PHD3 of KMT2A exhibit H3K4me3 binding properties [236]. The other PHD fingers appear not to recognize methylated H3K4 and thus might have other functions [239], including the recognition of other PTMs [240], as protein interaction domain, and ubiquitin ligase [241].
In mixed lineage leukemia, chromosomal translocations between the gene encoding KMT2A/MLL1 and one of more than 80 partner genes have been detected. These translocations result in oncogenic fusion proteins involving the N-terminal one-third of KMT2A [242]. These fusion proteins do not contain any of the PHD fingers of KMT2A. In fact, their loss appears to be important for the transforming potential of the fusion proteins, supporting the importance of PHDs for the correct recruitment to target loci [243,244]. PHD2 of KMT2A and B have E3 ubiquitin ligase activity and can ubiquitinate the core histones H3 and H4 in vitro [241]. Whether this occurs in cells or whether other substrates are targeted and what the consequences are is still poorly understood. Furthermore, PHD3 of KMT2A was reported to interact with the RRM of cyclophilin 33 (Cyp33 or PPIE) [240]. Cyp33 competes for binding with H3K4me3, thereby promoting target gene repression [245].
The function of the KMT2C and D PHD fingers are less well explored (Figure 1). A mass spectrometry screen for methylation readers did not identify any of these PHDs as direct interactors of methylated H3K4 [246]. Instead, PHD4 and 5 of KMT2D are involved in recognizing non-methylated or asymmetrically methylated arginine 3 of histone H4 (H4R3me0 or H4R3me2a, respectively). The binding to H4R3me2a is important for KMT2D localization and activity, while symmetrically methylated H4R3 (H3R3me2s) interferes with KMT2D function [239]. Finally, it appears that KMT2F and G do not to contain domains that recognize histone modifications. However, instead, they form a complex with CFP1, which possesses a CXXC DNA binding domain as mentioned before. CFP1, conserved also in yeast (Spp1), contains additionally two PHD fingers recognizing all three states of H3K4 methylation in vitro. Depletion of CFP1 reduces H3K4me3 with no apparent effect on the other methylation states [247].
KMT2A and B possess bromodomains (Figure 1). Such domains have been described as readers of acetylated lysine residues. This histone modification neutralizes the positive charge of lysine, which affects protein binding and is associated with open chromatin and active genes [248]. However, the KMT2A bromodomain, located between PHD3 and 4, does not bind acetylated lysine residues but rather seems to modulate the activity of the PHD fingers [243]. Beyond these bromodomains no additional domains in KMT2 complex subunits have been identified that recognize acetylation marks. KMT2 can cooperate with the histone acetyl transferases (HATs) CBP/p300, which possess bromodomains [143]. Nevertheless, the inability to recognize acetylation might suggest that typically acetylation is not important for recruiting KMT2 complexes. Of note is that mutations in the p300 PHD domain affect acetylation [249]. Whether this is because of altered binding to methylated histones remains to be determined. The KMT2A fusions with CBP or p300 are transforming and promote acute myeloid leukemia [250,251,252], supporting the importance of KMT2 interaction with HATs.
WD40 domains are highly conserved structures that have been recognized to be involved in functions associated with chromatin as well as many other activities in cells [253,254]. These domains are found in WDR5 and in RBBP5. At present it is not known whether the WD40 domain in RBBP5 is involved in binding to histones. For WDR5, the WD40 domain interacts with the N-terminal end of the histone H3 tail [255]. This interaction is controlled by methylation of H3R2. While non-methylated and symmetrically di-methylated R2 (H3R2me2s) allow WDR5 binding, asymmetrically methylated R2 (H3R2me2a) prevents binding. This latter observation is consistent with H3R2me2a being selectively depleted from active promoters [256,257,258]. The pocket of WDR5 that interacts with H3R2me2s binds also to the so-called WIN motif of the SET domain of KMT2 enzymes [254]. The WIN motif and the histone H3 tail compete for binding to WDR5 [259]. H3R2me2s correlates well with H3K4me3, while H3R2me2a is depleted from H3K4me3 marked promoters [256,260,261]. Thus, it remains to be clarified what the role of WDR5 bound to H3R2me2s modified tails is in promoting H3K4 methylation. It might be that in larger KMT2 complexes structural alterations in WDR5 allow binding of both H3R2me2s tails and SET domains. WDR5 participates in other protein complexes, as discussed above, and thus may exert a function at the H3 tails independently of KMT2 complexes. An additional function of WDR5 in promoting H3K4me3 was identified by studying androgen signaling. This stimulates phosphorylation of threonine 11 of histone H3. In turn H3T11Ph is recognized by WDR5, which recruits KMT2 complexes to enhance gene expression [262]. Together these findings reveal that WDR5 is cross-talking to both methylation and phosphorylation of histone H3 tails. However, clearly, more will have to be learned about the many functions WDR5 seems to possess in controlling different aspects of gene expression.
Ubiquitination has also been known to cross-talk with H3K4 methylation. Mono-ubiquitination of several sites in the core histones H2A and H2B have been noted, with H2BK120ub1 being particularly relevant for the discussion here [263,264]. Ubiquitination of H2BK120, which is catalyzed preferentially by RNF20/hBRE1, is associated with transcribed regions, and promotes gene transcription (e.g., [265,266,267,268,269]). H2BK120ub1 stimulates the tri-methylation of H3K4 [270]. Various mechanisms are being discussed to explain the cross-talk between H2BK120ub1 and H3K4me3 [264]. In one study, ASH2L was found to mediate this cross-talk by interacting with mono-ubiquitinated nucleosomes thereby stimulating MTase activity towards H3K4 [271]. Whether ASH2L is also important to mediate recruitment of KMT2 complexes to H2BK120ub1 chromatin in cells remains to be determined. Of note is that under some conditions changes in H2BK120ub1 do not correlate with modulation of H3K4me3 [272]. Together, the findings support the notion that H2BK120ub1 can promote H3K4me3, but to define the underlying molecular mechanism will require more work.
5. Functional Consequences of H3K4 Methylation
H3K4 methylation correlates with open, accessible chromatin and polymerase loading. This suggests that these histone marks, mono-, di- and tri-methylation of H3K4, are read by factors that translate the information of these PTMs into polymerase recruitment and activity. Different domains, including Tudor and PHD, have been defined to bind to methylated H3K4 [238,246,273]. Recently, a novel reader domain for methylated H3K4 has been described, which is referred to as CryptoTudor [274]. While other domains are specific binders of different methylation states [238,275], CryptoTudor interacts equally well with H3K4me1 and me3. CryptoTudor in BRWD2/PHIP appears to be involved in controlling H3K27ac [274], which cooperates with H3K4me3 in forming bivalent chromatin [71].
One of the first observations with a clear functional effect was the identification of TFIID as a reader of H3K4me3 [276]. TFIID is a multi-subunit complex that contains TBP (TATA-box binding protein) and thus is a key complex to position the RNAP II complex on core promoters [277,278]. TAF3, TBP-associated factor 3, was identified to bind to H3K4me3 through its PHD finger and to promote gene transcription, consistent with the high prevalence of this modification at core promoters [276,279]. The effect was enhanced when H3K9 and H3K14 were acetylated (ac), but inhibited by H3R2me2a, suggesting histone mark cross-talk.
Another H3K4me3 interactor is the nucleosome remodeling factor NURF [280], an essential chromatin remodeling complex [281]. Mapping experiments revealed that one of the PHD fingers in BPTF/NURF301 was responsible for H3K4me3 binding [280]. The knockdown of BPTF and of WDR5 were phenotypically similar in developing Xenopus embryos, supporting the notion that H3K4me3 is tightly linked to NURF-dependent chromatin remodeling. Moreover, the inhibitor of growth (ING) family of proteins, which possess tumor suppressor functions, sense histone marks [282]. The PHD fingers of ING proteins interact with H3K4me3 (e.g., [283,284,285]). They can recruit HATs as well as histone deacetylase (HDAC) complexes and affect chromatin compaction [286,287]. PHF13/SPOC1 is a chromatin associated protein with a PHD domain that controls cell proliferation and differentiation [288,289]. A recent study defines PHF13 as a reader of H3K4me2/3 and, because of its ability to also interact with RNAP II that is phosphorylated at serine 5, seems to be involved in transcriptional activation and progression. Interestingly, it can also interact with bivalent chromatin and PRC2, promoting gene repression under these conditions [290].
Recent findings suggest a role for H3K4 methylation in mitosis. It is thought that H3K4me3, which is a relatively stable histone mark, is maintained during the whole cell cycle, including mitosis when the chromatin is compacted [291,292,293]. This is consistent with the finding that KMT2 complexes appear to be associated with at least some promoters during mitosis [294], and the demonstration of low level transcription during mitosis [295]. Thus, maintaining H3K4 methylation is thought to be important to preserve the gene expression pattern from the mother cell to daughter cells and KMT2 complexes behave as mitotic “bookmarking factors” [296]. As many transcription factors are released from chromatin in mitosis, after reforming the nuclear envelope and the import of TFs, H3K4 methylation marks are thought to provide information for the appropriate localization of TFs [297,298]. H3K4me2/3 is shielded in mitosis by phosphorylation of threonine 3 and 6 of histone H3 (H3T3Ph and H3T6Ph, respectively), two sites near H3K4. H3T3Ph and H3T6Ph are thought to prevent access of demethylases and more generally of PHD finger containing proteins to H3K4me3 marks [299,300,301]. Also, H3T3Ph prevents binding of TFIID [302], which is recruited by H3K4me3 [276]. Unlike H3K4me3, the enhancer mark H3K4me1 is not shielded by H3T3Ph [302,303]. Together with other histone marks, including H3K27ac and upon dephosphorylation of the neighboring threonine residues, H3K4me2/3 will guide transcription early in G1 after exiting mitosis [293].
6. Conclusions
The studies summarized here suggest that H3K4 methylation is important for regulating gene transcription. H3K4me3 and H3K4me1 correlate well with open chromatin at promoters and enhancers, respectively. Indeed, numerous studies have provided evidence for this, which could not all be mentioned here. Instead, we concentrated exemplarily on some of those processes, for which direct interaction with H3K4 methylated histone H3 was demonstrated. The recent findings indicate that the targeting of the key enzymes that methylate H3K4, the KMT2 or COMPASS complexes, occurs through their recruitment by sTFs, by certain chromatin marks and possibly by lncRNAs. In addition, some evidence for direct binding to DNA has been obtained. Thus, these findings make KMT2 complexes to very broadly recruited key chromatin regulators strongly associated with transcribed genes and their enhancers. Still, relatively few sTFs have been described as recruiters. It will be interesting to see whether sTFs are the main anchors for KMT2 complexes on chromatin or whether other mechanisms, including the combinations of other histone marks, are used to position these complexes onto specific loci. Although some information is available about crosstalk with other histone modifications, certainly much more will have to be learned about the interaction of H3K4 methylation with the many other histone marks that have been identified. Thus, it will be interesting to see the results of future studies that will certainly provide a more complete understanding of the key roles KMT2 complexes and H3K4 methylation play in controlling gene transcription.
Acknowledgments
The work in our laboratory is supported by a START grant of the Medical School of the RWTH Aachen University (to J.L.-F.), a grant from the German Cancer Aid (109061 to B.L.), and a grant from the German Research Foundation DFG (LU466/17-1 to B.L.).
