Next Generation Sequencing: Advances in Characterizing the Methylome

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. Cutting-edge Technological Advances
2.1. Roche (454) genome sequencer FLX
2.2. Illumina genome analyzer
2.3. Applied biosystems (SOLiDTM)
2.4. Other sequencing technologies.
2.5. Third generation sequencing technologies.
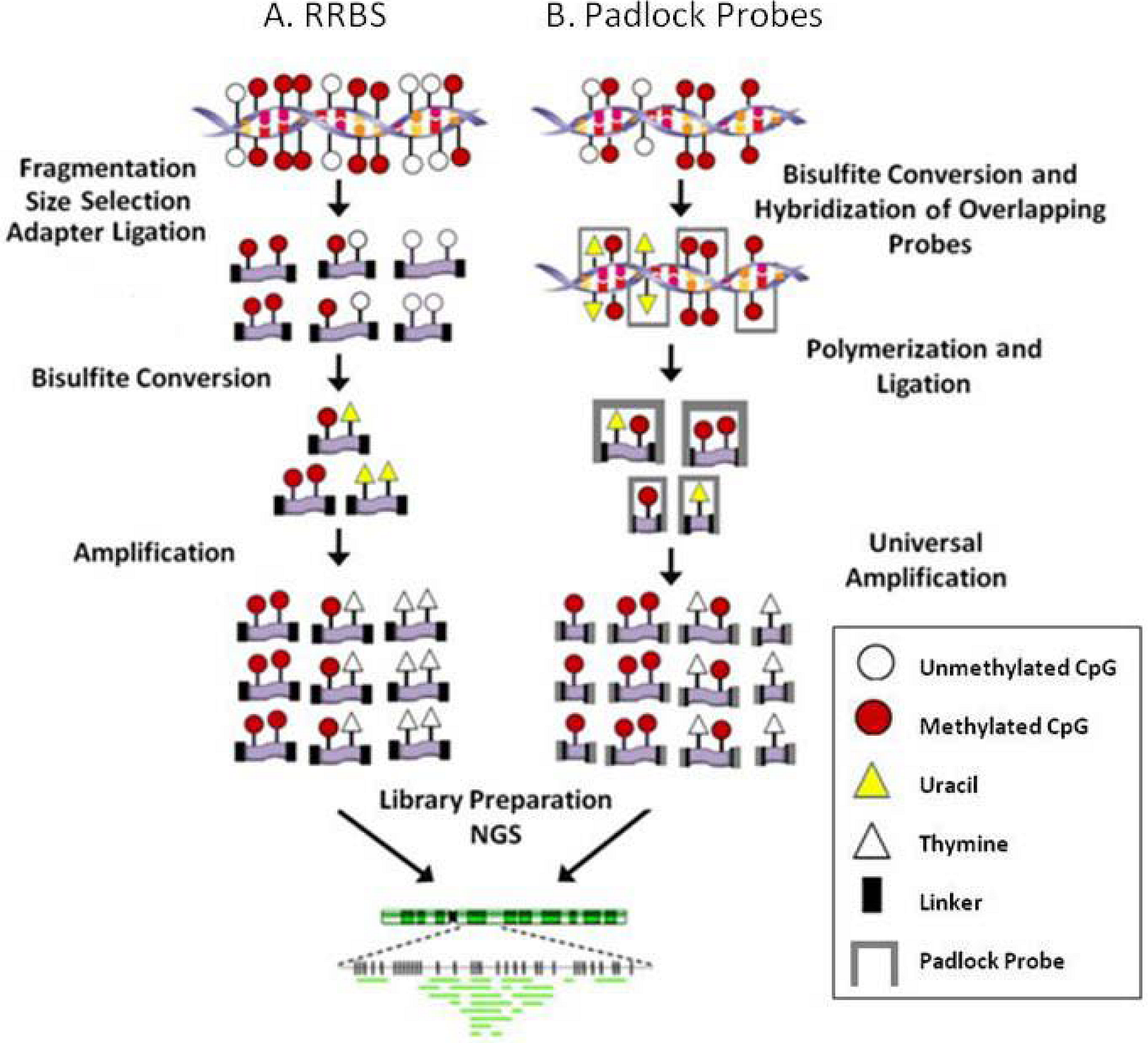
3. Methylome Methodologies
3.1. Site specific

3.2. Regional

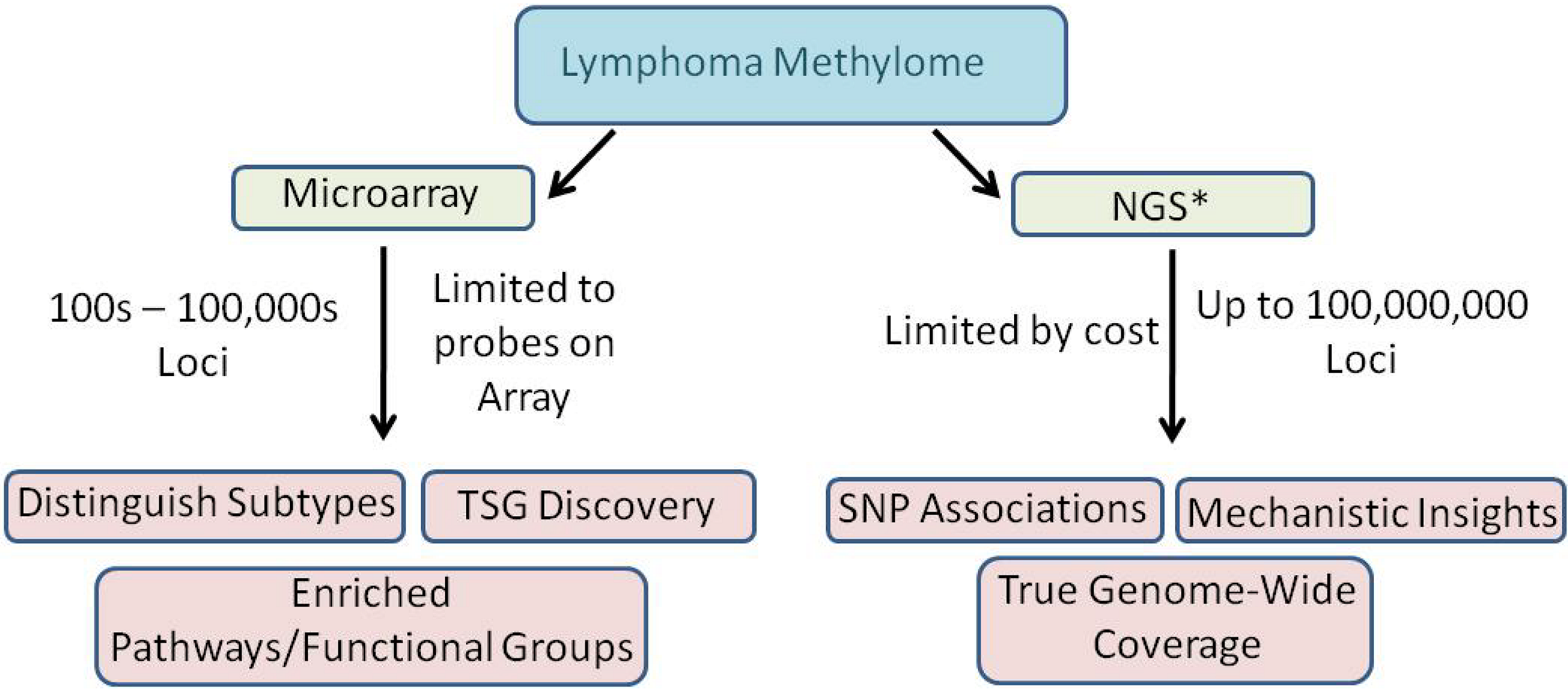
4. Progress Toward Characterizing the Methylome of Lymphoma and Leukemia
4.1. Genome-wide microarrays
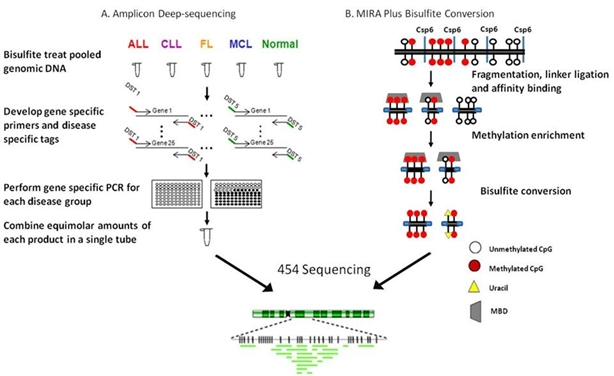
4.2. Ultradeep bisulfite NGS of amplicons in B-cell lymphomas
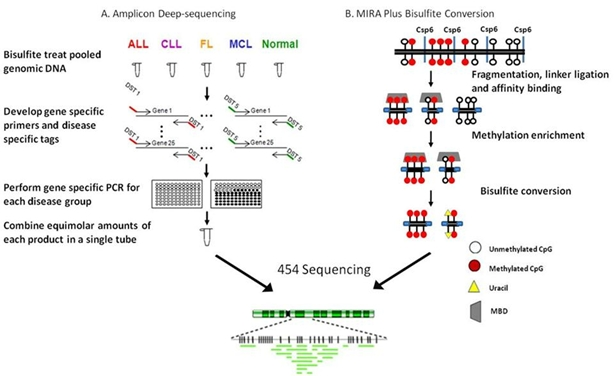
4.3. RRBS/MIRA plus bisulfite sequencing in a follicular lymphoma cell line:


5. Beyond the Methylome
6. On the Horizon
Acknowledgments
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in Human Disease and Prospects for Epigenetic Therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The Epigenetic Progenitor Origin of Human Cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Laird, P.W. The Power and the Promise of DNA Methylation Markers. Nat. Rev. Cancer 2003, 3, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA Methylation Patterns and Epigenetic Memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Robertson, K.D.; Jones, P.A. DNA Methylation: Past, Present and Future Directions. Carcinogenesis 2000, 21, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Craig, J.M.; Bickmore, W.A. The Distribution of CpG Islands in Mammalian Chromosomes. Nat. Genet. 1994, 7, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Monk, M. Regulation of X-Chromosome Inactivation in Development in Mice and Humans. Microbiol. Mol. Biol. Rev. 1998, 62, 362–378. [Google Scholar] [PubMed]
- Li, E.; Beard, C.; Jaenisch, R. Role for DNA Methylation in Genomic Imprinting. Nature 1993, 366, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Razin, A.; Cedar, H. DNA Methylation and Genomic Imprinting. Cell 1994, 77, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Cancer Epigenomics: DNA Methylomes and Histone-Modification Maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef]
