OGT (O-GlcNAc Transferase) Selectively Modifies Multiple Residues Unique to Lamin A

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Nuclear Extraction, Immunoprecipitation, and Western Blotting
2.3. Succinylated Wheat Germ Agglutinin (sWGA) Pulldown
2.4. Mice
2.5. Peptide Arrays and Antibody Epitope Mapping
2.6. Purification of Recombinant Lamin Tails
2.7. In Vitro O-GlcNAcylation Reactions
2.8. Mass Spectrometry
3. Results
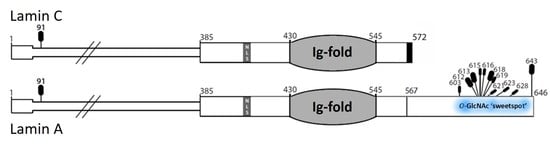
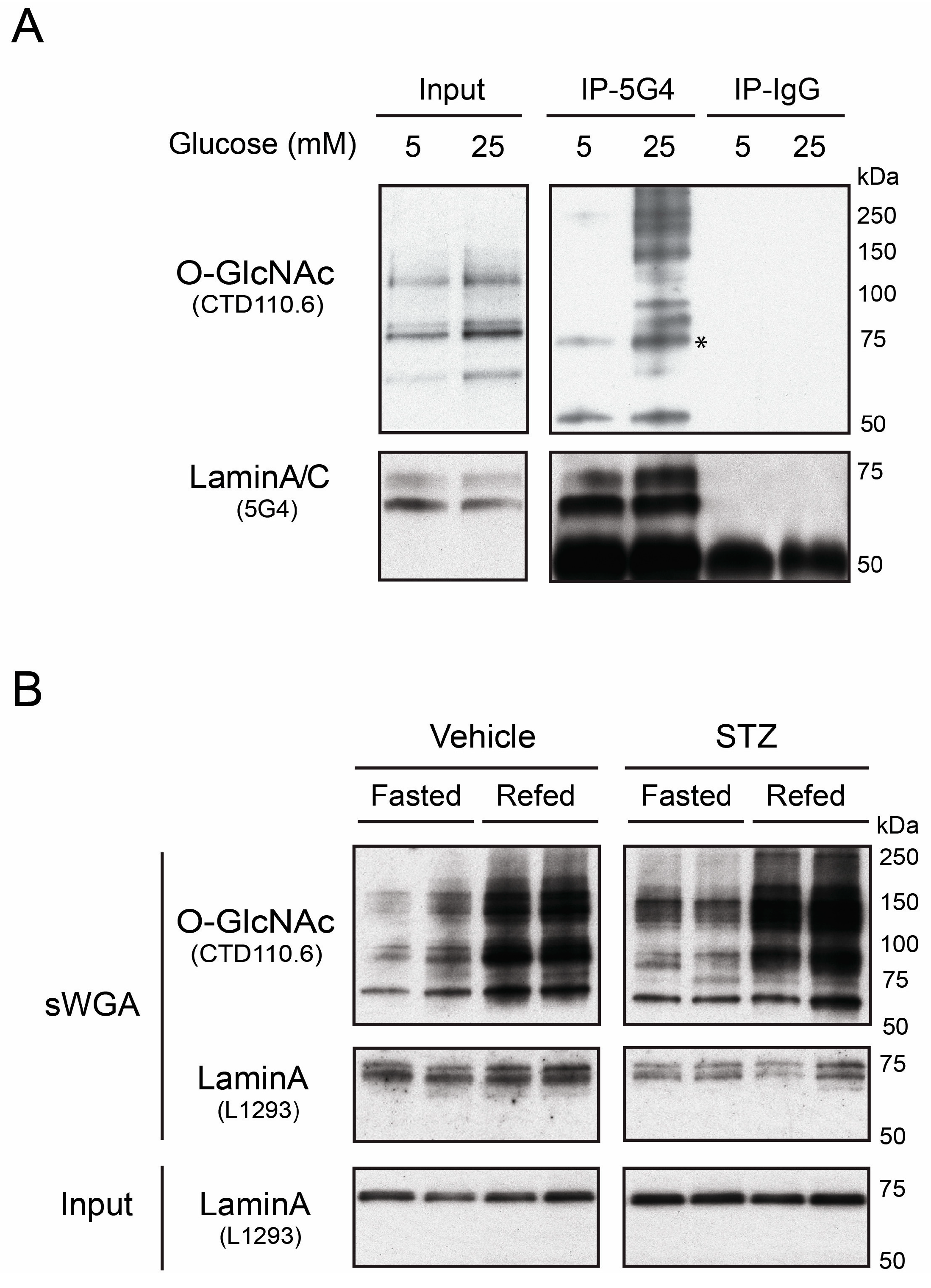
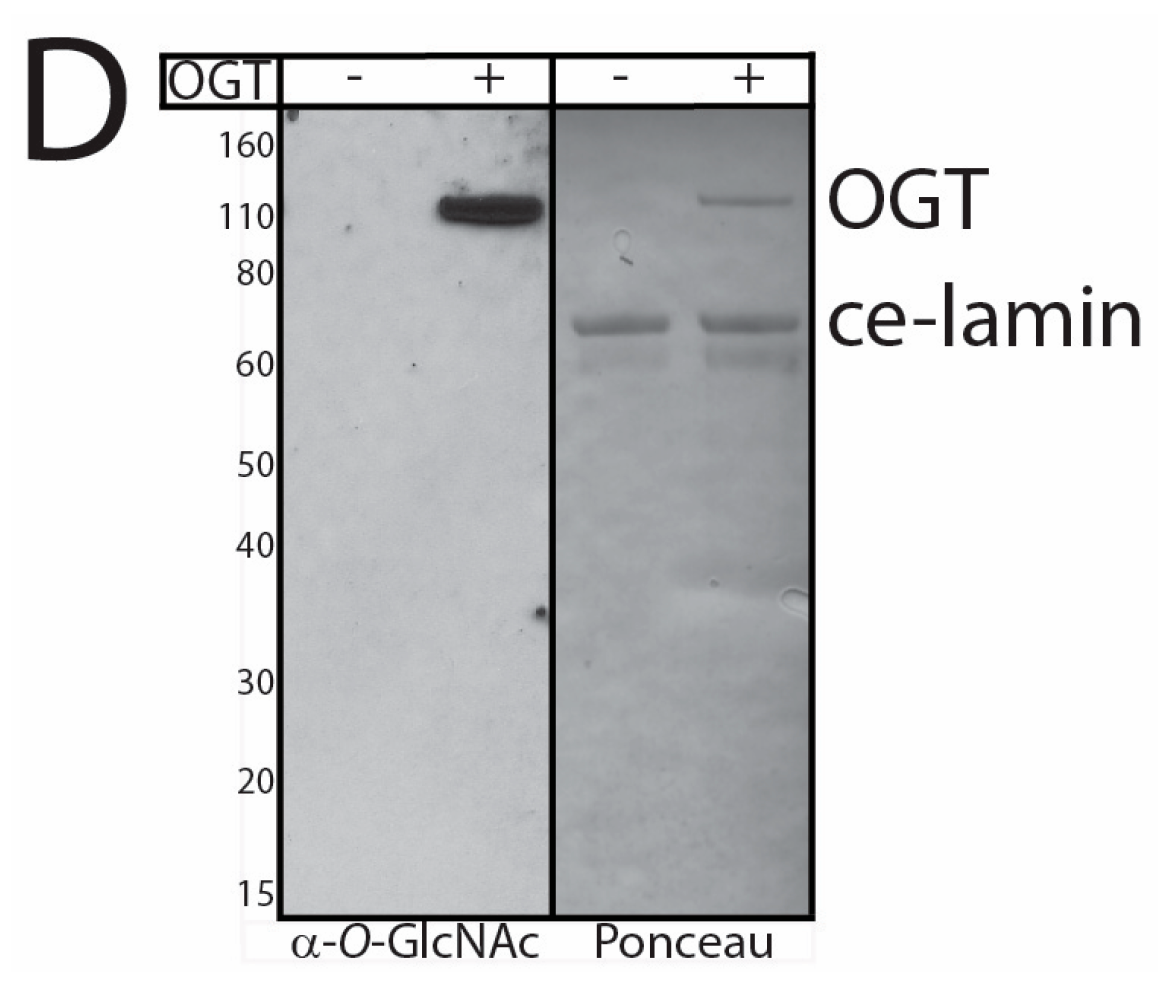
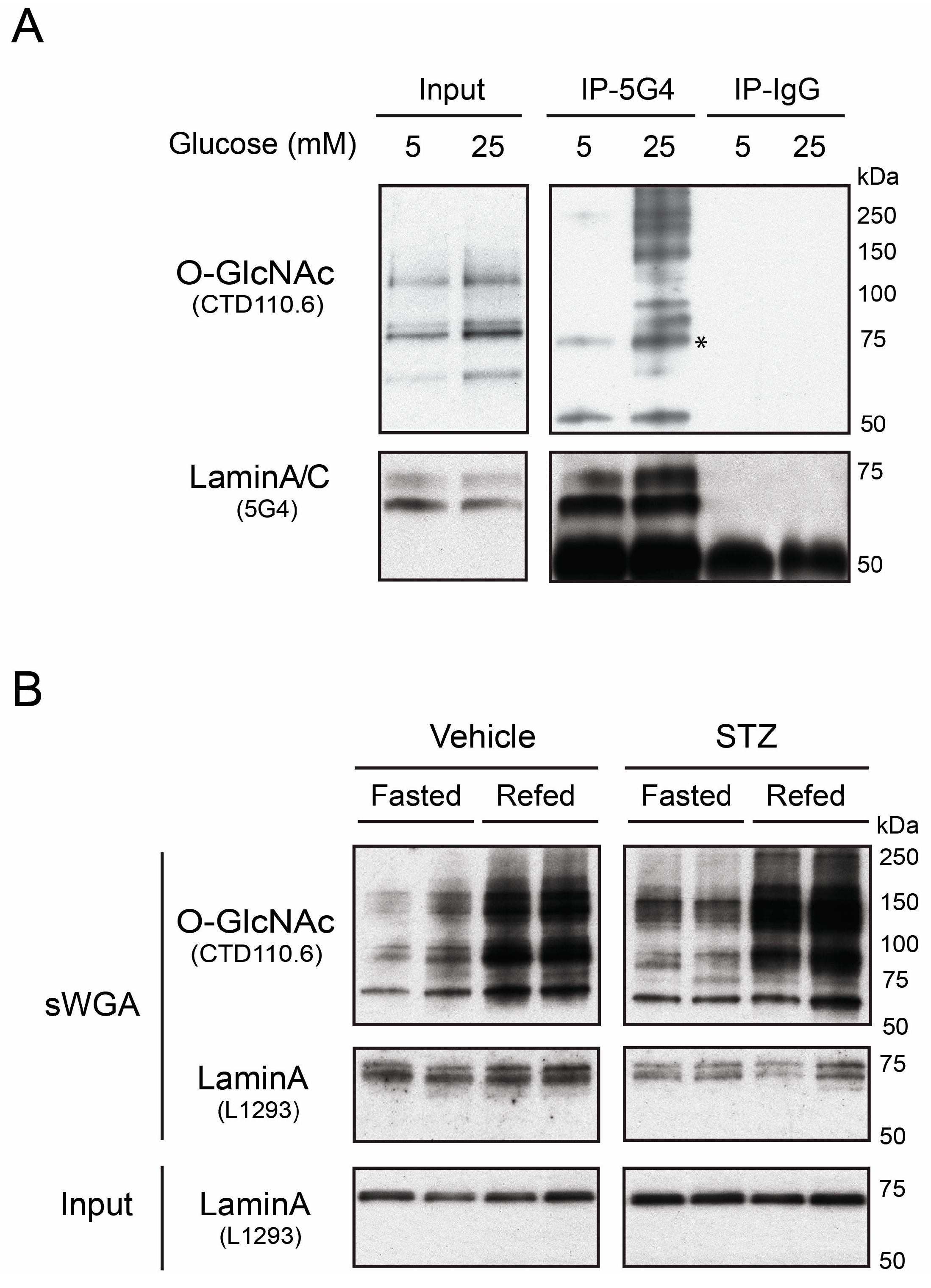
3.1. Native Lamin A is O-GlcNAcylated in Human Hepatoma Cells and Mouse Liver
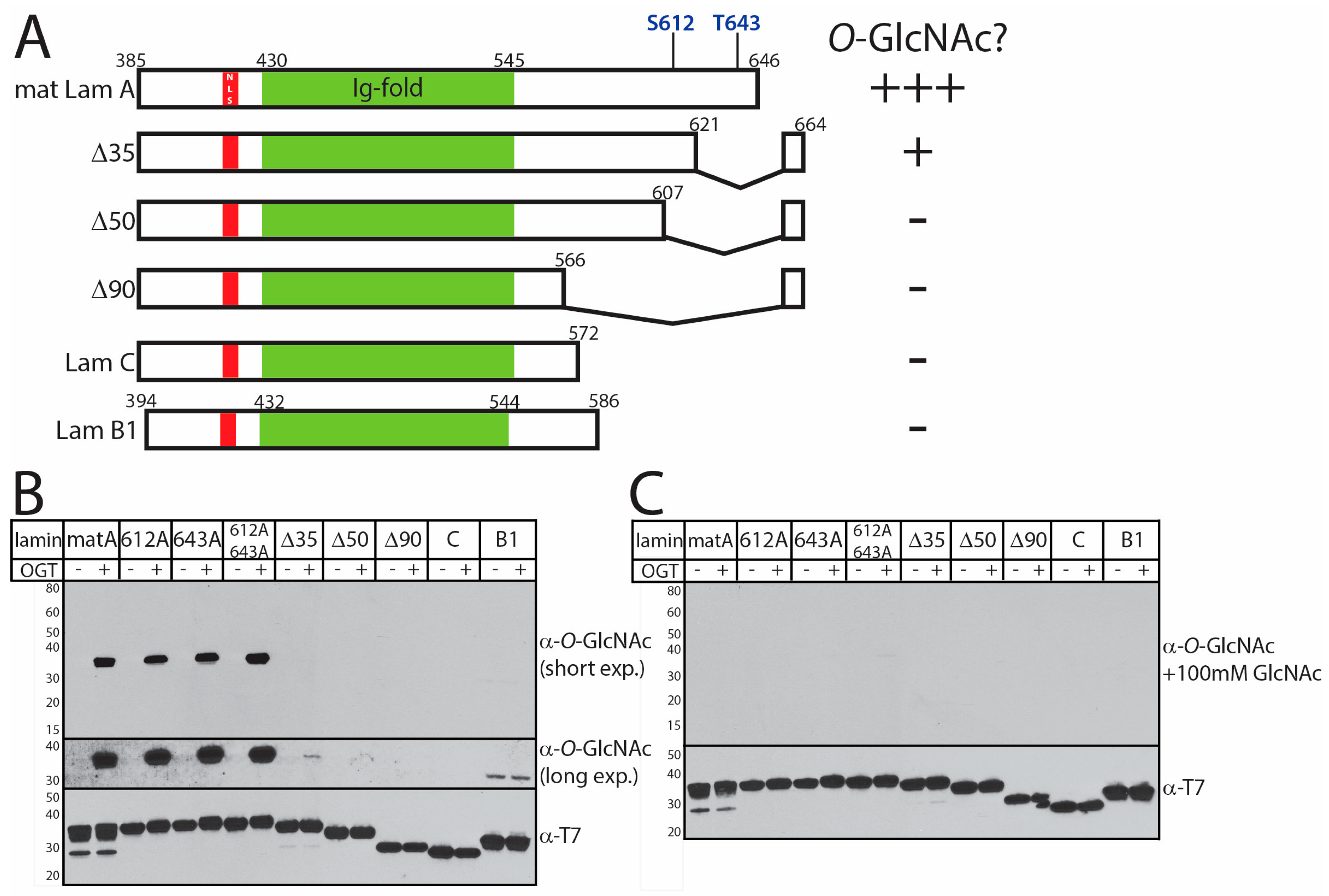
3.2. In Vitro O-GlcNAcylation of Recombinant Lamin Tails
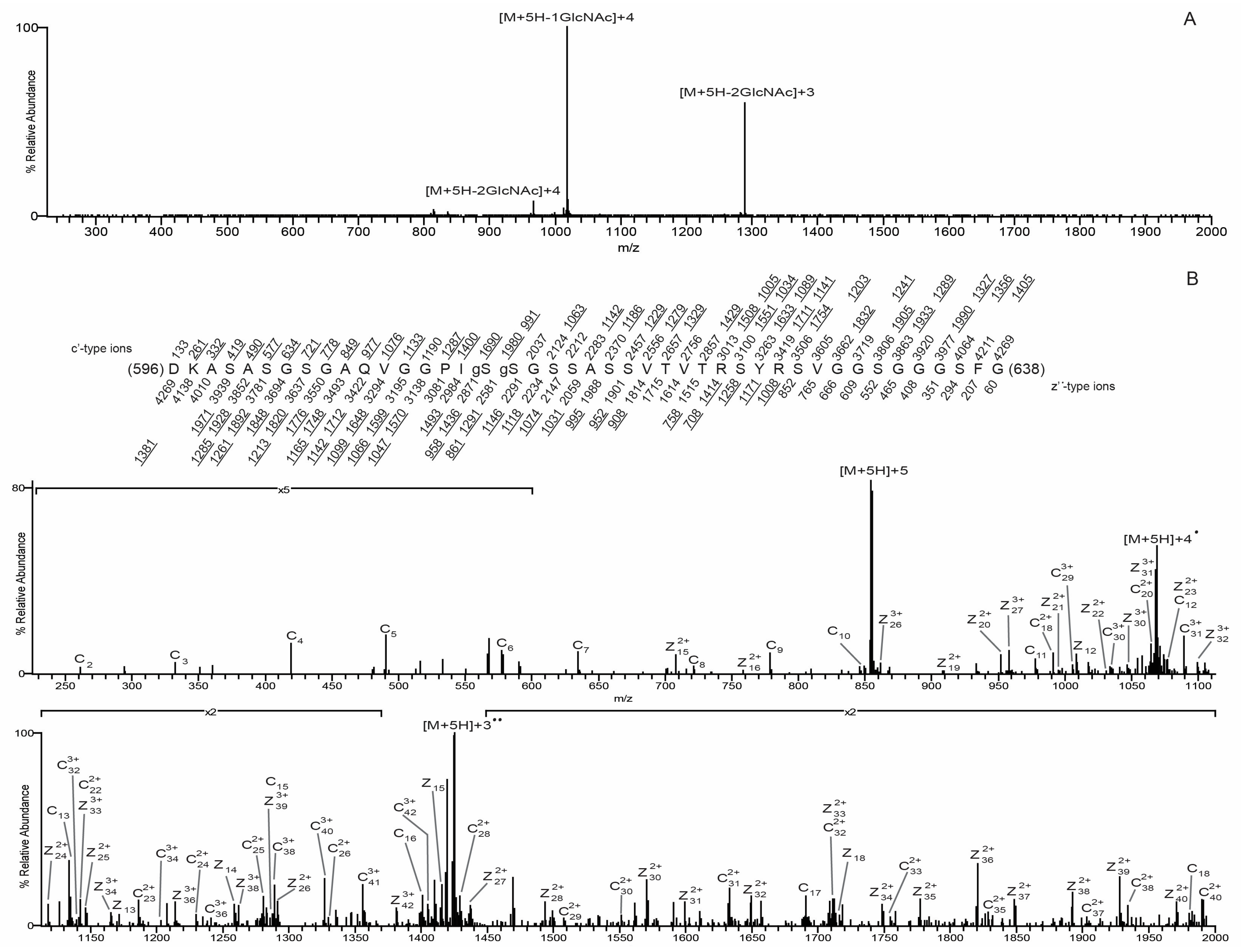
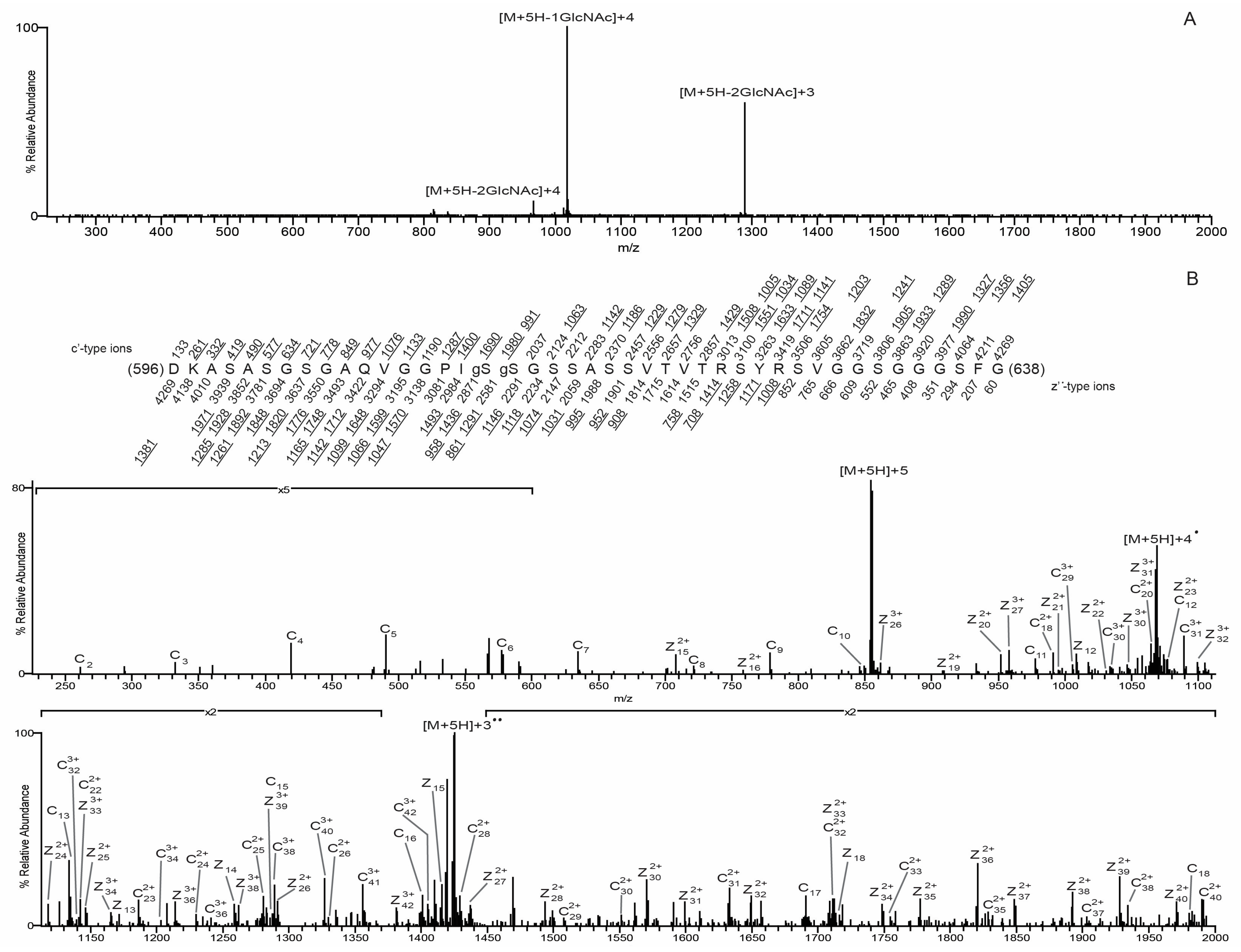
3.3. O-GlcNAc Site Identification by Mass Spectrometry
4. Discussion
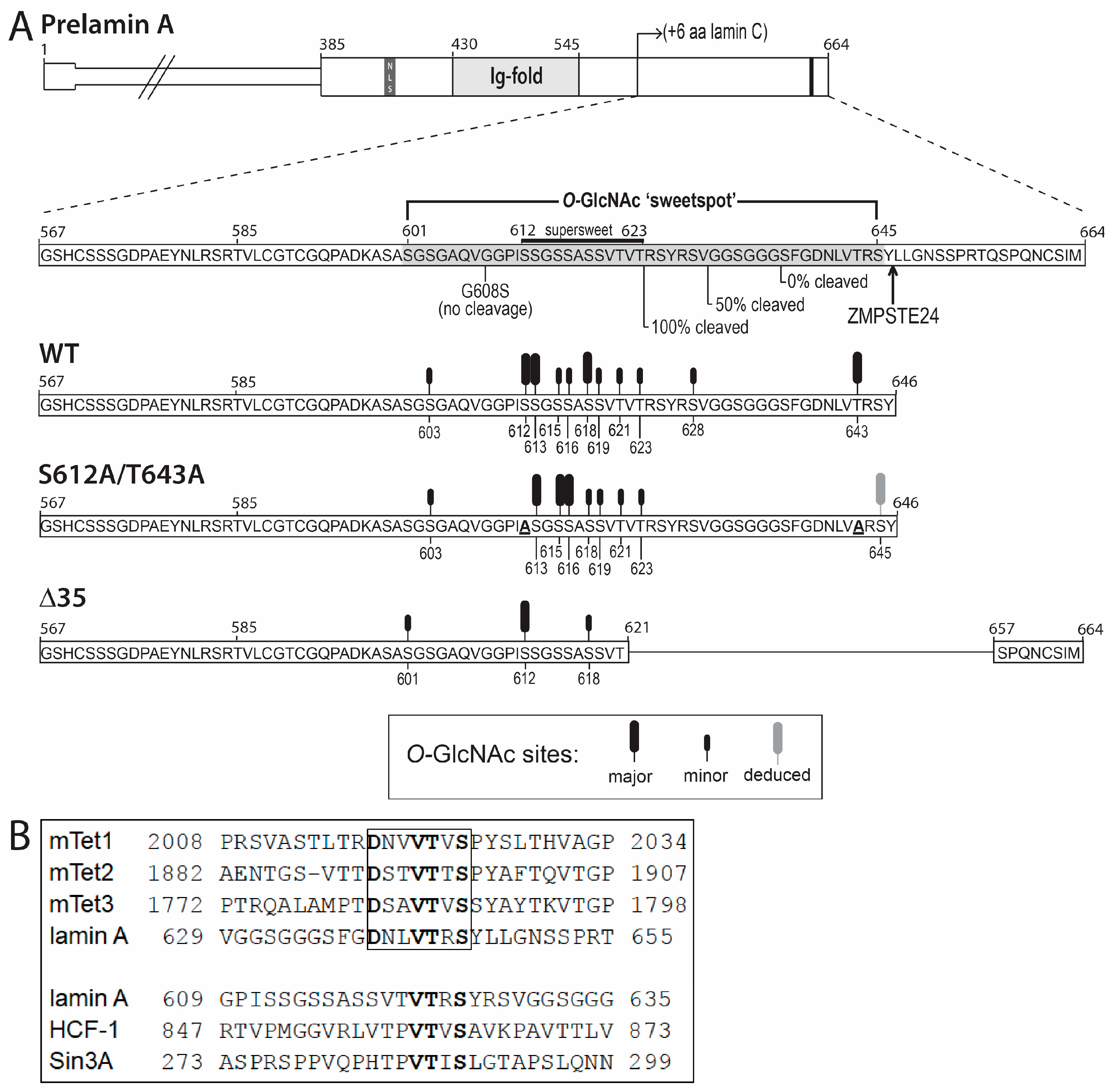
4.1. Most O-GlcNAc Sites in Lamin A Are Not Predicted by Current Algorithms
4.2. The ‘Sweet Spot’ in Relation to the Site of ZPMSTE24-Dependent Cleavage
4.3. Second Role for Residues Near the ZMPSTE24-Dependent Cleavage Site
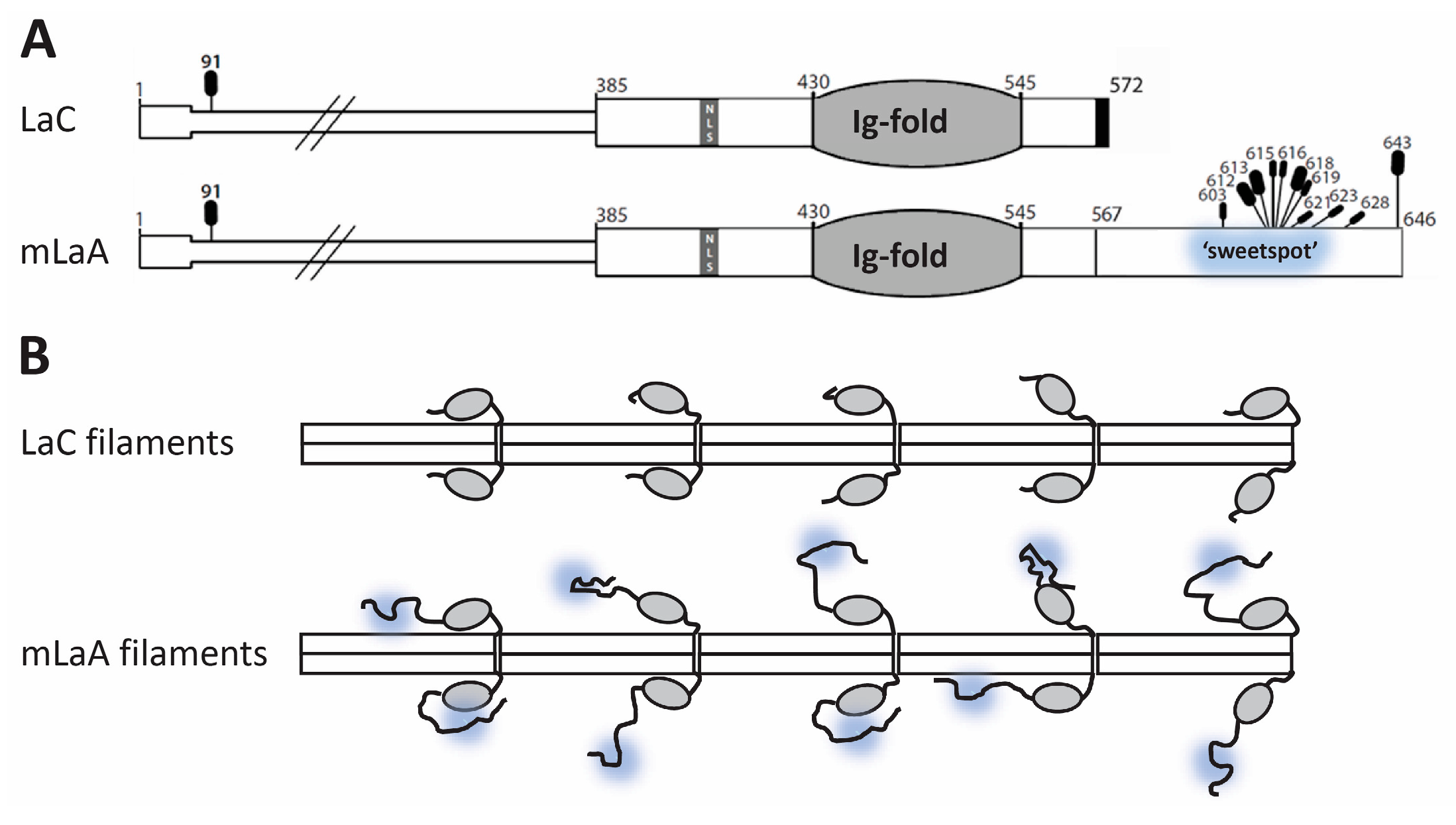
4.4. Selective OGT Targeting of Lamin A as a Mechanism for Separate Functions and Differential Regulation of Lamin A versus Lamin C
4.5. Potential Impacts of O-GlcNAc on Mature Lamin A
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Simon, D.N.; Wilson, K.L. The nucleoskeleton as a genome-associated dynamic ‘network of networks’. Nat. Rev. Mol. Cell Biol. 2011, 12, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J.; Schirmer, E.C. Nuclear membrane diversity: Underlying tissue-specific pathologies in disease? Curr. Opin. Cell Biol. 2015, 34, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Gruenbaum, Y.; Foisner, R. Lamins: Nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu. Rev. Biochem. 2015, 84, 131–164. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, T.A.; Misteli, T. The lamin protein family. Genome Biol. 2011, 12, 222. [Google Scholar] [CrossRef] [PubMed]
- Peter, A.; Stick, R. Evolution of the lamin protein family: What introns can tell. Nucleus 2012, 3, 44–59. [Google Scholar] [CrossRef] [PubMed]
- Koreny, L.; Field, M.C. Ancient eukaryotic origin and evolutionary plasticity of nuclear lamina. Genome Biol. Evol. 2016, 8, 2663–2671. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.; Stewart, C.L. Functional architecture of the cell’s nucleus in development, aging, and disease. Curr. Top. Dev. Biol. 2014, 109, 1–52. [Google Scholar] [PubMed]
- Young, J.; Morbois-Trabut, L.; Couzinet, B.; Lascols, O.; Dion, E.; Bereziat, V.; Feve, B.; Richard, I.; Capeau, J.; Chanson, P.; et al. Type A insulin resistance syndrome revealing a novel lamin A mutation. Diabetes 2005, 54, 1873–1878. [Google Scholar] [CrossRef] [PubMed]
