Role of Cell Division Autoantigen 1 (CDA1) in Cell Proliferation and Fibrosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Molecular Cloning of CDA1 Using an Autoimmune DLE Serum
3. CDA1 in Man, Mouse
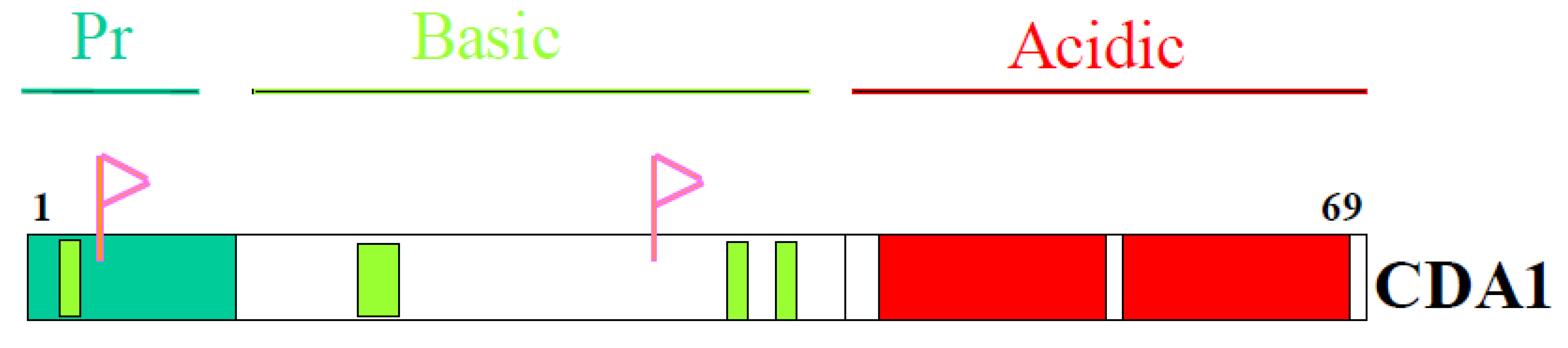
4. Domain Structure of CDA1

5. CDA1 Requires its C-Terminal Domain for Anti-Proliferative Activity
6. Proline-Rich N Terminal Domain of CDA1
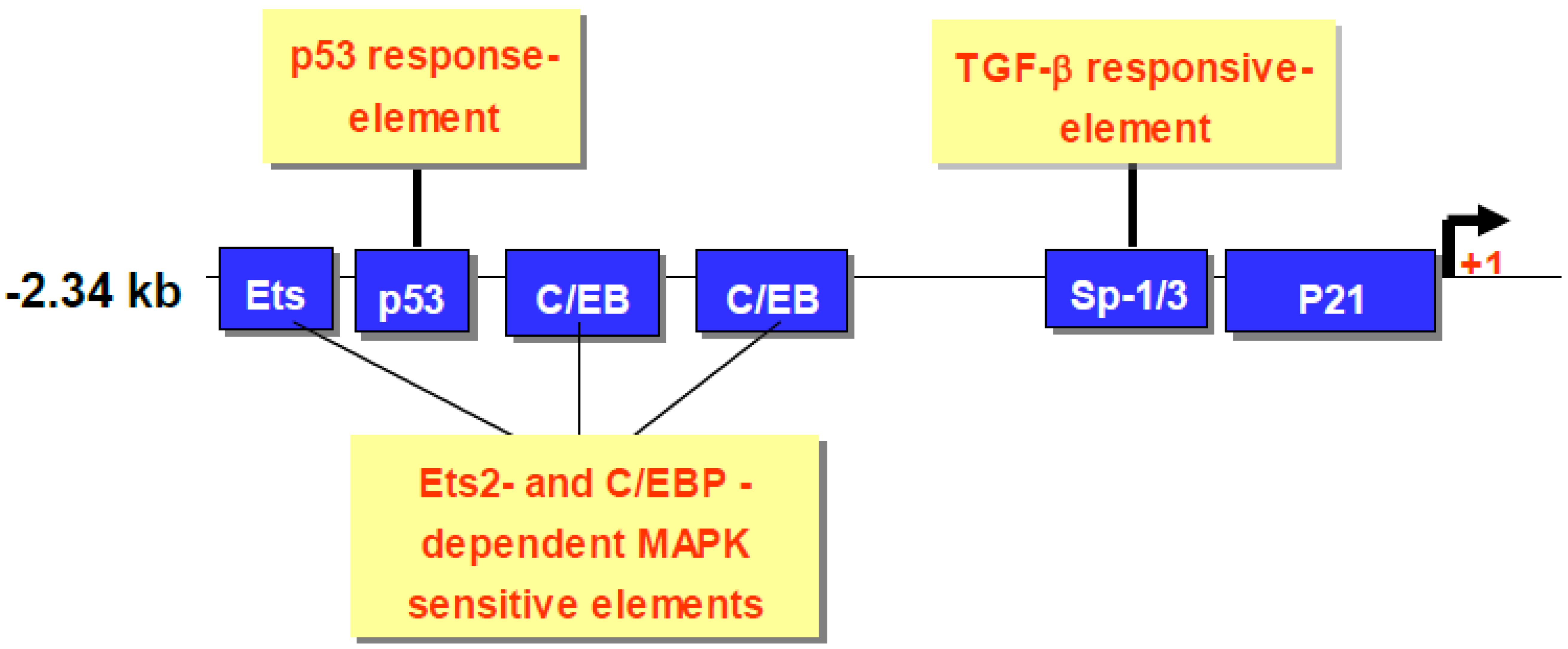
7. CDA1 Regulates p21 Expression
8. CDA1 is an Upstream Regulator of p53 Expression
9. CDA1 Activates ERK1/MAPK Pathways


10. CDA1 Inhibits Cyclin-Dependent Kinases Through Upregulation of CDK Inhibitors