- Gal-Yam, E.N.; Saito, Y.; Egger, G.; Jones, P.A. Cancer Epigenetics: Modifications, Screening, and Therapy. Annu. Rev. Med. 2008, 59, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The Human Colon Cancer Methylome Shows Similar Hypo- and Hypermethylation at Conserved Tissue-Specific CpG Island Shores . Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Dietrich, N.; Pasini, D.; Hansen, K.H.; Helin, K. Genome-Wide Mapping of Polycomb Target Genes Unravels Their Roles in Cell Fate Transitions. Genes Dev. 2006, 20, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Kirmizis, A.; Bartley, S.M.; Kuzmichev, A.; Margueron, R.; Reinberg, D.; Green, R.; Farnham, P.J. Silencing of Human Polycomb Target Genes Is Associated With Methylation of Histone H3 Lys 27. Genes Dev. 2004, 18, 1592–1605. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Villa, R.; Trojer, P.; Norman, J.; Yan, K.P.; Reinberg, D.; Di, C.L.; Shiekhattar, R. Demethylation of H3K27 Regulates Polycomb Recruitment and H2A Ubiquitination. Science 2007, 318, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Raaphorst, F.M. Deregulated Expression of Polycomb-Group Oncogenes in Human Malignant Lymphomas and Epithelial Tumors . Hum. Mol. Genet. 2005, 14, R93–R100. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; Bergman, Y.; Simon, I.; Cedar, H. Polycomb-Mediated Methylation on Lys27 of Histone H3 Pre-Marks Genes for De Novo Methylation in Cancer. Nat. Genet. 2007, 39, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van, E.A.; Bernard, D.; Vanderwinden, J.M.; et al. The Polycomb Group Protein EZH2 Directly Controls DNA Methylation . Nature 2005, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Ng, E.K.; Tsang, W.P.; Ng, S.S.; Jin, H.C.; Yu, J.; Li, J.J.; Rocken, C.; Ebert, M.P.; Kwok, T.T.; Sung, J.J. MicroRNA-143 Targets DNA Methyltransferases 3A in Colorectal Cancer . Br. J. Cancer 2009, 101, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Garzon, R.; Cimmino, A.; Liu, Z.; Zanesi, N.; Callegari, E.; Liu, S.; Alder, H.; Costinean, S.; Fernandez-Cymering, C.; et al. MicroRNA-29 Family Reverts Aberrant Methylation in Lung Cancer by Targeting DNA Methyltransferases 3A and 3B . Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 15805–15810. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Liu, S.; Fabbri, M.; Liu, Z.; Heaphy, C.E.; Callegari, E.; Schwind, S.; Pang, J.; Yu, J.; Muthusamy, N.; et al. MicroRNA-29b Induces Global DNA Hypomethylation and Tumor Suppressor Gene Reexpression in Acute Myeloid Leukemia by Targeting Directly DNMT3A and 3B and Indirectly DNMT1 . Blood 2009, 113, 6411–6418. [Google Scholar] [CrossRef] [PubMed]