- Guenantin, A.C.; Briand, N.; Bidault, G.; Afonso, P.; Bereziat, V.; Vatier, C.; Lascols, O.; Caron-Debarle, M.; Capeau, J.; Vigouroux, C. Nuclear envelope-related lipodystrophies. Semin. Cell Dev. Biol. 2014, 29, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Vigouroux, C.; Magre, J.; Vantyghem, M.C.; Bourut, C.; Lascols, O.; Shackleton, S.; Lloyd, D.J.; Guerci, B.; Padova, G.; Valensi, P.; et al. Lamin A/C gene: Sex-determined expression of mutations in Dunnigan-type familial partial lipodystrophy and absence of coding mutations in congenital and acquired generalized lipoatrophy. Diabetes 2000, 49, 1958–1962. [Google Scholar] [CrossRef] [PubMed]
- Rizza, R.A. Pathogenesis of fasting and postprandial hyperglycemia in type 2 diabetes: Implications for therapy. Diabetes 2010, 59, 2697–2707. [Google Scholar] [CrossRef] [PubMed]
- Dutour, A.; Roll, P.; Gaborit, B.; Courrier, S.; Alessi, M.C.; Tregouet, D.A.; Angelis, F.; Robaglia-Schlupp, A.; Lesavre, N.; Cau, P.; et al. High prevalence of laminopathies among patients with metabolic syndrome. Hum. Mol. Genet. 2011, 20, 3779–3786. [Google Scholar] [CrossRef] [PubMed]
- Decaudain, A.; Vantyghem, M.C.; Guerci, B.; Hecart, A.C.; Auclair, M.; Reznik, Y.; Narbonne, H.; Ducluzeau, P.H.; Donadille, B.; Lebbe, C.; et al. New metabolic phenotypes in laminopathies: LMNA mutations in patients with severe metabolic syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 4835–4844. [Google Scholar] [CrossRef] [PubMed]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, K.; Katsuya, T.; Sugimoto, K.; Kuremura, M.; Kim, H.D.; Li, L.; Ogihara, T. LMNA mutation in a 45 year old Japanese subject with Hutchinson-Gilford progeria syndrome. J. Med. Genet. 2004, 41, e67. [Google Scholar] [CrossRef] [PubMed]
- Vidak, S.; Foisner, R. Molecular insights into the premature aging disease progeria. Histochem. Cell Biol. 2016, 145, 401–417. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Kleinman, M.E.; Miller, D.T.; Neuberg, D.S.; Giobbie-Hurder, A.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.; Snyder, B.D.; et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, 16666–16671. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Udeshi, N.D.; Slawson, C.; Compton, P.D.; Sakabe, K.; Cheung, W.D.; Shabanowitz, J.; Hunt, D.F.; Hart, G.W. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates cytokinesis. Sci. Signal. 2010, 3, ra2. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.W.; Copeland, R.J. Glycomics hits the big time. Cell 2010, 143, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Shafi, R.; Iyer, S.P.; Ellies, L.G.; O’Donnell, N.; Marek, K.W.; Chui, D.; Hart, G.W.; Marth, J.D. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc. Natl. Acad. Sci. USA 2000, 97, 5735–5739. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.W. Minireview series on the thirtieth anniversary of research on O-GlcNAcylation of nuclear and cytoplasmic proteins: Nutrient regulation of cellular metabolism and physiology by O-GlcNAcylation. J. Biol. Chem. 2014, 289, 34422–34423. [Google Scholar] [CrossRef] [PubMed]