11. CDA1 is Upregulated in Diabetes-Associated Atherosclerosis
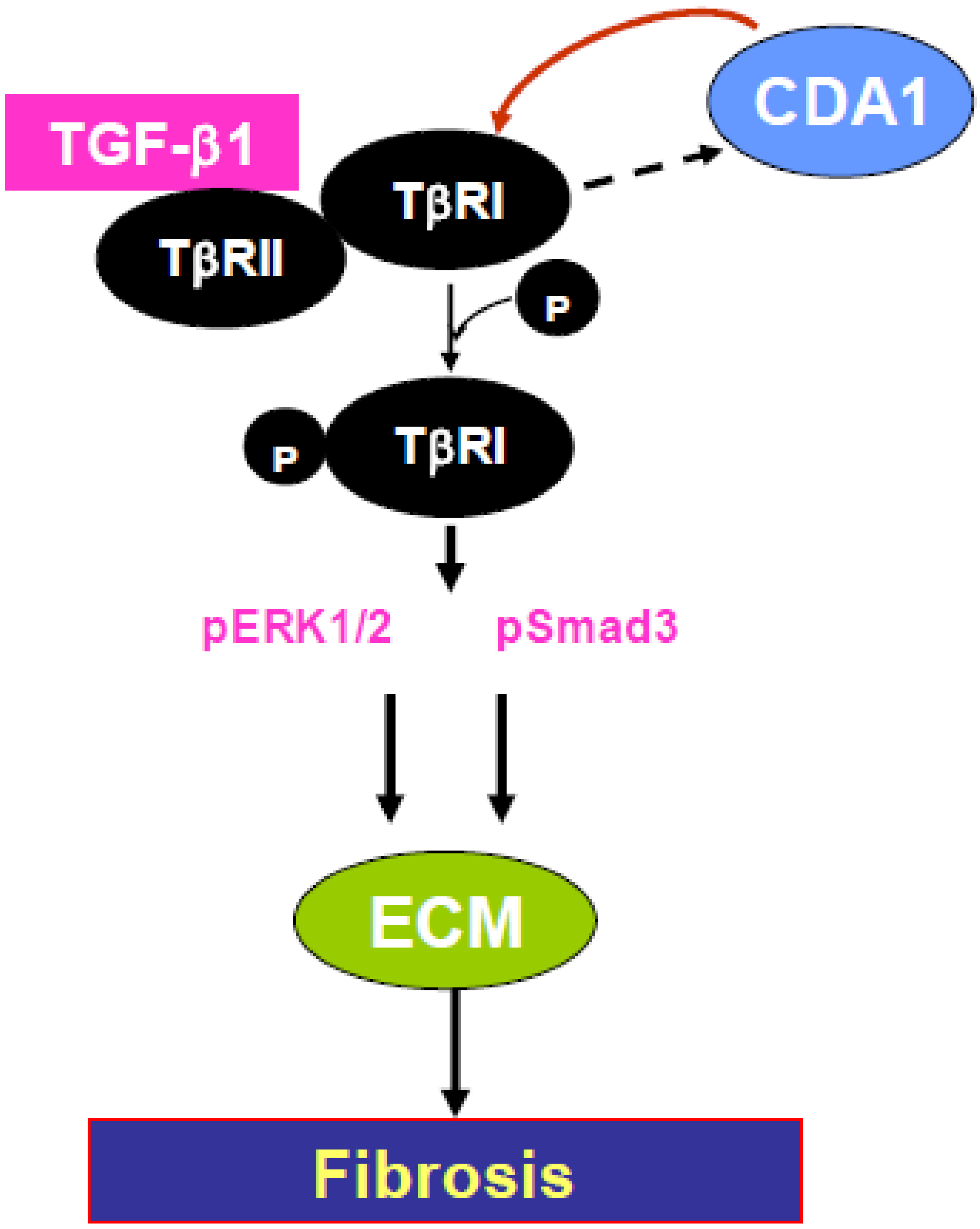
12. CDA1, TGF Cross-Talk

13. Conclusions
References and Notes
- Tan, E.M. Antinuclear antibodies: Diagnostic markers for autoimmune diseases, probes for cell biology. Adv. Immunol. 1989, 44, 93–151. [Google Scholar] [CrossRef] [PubMed]
- Callen, J.P. Chronic cutaneous lupus erythematosus. Clinical, laboratory, therapeutic, prognostic examination of 62 patients. Arch. Dermatol. 1982, 118, 412–416. [Google Scholar] [PubMed]
- Hirota, T.; Lipp, J.J.; Toh, B.H.; Peters, J.M. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature 2005, 438, 1176–1180. [Google Scholar] [CrossRef] [PubMed]
- Chai, Z.; Sarcevic, B.; Mawson, A.; Toh, B.H. SET-related cell division autoantigen-1 (CDA1) arrests cell growth. J. Biol. Chem. 2001, 276, 33665–33674. [Google Scholar] [CrossRef] [PubMed]
- Ueki, N.; Oda, T.; Kondo, M.; Yano, K.; Noguchi, T.; Muramatsu, M. Selection system for genes encoding nuclear-targeted proteins. Nat. Biotechnol. 1998, 16, 1338–1342. [Google Scholar] [CrossRef] [PubMed]
- HGNC: 24358; GeneID: 64061 . Available online: http://www.genenames.org/data/hgnc_data.php?hgnc_id=24358 (accessed on 1 September 2010).