- Laird, P.W. Principles and Challenges of Genome-Wide DNA Methylation Analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA Methylomes at Base Resolution Show Widespread Epigenomic Differences . Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing Technologies - the Next Generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. The Impact of Next-Generation Sequencing Technology on Genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar] [PubMed]
- Korshunova, Y.; Maloney, R.K.; Lakey, N.; Citek, R.W.; Bacher, B.; Budiman, A.; Ordway, J.M.; McCombie, W.R.; Leon, J.; Jeddeloh, J.A.; McPherson, J.D. Massively Parallel Bisulphite Pyrosequencing Reveals the Molecular Complexity of Breast Cancer-Associated Cytosine-Methylation Patterns Obtained From Tissue and Serum DNA. Genome Res. 2008, 18, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Ordway, J.M.; Budiman, M.A.; Korshunova, Y.; Maloney, R.K.; Bedell, J.A.; Citek, R.W.; Bacher, B.; Peterson, S.; Rohlfing, T.; Hall, J.; Brown, R.; Lakey, N.; Doerge, R.W.; Martienssen, R.A.; Leon, J.; McPherson, J.D.; Jeddeloh, J.A. Identification of Novel High-Frequency DNA Methylation Changes in Breast Cancer . PLoS. One 2007, 2, e1314. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.H.; Kramer, R.S.; Davis, J.W.; Guo, J.; Duff, D.J.; Xu, D.; Caldwell, C.W.; Shi, H. Ultradeep Bisulfite Sequencing Analysis of DNA Methylation Patterns in Multiple Gene Promoters by 454 Sequencing. Cancer Res. 2007, 67, 8511–8518. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Wang, M.; Bonaldo, M.F.; Rajaram, V.; Stellpflug, W.; Smith, C.; Arndt, K.; Goldman, S.; Tomita, T.; Soares, M.B. Epigenomic Analysis of Alu Repeats in Human Ependymomas. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 6952–6957. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Shoemaker, R.; Xie, B.; Gore, A.; LeProust, E.M.; ntosiewicz-Bourget, J.; Egli, D.; Maherali, N.; Park, I.H.; Yu, J.; Daley, G.Q.; Eggan, K.; Hochedlinger, K.; Thomson, J.; Wang, W.; Gao, Y.; Zhang, K. Targeted Bisulfite Sequencing Reveals Changes in DNA Methylation Associated With Nuclear Reprogramming. Nat. Biotechnol. 