- Levine, Z.G.; Walker, S. The biochemistry of O-GlcNAc transferase: Which functions make it essential in mammalian cells? Annu. Rev. Biochem. 2016, 85, 631–657. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.R.; Hanover, J.A. O-GlcNAc cycling: A link between metabolism and chronic disease. Annu. Rev. Nutr. 2013, 33, 205–229. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.J.; Bullen, J.W.; Hart, G.W. Cross-talk between GlcNAcylation and phosphorylation: Roles in insulin resistance and glucose toxicity. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E17–E28. [Google Scholar] [CrossRef] [PubMed]
- Hardiville, S.; Hart, G.W. Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metab. 2014, 20, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.V.; Zachara, N.E.; Nielsen, P.H.; Kimose, H.H.; Kristiansen, S.B.; Botker, H.E. Impact of O-GlcNAc on cardioprotection by remote ischaemic preconditioning in non-diabetic and diabetic patients. Cardiovasc. Res. 2013, 97, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Correa, G.A.; Jin, W.; Wang, Z.; Zhong, X.; Gao, W.D.; Dias, W.B.; Vecoli, C.; Hart, G.W.; Murphy, A.M. O-linked GlcNAc modification of cardiac myofilament proteins: A novel regulator of myocardial contractile function. Circ. Res. 2008, 103, 1354–1358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Yin, R.; Yang, X. O-GlcNAc: A bittersweet switch in liver. Front. Endocrinol. (Lausanne) 2014, 5, 221. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.; Overwin, H. Spot synthesis. Epitope analysis with arrays of synthetic peptides prepared on cellulose membranes. Methods Mol. Biol. 1996, 66, 149–169. [Google Scholar] [PubMed]
- Simon, D.N.; Zastrow, M.S.; Wilson, K.L. Direct actin binding to A- and B-type lamin tails and actin filament bundling by the lamin a tail. Nucleus 2010, 1, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, M.B.; Nam, Y.; Jiang, J.; Sliz, P.; Walker, S. Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature 2011, 469, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Udeshi, N.D.; Compton, P.D.; Shabanowitz, J.; Hunt, D.F.; Rose, K.L. Methods for analyzing peptides and proteins on a chromatographic timescale by electron-transfer dissociation mass spectrometry. Nat. Protoc. 2008, 3, 1709–1717. [Google Scholar] [CrossRef] [PubMed]
- Earley, L.; Anderson, L.C.; Bai, D.L.; Mullen, C.; Syka, J.E.; English, A.M.; Dunyach, J.J.; Stafford, G.C., Jr.; Shabanowitz, J.; Hunt, D.F.; et al. Front-end electron transfer dissociation: A new ionization source. Anal. Chem. 2013, 85, 8385–8390. [Google Scholar] [CrossRef] [PubMed]
- Geer, L.Y.; Markey, S.P.; Kowalak, J.A.; Wagner, L.; Xu, M.; Maynard, D.M.; Yang, X.; Shi, W.; Bryant, S.H. Open mass spectrometry search algorithm. J. Proteome Res. 2004, 3, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Like, A.A.; Rossini, A.A. Streptozotocin-induced pancreatic insulitis: New model of diabetes mellitus. Science 1976, 193, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.N.; Wilson, K.L. Partners and post-translational modifications of nuclear lamins. Chromosoma 2013, 122, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Kochin, V.; Shimi, T.; Torvaldson, E.; Adam, S.A.; Goldman, A.; Pack, C.G.; Melo-Cardenas, J.; Imanishi, S.Y.; Goldman, R.D.; Eriksson, J.E. Interphase phosphorylation of lamin A. J. Cell Sci. 2014, 127, 2683–2696. [Google Scholar] [CrossRef] [PubMed]