Author Contributions
A.B. and B.L. wrote the first draft and all authors revised the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Valentine, J.W.; Collins, A.G.; Meyer, C.P. Morphological Complexity Increase in Metazoans. Paleobiology 1994, 2, 131–142. [Google Scholar] [CrossRef]
- Kornberg, R.D. Structure of Chromatin. Annu. Rev. Biochem. 1977, 46, 931–954. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Sabari, B.R.; Garcia, B.A.; Allis, C.D.; Zhao, Y. Snapshot: Histone Modifications. Cell 2014, 2, 458–458. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Garcia, B.A. Comprehensive Catalog of Currently Documented Histone Modifications. Cold Spring Harb. Perspect. Biol. 2015, 9, a025064. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, M.S.; Boeke, J.D.; Wolberger, C. Regulated Nucleosome Mobility and the Histone Code. Nat. Struct. Mol. Biol. 2004, 11, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.; Gurard-Levin, Z.A.; Almouzni, G.; Loyola, A. Histone Lysine Methylation and Chromatin Replication. Biochim. Biophys. Acta 2014, 12, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, B.J.; Rall, N.A.; Ostwal, Y.; Kruitwagen, T.; Hiragami-Hamada, K.; Winkler, M.; Barral, Y.; Fischle, W.; Neumann, H. A Cascade of Histone Modifications Induces Chromatin Condensation in Mitosis. Science 2014, 6166, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhu, W.G. Biological Function and Regulation of Histone and Non-Histone Lysine Methylation in Response to DNA Damage. Acta Biochim. Biophys. Sin. 2016, 7, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Ning, B.; Li, W.; Zhao, W.; Wang, R. Targeting Epigenetic Regulations in Cancer. Acta Biochim. Biophys. Sin. 2016, 1, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, R. Epigenetics in Autoimmune Diseases: Pathogenesis and Prospects for Therapy. Autoimmun. Rev. 2015, 10, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Landgrave-Gomez, J.; Mercado-Gomez, O.; Guevara-Guzman, R. Epigenetic Mechanisms in Neurological and Neurodegenerative Diseases. Front. Cell. Neurosci. 2015, 9, 58. [Google Scholar] [PubMed]
- Strahl, B.D.; Allis, C.D. The Language of Covalent Histone Modifications. Nature 2000, 6765, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Encode Project Consortium. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 7414, 57–74. [Google Scholar]
- Murray, K. The Occurrence of Iε-N-Methyl Lysine in Histones. Biochemistry 1964, 3, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.W.; Schmid, M.; Opravil, S.; Mechtler, K.; Ponting, C.P.; Allis, C.D.; et al. Regulation of Chromatin Structure by Site-Specific Histone H3 Methyltransferases. Nature 2000, 6796, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.G.; Tsukada, Y. The Discovery of Histone Demethylases. Cold Spring Harb. Perspect. Biol. 2013, 5, a017947. [Google Scholar] [CrossRef] [PubMed]
- Morera, L.; Lubbert, M.; Jung, M. Targeting Histone Methyltransferases and Demethylases in Clinical Trials for Cancer Therapy. Clin. Epigenet. 2016, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, E.; Turberfield, A.H.; Klose, R.J. Histone Demethylases in Chromatin Biology and Beyond. EMBO Rep. 2015, 12, 1620–1639. [Google Scholar] [CrossRef] [PubMed]
- Snowden, A.W.; Gregory, P.D.; Case, C.C.; Pabo, C.O. Gene-Specific Targeting of H3k9 Methylation is Sufficient for Initiating Repression In Vivo. Curr. Biol. 2002, 24, 2159–2166. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science 2002, 5595, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Humphrey, E.L.; Erlich, R.L.; Schneider, R.; Bouman, P.; Liu, J.S.; Kouzarides, T.; Schreiber, S.L. Methylation of Histone H3 Lys 4 in Coding Regions of Active Genes. Proc. Natl. Acad. Sci. USA 2002, 13, 8695–8700. [Google Scholar] [CrossRef] [PubMed]
- Chantalat, S.; Depaux, A.; Hery, P.; Barral, S.; Thuret, J.Y.; Dimitrov, S.; Gerard, M. Histone H3 Trimethylation at Lysine 36 Is Associated with Constitutive and Facultative Heterochromatin. Genome Res. 2011, 9, 1426–1437. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Berger, S.L.; Cote, J.; Dent, S.; Jenuwien, T.; Kouzarides, T.; Pillus, L.; Reinberg, D.; Shi, Y.; Shiekhattar, R.; et al. New Nomenclature for Chromatin-Modifying Enzymes. Cell 2007, 4, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.; Krogan, N.J.; Dover, J.; Erdjument-Bromage, H.; Tempst, P.; Johnston, M.; Greenblatt, J.F.; Shilatifard, A. Compass: A Complex of Proteins Associated with a Trithorax-Related Set Domain Protein. Proc. Natl. Acad. Sci. USA 2001, 23, 12902–12907. [Google Scholar] [CrossRef] [PubMed]
- Ruthenburg, A.J.; Allis, C.D.; Wysocka, J. Methylation of Lysine 4 on Histone H3: Intricacy of Writing and Reading a Single Epigenetic Mark. Mol. Cell 2007, 1, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Tschiersch, B.; Hofmann, A.; Krauss, V.; Dorn, R.; Korge, G.; Reuter, G. The Protein Encoded by the Drosophila Position-Effect Variegation Suppressor Gene Su(Var)3-9 Combines Domains of Antagonistic Regulators of Homeotic Gene Complexes. EMBO J. 1994, 16, 3822–3831. [Google Scholar]
- Schubert, H.L.; Blumenthal, R.M.; Cheng, X. Many Paths to Methyltransfer: A Chronicle of Convergence. Trends Biochem. Sci. 2003, 6, 329–335. [Google Scholar] [CrossRef]
- Shilatifard, A. The Compass Family of Histone H3k4 Methylases: Mechanisms of Regulation in Development and Disease Pathogenesis. Annu. Rev. Biochem. 2012, 81, 65–95. [Google Scholar] [CrossRef] [PubMed]
- Varier, R.A.; Timmers, H.T. Histone Lysine Methylation and Demethylation Pathways in Cancer. Biochim. Biophys. Acta 2011, 1, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.C.; Dou, Y. Hijacked in Cancer: The Kmt2 (Mll) Family of Methyltransferases. Nat. Rev. Cancer 2015, 6, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Byrd, K.N.; Shearn, A. Ash1, a Drosophila Trithorax Group Protein, Is Required for Methylation of Lysine 4 Residues on Histone H3. Proc. Natl. Acad. Sci. USA 2003, 20, 11535–11540. [Google Scholar] [CrossRef] [PubMed]
- Gregory, G.D.; Vakoc, C.R.; Rozovskaia, T.; Zheng, X.; Patel, S.; Nakamura, T.; Canaani, E.; Blobel, G.A. Mammalian Ash1l Is a Histone Methyltransferase That Occupies the Transcribed Region of Active Genes. Mol. Cell. Biol. 2007, 24, 8466–8479. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Xu, M.; Huang, C.; Liu, N.; Chen, S.; Zhu, B. H3k36 Methylation Antagonizes Prc2-Mediated H3k27 Methylation. J. Biol. Chem. 2011, 10, 7983–7989. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Yoshida, K.; Matsui, Y. A Histone H3 Methyltransferase Controls Epigenetic Events Required for Meiotic Prophase. Nature 2005, 7066, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Segurel, L.; Leffler, E.M.; Przeworski, M. The Case of the Fickle Fingers: How the Prdm9 Zinc Finger Protein Specifies Meiotic Recombination Hotspots in Humans. PLoS Biol. 2011, 12, e1001211. [Google Scholar] [CrossRef] [PubMed]
- Dhayalan, A.; Kudithipudi, S.; Rathert, P.; Jeltsch, A. Specificity Analysis-Based Identification of New Methylation Targets of the Set7/9 Protein Lysine Methyltransferase. Chem. Biol. 2011, 1, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Spellmon, N.; Holcomb, J.; Trescott, L.; Sirinupong, N.; Yang, Z. Structure and Function of Set and Mynd Domain-Containing Proteins. Int. J. Mol. Sci. 2015, 1, 1406–1428. [Google Scholar] [CrossRef] [PubMed]
- Giakountis, A.; Moulos, P.; Sarris, M.E.; Hatzis, P.; Talianidis, I. Smyd3-Associated Regulatory Pathways in Cancer. Semin. Cancer Biol. 2017, 42, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Kampranis, S.C.; Tsichlis, P.N. Histone Demethylases and Cancer. Adv. Cancer Res. 2009, 102, 103–169. [Google Scholar] [PubMed]
- Alicea-Velazquez, N.L.; Shinsky, S.A.; Loh, D.M.; Lee, J.H.; Skalnik, D.G.; Cosgrove, M.S. Targeted Disruption of the Interaction between Wd-40 Repeat Protein 5 (Wdr5) and Mixed Lineage Leukemia (Mll)/Set1 Family Proteins Specifically Inhibits Mll1 and Setd1a Methyltransferase Complexes. J. Biol. Chem. 2016, 43, 22357–22372. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Lin, C.; Smith, E.R.; Guo, H.; Sanderson, B.W.; Wu, M.; Gogol, M.; Alexander, T.; Seidel, C.; Wiedemann, L.M.; et al. Global Analysis of H3k4 Methylation Defines Mll Family Member Targets and Points to a Role for Mll1-Mediated H3k4 Methylation in the Regulation of Transcriptional Initiation by Rna Polymerase Ii. Mol. Cell. Biol. 2009, 22, 6074–6085. [Google Scholar] [CrossRef] [PubMed]
- Andreu-Vieyra, C.V.; Chen, R.; Agno, J.E.; Glaser, S.; Anastassiadis, K.; Stewart, A.F.; Matzuk, M.M. Mll2 Is Required in Oocytes for Bulk Histone 3 Lysine 4 Trimethylation and Transcriptional Silencing. PLoS Biol. 2010, 8, e1000453. [Google Scholar] [CrossRef] [PubMed]
- Placek, K.; Hu, G.; Cui, K.; Zhang, D.; Ding, Y.; Lee, J.E.; Jang, Y.; Wang, C.; Konkel, J.E.; Song, J.; et al. Mll4 Prepares the Enhancer Landscape for Foxp3 Induction Via Chromatin Looping. Nat. Immunol. 2017, 9, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Wang, C.; Zhuang, L.; Liu, C.; Ge, K. H3k4 Methyltransferase Activity Is Required for Mll4 Protein Stability. J. Mol. Biol. 2017, 13, 2046–2054. [Google Scholar] [CrossRef] [PubMed]
- Bledau, A.S.; Schmidt, K.; Neumann, K.; Hill, U.; Ciotta, G.; Gupta, A.; Torres, D.C.; Fu, J.; Kranz, A.; Stewart, A.F.; et al. The H3k4 Methyltransferase Setd1a Is First Required at the Epiblast Stage, Whereas Setd1b Becomes Essential after Gastrulation. Development 2014, 5, 1022–1035. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, R.; Furukawa, Y.; Morita, M.; Iimura, Y.; Silva, F.P.; Li, M.; Yagyu, R.; Nakamura, Y. Smyd3 Encodes a Histone Methyltransferase Involved in the Proliferation of Cancer Cells. Nat. Cell Biol. 2004, 8, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cao, R.; Xia, L.; Erdjument-Bromage, H.; Borchers, C.; Tempst, P.; Zhang, Y. Purification and Functional Characterization of a Histone H3-Lysine 4-Specific Methyltransferase. Mol. Cell 2001, 6, 1207–1217. [Google Scholar] [CrossRef]
- Kim, J.M.; Kim, K.; Schmidt, T.; Punj, V.; Tucker, H.; Rice, J.C.; Ulmer, T.S.; An, W. Cooperation between Smyd3 and Pc4 Drives a Distinct Transcriptional Program in Cancer Cells. Nucleic Acids Res. 2015, 18, 8868–8883. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Sims, R.J., 3rd; Gottlieb, P.D.; Tucker, P.W. Identification and Characterization of Smyd2: A Split Set/Mynd Domain-Containing Histone H3 Lysine 36-Specific Methyltransferase That Interacts with the Sin3 Histone Deacetylase Complex. Mol. Cancer 2006, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Van Nuland, R.; Smits, A.H.; Pallaki, P.; Jansen, P.W.; Vermeulen, M.; Timmers, H.T. Quantitative Dissection and Stoichiometry Determination of the Human Set1/Mll Histone Methyltransferase Complexes. Mol. Cell. Biol. 2013, 10, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Couture, J.F.; Skiniotis, G. Assembling a Compass. Epigenetics 2013, 4, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Dharmarajan, V.; Vought, V.E.; Cosgrove, M.S. On the Mechanism of Multiple Lysine Methylation by the Human Mixed Lineage Leukemia Protein-1 (Mll1) Core Complex. J. Biol. Chem. 2009, 36, 24242–24256. [Google Scholar] [CrossRef] [PubMed]
- Southall, S.M.; Wong, P.S.; Odho, Z.; Roe, S.M.; Wilson, J.R. Structural Basis for the Requirement of Additional Factors for Mll1 Set Domain Activity and Recognition of Epigenetic Marks. Mol. Cell 2009, 2, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, M.; Timmers, H.T. Grasping Trimethylation of Histone H3 at Lysine 4. Epigenomics 2010, 3, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Avdic, V.; Zhang, P.; Lanouette, S.; Groulx, A.; Tremblay, V.; Brunzelle, J.; Couture, J.F. Structural and Biochemical Insights into Mll1 Core Complex Assembly. Structure 2011, 1, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Chaturvedi, C.P.; Tremblay, V.; Cramet, M.; Brunzelle, J.S.; Skiniotis, G.; Brand, M.; Shilatifard, A.; Couture, J.F. A Phosphorylation Switch on Rbbp5 Regulates Histone H3 Lys4 Methylation. Genes Dev. 2015, 2, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Lee, H.; Brunzelle, J.S.; Couture, J.F. The Plasticity of Wdr5 Peptide-Binding Cleft Enables the Binding of the Set1 Family of Histone Methyltransferases. Nucleic Acids Res. 2012, 9, 4237–4246. [Google Scholar] [CrossRef] [PubMed]