2009, 27, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Bock, C.; Mikkelsen, T.S.; Jager, N.; Smith, Z.D.; Tomazou, E.; Gnirke, A.; Lander, E.S.; Meissner, A. Genome-Scale DNA Methylation Mapping of Clinical Samples at Single-Nucleotide Resolution. Nat. Methods 2010, 7, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Hodges, E.; Smith, A.D.; Kendall, J.; Xuan, Z.; Ravi, K.; Rooks, M.; Zhang, M.Q.; Ye, K.; Bhattacharjee, A.; Brizuela, L.; et al. High Definition Profiling of Mammalian DNA Methylation by Array Capture and Single Molecule Bisulfite Sequencing . Genome Res. 2009, 19, 1593–1605. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun Bisulphite Sequencing of the Arabidopsis Genome Reveals DNA Methylation Patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; Ukomadu, C.; Sadler, K.C.; Pradhan, S.; Pellegrini, M.; Jacobsen, S.E. Conservation and Divergence of Methylation Patterning in Plants and Animals. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O'Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly Integrated Single-Base Resolution Maps of the Epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.H.; LeProust, E.M.; Park, I.H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and Genome-Scale Strategies Reveal Gene-Body Methylation Signatures in Human Cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gao, F.; Li, N.; Li, S.; Yin, G.; Tian, G.; Jia, S.; Wang, K.; Zhang, X.; Yang, H.; Nielsen, A.L.; Bolund, L. An Improved Method for Genome Wide DNA Methylation Profiling Correlated to Transcription and Genomic Instability in Two Breast Cancer Cell Lines. BMC Genomics 2009, 10, 223. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Porreca, G.J.; Reppas, N.B.; Lin, X.; McCutcheon, J.P.; Rosenbaum, A.M.; Wang, M.D.; Zhang, K.; Mitra, R.D.; Church, G.M. Accurate Multiplex Polony Sequencing of an Evolved Bacterial Genome. Science 2005, 309, 1728–1732. [Google Scholar] [CrossRef] [PubMed]
- Bormann Chung, C.A.; Boyd, V.L.; McKernan, K.J.; Fu, Y.; Monighetti, C.; Peckham, H.E.; Barker, M. Whole Methylome Analysis by Ultra-Deep Sequencing Using Two-Base Encoding . PLoS One 2010, 5, e9320. [Google Scholar] [CrossRef] [PubMed]
- Drmanac, R.; Sparks, A.B.; Callow, M.J.; Halpern, A.L.; Burns, N.L.; Kermani, B.G.; Carnevali, P.; Nazarenko, I.; Nilsen, G.B.; Yeung, G.; et al. Human Genome Sequencing Using Unchained Base Reads on Self-Assembling DNA Nanoarrays . Science 2010, 327, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Kin Sung, K.W.; Rigoutsos, I.; Loring, J.; Wei, C.L. Dynamic Changes in the Human Methylome During Differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Gnirke, A.; Bell, G.W.; Ramsahoye, B.; Lander, E.S.; Jaenisch, R. Reduced Representation Bisulfite Sequencing for Comparative High-Resolution DNA Methylation Analysis. Nucleic Acids Res. 2005, 33, 5868–5877. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Gu, H.; Bock, C.; Gnirke, A.; Meissner, A. High-Throughput Bisulfite Sequencing in Mammalian Genomes. Methods 2009, 48, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Zeschnigk, M.; Martin, M.; Betzl, G.; Kalbe, A.; Sirsch, C.; Buiting, K.; Gross, S.; Fritzilas, E.; Frey, B.; Rahmann, S.; Horsthemke, B. Massive Parallel Bisulfite Sequencing of CG-Rich DNA Fragments Reveals That Methylation of Many X-Chromosomal CpG Islands in Female Blood DNA Is Incomplete. Hum. Mol. Genet. 2009, 18, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and Analysis of Functional Elements in 1% of the Human Genome by the ENCODE Pilot Project . Nature 2007, 447, 799–816. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Glass, J.L.; Thompson, R.F.; Mo, Y.; Olivier, E.N.; Figueroa, M.E.; Selzer, R.R.; Richmond, T.A.; Zhang, X.; Dannenberg, L.; et al. High-Resolution Genome-Wide Cytosine Methylation Profiling With Simultaneous Copy Number Analysis and Optimization for Limited Cell Numbers . Nucleic Acids Res. 2009, 37, 3829–3839. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Elling, A.A.; Li, X.; Li, N.; Peng, Z.; He, G.; Sun, H.; Qi, Y.; Liu, X.S.; Deng, X.W. Genome-Wide and Organ-Specific Landscapes of Epigenetic Modifications and Their Relationships to MRNA and Small RNA Transcriptomes in Maize. Plant Cell 2009, 21, 1053–1069. [Google Scholar] [CrossRef] [PubMed]
- Rauch, T.; Pfeifer, G.P. Methylated-CpG Island Recovery Assay: a New Technique for the Rapid Detection of Methylated-CpG Islands in Cancer. Lab. Invest. 2005, 85, 1172–1180. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Davies, J.J.; Wittig, D.; Oakeley, E.J.; Haase, M.; Lam, W.L.; Schubeler, D. Chromosome-Wide and Promoter-Specific Analyses Identify Sites of Differential DNA Methylation in Normal and Transformed Human Cells. Nat. Genet. 2005, 37, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Butcher, L.M.; Beck, S. AutoMeDIP-Seq: A High-Throughput, Whole Genome, DNA Methylation Assay . Methods. 2010. Available online: http://www.ncbi.nlm.nih.gov/pubmed/20385236/.