- Navarro, C.L.; De Sandre-Giovannoli, A.; Bernard, R.; Boccaccio, I.; Boyer, A.; Genevieve, D.; Hadj-Rabia, S.; Gaudy-Marqueste, C.; Smitt, H.S.; Vabres, P.; et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum. Mol. Genet. 2004, 13, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Krimm, I.; Ostlund, C.; Gilquin, B.; Couprie, J.; Hossenlopp, P.; Mornon, J.P.; Bonne, G.; Courvalin, J.C.; Worman, H.J.; Zinn-Justin, S. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure 2002, 10, 811–823. [Google Scholar] [CrossRef]
- Barrowman, J.; Hamblet, C.; Kane, M.S.; Michaelis, S. Requirements for efficient proteolytic cleavage of prelamin a by ZMPSTE24. PLoS ONE 2012, 7, e32120. [Google Scholar] [CrossRef] [PubMed]
- Casasola, A.; Scalzo, D.; Nandakumar, V.; Halow, J.; Recillas-Targa, F.; Groudine, M.; Rincon-Arano, H. Prelamin A processing, accumulation and distribution in normal cells and laminopathy disorders. Nucleus 2016, 7, 84–102. [Google Scholar] [CrossRef] [PubMed]
- Hrit, J.; Li, C.; Martin, E.A.; Simental, E.; Goll, M.; Panning, B. OGT binds a conserved c-terminal domain of TET1 to regulate TET1 activity and function in development. bioRxiv 2017, 125419. [Google Scholar] [CrossRef]
- Ferraro, A.; Eufemi, M.; Cervoni, L.; Marinetti, R.; Turano, C. Glycosylated forms of nuclear lamins. FEBS Lett. 1989, 257, 241–246. [Google Scholar] [CrossRef]
- Alfaro, J.F.; Gong, C.X.; Monroe, M.E.; Aldrich, J.T.; Clauss, T.R.; Purvine, S.O.; Wang, Z.; Camp, D.G., 2nd; Shabanowitz, J.; Stanley, P.; et al. Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc. Natl. Acad. Sci. USA 2012, 109, 7280–7285. [Google Scholar] [CrossRef] [PubMed]
- Pathak, S.; Alonso, J.; Schimpl, M.; Rafie, K.; Blair, D.E.; Borodkin, V.S.; Schuttelkopf, A.W.; Albarbarawi, O.; van Aalten, D.M. The active site of O-GlcNAc transferase imposes constraints on substrate sequence. Nat. Struct. Mol. Biol. 2015, 22, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Kreplak, L.; Aebi, U. Isolation, characterization, and in vitro assembly of intermediate filaments. Methods Cell Biol. 2004, 78, 3–24. [Google Scholar] [PubMed]
- Zhao, P.; Schulz, T.C.; Sherrer, E.S.; Weatherly, D.B.; Robins, A.J.; Wells, L. The human embryonic stem cell proteome revealed by multidimensional fractionation followed by tandem mass spectrometry. Proteomics 2015, 15, 554–566. [Google Scholar] [CrossRef] [PubMed]
- Jochmann, R.; Holz, P.; Sticht, H.; Sturzl, M. Validation of the reliability of computational O-GlcNAc prediction. Biochim. Biophys. Acta 2014, 1844, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Schutkowski, M.; Reimer, U.; Panse, S.; Dong, L.; Lizcano, J.M.; Alessi, D.R.; Schneider-Mergener, J. High-content peptide microarrays for deciphering kinase specificity and biology. Angew. Chem. Int. Ed. Engl. 2004, 43, 2671–2674. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, L.; Wang, Y.; Yan, H.; Ma, X.; Wang, P.G.; Zhang, L. A peptide panel investigation reveals the acceptor specificity of O-GlcNAc transferase. FASEB J. 2014, 28, 3362–3372. [Google Scholar] [CrossRef] [PubMed]