- Steward, M.M.; Lee, J.S.; O’Donovan, A.; Wyatt, M.; Bernstein, B.E.; Shilatifard, A. Molecular Regulation of H3k4 Trimethylation by Ash2l, a Shared Subunit of Mll Complexes. Nat. Struct. Mol. Biol. 2006, 9, 852–854. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Milne, T.A.; Ruthenburg, A.J.; Lee, S.; Lee, J.W.; Verdine, G.L.; Allis, C.D.; Roeder, R.G. Regulation of Mll1 H3k4 Methyltransferase Activity by Its Core Components. Nat. Struct. Mol. Biol. 2006, 8, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Vought, V.E.; Dharmarajan, V.; Cosgrove, M.S. A Novel Non-Set Domain Multi-Subunit Methyltransferase Required for Sequential Nucleosomal Histone H3 Methylation by the Mixed Lineage Leukemia Protein-1 (Mll1) Core Complex. J. Biol. Chem. 2011, 5, 3359–3369. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Han, J.; Zhang, Y.; Cao, F.; Liu, Z.; Li, S.; Wu, J.; Hu, C.; Wang, Y.; Shuai, J.; et al. Structural Basis for Activity Regulation of Mll Family Methyltransferases. Nature 2016, 7591, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cao, F.; Wan, B.; Dou, Y.; Lei, M. Structure of the Spry Domain of Human Ash2l and Its Interactions with Rbbp5 and Dpy30. Cell Res. 2012, 3, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Odho, Z.; Southall, S.M.; Wilson, J.R. Characterization of a Novel Wdr5-Binding Site That Recruits Rbbp5 through a Conserved Motif to Enhance Methylation of Histone H3 Lysine 4 by Mixed Lineage Leukemia Protein-1. J. Biol. Chem. 2010, 43, 32967–32976. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.M.; Rozenblatt-Rosen, O.; Milne, T.A.; Copeland, T.D.; Levine, S.S.; Lee, J.C.; Hayes, D.N.; Shanmugam, K.S.; Bhattacharjee, A.; Biondi, C.A.; et al. Menin Associates with a Trithorax Family Histone Methyltransferase Complex and with the Hoxc8 Locus. Mol. Cell 2004, 4, 587–597. [Google Scholar] [CrossRef]
- Yokoyama, A.; Cleary, M.L. Menin Critically Links Mll Proteins with Ledgf on Cancer-Associated Target Genes. Cancer Cell 2008, 1, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Murai, M.J.; Pollock, J.; He, S.; Miao, H.; Purohit, T.; Yokom, A.; Hess, J.L.; Muntean, A.G.; Grembecka, J.; Cierpicki, T. The Same Site on the Integrase-Binding Domain of Lens Epithelium-Derived Growth Factor is a Therapeutic Target for Mll Leukemia and Hiv. Blood 2014, 25, 3730–3737. [Google Scholar] [CrossRef] [PubMed]
- Goo, Y.H.; Sohn, Y.C.; Kim, D.H.; Kim, S.W.; Kang, M.J.; Jung, D.J.; Kwak, E.; Barlev, N.A.; Berger, S.L.; Chow, V.T.; et al. Activating Signal Cointegrator 2 Belongs to a Novel Steady-State Complex That Contains a Subset of Trithorax Group Proteins. Mol. Cell. Biol. 2003, 1, 140–149. [Google Scholar] [CrossRef]
- Cho, Y.W.; Hong, T.; Hong, S.; Guo, H.; Yu, H.; Kim, D.; Guszczynski, T.; Dressler, G.R.; Copeland, T.D.; Kalkum, M.; et al. Ptip Associates with Mll3- and Mll4-Containing Histone H3 Lysine 4 Methyltransferase Complex. J. Biol. Chem. 2007, 28, 20395–20406. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.R.; Kim, D.; Levitan, I.; Dressler, G.R. The Brct-Domain Containing Protein Ptip Links Pax2 to a Histone H3, Lysine 4 Methyltransferase Complex. Dev. Cell 2007, 4, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Tate, C.M.; You, J.S.; Skalnik, D.G. Identification and Characterization of the Human Set1b Histone H3-Lys4 Methyltransferase Complex. J. Biol. Chem. 2007, 18, 13419–13428. [Google Scholar] [CrossRef] [PubMed]
- Voigt, P.; Tee, W.W.; Reinberg, D. A Double Take on Bivalent Promoters. Genes Dev. 2013, 12, 1318–1338. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Vought, V.E.; Dharmarajan, V.; Cosgrove, M.S. A Conserved Arginine-Containing Motif Crucial for the Assembly and Enzymatic Activity of the Mixed Lineage Leukemia Protein-1 Core Complex. J. Biol. Chem. 2008, 47, 32162–32175. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Ernst, P. Distinct Functions of Histone H3, Lysine 4 Methyltransferases in Normal and Malignant Hematopoiesis. Curr. Opin. Hematol. 2017, 4, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Wang, C.; Xu, S.; Cho, Y.W.; Wang, L.; Feng, X.; Baldridge, A.; Sartorelli, V.; Zhuang, L.; Peng, W.; et al. H3k4 Mono- and Di-Methyltransferase Mll4 Is Required for Enhancer Activation During Cell Differentiation. eLife 2013, 2, e01503. [Google Scholar] [CrossRef] [PubMed]
- Dorighi, K.M.; Swigut, T.; Henriques, T.; Bhanu, N.V.; Scruggs, B.S.; Nady, N.; Still, C.D., 2nd; Garcia, B.A.; Adelman, K.; Wysocka, J. Mll3 and Mll4 Facilitate Enhancer Rna Synthesis and Transcription from Promoters Independently of H3k4 Monomethylation. Mol. Cell 2017, 4, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Rickels, R.; Herz, H.M.; Sze, C.C.; Cao, K.; Morgan, M.A.; Collings, C.K.; Gause, M.; Takahashi, Y.H.; Wang, L.; Rendleman, E.J.; et al. Histone H3k4 Monomethylation Catalyzed by Trr and Mammalian Compass-Like Proteins at Enhancers is Dispensable for Development and Viability. Nat. Genet. 2017, 11, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Chen, L.H.; Huang, Y.; Chang, C.C.; Wang, P.; Pirozzi, C.J.; Qin, X.; Bao, X.; Greer, P.K.; McLendon, R.E.; et al. Kmt2d Maintains Neoplastic Cell Proliferation and Global Histone H3 Lysine 4 Monomethylation. Oncotarget 2013, 11, 2144–2153. [Google Scholar] [CrossRef] [PubMed]
- Herz, H.M.; Mohan, M.; Garruss, A.S.; Liang, K.; Takahashi, Y.H.; Mickey, K.; Voets, O.; Verrijzer, C.P.; Shilatifard, A. Enhancer-Associated H3k4 Monomethylation by Trithorax-Related, the Drosophila Homolog of Mammalian Mll3/Mll4. Genes Dev. 2012, 23, 2604–2620. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Gao, X.; Morgan, M.A.; Herz, H.M.; Smith, E.R.; Shilatifard, A. The Mll3/Mll4 Branches of the Compass Family Function as Major Histone H3k4 Monomethylases at Enhancers. Mol. Cell. Biol. 2013, 23, 4745–4754. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wang, P.F.; Lee, J.S.; Martin-Brown, S.; Florens, L.; Washburn, M.; Shilatifard, A. Molecular Regulation of H3k4 Trimethylation by Wdr82, a Component of Human Set1/Compass. Mol. Cell. Biol. 2008, 24, 7337–7344. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Li, Y.; Liang, S.; Cui, K.; Salz, T.; Yang, H.; Tang, Z.; Gallagher, P.G.; Qiu, Y.; Roeder, R.; et al. Usf1 and Hset1a Mediated Epigenetic Modifications Regulate Lineage Differentiation and Hoxb4 Transcription. PLoS Genet. 2013, 6, e1003524. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Zhang, J.; Zhang, H.; Yang, X.; Jin, X.; Zhang, L.; Skalnik, D.G.; Jin, Y.; Zhang, Y.; Huang, X.; et al. H3k4 Methyltransferase Set1a Is a Key Oct4 Coactivator Essential for Generation of Oct4 Positive Inner Cell Mass. Stem Cells 2016, 3, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Kaikkonen, M.U.; Spann, N.J.; Heinz, S.; Romanoski, C.E.; Allison, K.A.; Stender, J.D.; Chun, H.B.; Tough, D.F.; Prinjha, R.K.; Benner, C.; et al. Remodeling of the Enhancer Landscape During Macrophage Activation is Coupled to Enhancer Transcription. Mol. Cell 2013, 3, 310–325. [Google Scholar] [CrossRef] [PubMed]
- Bulger, M.; Groudine, M. Functional and Mechanistic Diversity of Distal Transcription Enhancers. Cell 2011, 3, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Scacheri, P.C. The Chromatin Fingerprint of Gene Enhancer Elements. J. Biol. Chem. 2012, 37, 30888–30896. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Blum, R.; Bowman, C.; Hu, D.; Shilatifard, A.; Shen, S.; Dynlacht, B.D. A Role for H3k4 Monomethylation in Gene Repression and Partitioning of Chromatin Readers. Mol. Cell 2014, 6, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.; Agger, K.; Cloos, P.A.; Pasini, D.; Rose, S.; Sennels, L.; Rappsilber, J.; Hansen, K.H.; Salcini, A.E.; Helin, K. Rbp2 Belongs to a Family of Demethylases, Specific for Tri-and Dimethylated Lysine 4 on Histone 3. Cell 2007, 6, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Kallin, E.M.; Zhang, Y. Jmjc-Domain-Containing Proteins and Histone Demethylation. Nat. Rev. Genet. 2006, 9, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Yan, Q.; Tothova, Z.; Yamane, K.; Erdjument-Bromage, H.; Tempst, P.; Gilliland, D.G.; Zhang, Y.; Kaelin, W.G., Jr. The Retinoblastoma Binding Protein Rbp2 Is an H3k4 Demethylase. Cell 2007, 5, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Zhang, J.; Klose, R.J.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. The Trithorax-Group Protein Lid Is a Histone H3 Trimethyl-Lys4 Demethylase. Nat. Struct. Mol. Biol. 2007, 4, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, T.; Yonezawa, M.; Lein, S.; Heidrich, K.; Kubicek, S.; Schafer, C.; Phalke, S.; Walther, M.; Schmidt, A.; Jenuwein, T.; et al. Heterochromatin Formation in Drosophila Is Initiated through Active Removal of H3k4 Methylation by the Lsd1 Homolog Su(Var)3-3. Mol. Cell 2007, 1, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Wissmann, M.; Yin, N.; Muller, J.M.; Greschik, H.; Fodor, B.D.; Jenuwein, T.; Vogler, C.; Schneider, R.; Gunther, T.; Buettner, R.; et al. Cooperative Demethylation by Jmjd2c and Lsd1 Promotes Androgen Receptor-Dependent Gene Expression. Nat. Cell Biol. 2007, 3, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Yamane, K.; Tateishi, K.; Klose, R.J.; Fang, J.; Fabrizio, L.A.; Erdjument-Bromage, H.; Taylor-Papadimitriou, J.; Tempst, P.; Zhang, Y. Plu-1 is an H3k4 Demethylase Involved in Transcriptional Repression and Breast Cancer Cell Proliferation. Mol. Cell 2007, 6, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Yagi, H.; Deguchi, K.; Aono, A.; Tani, Y.; Kishimoto, T.; Komori, T. “Growth Disturbance in Fetal Liver Hematopoiesis of Mll-Mutant Mice”. Blood 1998, 1, 108–117. [Google Scholar]
- Yu, B.D.; Hess, J.L.; Horning, S.E.; Brown, G.A.; Korsmeyer, S.J. Altered Hox Expression and Segmental Identity in Mll-Mutant Mice. Nature 1995, 6556, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Ernst, P. Set/Mll Family Proteins in Hematopoiesis and Leukemia. Int. J. Hematol. 2017, 1, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Gan, T.; Jude, C.D.; Zaffuto, K.; Ernst, P. Developmentally Induced Mll1 Loss Reveals Defects in Postnatal Haematopoiesis. Leukemia 2010, 10, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Ernst, P.; Fisher, J.K.; Avery, W.; Wade, S.; Foy, D.; Korsmeyer, S.J. Definitive Hematopoiesis Requires the Mixed-Lineage Leukemia Gene. Dev. Cell 2004, 3, 437–443. [Google Scholar] [CrossRef]
- Jude, C.D.; Climer, L.; Xu, D.; Artinger, E.; Fisher, J.K.; Ernst, P. Unique and Independent Roles for Mll in Adult Hematopoietic Stem Cells and Progenitors. Cell Stem Cell 2007, 3, 324–337. [Google Scholar] [CrossRef] [PubMed]
- McMahon, K.A.; Hiew, S.Y.; Hadjur, S.; Veiga-Fernandes, H.; Menzel, U.; Price, A.J.; Kioussis, D.; Williams, O.; Brady, H.J. Mll Has a Critical Role in Fetal and Adult Hematopoietic Stem Cell Self-Renewal. Cell Stem Cell 2007, 3, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.; Schaft, J.; Lubitz, S.; Vintersten, K.; van der Hoeven, F.; Tufteland, K.R.; Aasland, R.; Anastassiadis, K.; Ang, S.L.; Stewart, A.F. Multiple Epigenetic Maintenance Factors Implicated by the Loss of Mll2 in Mouse Development. Development 2006, 8, 1423–1432. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.; Lubitz, S.; Loveland, K.L.; Ohbo, K.; Robb, L.; Schwenk, F.; Seibler, J.; Roellig, D.; Kranz, A.; Anastassiadis, K.; et al. The Histone 3 Lysine 4 Methyltransferase, Mll2, Is Only Required Briefly in Development and Spermatogenesis. Epigenet. Chromatin 2009, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Stoller, J.Z.; Huang, L.; Tan, C.C.; Huang, F.; Zhou, D.D.; Yang, J.; Gelb, B.D.; Epstein, J.A. Ash2l Interacts with Tbx1 and Is Required During Early Embryogenesis. Exp. Biol. Med. 2010, 5, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.L.; Luscher-Firzlaff, J.; Ullius, A.; Schneider, U.; Longerich, T.; Luscher, B. Loss of the Epigenetic Regulator Ash2l Results in Desintegration of Hepatocytes and Liver Failure. Int. J. Clin. Exp. Pathol. 2016, 5, 5167–5175. [Google Scholar]