- Serre, D.; Lee, B.H.; Ting, A.H. MBD-Isolated Genome Sequencing Provides a High-Throughput and Comprehensive Survey of DNA Methylation in the Human Genome. Nucleic Acids Res. 2010, 38, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Heisler, L.E.; Torti, D.; Boutros, P.C.; Watson, J.; Chan, C.; Winegarden, N.; Takahashi, M.; Yau, P.; Huang, T.H.; Farnham, P.J.; Jurisica, I.; Woodgett, J.R.; Bremner, R.; Penn, L.Z.; Der, S.D. CpG Island Microarray Probe Sequences Derived From a Physical Library Are Representative of CpG Islands Annotated on the Human Genome. Nucleic Acids Res. 2005, 33, 2952–2961. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.S.; Chen, C.M.; Shi, H.; Rahmatpanah, F.; Wei, S.H.; Caldwell, C.W.; Huang, T.H. Dissecting Complex Epigenetic Alterations in Breast Cancer Using CpG Island Microarrays. Cancer Res. 2001, 61, 8375–8380. [Google Scholar] [PubMed]
- Guo, J.; Burger, M.; Nimmrich, I.; Maier, S.; Becker, E.; Genc, B.; Duff, D.; Rahmatpanah, F.; Chitma-Matsiga, R.; Shi, H.; Berlin, K.; Huang, T.H.; Caldwell, C.W. Differential DNA Methylation of Gene Promoters in Small B-Cell Lymphomas. Am. J. Clin. Pathol. 2005, 124, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Guo, J.; Duff, D.J.; Rahmatpanah, F.; Chitima-Matsiga, R.; Al-Kuhlani, M.; Taylor, K.H.; Sjahputera, O.; Andreski, M.; Wooldridge, J.E.; Caldwell, C.W. Discovery of Novel Epigenetic Markers in Non-Hodgkin's Lymphoma. Carcinogenesis 2007, 28, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.H.; Pena-Hernandez, K.E.; Davis, J.W.; Arthur, G.L.; Duff, D.J.; Shi, H.; Rahmatpanah, F.B.; Sjahputera, O.; Caldwell, C.W. Large-Scale CpG Methylation Analysis Identifies Novel Candidate Genes and Reveals Methylation Hotspots in Acute Lymphoblastic Leukemia. Cancer Res. 2007, 67, 2617–2625. [Google Scholar] [CrossRef] [PubMed]
- van, D.R.; Zoutman, W.H.; Dijkman, R.; de Menezes, R.X.; Commandeur, S.; Mulder, A.A.; van, d.; Vermeer, M.H.; Willemze, R.; Yan, P.S.; Huang, T.H.; Tensen, C.P. Epigenetic Profiling of Cutaneous T-Cell Lymphoma: Promoter Hypermethylation of Multiple Tumor Suppressor Genes Including BCL7a, PTPRG, and P73 . J. Clin. Oncol. 2005, 23, 3886–3896. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.; Nimmrich, I.; Burger, M.; Becker, E.; Dorken, B.; Ludwig, W.D.; Maier, S. Distinction of Acute Lymphoblastic Leukemia From Acute Myeloid Leukemia Through Microarray-Based DNA Methylation Analysis. Ann. Hematol. 2005, 84, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Martin-Subero, J.I.; Ammerpohl, O.; Bibikova, M.; Wickham-Garcia, E.; Agirre, X.; Alvarez, S.; Bruggemann, M.; Bug, S.; Calasanz, M.J.; Deckert, M.; et al. A Comprehensive Microarray-Based DNA Methylation Study of 367 Hematological Neoplasms . PLoS. One. 2009, 4, e6986. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.H.; Rahmatpanah, F.; Davis, J.W.; Caldwell, C.W. Chromosomal Localization of DNA Methylation in Small B-Cell Lymphoma. Leukemia 2007, 22, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Davidsson, J.; Lilljebjorn, H.; Andersson, A.; Veerla, S.; Heldrup, J.; Behrendtz, M.; Fioretos, T.; Johansson, B. The DNA Methylome of Pediatric Acute Lymphoblastic Leukemia. Hum. Mol. Genet. 2009, 18, 4054–4065. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Reimers, M.; Thompson, R.F.; Ye, K.; Li, Y.; Selzer, R.R.; Fridriksson, J.; Paietta, E.; Wiernik, P.; Green, R.D.; Greally, J.M.; Melnick, A. An Integrative Genomic and Epigenomic Approach for the Study of Transcriptional Regulation . PLoS One 2008, 3, e1882. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Wouters, B.J.; Skrabanek, L.; Glass, J.; Li, Y.; Erpelinck-Verschueren, C.A.; Langerak, A.W.; Lowenberg, B.; Fazzari, M.; Greally, J.M.; Valk, P.J.; Melnick, A.; Delwel, R. Genome-Wide Epigenetic Analysis Delineates a Biologically Distinct Immature Acute Leukemia With Myeloid/T-Lymphoid Features. Blood 2009, 113, 2795–2804. [Google Scholar] [CrossRef] [PubMed]
- Kanduri, M.; Cahill, N.; Goransson, H.; Enstrom, C.; Ryan, F.; Isaksson, A.; Rosenquist, R. Differential Genome-Wide Array-Based Methylation Profiles in Prognostic Subsets of Chronic Lymphocytic Leukemia. Blood 2010, 115, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Martin-Subero, J.I.; Kreuz, M.; Bibikova, M.; Bentink, S.; Ammerpohl, O.; Wickham-Garcia, E.; Rosolowski, M.; Richter, J.; Lopez-Serra, L.; Ballestar, E.; et al. New Insights into the Biology and Origin of Mature Aggressive B-Cell Lymphomas by Combined Epigenomic, Genomic, and Transcriptional Profiling . Blood 2009, 113, 2488–2497. [Google Scholar] [CrossRef] [PubMed]
- Deneberg, S.; Grovdal, M.; Karimi, M.; Jansson, M.; Nahi, H.; Corbacioglu, A.; Gaidzik, V.; Dohner, K.; Paul, C.; Ekstrom, T.J.; Hellstrom-Lindberg, E.; Lehmann, S. Gene-Specific and Global Methylation Patterns Predict Outcome in Patients With Acute Myeloid Leukemia. Leukemia 2010, 24, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Dunwell, T.; Hesson, L.; Rauch, T.A.; Wang, L.; Clark, R.E.; Dallol, A.; Gentle, D.; Catchpoole, D.; Maher, E.R.; Pfeifer, G.P.; Latif, F. A Genome-Wide Screen Identifies Frequently Methylated Genes in Haematological and Epithelial Cancers. Mol. Cancer 2010, 9, 44. [Google Scholar] [CrossRef]
- Schafer, E.; Irizarry, R.; Negi, S.; McIntyre, E.; Small, D.; Figueroa, M.E.; Melnick, A.; Brown, P. Promoter Hypermethylation in MLL-r Infant Acute Lymphoblastic Leukemia: Biology and Therapeutic Targeting. Blood 2010, 115, 4798–4809. [Google Scholar] [CrossRef] [PubMed]
- Shaknovich, R.; Figueroa, M.E.; Melnick, A. HELP (HpaII Tiny Fragment Enrichment by Ligation-Mediated PCR) Assay for DNA Methylation Profiling of Primary Normal and Malignant B Lymphocytes. Methods Mol. Biol. 2010, 632, 191–201. [Google Scholar] [PubMed]
- Bennett, L.B.; Schnabel, J.L.; Kelchen, J.M.; Taylor, K.H.; Guo, J.; Arthur, G.L.; Papageorgio, C.N.; Shi, H.; Caldwell, C.W. DNA Hypermethylation Accompanied by Transcriptional Repression in Follicular Lymphoma. Genes Chromosomes Cancer 2009, 48, 828–841. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.Q.; Bai, H.; Fang, Z.H.; Lopez, G.; Yang, H.; Tong, W.; Wang, Z.Z.; Garcia-Manero, G. Aberrant DNA Methylation and Epigenetic Inactivation of Eph Receptor Tyrosine Kinases and Ephrin Ligands in Acute Lymphoblastic Leukemia. Blood 2010, 115, 2412–2419. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Profiling Aberrant DNA Methylation in Hematologic Neoplasms: a View From the Tip of the Iceberg. Clin. Immunol. 