- Kao, H.J.; Huang, C.H.; Bretana, N.A.; Lu, C.T.; Huang, K.Y.; Weng, S.L.; Lee, T.Y. A two-layered machine learning method to identify protein O-GlcNAcylation sites with O-GlcNAc transferase substrate motifs. BMC Bioinform. 2015, 16 (Suppl. 18), S10. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Brunak, S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac. Symp. Biocomput. 2002, 310–322. [Google Scholar] [CrossRef]
- Jia, C.Z.; Liu, T.; Wang, Z.P. O-GlcNAcPRED: A sensitive predictor to capture protein O-GlcNAcylation sites. Mol. Biosyst. 2013, 9, 2909–2913. [Google Scholar] [CrossRef] [PubMed]
- Berk, J.M.; Maitra, S.; Dawdy, A.W.; Shabanowitz, J.; Hunt, D.F.; Wilson, K.L. O-linked beta-N-acetylglucosamine (O-GlcNAc) regulates emerin binding to barrier to autointegration factor (BAF) in a chromatin- and lamin B-enriched “niche”. J. Biol. Chem. 2013, 288, 30192–30209. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Wang, Y.; Li, W. Ppi finder: A mining tool for human protein-protein interactions. PLoS ONE 2009, 4, e4554. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Chen, Y.; Bian, C.; Fujiki, R.; Yu, X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 2013, 493, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, X.; Gao, W.; Li, P.; Hou, J.; Li, J.; Wong, J. Differential regulation of the ten-eleven translocation (TET) family of dioxygenases by O-linked beta-N-acetylglucosamine transferase (OGT). J. Biol. Chem. 2014, 289, 5986–5996. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, M.B.; Jiang, J.; Kapuria, V.; Bhuiyan, T.; Janetzko, J.; Zandberg, W.F.; Vocadlo, D.J.; Herr, W.; Walker, S. HCF-1 is cleaved in the active site of O-GlcNAc transferase. Science 2013, 342, 1235–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Zhang, F.; Kudlow, J.E. Recruitment of O-GlcNAc transferase to promoters by corepressor mSin3A: Coupling protein O-GlcNAcylation to transcriptional repression. Cell 2002, 110, 69–80. [Google Scholar] [CrossRef]
- Shimi, T.; Kittisopikul, M.; Tran, J.; Goldman, A.E.; Adam, S.A.; Zheng, Y.; Jaqaman, K.; Goldman, R.D. Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Mol. Biol. Cell 2015, 26, 4075–4086. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Chojnowski, A.; Boudier, T.; Lim, J.S.; Ahmed, S.; Ser, Z.; Stewart, C.; Burke, B. A-type lamins form distinct filamentous networks with differential nuclear pore complex associations. Curr. Biol. 2016, 26, 2651–2658. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Coffinier, C.; Choe, Y.; Beigneux, A.P.; Davies, B.S.; Yang, S.H.; Barnes, R.H., 2nd; Hong, J.; Sun, T.; Pleasure, S.J.; et al. Regulation of prelamin A but not lamin c by miR-9, a brain-specific microRNA. Proc. Natl. Acad. Sci. USA 2012, 109, E423–E431. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Mejia, I.C.; de Toledo, M.; Chavey, C.; Lapasset, L.; Cavelier, P.; Lopez-Herrera, C.; Chebli, K.; Fort, P.; Beranger, G.; Fajas, L.; et al. Antagonistic functions of LMNA isoforms in energy expenditure and lifespan. EMBO Rep. 2014, 15, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Coffinier, C.; Jung, H.J.; Li, Z.; Nobumori, C.; Yun, U.J.; Farber, E.A.; Davies, B.S.; Weinstein, M.M.; Yang, S.H.; Lammerding, J.; et al. Direct synthesis of lamin A, bypassing prelamin a processing, causes misshapen nuclei in fibroblasts but no detectable pathology in mice. J. Biol. Chem. 2010, 285, 20818–20826. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, C.; Cenni, V.; Lattanzi, G. Potential therapeutic effects of the MTOR inhibitors for preventing ageing and progeria-related disorders. Br. J. Clin. Pharmacol. 2016, 82, 1229–1244. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, A.R.; Larrick, J.W. Rapamycin as an antiaging therapeutic?: Targeting mammalian target of rapamycin to treat Hutchinson-Gilford progeria and neurodegenerative diseases. Rejuvenation Res. 2011, 14, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.E.; Hayashi, Y.K.; Bonne, G.; Arimura, T.; Noguchi, S.; Nonaka, I.; Nishino, I. Autophagic degradation of nuclear components in mammalian cells. Autophagy 2009, 5, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Ramos, F.J.; Chen, S.C.; Garelick, M.G.; Dai, D.F.; Liao, C.Y.; Schreiber, K.H.; MacKay, V.L.; An, E.H.; Strong, R.; Ladiges, W.C.; et al. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med. 2012, 4, 144ra103. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.C.; Muchir, A.; Wu, W.; Iwata, S.; Homma, S.; Morrow, J.P.; Worman, H.J. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med. 2012, 4, 144ra102. [Google Scholar] [CrossRef] [PubMed]
- Cattin, M.E.; Wang, J.; Weldrick, J.J.; Roeske, C.L.; Mak, E.; Thorn, S.L.; DaSilva, J.N.; Wang, Y.; Lusis, A.J.; Burgon, P.G. Deletion of MLIP (muscle-enriched A-type lamin-interacting protein) leads to cardiac hyperactivation of Akt/mammalian target of rapamycin (mTOR) and impaired cardiac adaptation. J. Biol. Chem. 2015, 290, 26699–26714. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.C.; Worman, H.J. Reactivation of autophagy ameliorates LMNA cardiomyopathy. Autophagy 2013, 9, 110–111. [Google Scholar] [CrossRef] [PubMed]
- Ramos, F.J.; Kaeberlein, M.; Kennedy, B.K. Elevated MTORC1 signaling and impaired autophagy. Autophagy 2013, 9, 108–109. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, N.J.; Kieran, M.W.; Miller, D.T.; Gordon, L.B.; Cho, Y.J.; Silvera, V.M.; Giobbie-Hurder, A.; Neuberg, D.; Kleinman, M.E. Neurologic features of Hutchinson-Gilford progeria syndrome after lonafarnib treatment. Neurology 2013, 81, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Florwick, A.; Dharmaraj, T.; Jurgens, J.; Valle, D.; Wilson, K.L. LMNA sequences of 60,706 unrelated individuals reveal 132 novel missense variants in A-type lamins and suggest a link between variant p.G602S and type 2 diabetes. Front. Genet. 2017, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Rankin, J.; Auer-Grumbach, M.; Bagg, W.; Colclough, K.; Nguyen, T.D.; Fenton-May, J.; Hattersley, A.; Hudson, J.; Jardine, P.; Josifova, D.; et al. Extreme phenotypic diversity and nonpenetrance in families with the LMNA gene mutation R644C. Am. J. Med. Genet. A 2008, 146A, 1530–1542. [Google Scholar] [CrossRef] [PubMed]