- Yang, Z.; Shah, K.; Khodadadi-Jamayran, A.; Jiang, H. Dpy30 Is Critical for Maintaining the Identity and Function of Adult Hematopoietic Stem Cells. J. Exp. Med. 2016, 11, 2349–2364. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Metzger, E.; Schule, R.; Kirfel, J.; Buettner, R. Epigenetic Regulation of Cancer Growth by Histone Demethylases. Int. J. Cancer 2010, 9, 1991–1998. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Hofmann, J.; Burmeister, T.; Groger, D.; Park, T.S.; Emerenciano, M.; de Oliveira, M.P.; Renneville, A.; Villarese, P.; Macintyre, E.; et al. The Mll Recombinome of Acute Leukemias in 2013. Leukemia 2013, 11, 2165–2176. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.M.; Karemaker, D.; van Leeuwen, F.I. The Emerging Roles of Dot1l in Leukemia and Normal Development. Leukemia 2014, 11, 2131–2138. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Hoshii, T.; Armstrong, S.A. Mixed-Lineage Leukemia Fusions and Chromatin in Leukemia. Cold Spring Harb. Perspect. Med. 2017, 7, a026658. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Lin, C.; Shilatifard, A. The Super Elongation Complex (Sec) Family in Transcriptional Control. Nat. Rev. Mol. Cell Biol. 2012, 9, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Winters, A.C.; Bernt, K.M. Mll-Rearranged Leukemias-an Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.D.; Dafou, D.; McEntagart, M.; Woollard, W.J.; Elmslie, F.V.; Holder-Espinasse, M.; Irving, M.; Saggar, A.K.; Smithson, S.; Trembath, R.C.; et al. De Novo Mutations in Mll Cause Wiedemann-Steiner Syndrome. Am. J. Hum. Genet. 2012, 2, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Strom, S.P.; Lozano, R.; Lee, H.; Dorrani, N.; Mann, J.; O’Lague, P.F.; Mans, N.; Deignan, J.L.; Vilain, E.; Nelson, S.F.; et al. De Novo Variants in the Kmt2a (Mll) Gene Causing Atypical Wiedemann-Steiner Syndrome in Two Unrelated Individuals Identified by Clinical Exome Sequencing. BMC Med. Genet. 2014, 15, 49. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, B.A.; Pronold, M.; Long, R.; Smaoui, N.; Slavotinek, A.M. Advanced Bone Age in a Girl with Wiedemann-Steiner Syndrome and an Exonic Deletion in Kmt2a (Mll). Am. J. Med. Genet. A 2014, 8, 2079–2083. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, Y.; Rappaport, A.R.; Kitzing, T.; Schultz, N.; Zhao, Z.; Shroff, A.S.; Dickins, R.A.; Vakoc, C.R.; Bradner, J.E.; et al. Mll3 Is a Haploinsufficient 7q Tumor Suppressor in Acute Myeloid Leukemia. Cancer Cell 2014, 5, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Weeraratne, S.D.; Archer, T.C.; Krummel, D.A.P.; Auclair, D.; Bochicchio, J.; Carneiro, M.O.; Carter, S.L.; Cibulskis, K.; Erlich, R.L.; et al. Medulloblastoma Exome Sequencing Uncovers Subtype-Specific Somatic Mutations. Nature 2012, 7409, 106–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, F.; Lan, R.; Zhang, X.; Zhu, L.; Chen, F.; Xu, Z.; Liu, Y.; Ye, T.; Sun, H.; Lu, F.; et al. Lsd1 Regulates Pluripotency of Embryonic Stem/Carcinoma Cells through Histone Deacetylase 1-Mediated Deacetylation of Histone H4 at Lysine 16. Mol. Cell. Biol. 2014, 2, 158–179. [Google Scholar] [CrossRef] [PubMed]
- Schulz, Y.; Freese, L.; Manz, J.; Zoll, B.; Volter, C.; Brockmann, K.; Bogershausen, N.; Becker, J.; Wollnik, B.; Pauli, S. Charge and Kabuki Syndromes: A Phenotypic and Molecular Link. Hum. Mol. Genet. 2014, 16, 4396–4405. [Google Scholar] [CrossRef] [PubMed]
- Dreijerink, K.M.; JHoppener, W.; Timmers, H.M.; Lips, C.J. Mechanisms of Disease: Multiple Endocrine Neoplasia Type 1-Relation to Chromatin Modifications and Transcription Regulation. Nat. Clin. Pract. Endocrinol. MeTab. 2006, 10, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Matkar, S.; Thiel, A.; Hua, X. Menin: A Scaffold Protein That Controls Gene Expression and Cell Signaling. Trends Biochem. Sci. 2013, 8, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Luscher-Firzlaff, J.; Gawlista, I.; Vervoorts, J.; Kapelle, K.; Braunschweig, T.; Walsemann, G.; Rodgarkia-Schamberger, C.; Schuchlautz, H.; Dreschers, S.; Kremmer, E.; et al. The Human Trithorax Protein Hash2 Functions as an Oncoprotein. Cancer Res. 2008, 3, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Magerl, C.; Ellinger, J.; Braunschweig, T.; Kremmer, E.; Koch, L.K.; Holler, T.; Buttner, R.; Luscher, B.; Gutgemann, I. H3k4 Dimethylation in Hepatocellular Carcinoma Is Rare Compared with Other Hepatobiliary and Gastrointestinal Carcinomas and Correlates with Expression of the Methylase Ash2 and the Demethylase Lsd1. Hum. Pathol. 2010, 2, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.S.; Qiu, Y.H.; Zhang, N.; Yoo, S.Y.; Coombes, K.R.; Dent, S.Y.; Kornblau, S.M. Low Expression of Ash2l Protein Correlates with a Favorable Outcome in Acute Myeloid Leukemia. Leuk. Lymphoma 2017, 5, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Song, E.J.; Kawasawa, Y.I.; Li, J.; Dovat, S.; Song, C. Wdr5 High Expression and Its Effect on Tumorigenesis in Leukemia. Oncotarget 2016, 25, 37740–37754. [Google Scholar] [CrossRef] [PubMed]
- Steinhilber, D.; Marschalek, R. How to Effectively Treat Acute Leukemia Patients Bearing Mll-Rearrangements ? Biochem. Pharmacol. 2018, 147, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Grebien, F.; Vedadi, M.; Getlik, M.; Giambruno, R.; Grover, A.; Avellino, R.; Skucha, A.; Vittori, S.; Kuznetsova, E.; Smil, D.; et al. Pharmacological Targeting of the Wdr5-Mll Interaction in C/Ebpalpha N-Terminal Leukemia. Nat. Chem. Biol. 2015, 8, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Senisterra, G.; Wu, H.; Allali-Hassani, A.; Wasney, G.A.; Barsyte-Lovejoy, D.; Dombrovski, L.; Dong, A.; Nguyen, K.T.; Smil, D.; Bolshan, Y.; et al. Small-Molecule Inhibition of Mll Activity by Disruption of Its Interaction with Wdr5. Biochem. J. 2013, 1, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.W.; Armstrong, S.A. Designed to Kill: Novel Menin-Mll Inhibitors Target Mll-Rearranged Leukemia. Cancer Cell 2015, 4, 431–433. [Google Scholar] [CrossRef] [PubMed]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J.; et al. Pharmacologic Inhibition of the Menin-Mll Interaction Blocks Progression of Mll Leukemia In Vivo. Cancer Cell 2015, 4, 589–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brondfield, S.; Umesh, S.; Corella, A.; Zuber, J.; Rappaport, A.R.; Gaillard, C.; Lowe, S.W.; Goga, A.; Kogan, S.C. Direct and Indirect Targeting of Myc to Treat Acute Myeloid Leukemia. Cancer Chemother. Pharmacol. 2015, 1, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent Inhibition of Dot1l as Treatment of Mll-Fusion Leukemia. Blood 2013, 6, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Deng, L.; Song, Y.; Redell, M. Dot1l Inhibition Sensitizes Mll-Rearranged Aml to Chemotherapy. PLoS ONE 2014, 5, e98270. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Yao, Y.; Zhou, C.; Chen, F.; Wu, F.; Wei, L.; Liu, W.; Dong, S.; Redell, M.; Mo, Q.; et al. Pharmacological Inhibition of Lsd1 for the Treatment of Mll-Rearranged Leukemia. J. Hematol. Oncol. 2016, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of Bet Recruitment to Chromatin as an Effective Treatment for Mll-Fusion Leukaemia. Nature 2011, 7370, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.; Volk, A.G.; Haug, J.S.; Marshall, S.A.; Woodfin, A.R.; Bartom, E.T.; Gilmore, J.M.; Florens, L.; Washburn, M.P.; Sullivan, K.D.; et al. Therapeutic Targeting of Mll Degradation Pathways in Mll-Rearranged Leukemia. Cell 2017, 168, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Sandhofer, N.; Metzeler, K.H.; Rothenberg, M.; Herold, T.; Tiedt, S.; Groiss, V.; Carlet, M.; Walter, G.; Hinrichsen, T.; Wachter, O.; et al. Dual Pi3k/Mtor Inhibition Shows Antileukemic Activity in Mll-Rearranged Acute Myeloid Leukemia. Leukemia 2015, 4, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Chang, C.C.; Wortham, M.; Chen, L.H.; Kernagis, D.N.; Qin, X.; Cho, Y.W.; Chi, J.T.; Grant, G.A.; McLendon, R.E.; et al. Global Identification of Mll2-Targeted Loci Reveals Mll2’s Role in Diverse Signaling Pathways. Proc. Natl. Acad. Sci. USA 2012, 43, 17603–17608. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The Role of Chromatin During Transcription. Cell 2007, 4, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.A.; Frey, B.L.; Smith, L.M.; Auble, D.T. Formaldehyde Crosslinking: A Tool for the Study of Chromatin Complexes. J. Biol. Chem. 2015, 44, 26404–26411. [Google Scholar] [CrossRef] [PubMed]
- Ang, Y.S.; Tsai, S.Y.; Lee, D.F.; Monk, J.; Su, J.; Ratnakumar, K.; Ding, J.; Ge, Y.; Darr, H.; Chang, B.; et al. Wdr5 Mediates Self-Renewal and Reprogramming Via the Embryonic Stem Cell Core Transcriptional Network. Cell 2011, 2, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Augustin, J.; Hu, J.; Jiang, H. Physical Interactions and Functional Coordination between the Core Subunits of Set1/Mll Complexes and the Reprogramming Factors. PLoS ONE 2015, 12, e0145336. [Google Scholar] [CrossRef] [PubMed]
- Bertero, A.; Madrigal, P.; Galli, A.; Hubner, N.C.; Moreno, I.; Burks, D.; Brown, S.; Pedersen, R.A.; Gaffney, D.; Mendjan, S.; et al. Activin/Nodal Signaling and Nanog Orchestrate Human Embryonic Stem Cell Fate Decisions by Controlling the H3k4me3 Chromatin Mark. Genes Dev. 2015, 7, 702–717. [Google Scholar] [CrossRef] [PubMed]
- Ullius, A.; Luscher-Firzlaff, J.; Costa, I.G.; Walsemann, G.; Forst, A.H.; Gusmao, E.G.; Kapelle, K.; Kleine, H.; Kremmer, E.; Vervoorts, J.; et al. The Interaction of Myc with the Trithorax Protein Ash2l Promotes Gene Transcription by Regulating H3k27 Modification. Nucleic Acids Res. 2014, 11, 6901–6920. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.R.; Wang, Q.; Grieb, B.C.; Phan, J.; Foshage, A.M.; Sun, Q.; Olejniczak, E.T.; Clark, T.; Dey, S.; Lorey, S.; et al. Interaction with Wdr5 Promotes Target Gene Recognition and Tumorigenesis by Myc. Mol. Cell 2015, 3, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Rampalli, S.; Li, L.; Mak, E.; Ge, K.; Brand, M.; Tapscott, S.J.; Dilworth, F.J. P38 Mapk Signaling Regulates Recruitment of Ash2l-Containing Methyltransferase Complexes to Specific Genes During Differentiation. Nat. Struct. Mol. Biol. 2007, 12, 1150–1156. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.L.; Loffler, K.A.; Chen, D.; Stallcup, M.R.; Muscat, G.E. The Coactivator-Associated Arginine Methyltransferase Is Necessary for Muscle Differentiation: Carm1 Coactivates Myocyte Enhancer Factor-2. J. Biol. Chem. 2002, 6, 4324–4333. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Pan, W.S.; Dai, H.; Zhang, Y.; Wu, N.H.; Shen, Y.F. Carm1 Activates Myogenin Gene Via Pcaf in the Early Differentiation of Tpa-Induced Rhabdomyosarcoma-Derived Cells. J. Cell Biochem. 2010, 1, 162–170. [Google Scholar]
- Kawabe, Y.; Wang, Y.X.; McKinnell, I.W.; Bedford, M.T.; Rudnicki, M.A. Carm1 Regulates Pax7 Transcriptional Activity through Mll1/2 Recruitment During Asymmetric Satellite Stem Cell Divisions. Cell Stem Cell 2012, 3, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Fossati, A.; Dolfini, D.; Donati, G.; Mantovani, R. Nf-Y Recruits Ash2l to Impart H3k4 Trimethylation on Ccaat Promoters. PLoS ONE 2011, 3, e17220. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Song, S.H.; Brand, M.; Dean, A. Nucleosome and Transcription Activator Antagonism at Human Beta-Globin Locus Control Region Dnase I Hypersensitive Sites. Nucleic Acids Res. 2007, 17, 5831–5838. [Google Scholar] [CrossRef] [PubMed]
- Demers, C.; Chaturvedi, C.P.; Ranish, J.A.; Juban, G.; Lai, P.; Morle, F.; Aebersold, R.; Dilworth, F.J.; Groudine, M.; Brand, M. Activator-Mediated Recruitment of the Mll2 Methyltransferase Complex to the Beta-Globin Locus. Mol. Cell 2007, 4, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Monckton, E.A.; Godbout, R. Ectopic Expression of Transcription Factor Ap-2delta in Developing Retina: Effect on Psa-Ncam and Axon Routing. J. Neurochem. 2014, 1, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.C.; Sindhu, K.V.; Li, S.; Nishio, H.; Stoller, J.Z.; Oishi, K.; Puttreddy, S.; Lee, T.J.; Epstein, J.A.; Walsh, M.J.; et al. Transcription Factor Ap2delta Associates with Ash2l and Alr, a Trithorax Family Histone Methyltransferase, to Activate Hoxc8 Transcription. Proc. Natl. Acad. Sci. USA 2008, 21, 7472–7477. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.C.; Walsh, M.J.; Gelb, B.D. Fgfr3 Is a Transcriptional Target of Ap2delta and Ash2l-Containing Histone Methyltransferase Complexes. PLoS ONE 2009, 12, e8535. [Google Scholar]
- Schwab, K.R.; Patel, S.R.; Dressler, G.R. Role of Ptip in Class Switch Recombination and Long-Range Chromatin Interactions at the Immunoglobulin Heavy Chain Locus. Mol. Cell. Biol. 2011, 7, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- McManus, S.; Ebert, A.; Salvagiotto, G.; Medvedovic, J.; Sun, Q.; Tamir, I.; Jaritz, M.; Tagoh, H.; Busslinger, M. The Transcription Factor Pax5 Regulates Its Target Genes by Recruiting Chromatin-Modifying Proteins in Committed B Cells. EMBO J. 2011, 12, 2388–2404. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, D.K.; Dou, Y.; Lee, J.; Lee, B.; Kwak, E.; Kong, Y.Y.; Lee, S.K.; Roeder, R.G.; Lee, J.W. Coactivator as a Target Gene Specificity Determinant for Histone H3 Lysine 4 Methyltransferases. Proc. Natl. Acad. Sci. USA 2006, 42, 15392–15397. [Google Scholar] [CrossRef] [PubMed]