2003, 109, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Rahmatpanah, F.B.; Carstens, S.; Guo, J.; Sjahputera, O.; Taylor, K.H.; Duff, D.; Shi, H.; Davis, J.W.; Hooshmand, S.I.; Chitma-Matsiga, R.; Caldwell, C.W. Differential DNA Methylation Patterns of Small B-Cell Lymphoma Subclasses With Different Clinical Behavior. Leukemia 2006, 20, 1855–1862. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.A.; Chabner, B.A. Histone Deacetylase Inhibitors in Cancer Therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Kamal, M.; Lindblad-Toh, K.; Bekiranov, S.; Bailey, D.K.; Huebert, D.J.; McMahon, S.; Karlsson, E.K.; Kulbokas III, E.J.; Gingeras, T.R.; Schreiber, S.L.; Lander, E.S. Genomic Maps and Comparative Analysis of Histone Modifications in Human and Mouse . Cell 2005, 120, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The Mammalian Epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.K.; Koche, R.P.; et al. Genome-Wide Maps of Chromatin State in Pluripotent and Lineage-Committed Cells . Nature 2007, 448, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Rank, G.; Tan, Y.T.; Li, H.; Moritz, R.L.; Simpson, R.J.; Cerruti, L.; Curtis, D.J.; Patel, D.J.; Allis, C.D.; Cunningham, J.M.; Jane, S.M. PRMT5-Mediated Methylation of Histone H4R3 Recruits DNMT3A, Coupling Histone and DNA Methylation in Gene Silencing. Nat. Struct. Mol. Biol. 2009, 16, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Ruan, K.; Fang, X.; Ouyang, G. MicroRNAs: Novel Regulators in the Hallmarks of Human Cancer. Cancer Lett. 2009, 285, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Calin, G.A.; Croce, C.M. MicroRNAs in Cancer. Annu. Rev. Med. 2009, 60, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Sotiropoulou, G.; Pampalakis, G.; Lianidou, E.; Mourelatos, Z. Emerging Roles of MicroRNAs As Molecular Switches in the Integrated Circuit of the Cancer Cell. RNA 2009, 15, 1443–1461. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Stresemann, C.; Brueckner, B.; Lyko, F. Methylation of Human MicroRNA Genes in Normal and Neoplastic Cells. Cell Cycle 2007, 6, 1001–1005. [Google Scholar]
- Guil, S.; Esteller, M. DNA Methylomes, Histone Codes and MiRNAs: Tying It All Together. Int. J. Biochem. Cell Biol. 2009, 41, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Giresi, P.G.; Kim, J.; McDaniell, R.M.; Iyer, V.R.; Lieb, J.D. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) Isolates Active Regulatory Elements From Human Chromatin. Genome Res. 2007, 17, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, K.J.; Nammo, T.; Pasquali, L.; Simon, J.M.; Giresi, P.G.; Fogarty, M.P.; Panhuis, T.M.; Mieczkowski, P.; Secchi, A.; Bosco, D.; Berney, T.; Montanya, E.; Mohlke, K.L.; Lieb, J.D.; Ferrer, J. A Map of Open Chromatin in Human Pancreatic Islets. Nat. Genet. 2010, 42, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome . Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Taylor, K.H.; Shi, H.; Caldwell, C.W. Next Generation Sequencing: Advances in Characterizing the Methylome. Genes 2010, 1, 143-165. https://doi.org/10.3390/genes1020143
Taylor KH, Shi H, Caldwell CW. Next Generation Sequencing: Advances in Characterizing the Methylome. Genes. 2010; 1(2):143-165. https://doi.org/10.3390/genes1020143
Chicago/Turabian StyleTaylor, Kristen H., Huidong Shi, and Charles W. Caldwell. 2010. "Next Generation Sequencing: Advances in Characterizing the Methylome" Genes 1, no. 2: 143-165. https://doi.org/10.3390/genes1020143