- Harwood, K.R.; Hanover, J.A. Nutrient-driven O-GlcNAc cycling—Think globally but act locally. J. Cell Sci. 2014, 127, 1857–1867. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, R.; Gruenbaum, Y.; Medalia, O. Nuclear lamins: Thin filaments with major functions. Trends Cell Biol. 2017, 28, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Samwer, M.; Schneider, M.W.G.; Hoefler, R.; Schmalhorst, P.S.; Jude, J.G.; Zuber, J.; Gerlich, D.W. DNA cross-bridging shapes a single nucleus from a set of mitotic chromosomes. Cell 2017, 170, 956–972. [Google Scholar] [CrossRef] [PubMed]
- Naetar, N.; Ferraioli, S.; Foisner, R. Lamins in the nuclear interior—Life outside the lamina. J. Cell Sci. 2017, 130, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lee, K.K.; Segura-Totten, M.; Neufeld, E.; Wilson, K.L.; Gruenbaum, Y. MAN1 and emerin have overlapping function(s) essential for chromosome segregation and cell division in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2003, 100, 4598–4603. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Rolef Ben-Shahar, T.; Riemer, D.; Treinin, M.; Spann, P.; Weber, K.; Fire, A.; Gruenbaum, Y. Essential roles for Caenorhabditis elegans lamin gene in nuclear organization, cell cycle progression, and spatial organization of nuclear pore complexes. Mol. Biol. Cell 2000, 11, 3937–3947. [Google Scholar] [CrossRef] [PubMed]
- Margalit, A.; Segura-Totten, M.; Gruenbaum, Y.; Wilson, K.L. Barrier-to-autointegration factor is required to segregate and enclose chromosomes within the nuclear envelope and assemble the nuclear lamina. Proc. Natl. Acad. Sci. USA 2005, 102, 3290–3295. [Google Scholar] [CrossRef] [PubMed]
- Snider, N.T.; Omary, M.B. Post-translational modifications of intermediate filament proteins: Mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.X.; Du, J.T.; Zhou, L.X.; Liu, X.H.; Zhao, Y.F.; Nakanishi, H.; Li, Y.M. Alternative O-GlcNAcylation/O-phosphorylation of Ser16 induce different conformational disturbances to the n terminus of murine estrogen receptor beta. Chem. Biol. 2006, 13, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Roque, A.; Ponte, I.; Suau, P. Post-translational modifications of the intrinsically disordered terminal domains of histone H1: Effects on secondary structure and chromatin dynamics. Chromosoma 2017, 126, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsic disorder here, there, and everywhere, and nowhere to escape from it. Cell. Mol. Life Sci. 2017, 74, 3065–3067. [Google Scholar] [CrossRef] [PubMed]