- Ananthanarayanan, M.; Li, Y.; Surapureddi, S.; Balasubramaniyan, N.; Ahn, J.; Goldstein, J.A.; Suchy, F.J. Histone H3k4 Trimethylation by Mll3 as Part of Ascom Complex Is Critical for Nr Activation of Bile Acid Transporter Genes and Is Downregulated in Cholestasis. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 5, G771–G781. [Google Scholar] [CrossRef] [PubMed]
- Scoville, D.W.; Cyphert, H.A.; Liao, L.; Xu, J.; Reynolds, A.; Guo, S.; Stein, R. Mll3 and Mll4 Methyltransferases Bind to the Mafa and Mafb Transcription Factors to Regulate Islet Beta-Cell Function. Diabetes 2015, 11, 3772–3783. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, D.H.; Lee, S.; Yang, Q.H.; Lee, D.K.; Lee, S.K.; Roeder, R.G.; Lee, J.W. A Tumor Suppressive Coactivator Complex of P53 Containing Asc-2 and Histone H3-Lysine-4 Methyltransferase Mll3 or Its Paralogue Mll4. Proc. Natl. Acad. Sci. USA 2009, 21, 8513–8518. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Chen, W.Y.; Shimada, M.; Nguyen, U.T.; Kim, J.; Sun, X.J.; Sengoku, T.; McGinty, R.K.; Fernandez, J.P.; Muir, T.W.; et al. Set1 and P300 Act Synergistically, through Coupled Histone Modifications, in Transcriptional Activation by P53. Cell 2013, 2, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Milne, T.A.; Tackett, A.J.; Smith, E.R.; Fukuda, A.; Wysocka, J.; Allis, C.D.; Chait, B.T.; Hess, J.L.; Roeder, R.G. Physical Association and Coordinate Function of the H3 K4 Methyltransferase Mll1 and the H4 K16 Acetyltransferase Mof. Cell 2005, 6, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Mungamuri, S.K.; Wang, S.; Manfredi, J.J.; Gu, W.; Aaronson, S.A. Ash2l Enables P53-Dependent Apoptosis by Favoring Stable Transcription Pre-Initiation Complex Formation on Its Pro-Apoptotic Target Promoters. Oncogene 2015, 19, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Mo, R.; Rao, S.M.; Zhu, Y.J. Identification of the Mll2 Complex as a Coactivator for Estrogen Receptor Alpha. J. Biol. Chem. 2006, 23, 15714–15720. [Google Scholar] [CrossRef] [PubMed]
- Ansari, K.I.; Hussain, I.; Shrestha, B.; Kasiri, S.; Mandal, S.S. Hoxc6 Is Transcriptionally Regulated Via Coordination of Mll Histone Methylase and Estrogen Receptor in an Estrogen Environment. J. Mol. Biol. 2011, 2, 334–349. [Google Scholar] [CrossRef] [PubMed]
- Ansari, K.I.; Shrestha, B.; Hussain, I.; Kasiri, S.; Mandal, S.S. Histone Methylases Mll1 and Mll3 Coordinate with Estrogen Receptors in Estrogen-Mediated Hoxb9 Expression. Biochemistry 2011, 17, 3517–3527. [Google Scholar] [CrossRef] [PubMed]
- Ansari, K.I.; Kasiri, S.; Hussain, I.; Bobzean, S.A.; Perrotti, L.I.; Mandal, S.S. Mll Histone Methylases Regulate Expression of Hdlr-Sr-B1 in Presence of Estrogen and Control Plasma Cholesterol In Vivo. Mol. Endocrinol. 2013, 1, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Dreijerink, K.M.; Mulder, K.W.; Winkler, G.S.; Hoppener, J.W.; Lips, C.J.; Timmers, H.T. Menin Links Estrogen Receptor Activation to Histone H3k4 Trimethylation. Cancer Res. 2006, 9, 4929–4935. [Google Scholar] [CrossRef] [PubMed]
- Imachi, H.; Murao, K.; Dobashi, H.; Bhuyan, M.M.; Cao, X.; Kontani, K.; Niki, S.; Murazawa, C.; Nakajima, H.; Kohno, N.; et al. Menin, a Product of the Meni Gene, Binds to Estrogen Receptor to Enhance Its Activity in Breast Cancer Cells: Possibility of a Novel Predictive Factor for Tamoxifen Resistance. Breast Cancer Res. Treat. 2010, 2, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Zhao, H.; Yi, Y.; Nakata, Y.; Kalota, A.; Gewirtz, A.M. C-Myb Binds Mll through Menin in Human Leukemia Cells and Is an Important Driver of Mll-Associated Leukemogenesis. J. Clin. Investig. 2010, 2, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Huo, L.; Zhu, Y.T.; Zhu, Y.J. Absent, Small or Homeotic 2-Like Protein (Ash2l) Enhances the Transcription of the Estrogen Receptor Alpha Gene through Gata-Binding Protein 3 (Gata3). J. Biol. Chem. 2014, 45, 31373–31381. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.S.; Liu, X.S.; Brodsky, A.S.; Li, W.; Meyer, C.A.; Szary, A.J.; Eeckhoute, J.; Shao, W.; Hestermann, E.V.; Geistlinger, T.R.; et al. Chromosome-Wide Mapping of Estrogen Receptor Binding Reveals Long-Range Regulation Requiring the Forkhead Protein Foxa1. Cell 2005, 1, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Jozwik, K.M.; Chernukhin, I.; Serandour, A.A.; Nagarajan, S.; Carroll, J.S. Foxa1 Directs H3k4 Monomethylation at Enhancers Via Recruitment of the Methyltransferase Mll3. Cell Rep. 2016, 10, 2715–2723. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Muller, G.A. Cell Cycle Transcription Control: Dream/Muvb and Rb-E2f Complexes. Crit. Rev. Biochem. Mol. Biol. 2017, 6, 638–662. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Chabes, A.L.; Wysocka, J.; Herr, W. E2f Activation of S Phase Promoters Via Association with Hcf-1 and the Mll Family of Histone H3k4 Methyltransferases. Mol. Cell 2007, 1, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Chen, D.Y.; Westergard, T.D.; Fisher, J.K.; Rubens, J.A.; Sasagawa, S.; Kan, J.T.; Korsmeyer, S.J.; Cheng, E.H.; Hsieh, J.J. Proteolysis of Mll Family Proteins Is Essential for Taspase1-Orchestrated Cell Cycle Progression. Genes Dev. 2006, 17, 2397–2409. [Google Scholar] [CrossRef] [PubMed]
- Boussouar, F.; Jamshidikia, M.; Morozumi, Y.; Rousseaux, S.; Khochbin, S. Malignant Genome Reprogramming by Atad2. Biochim. Biophys. Acta 2013, 10, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Revenko, A.S.; Kalashnikova, E.V.; Gemo, A.T.; Zou, J.X.; Chen, H.W. Chromatin Loading of E2f-Mll Complex by Cancer-Associated Coregulator Ancca Via Reading a Specific Histone Mark. Mol. Cell. Biol. 2010, 22, 5260–5272. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Herr, W. E2f1 Mediates DNA Damage and Apoptosis through Hcf-1 and the Mll Family of Histone Methyltransferases. EMBO J. 2009, 20, 3185–3195. [Google Scholar] [CrossRef] [PubMed]
- Zaret, K.S. Pioneering the Chromatin Landscape. Nat. Genet. 2018, 2, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Skalnik, D.G. Wdr82 Is a C-Terminal Domain-Binding Protein That Recruits the Setd1a Histone H3-Lys4 Methyltransferase Complex to Transcription Start Sites of Transcribed Human Genes. Mol. Cell. Biol. 2008, 2, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Shibata, Y.; Rao, B.; Laribee, R.N.; O’Rourke, R.; Buck, M.J.; Greenblatt, J.F.; Krogan, N.J.; Lieb, J.D.; Strahl, B.D. The Rna Polymerase Ii Kinase Ctk1 Regulates Positioning of a 5′ Histone Methylation Boundary Along Genes. Mol. Cell. Biol. 2007, 2, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.; Deb, S.; Moniaux, N.; Ponnusamy, M.P.; Batra, S.K. Human Rna Polymerase Ii-Associated Factor Complex: Dysregulation in Cancer. Oncogene 2007, 54, 7499–7507. [Google Scholar] [CrossRef] [PubMed]
- Muntean, A.G.; Tan, J.; Sitwala, K.; Huang, Y.; Bronstein, J.; Connelly, J.A.; Basrur, V.; Elenitoba-Johnson, K.S.; Hess, J.L. The Paf Complex Synergizes with Mll Fusion Proteins at Hox Loci to Promote Leukemogenesis. Cancer Cell 2010, 6, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Soares, L.M.; He, P.C.; Chun, Y.; Suh, H.; Kim, T.; Buratowski, S. Determinants of Histone H3k4 Methylation Patterns. Mol. Cell 2017, 4, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Ladopoulos, V.; Hofemeister, H.; Hoogenkamp, M.; Riggs, A.D.; Stewart, A.F.; Bonifer, C. The Histone Methyltransferase Kmt2b Is Required for Rna Polymerase Ii Association and Protection from DNA Methylation at the Magohb Cpg Island Promoter. Mol. Cell. Biol. 2013, 7, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Y.; Xie, S.; Liu, Y.; Xie, Z. Global and Cell-Type Specific Properties of Lincrnas with Ribosome Occupancy. Nucleic Acids Res. 2017, 5, 2786–2796. [Google Scholar] [CrossRef] [PubMed]
- Marchese, F.P.; Raimondi, I.; Huarte, M. The Multidimensional Mechanisms of Long Noncoding Rna Function. Genome Biol. 2017, 1, 206. [Google Scholar] [CrossRef] [PubMed]
- Bonasio, R.; Shiekhattar, R. Regulation of Transcription by Long Noncoding Rnas. Annu. Rev. Genet. 2014, 48, 433–455. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Dominguez, J.R.; Lodish, H.F. Emerging Mechanisms of Long Noncoding Rna Function During Normal and Malignant Hematopoiesis. Blood 2017, 18, 1965–1975. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.M.; Chang, H.Y. Long Noncoding Rnas in Cancer Pathways. Cancer Cell 2016, 4, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; Caffrey, D.R. Long Noncoding Rnas in Innate and Adaptive Immunity. Curr. Opin. Immunol. 2014, 26, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Wang, X.; Youmans, D.T.; Cech, T.R. How Do Lncrnas Regulate Transcription? Sci. Adv. 2017, 9, eaao2110. [Google Scholar] [CrossRef] [PubMed]
- Chedin, F. Nascent Connections: R-Loops and Chromatin Patterning. Trends Genet. 2016, 12, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Santos-Pereira, J.M.; Aguilera, A. R Loops: New Modulators of Genome Dynamics and Function. Nat. Rev. Genet. 2015, 10, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Yang, Y.W.; Liu, B.; Sanyal, A.; Corces-Zimmerman, R.; Chen, Y.; Lajoie, B.R.; Protacio, A.; Flynn, R.A.; Gupta, R.A.; et al. A Long Noncoding Rna Maintains Active Chromatin to Coordinate Homeotic Gene Expression. Nature 2011, 7341, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Malek, R.; Gajula, R.P.; Williams, R.D.; Nghiem, B.; Simons, B.W.; Nugent, K.; Wang, H.; Taparra, K.; Lemtiri-Chlieh, G.; Yoon, A.R.; et al. Twist1-Wdr5-Hottip Regulates Hoxa9 Chromatin to Facilitate Prostate Cancer Metastasis. Cancer Res. 2017, 12, 3181–3193. [Google Scholar] [CrossRef] [PubMed]
- Guenther, M.G.; Jenner, R.G.; Chevalier, B.; Nakamura, T.; Croce, C.M.; Canaani, E.; Young, R.A. Global and Hox-Specific Roles for the Mll1 Methyltransferase. Proc. Natl. Acad. Sci. USA 2005, 24, 8603–8608. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Chen, C.; Zhou, Q.; Wang, Y.; Zhao, Y.; Zhao, X.; Li, W.; Zheng, S.; Ye, H.; Wang, L.; et al. Lncrna Hottip Modulates Cancer Stem Cell Properties in Human Pancreatic Cancer by Regulating Hoxa9. Cancer Lett. 2017, 410, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Quagliata, L.; Matter, M.S.; Piscuoglio, S.; Arabi, L.; Ruiz, C.; Procino, A.; Kovac, M.; Moretti, F.; Makowska, Z.; Boldanova, T.; et al. Long Noncoding Rna Hottip/Hoxa13 Expression Is Associated with Disease Progression and Predicts Outcome in Hepatocellular Carcinoma Patients. Hepatology 2014, 3, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhao, X.; Zhou, Y.; Liu, Y.; Zhou, Q.; Ye, H.; Wang, Y.; Zeng, J.; Song, Y.; Gao, W.; et al. The Long Non-Coding Rna Hottip Promotes Progression and Gemcitabine Resistance by Regulating Hoxa13 in Pancreatic Cancer. J. Transl. Med. 2015, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Lian, Y.; Cai, Z.; Gong, H.; Xue, S.; Wu, D.; Wang, K. Hottip: A Critical Oncogenic Long Non-Coding Rna in Human Cancers. Mol. Biosyst. 2016, 11, 3247–3253. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.W.; Li, Y.; Zhou, L.; Cho, J.; Patel, B.; Terada, N.; Li, Y.Q.; Bungert, J.; Qiu, Y.; Huang, S.M. Hoxblinc Rna Recruits Set1/Mll Complexes to Activate Hox Gene Expression Patterns and Mesoderm Lineage Development. Cell Rep. 2016, 1, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Grote, P.; Wittler, L.; Hendrix, D.; Koch, F.; Wahrisch, S.; Beisaw, A.; Macura, K.; Blass, G.; Kellis, M.; Werber, M.; et al. The Tissue-Specific Lncrna Fendrr Is an Essential Regulator of Heart and Body Wall Development in the Mouse. Dev. Cell 2013, 2, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Wen, S.; Zhang, Y.; Shi, X.; Zhu, Y.; Xu, Y.; Lv, H.; Wang, G. Identification of Dysregulated Long Non-Coding Rnas/Micrornas/Mrnas in Tnm I Stage Lung Adenocarcinoma. Oncotarget 2017, 31, 51703–51718. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.J.; Tang, R.X.; He, R.Q.; Li, D.Y.; Liang, L.; Zeng, J.H.; Hu, X.H.; Ma, J.; Li, S.K.; Chen, G. Clinical Roles of the Aberrantly Expressed Lncrnas in Lung Squamous Cell Carcinoma: A Study Based on Rna-Sequencing and Microarray Data Mining. Oncotarget 2017, 37, 61282–61304. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.P.; Huang, M.D.; Xia, R.; Liu, X.X.; Sun, M.; Yin, L.; Chen, W.M.; Han, L.; Zhang, E.B.; Kong, R.; et al. Decreased Expression of the Long Non-Coding Rna Fendrr Is Associated with Poor Prognosis in Gastric Cancer and Fendrr Regulates Gastric Cancer Cell Metastasis by Affecting Fibronectin1 Expression. J. Hematol. Oncol. 2014, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Morales, D.R.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many Human Large Intergenic Noncoding Rnas Associate with Chromatin-Modifying Complexes and Affect Gene Expression. Proc. Natl. Acad. Sci. USA 2009, 28, 11667–11672. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.W.; Flynn, R.A.; Chen, Y.; Qu, K.; Wan, B.; Wang, K.C.; Lei, M.; Chang, H.Y. Essential Role of Lncrna Binding for Wdr5 Maintenance of Active Chromatin and Embryonic Stem Cell Pluripotency. eLife 2014, 3, e02046. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Notani, D.; Rosenfeld, M.G. Enhancers as Non-Coding Rna Transcription Units: Recent Insights and Future Perspectives. Nat. Rev. Genet. 2016, 4, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.; Lajoie, B.R.; Jain, G.; Dekker, J. The Long-Range Interaction Landscape of Gene Promoters. Nature 2012, 7414, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long Noncoding Rna as Modular Scaffold of Histone Modification Complexes. Science 2010, 5992, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Sayou, C.; Millan-Zambrano, G.; Santos-Rosa, H.; Petfalski, E.; Robson, S.; Houseley, J.; Kouzarides, T.; Tollervey, D. Rna Binding by Histone Methyltransferases Set1 and Set2. Mol. Cell. Biol. 2017, 37, e00165-17. [Google Scholar] [CrossRef] [PubMed]