- Crisp, M.; Liu, Q.; Roux, K.; Rattner, J.B.; Shanahan, C.; Burke, B.; Stahl, P.D.; Hodzic, D. Coupling of the nucleus and cytoplasm: Role of the LINC complex. J. Cell Biol. 2006, 172, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittmer, T.A.; Sahni, N.; Kubben, N.; Hill, D.E.; Vidal, M.; Burgess, R.C.; Roukos, V.; Misteli, T. Systematic identification of pathological lamin A interactors. Mol. Biol. Cell 2014, 25, 1493–1510. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Fang, P.; Kim, S.; Guo, M.; Young, N.L.; Marshall, A.G. Mapping the contact surfaces in the lamin A:AIMP3 complex by hydrogen/deuterium exchange FT-ICR mass spectrometry. PLoS ONE 2017, 12, e0181869. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Kim, D.G.; Kim, G.; Choi, E.C.; Kennedy, B.K.; Suh, Y.; Park, B.J.; Kim, S. Downregulation of lamin A by tumor suppressor AIMP3/p18 leads to a progeroid phenotype in mice. Aging Cell 2010, 9, 810–822. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Burke, B. Nuclear networking. Nucleus 2017, 8, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.R.; Hanover, J.A. A little sugar goes a long way: The cell biology of O-GlcNAc. J. Cell Biol. 2015, 208, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Hart, G.W. Protein O-GlcNAcylation in diabetes and diabetic complications. Expert Rev. Proteom. 2013, 10, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Bronshtein, I.; Kepten, E.; Kanter, I.; Berezin, S.; Lindner, M.; Redwood, A.B.; Mai, S.; Gonzalo, S.; Foisner, R.; Shav-Tal, Y.; et al. Loss of lamin A function increases chromatin dynamics in the nuclear interior. Nat. Commun. 2015, 6, 8044. [Google Scholar] [CrossRef] [PubMed]
- Cesarini, E.; Mozzetta, C.; Marullo, F.; Gregoretti, F.; Gargiulo, A.; Columbaro, M.; Cortesi, A.; Antonelli, L.; Di Pelino, S.; Squarzoni, S.; et al. Lamin A/C sustains PcG protein architecture, maintaining transcriptional repression at target genes. J. Cell Biol. 2015, 211, 533–551. [Google Scholar] [CrossRef] [PubMed]
- Ronningen, T.; Shah, A.; Oldenburg, A.R.; Vekterud, K.; Delbarre, E.; Moskaug, J.O.; Collas, P. Prepatterning of differentiation-driven nuclear lamin A/C-associated chromatin domains by GlcNAcylated histone H2B. Genome Res. 2015, 25, 1825–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perovanovic, J.; Dell’Orso, S.; Gnochi, V.F.; Jaiswal, J.K.; Sartorelli, V.; Vigouroux, C.; Mamchaoui, K.; Mouly, V.; Bonne, G.; Hoffman, E.P. Laminopathies disrupt epigenomic developmental programs and cell fate. Sci. Transl. Med. 2016, 8, 335ra358. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simon, D.N.; Wriston, A.; Fan, Q.; Shabanowitz, J.; Florwick, A.; Dharmaraj, T.; Peterson, S.B.; Gruenbaum, Y.; Carlson, C.R.; Grønning-Wang, L.M.; et al. OGT (O-GlcNAc Transferase) Selectively Modifies Multiple Residues Unique to Lamin A. Cells 2018, 7, 44. https://doi.org/10.3390/cells7050044
Simon DN, Wriston A, Fan Q, Shabanowitz J, Florwick A, Dharmaraj T, Peterson SB, Gruenbaum Y, Carlson CR, Grønning-Wang LM, et al. OGT (O-GlcNAc Transferase) Selectively Modifies Multiple Residues Unique to Lamin A. Cells. 2018; 7(5):44. https://doi.org/10.3390/cells7050044
Chicago/Turabian StyleSimon, Dan N., Amanda Wriston, Qiong Fan, Jeffrey Shabanowitz, Alyssa Florwick, Tejas Dharmaraj, Sherket B. Peterson, Yosef Gruenbaum, Cathrine R. Carlson, Line M. Grønning-Wang, and et al. 2018. "OGT (O-GlcNAc Transferase) Selectively Modifies Multiple Residues Unique to Lamin A" Cells 7, no. 5: 44. https://doi.org/10.3390/cells7050044