- Long, H.K.; Blackledge, N.P.; Klose, R.J. Zf-Cxxc Domain-Containing Proteins, Cpg Islands and the Chromatin Connection. Biochem. Soc. Trans. 2013, 3, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Deaton, A.M.; Bird, A. Cpg Islands and the Regulation of Transcription. Genes Dev. 2011, 10, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Cancer Epigenomics: DNA Methylomes and Histone-Modification Maps. Nat. Rev. Genet. 2007, 4, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Birke, M.; Schreiner, S.; Garcia-Cuellar, M.P.; Mahr, K.; Titgemeyer, F.; Slany, R.K. The Mt Domain of the Proto-Oncoprotein Mll Binds to Cpg-Containing DNA and Discriminates against Methylation. Nucleic Acids Res. 2002, 4, 958–965. [Google Scholar] [CrossRef]
- Ayton, P.M.; Chen, E.H.; Cleary, M.L. Binding to Nonmethylated Cpg DNA Is Essential for Target Recognition, Transactivation, and Myeloid Transformation by an Mll Oncoprotein. Mol. Cell. Biol. 2004, 23, 10470–10478. [Google Scholar] [CrossRef] [PubMed]
- Cierpicki, T.; Risner, L.E.; Grembecka, J.; Lukasik, S.M.; Popovic, R.; Omonkowska, M.; Shultis, D.D.; Zeleznik-Le, N.J.; Bushweller, J.H. Structure of the Mll Cxxc Domain-DNA Complex and Its Functional Role in Mll-Af9 Leukemia. Nat. Struct. Mol. Biol. 2010, 1, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Gao, X.; Cao, K.; Morgan, M.A.; Mas, G.; Smith, E.R.; Volk, A.G.; Bartom, E.T.; Crispino, J.D.; di Croce, L.; et al. Not All H3k4 Methylations Are Created Equal: Mll2/Compass Dependency in Primordial Germ Cell Specification. Mol. Cell 2017, 3, 460–475. [Google Scholar] [CrossRef] [PubMed]
- Erfurth, F.E.; Popovic, R.; Grembecka, J.; Cierpicki, T.; Theisler, C.; Xia, Z.B.; Stuart, T.; Diaz, M.O.; Bushweller, J.H.; Zeleznik-Le, N.J. Mll Protects Cpg Clusters from Methylation within the Hoxa9 Gene, Maintaining Transcript Expression. Proc. Natl. Acad. Sci. USA 2008, 21, 7517–7522. [Google Scholar] [CrossRef] [PubMed]
- Reeves, R.; Beckerbauer, L. Hmgi/Y Proteins: Flexible Regulators of Transcription and Chromatin Structure. Biochim. Biophys. Acta 2001, 1519, 13–29. [Google Scholar] [CrossRef]
- Benecke, A.G.; Eilebrecht, S. Rna-Mediated Regulation of Hmga1 Function. Biomolecules 2015, 2, 943–957. [Google Scholar] [CrossRef]
- Bustin, M. Revised Nomenclature for High Mobility Group (Hmg) Chromosomal Proteins. Trends Biochem. Sci. 2001, 3, 152–153. [Google Scholar] [CrossRef]
- Aravind, L.; Landsman, D. At-Hook Motifs Identified in a Wide Variety of DNA-Binding Proteins. Nucleic Acids Res. 1998, 19, 4413–4421. [Google Scholar] [CrossRef]
- Huth, J.R.; Bewley, C.A.; Nissen, M.S.; Evans, J.N.; Reeves, R.; Gronenborn, A.M.; Clore, G.M. The Solution Structure of an Hmg-I(Y)-DNA Complex Defines a New Architectural Minor Groove Binding Motif. Nat. Struct. Biol. 1997, 8, 657–665. [Google Scholar] [CrossRef]
- Brazda, V.; Laister, R.C.; Jagelska, E.B.; Arrowsmith, C. Cruciform Structures Are a Common DNA Feature Important for Regulating Biological Processes. BMC Mol. Biol. 2011, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Peckham, H.E.; Thurman, R.E.; Fu, Y.; Stamatoyannopoulos, J.A.; Noble, W.S.; Struhl, K.; Weng, Z. Nucleosome Positioning Signals in Genomic DNA. Genome Res. 2007, 8, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Grosschedl, R.; Giese, K.; Pagel, J. Hmg Domain Proteins: Architectural Elements in the Assembly of Nucleoprotein Structures. Trends Genet. 1994, 3, 94–100. [Google Scholar] [CrossRef]
- Sarvan, S.; Avdic, V.; Tremblay, V.; Chaturvedi, C.P.; Zhang, P.; Lanouette, S.; Blais, A.; Brunzelle, J.S.; Brand, M.; Couture, J.F. Crystal Structure of the Trithorax Group Protein Ash2l Reveals a Forkhead-Like DNA Binding Domain. Nat. Struct. Mol. Biol. 2011, 7, 857–859. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wan, B.; Wang, K.C.; Cao, F.; Yang, Y.; Protacio, A.; Dou, Y.; Chang, H.Y.; Lei, M. Crystal Structure of the N-Terminal Region of Human Ash2l Shows a Winged-Helix Motif Involved in DNA Binding. EMBO Rep. 2011, 8, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Liang, J.; Xiong, Y.; Shi, F.; Zhang, Y.; Lu, W.; He, Q.; Yang, D.; Chen, R.; Liu, D.; et al. The Trithorax Group Protein Ash2l Is Essential for Pluripotency and Maintaining Open Chromatin in Embryonic Stem Cells. J. Biol. Chem. 2013, 7, 5039–5048. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb Complex Prc2 and Its Mark in Life. Nature 2011, 7330, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Turker, M.S. Gene Silencing in Mammalian Cells and the Spread of DNA Methylation. Oncogene 2002, 35, 5388–5393. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA Methylation Patterns and Epigenetic Memory. Genes Dev. 2002, 1, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.; Zhou, M.M. The Phd Finger: A Versatile Epigenome Reader. Trends Biochem. Sci. 2011, 7, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, Erasing and Reading Histone Lysine Methylations. Exp. Mol. Med. 2017, 4, e324. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.; Wu, J.; Workman, J.L.; Li, B. Readers of Histone Modifications. Cell Res. 2011, 4, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.S.; Lee, S.H.; Kan, P.Y.; Voigt, P.; Ma, L.; Shi, X.; Reinberg, D.; Lee, M.G. Trans-Tail Regulation of Mll4-Catalyzed H3k4 Methylation by H4r3 Symmetric Dimethylation Is Mediated by a Tandem Phd of Mll4. Genes Dev. 2012, 24, 2749–2762. [Google Scholar] [CrossRef] [PubMed]
- Fair, K.; Anderson, M.; Bulanova, E.; Mi, H.; Tropschug, M.; Diaz, M.O. Protein Interactions of the Mll Phd Fingers Modulate Mll Target Gene Regulation in Human Cells. Mol. Cell. Biol. 2001, 10, 3589–3597. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Muntean, A.G.; Wu, L.; Hess, J.L. A Subset of Mixed Lineage Leukemia Proteins Has Plant Homeodomain (Phd)-Mediated E3 Ligase Activity. J. Biol. Chem. 2012, 52, 43410–43416. [Google Scholar] [CrossRef] [PubMed]
- Slany, R.K. The Molecular Mechanics of Mixed Lineage Leukemia. Oncogene 2016, 40, 5215–5223. [Google Scholar] [CrossRef] [PubMed]
- Muntean, A.G.; Giannola, D.; Udager, A.M.; Hess, J.L. The Phd Fingers of Mll Block Mll Fusion Protein-Mediated Transformation. Blood 2008, 12, 4690–4693. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Santillan, D.A.; Koonce, M.; Wei, W.; Luo, R.; Thirman, M.J.; Zeleznik-Le, N.J.; Diaz, M.O. Loss of Mll Phd Finger 3 Is Necessary for Mll-Enl-Induced Hematopoietic Stem Cell Immortalization. Cancer Res. 2008, 15, 6199–6207. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Osmers, U.; Raman, G.; Schwantes, R.H.; Diaz, M.O.; Bushweller, J.H. The Phd3 Domain of Mll Acts as a Cyp33-Regulated Switch between Mll-Mediated Activation and Repression. Biochemistry 2010, 31, 6576–6586. [Google Scholar] [CrossRef] [PubMed]
- Eberl, H.C.; Spruijt, C.G.; Kelstrup, C.D.; Vermeulen, M.; Mann, M. A Map of General and Specialized Chromatin Readers in Mouse Tissues Generated by Label-Free Interaction Proteomics. Mol. Cell 2013, 2, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.P.; Skene, P.J.; Selfridge, J.; Clouaire, T.; Guy, J.; Webb, S.; Kerr, A.R.; Deaton, A.; Andrews, R.; James, K.D.; et al. Cpg Islands Influence Chromatin Structure Via the Cpg-Binding Protein Cfp1. Nature 2010, 7291, 1082–1086. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E.; Ott, M. 50 Years of Protein Acetylation: From Gene Regulation to Epigenetics, Metabolism and Beyond. Nat. Rev. Mol. Cell Biol. 2015, 4, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Rack, J.G.; Lutter, T.; Bjerga, G.E.K.; Guder, C.; Ehrhardt, C.; Varv, S.; Ziegler, M.; Aasland, R. The Phd Finger of P300 Influences Its Ability to Acetylate Histone and Non-Histone Targets. J. Mol. Biol. 2014, 24, 3960–3972. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.D.; Reshmi, S.; Sobulo, O.; Musvee, T.; Anastasi, J.; Raimondi, S.; Schneider, N.R.; Barredo, J.C.; Cantu, E.S.; Schlegelberger, B.; et al. All Patients with the T(11;16)(Q23;P13.3) That Involves Mll and Cbp Have Treatment-Related Hematologic Disorders. Blood 1997, 2, 535–541. [Google Scholar]
- Ida, K.; Kitabayashi, I.; Taki, T.; Taniwaki, M.; Noro, K.; Yamamoto, M.; Ohki, M.; Hayashi, Y. Adenoviral E1a-Associated Protein P300 Is Involved in Acute Myeloid Leukemia with T(11;22)(Q23;Q13). Blood 1997, 12, 4699–4704. [Google Scholar]
- Sugita, K.; Taki, T.; Hayashi, Y.; Shimaoka, H.; Kumazaki, H.; Inoue, H.; Konno, Y.; Taniwaki, M.; Kurosawa, H.; Eguchi, M. Mll-Cbp Fusion Transcript in a Therapy-Related Acute Myeloid Leukemia with the T(11;16)(Q23;P13) Which Developed in an Acute Lymphoblastic Leukemia Patient with Fanconi Anemia. Genes Chromosom. Cancer 2000, 3, 264–269. [Google Scholar] [CrossRef]
- Suganuma, T.; Pattenden, S.G.; Workman, J.L. Diverse Functions of Wd40 Repeat Proteins in Histone Recognition. Genes Dev. 2008, 10, 1265–1268. [Google Scholar] [CrossRef] [PubMed]
- Migliori, V.; Mapelli, M.; Guccione, E. On Wd40 Proteins: Propelling Our Knowledge of Transcriptional Control? Epigenetics 2012, 8, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Wysocka, J.; Swigut, T.; Milne, T.A.; Dou, Y.; Zhang, X.; Burlingame, A.L.; Roeder, R.G.; Brivanlou, A.H.; Allis, C.D. Wdr5 Associates with Histone H3 Methylated at K4 and Is Essential for H3 K4 Methylation and Vertebrate Development. Cell 2005, 6, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Guccione, E.; Bassi, C.; Casadio, F.; Martinato, F.; Cesaroni, M.; Schuchlautz, H.; Luscher, B.; Amati, B. Methylation of Histone H3r2 by Prmt6 and H3k4 by an Mll Complex Are Mutually Exclusive. Nature 2007, 7164, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Guo, L.; Wang, H.; Shen, Y.; Deng, X.W.; Chai, J. Structural Basis for the Specific Recognition of Methylated Histone H3 Lysine 4 by the Wd-40 Protein Wdr5. Mol. Cell 2006, 1, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Couture, J.F.; Collazo, E.; Trievel, R.C. Molecular Recognition of Histone H3 by the Wd40 Protein Wdr5. Nat. Struct. Mol. Biol. 2006, 8, 698–703. [Google Scholar] [CrossRef] [PubMed]
- Dharmarajan, V.; Lee, J.H.; Patel, A.; Skalnik, D.G.; Cosgrove, M.S. Structural Basis for Wdr5 Interaction (Win) Motif Recognition in Human Set1 Family Histone Methyltransferases. J. Biol. Chem. 2012, 33, 27275–27289. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.C.; Matthews, A.G.; Jin, Y.; Chen, C.F.; Chapman, B.A.; Ohsumi, T.K.; Glass, K.C.; Kutateladze, T.G.; Borowsky, M.L.; Struhl, K.; et al. Histone H3r2 Symmetric Dimethylation and Histone H3k4 Trimethylation Are Tightly Correlated in Eukaryotic Genomes. Cell Rep. 2012, 2, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.; Zielinska, A.E.; Shaaban, A.M.; Sanchez-Bailon, M.P.; Jarrold, J.; Clarke, T.L.; Zhang, J.; Francis, A.; Jones, L.J.; Smith, S.; et al. Prmt5 is a Critical Regulator of Breast Cancer Stem Cell Function Via Histone Methylation and Foxp1 Expression. Cell Rep. 2017, 12, 3498–3513. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Banerjee, T.; Vinckevicius, A.; Luo, Q.; Parker, J.B.; Baker, M.R.; Radhakrishnan, I.; Wei, J.J.; Barish, G.D.; Chakravarti, D. A Role for Wdr5 in Integrating Threonine 11 Phosphorylation to Lysine 4 Methylation on Histone H3 During Androgen Signaling and in Prostate Cancer. Mol. Cell 2014, 4, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.J.; Clifton-Bligh, R.; Marsh, D.J. Histone H2b Monoubiquitination: Roles to Play in Human Malignancy. Endocr. Relat. Cancer 2015, 1, T19–T33. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, G.; Oren, M. Writing and Reading H2b Monoubiquitylation. Biochim. Biophys. Acta 2014, 8, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Zheng, Y.; Pham, A.D.; Mandal, S.S.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Monoubiquitination of Human Histone H2b: The Factors Involved and Their Roles in Hox Gene Regulation. Mol. Cell 2005, 4, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Shema, E.; Tirosh, I.; Aylon, Y.; Huang, J.; Ye, C.; Moskovits, N.; Raver-Shapira, N.; Minsky, N.; Pirngruber, J.; Tarcic, G.; et al. The Histone H2b-Specific Ubiquitin Ligase Rnf20/Hbre1 Acts as a Putative Tumor Suppressor through Selective Regulation of Gene Expression. Genes Dev. 2008, 19, 2664–2676. [Google Scholar] [CrossRef] [PubMed]
- Minsky, N.; Shema, E.; Field, Y.; Schuster, M.; Segal, E.; Oren, M. Monoubiquitinated H2b Is Associated with the Transcribed Region of Highly Expressed Genes in Human Cells. Nat. Cell Biol. 2008, 4, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.K.; Heath, C.; Hair, A.; West, A.G. Histone Crosstalk Directed by H2b Ubiquitination Is Required for Chromatin Boundary Integrity. PLoS Genet. 2011, 7, e1002175. [Google Scholar] [CrossRef] [PubMed]
- Batta, K.; Zhang, Z.; Yen, K.; Goffman, D.B.; Pugh, B.F. Genome-Wide Function of H2b Ubiquitylation in Promoter and Genic Regions. Genes Dev. 2011, 21, 2254–2265. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Guermah, M.; McGinty, R.K.; Lee, J.S.; Tang, Z.; Milne, T.A.; Shilatifard, A.; Muir, T.W.; Roeder, R.G. Rad6-Mediated Transcription-Coupled H2b Ubiquitylation Directly Stimulates H3k4 Methylation in Human Cells. Cell 2009, 3, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Lee, S.Y.; Zhou, B.; Nguyen, U.T.; Muir, T.W.; Tan, S.; Dou, Y. Ash2l Regulates Ubiquitylation Signaling to Mll: Trans-Regulation of H3 K4 Methylation in Higher Eukaryotes. Mol. Cell 2013, 6, 1108–1120. [Google Scholar] [CrossRef] [PubMed]
- Vethantham, V.; Yang, Y.; Bowman, C.; Asp, P.; Lee, J.H.; Skalnik, D.G.; Dynlacht, B.D. Dynamic Loss of H2b Ubiquitylation without Corresponding Changes in H3k4 Trimethylation During Myogenic Differentiation. Mol. Cell. Biol. 2012, 6, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Daniel, J.; Espejo, A.; Lake, A.; Krishna, M.; Xia, L.; Zhang, Y.; Bedford, M.T. Tudor, Mbt and Chromo Domains Gauge the Degree of Lysine Methylation. EMBO Rep. 2006, 4, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.J.; Rickels, R.A.; Collings, C.K.; He, X.; Cao, K.; Herz, H.M.; Cozzolino, K.A.; Abshiru, N.A.; Marshall, S.A.; Rendleman, E.J.; et al. A Cryptic Tudor Domain Links Brwd2/Phip to Compass-Mediated Histone H3k4 Methylation. Genes Dev. 2017, 19, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.J.; Wang, Z. Readout of Epigenetic Modifications. Annu. Rev. Biochem. 2013, 82, 81–118. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, M.; Mulder, K.W.; Denissov, S.; Pijnappel, W.W.; van Schaik, F.M.; Varier, R.A.; Baltissen, M.P.; Stunnenberg, H.G.; Mann, M.; Timmers, H.T. Selective Anchoring of Tfiid to Nucleosomes by Trimethylation of Histone H3 Lysine 4. Cell 2007, 1, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Nogales, E.; Patel, A.B.; Louder, R.K. Towards a Mechanistic Understanding of Core Promoter Recognition from Cryo-Em Studies of Human Tfiid. Curr. Opin. Struct. Biol. 2017, 47, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R. Tbp-Associated Factors (Tafiis): Multiple, Selective Transcriptional Mediators in Common Complexes. Trends Biochem. Sci. 2000, 2, 59–63. [Google Scholar] [CrossRef]
- Van Ingen, H.; van Schaik, F.M.; Wienk, H.; Ballering, J.; Rehmann, H.; Dechesne, A.C.; Kruijzer, J.A.; Liskamp, R.M.; Timmers, H.T.; Boelens, R. Structural Insight into the Recognition of the H3k4me3 Mark by the Tfiid Subunit Taf3. Structure 2008, 8, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Wysocka, J.; Swigut, T.; Xiao, H.; Milne, T.A.; Kwon, S.Y.; Landry, J.; Kauer, M.; Tackett, A.J.; Chait, B.T.; Badenhorst, P.; et al. A Phd Finger of Nurf Couples Histone H3 Lysine 4 Trimethylation with Chromatin Remodelling. Nature 2006, 7098, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Alkhatib, S.G.; Landry, J.W. The Nucleosome Remodeling Factor. FEBS Lett. 2011, 20, 3197–3207. [Google Scholar] [CrossRef] [PubMed]
- Tallen, G.; Riabowol, K. Keep-Ing Balance: Tumor Suppression by Epigenetic Regulation. FEBS Lett. 2014, 16, 2728–2742. [Google Scholar] [CrossRef] [PubMed]
- Champagne, K.S.; Saksouk, N.; Pena, P.V.; Johnson, K.; Ullah, M.; Yang, X.J.; Cote, J.; Kutateladze, T.G. The Crystal Structure of the Ing5 Phd Finger in Complex with an H3k4me3 Histone Peptide. Proteins 2008, 4, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Pena, P.V.; Davrazou, F.; Shi, X.; Walter, K.L.; Verkhusha, V.V.; Gozani, O.; Zhao, R.; Kutateladze, T.G. Molecular Mechanism of Histone H3k4me3 Recognition by Plant Homeodomain of Ing2. Nature 2006, 7098, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Hong, T.; Walter, K.L.; Ewalt, M.; Michishita, E.; Hung, T.; Carney, D.; Pena, P.; Lan, F.; Kaadige, M.R.; et al. Ing2 Phd Domain Links Histone H3 Lysine 4 Methylation to Active Gene Repression. Nature 2006, 7098, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; Cayrou, C.; Ullah, M.; Landry, A.J.; Cote, V.; Selleck, W.; Lane, W.S.; Tan, S.; Yang, X.J.; Cote, J. Ing Tumor Suppressor Proteins Are Critical Regulators of Chromatin Acetylation Required for Genome Expression and Perpetuation. Mol. Cell 2006, 1, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Palacios, A.; Moreno, A.; Oliveira, B.L.; Rivera, T.; Prieto, J.; Garcia, P.; Fernandez-Fernandez, M.R.; Bernado, P.; Palmero, I.; Blanco, F.J. The Dimeric Structure and the Bivalent Recognition of H3k4me3 by the Tumor Suppressor Ing4 Suggests a Mechanism for Enhanced Targeting of the Hbo1 Complex to Chromatin. J. Mol. Biol. 2010, 4, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Kinkley, S.; Staege, H.; Mohrmann, G.; Rohaly, G.; Schaub, T.; Kremmer, E.; Winterpacht, A.; Will, H. Spoc1: A Novel Phd-Containing Protein Modulating Chromatin Structure and Mitotic Chromosome Condensation. J. Cell Sci. 2009, 122, 2946–2956. [Google Scholar] [CrossRef] [PubMed]
- Bordlein, A.; Scherthan, H.; Nelkenbrecher, C.; Molter, T.; Bosl, M.R.; Dippold, C.; Birke, K.; Kinkley, S.; Staege, H.; Will, H.; et al. Spoc1 (Phf13) Is Required for Spermatogonial Stem Cell Differentiation and Sustained Spermatogenesis. J. Cell Sci. 2011, 124, 3137–3148. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.R.; Xu, C.; Fuchs, A.; Mund, A.; Lange, M.; Staege, H.; Schubert, T.; Bian, C.; Dunkel, I.; Eberharter, A.; et al. Phf13 Is a Molecular Reader and Transcriptional Co-Regulator of H3k4me2/3. eLife 2016, 5, e10607. [Google Scholar] [CrossRef] [PubMed]
- Barth, T.K.; Imhof, A. Fast Signals and Slow Marks: The Dynamics of Histone Modifications. Trends Biochem. Sci. 2010, 11, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Higgins, J.M. Histone Modifications and Mitosis: Countermarks, Landmarks, and Bookmarks. Trends Cell Biol. 2013, 4, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Oomen, M.E.; Dekker, J. Epigenetic Characteristics of the Mitotic Chromosome in 1d and 3d. Crit. Rev. Biochem. Mol. Biol. 2017, 2, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Blobel, G.A.; Kadauke, S.; Wang, E.; Lau, A.W.; Zuber, J.; Chou, M.M.; Vakoc, C.R. A Reconfigured Pattern of Mll Occupancy within Mitotic Chromatin Promotes Rapid Transcriptional Reactivation Following Mitotic Exit. Mol. Cell 2009, 6, 970–983. [Google Scholar] [CrossRef] [PubMed]
- Palozola, K.C.; Donahue, G.; Liu, H.; Grant, G.R.; Becker, J.S.; Cote, A.; Yu, H.; Raj, A.; Zaret, K.S. Mitotic Transcription and Waves of Gene Reactivation During Mitotic Exit. Science 2017, 6359, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Festuccia, N.; Gonzalez, I.; Owens, N.; Navarro, P. Mitotic Bookmarking in Development and Stem Cells. Development 2017, 20, 3633–3645. [Google Scholar] [CrossRef] [PubMed]
- Schellhaus, A.K.; de Magistris, P.; Antonin, W. Nuclear Reformation at the End of Mitosis. J. Mol. Biol. 2016, 428, 1962–1985. [Google Scholar] [CrossRef] [PubMed]
- Raccaud, M.; Suter, D.M. Transcription Factor Retention on Mitotic Chromosomes: Regulatory Mechanisms and Impact on Cell Fate Decisions. FEBS Lett. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gatchalian, J.; Gallardo, C.M.; Shinsky, S.A.; Ospina, R.R.; Liendo, A.M.; Krajewski, K.; Klein, B.J.; Andrews, F.H.; Strahl, B.D.; van Wely, M.; et al. Chromatin Condensation and Recruitment of Phd Finger Proteins to Histone H3k4me3 Are Mutually Exclusive. Nucleic Acids Res. 2016, 13, 6102–6112. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Rincon-Arano, H.; Zhao, W.; Rothbart, S.B.; Tong, Q.; Parkhurst, S.M.; Strahl, B.D.; Deng, L.W.; Groudine, M.; Kutateladze, T.G. Molecular Basis for Chromatin Binding and Regulation of Mll5. Proc. Natl. Acad. Sci. USA 2013, 28, 11296–11301. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Dai, J.; Daum, J.R.; Niedzialkowska, E.; Banerjee, B.; Stukenberg, P.T.; Gorbsky, G.J.; Higgins, J.M. Histone H3 Thr-3 Phosphorylation by Haspin Positions Aurora B at Centromeres in Mitosis. Science 2010, 6001, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Varier, R.A.; Outchkourov, N.S.; de Graaf, P.; van Schaik, F.M.; Ensing, H.J.; Wang, F.; Higgins, J.M.; Kops, G.J.; Timmers, H.T. A Phospho/Methyl Switch at Histone H3 Regulates Tfiid Association with Mitotic Chromosomes. EMBO J. 2010, 23, 3967–3978. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Yuan, Z.F.; Han, Y.; Marchione, D.M.; Garcia, B.A. Preferential Phosphorylation on Old Histones During Early Mitosis in Human Cells. J. Biol. Chem. 2016, 29, 15342–15357. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Domain organization of human KMT2 enzymes and WRAD complex proteins. AT-hooks, adenosine-thymidine-hook; CXXC, Zinc finger-CXXC domain; FYRN/C, phenylalanine and tyrosine rich region (N- and C-terminal); HMG, high mobility group; HWH, helix-wing-helix domain; N-SET, N-terminal of SET; PHD, plant homeodomain; Post-SET, C-terminal of SET; RRM RNA recognition motive; SDI, Sdc1-Dpy-30 interaction; SET, Su(var)3-9, Enhancer-of-zeste and Trithorax; SPRY, spla and the ryanodine receptor domain; WD repeat, tryptophan-aspartic acid repeat.
Figure 1.
Domain organization of human KMT2 enzymes and WRAD complex proteins. AT-hooks, adenosine-thymidine-hook; CXXC, Zinc finger-CXXC domain; FYRN/C, phenylalanine and tyrosine rich region (N- and C-terminal); HMG, high mobility group; HWH, helix-wing-helix domain; N-SET, N-terminal of SET; PHD, plant homeodomain; Post-SET, C-terminal of SET; RRM RNA recognition motive; SDI, Sdc1-Dpy-30 interaction; SET, Su(var)3-9, Enhancer-of-zeste and Trithorax; SPRY, spla and the ryanodine receptor domain; WD repeat, tryptophan-aspartic acid repeat.
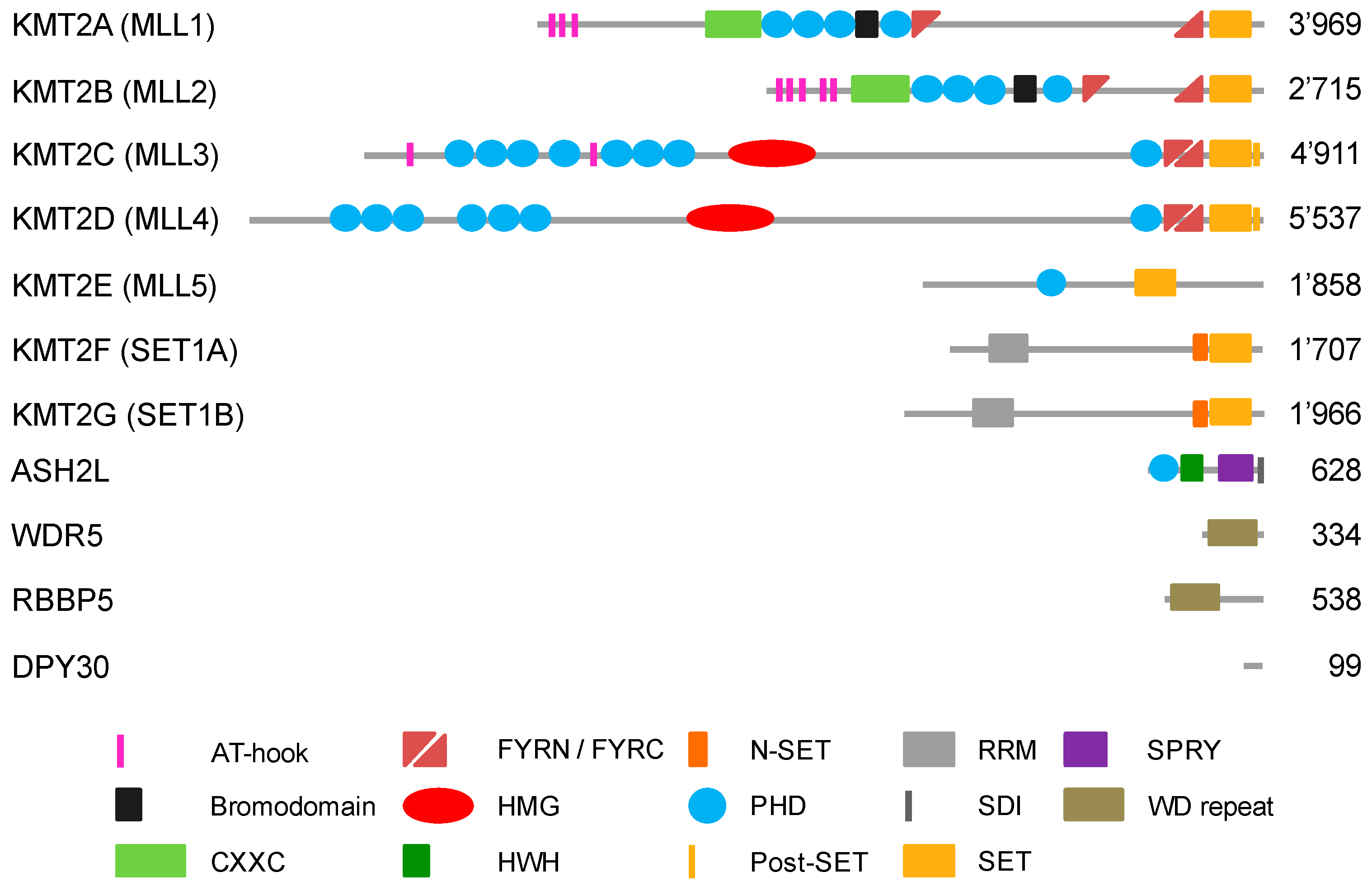
Figure 2.
Schematic representation of the KMT2 complex. The interactions of the KMT2 enzymes with the subunits of the WRAD complex are shown by red lines [61,62,63]. For details see the text.

Figure 3.
Multiple ways of recruiting KMT2s complexes to chromatin. KMT2 complex recruitment includes direct and indirect DNA interactions, possibly by more than one of these occurring cooperatively: (1) AT-rich regions recognized by AT-hooks; (2) un-methylated CpG islands by CXXC domains; (3) DNA structures by AT-hooks and HMG-I domains; (4) nucleosomes by WDR5 that interacts with the histone H3 N-terminal region; (5) nucleosomes with distinct histone marks by PHD fingers; (6) through binding to sequence specific transcription factors (sTF); (7) recruited by co-factors that bind to sTFs; (8) through long noncoding RNAs; (9) through interaction with basal transcription factors (bTF); (10) indirectly through binding to RNA polymerase II (RNAP II).
Figure 3.
Multiple ways of recruiting KMT2s complexes to chromatin. KMT2 complex recruitment includes direct and indirect DNA interactions, possibly by more than one of these occurring cooperatively: (1) AT-rich regions recognized by AT-hooks; (2) un-methylated CpG islands by CXXC domains; (3) DNA structures by AT-hooks and HMG-I domains; (4) nucleosomes by WDR5 that interacts with the histone H3 N-terminal region; (5) nucleosomes with distinct histone marks by PHD fingers; (6) through binding to sequence specific transcription factors (sTF); (7) recruited by co-factors that bind to sTFs; (8) through long noncoding RNAs; (9) through interaction with basal transcription factors (bTF); (10) indirectly through binding to RNA polymerase II (RNAP II).
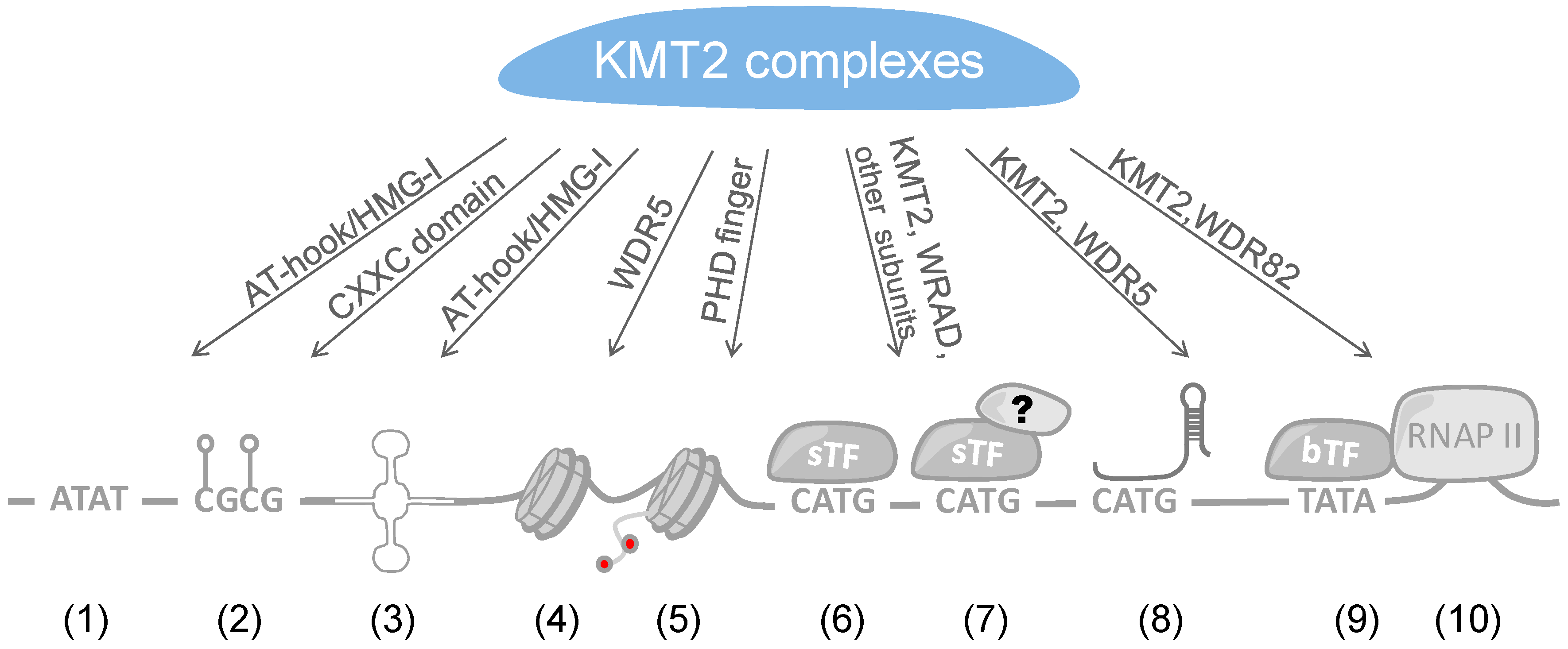

{kind=link}
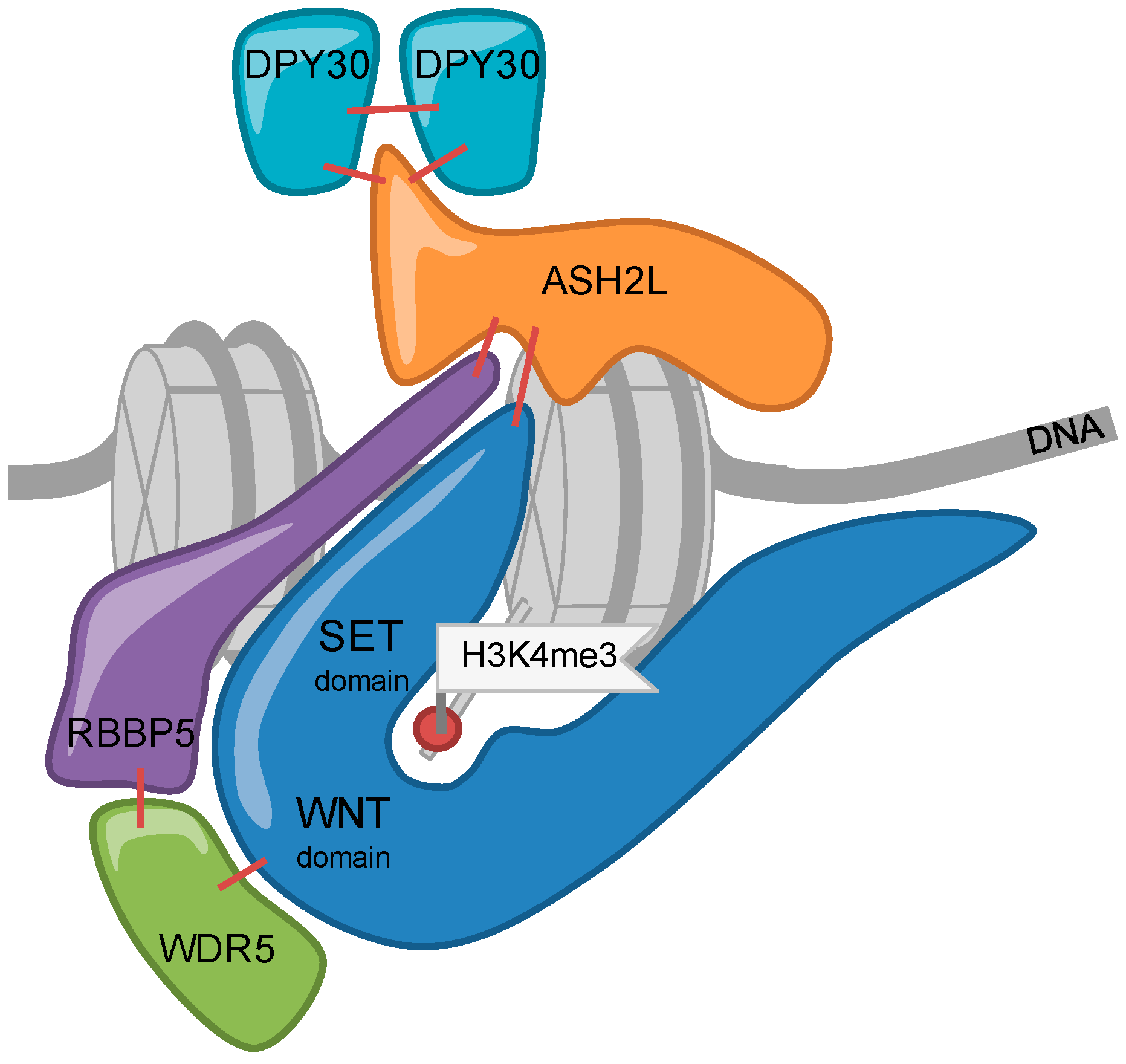{kind=link}
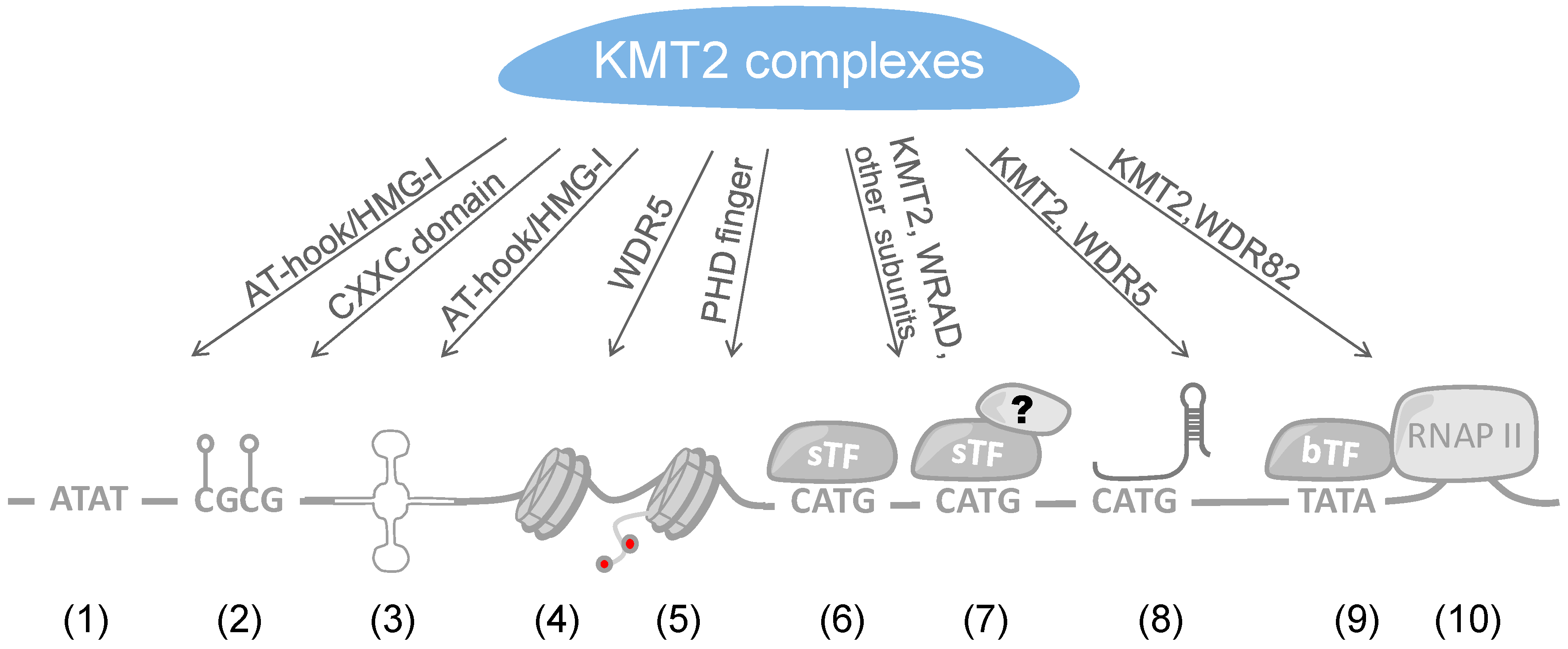{kind=link}
Table 1.
Overview of enzymes with methyltransferase activity in vitro (gray) or in cells (dark grey), and demethylase activity.
Table 1.
Overview of enzymes with methyltransferase activity in vitro (gray) or in cells (dark grey), and demethylase activity.
| Writers Able to Methylate H3K4 (Gene ID) | Me1 | Me2 | Me3 | Erasers Able to De-Methylate H3K4 (Gene ID) [39] | Me1 | Me2 | Me3 | |||
|---|---|---|---|---|---|---|---|---|---|---|
| MLL1 (KMT2A) (#4297) | [40] * | [41] | LSD1(KDM1A) (#23028) | |||||||
| MLL2 (KMT2B) (#9757) | [40] | [42] | LSD2 (KDM1B) (#221656) | |||||||
| MLL3 (KMT2C) (#58508) | [40] | [43] | JHDM1B (KDM2B) (#84678) | |||||||
| MLL4 (KMT2D) (#8085) | [40] | [44] | JARID1A (KDM5A) (#5927) | |||||||
| SET1A (KMT2F) (#9739) | [40] | [45] | JARID1B (KDM5B) (#10765) | |||||||
| SET1B (KMT2G) (#23067) | [40] | [45] | JARID1C (KDM5C) (#8242) | |||||||
| PRDM9 (Meisetz) (#56979) | [34] | [34] | JARID1D (KDM5D) (#8284) | |||||||
| SET7/9 (KMT7) (#80854) | [46,47] | |||||||||
| SMYD3 (KMT3E) (#64754) | [46] | [48] | ||||||||
| SMYD1/2 (#150572/56950) | [49] | |||||||||
* If a reference is given for di- or tri-methylation, lower levels of methylation can potentially also occur by this enzyme.
Table 2.
Summary of sequence-specific transcription factors known to recruit KMT2 complexes.
| Transcription Factor | References |
|---|---|
| OCT4 | [140,141] |
| ANCCA/ATAD2 | [175,178,179] |
| Ap2delta | [152,153] |
| c-MYB | [170] |
| E2F | [175,176] |
| ERα | [164] |
| FOXA1 | [173] |
| FXR | [158] |
| MAFA and B | [159] |
| Mef2d | [140] |
| MYC/MAX | [143,144] |
| NANOG | [140] |
| NF-Y | [149] |
| NF-E2 | [150,151] |
| p53 | [160] |
| Pax2 and Pax5 | [50,68,69,155,156] |
| Pax7 | [148,173] |
| PRMT4 | [146,147] |
| SOX2 | [140,141] |
| USF1 | [81] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bochyńska, A.; Lüscher-Firzlaff, J.; Lüscher, B. Modes of Interaction of KMT2 Histone H3 Lysine 4 Methyltransferase/COMPASS Complexes with Chromatin. Cells 2018, 7, 17. https://doi.org/10.3390/cells7030017
AMA Style
Bochyńska A, Lüscher-Firzlaff J, Lüscher B. Modes of Interaction of KMT2 Histone H3 Lysine 4 Methyltransferase/COMPASS Complexes with Chromatin. Cells. 2018; 7(3):17. https://doi.org/10.3390/cells7030017
Chicago/Turabian StyleBochyńska, Agnieszka, Juliane Lüscher-Firzlaff, and Bernhard Lüscher. 2018. "Modes of Interaction of KMT2 Histone H3 Lysine 4 Methyltransferase/COMPASS Complexes with Chromatin" Cells 7, no. 3: 17. https://doi.org/10.3390/cells7030017
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.