- Eichmuller, S.; Usener, D.; Dummer, R.; Stein, A.; Thiel, D.; Schadendorf, D. Serological detection of cutaneous T-cell lymphoma-associated antigens. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Ozbun, L.L.; You, L.; Kiang, S.; Angdisen, J.; Martinez, A.; Jakowlew, S.B. Identification of differentially expressed nucleolar TGF-beta1 target (DENTT) in human lung cancer cells that is a new member of the TSPY/SET/NAP-1 superfamily. Genomics 2001, 73, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Yuen, C.S.; Yuan, Y.; Wang, C.K.; Qiu, G.; Sun, K.; Ping, L.M. Isolation of differentially expressed genes in human heart tissues . Biochim. Biophys. Acta 2002, 1588, 241–246. [Google Scholar] [PubMed]
- Wang, G.S.; Hong, C.J.; Yen, T.Y.; Huang, H.Y.; Ou, Y.; Huang, T.N.; Jung, W.G.; Kuo, T.Y.; Sheng, M.; Wang, T.F.; Hsueh, Y.P. Transcriptional modification by a CASK-interacting nucleosome assembly protein. Neuron 2004, 42, 113–128. [Google Scholar] [CrossRef] [PubMed]
- von Lindern, M.; van Baal, S.; Wiegant, J.; Raap, A.; Hagemeijer, A.; Grosveld, G. Can, a putative oncogene associated with myeloid leukemogenesis, may be activated by fusion of its 3’ half to different genes: Characterization of the set gene. Mol. Cell Biol. 1992, 12, 3346–3355. [Google Scholar] [PubMed]
- von Lindern, M.; Fornerod, M.; Soekarman, N.; van Baal, S.; Jaegle, M.; Hagemeijer, A.; Bootsma, D.; Grosveld, G. Translocation t (6;9) in acute non-lymphocytic leukaemia results in the formation of a DEK-CAN fusion gene. Baillieres Clin. Haematol. 1992, 5, 857–879. [Google Scholar]
- Li, M.; Makkinje, A.; Damuni, Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J. Biol. Chem. 1996, 271, 11059–11062. [Google Scholar] [CrossRef] [PubMed]
- Kandalaft, L.E.; Zudaire, E.; Portal-Nunez, S.; Cuttitta, F.; Jakowlew, S.B. Differentially Expressed Nucleolar TGF-{beta}1 Target (DENTT) exhibits an inhibitory role on tumorigenesis. Carcinog. 2008, 29, 1282–1289. [Google Scholar] [CrossRef]
- Ito, T.; Tsukumo, S.; Suzuki, N.; Motohashi, H.; Yamamoto, M.; Fujii-Kuriyama, Y.; Mimura, J.; Lin, T. M.; Peterson, R.E.; Tohyama, C.; Nohara, K. A Constitutively Active Arylhydrocarbon Receptor Induces Growth Inhibition of Jurkat T Cells through Changes in the Expression of Genes Related to Apoptosis, Cell Cycle Arrest. J. Biol. Chem. 2004, 279, 25204–25210. [Google Scholar] [CrossRef] [PubMed]
- Radke, J.R.; Donald, R.G.; Eibs, A.; Jerome, M.E.; Behnke, M.S.; Liberator, P.; White, M.W. Changes in the expression of human cell division autoantigen-1 influence Toxoplasma gondii growth, development . PLoS Pathog. 2006, 2, e105. [Google Scholar] [CrossRef] [PubMed]
- Morton, C.J.; Campbell, I.D. SH3 domains. Molecular ‘Velcro’. Curr. Biol. 1994, 4, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Birge, R.B.; Knudsen, B.S.; Besser, D.; Hanafusa, H. SH2, SH3-containing adaptor proteins: Redundant or independent mediators of intracellular signal transduction. Genes Cells 1996, 1, 595–613. [Google Scholar] [PubMed]
- Kaneko, T.; Li, L.; Li, S.S. The SH3 domain—a family of versatile peptide-, protein-recognition module. Front. Biosci. 2008, 13, 4938–4952. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive, negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Hannon, G.J.; Zhang, H.; Casso, D.; Kobayashi, R.; Beach, D. p21 is a universal inhibitor of cyclin kinases. Nature 1993, 366, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Zhang, H.; Beach, D. D type cyclins associate with multiple protein kinases, the DNA replication, repair factor PCNA. Cell 1992, 71, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Dulic, V.; Kaufmann, W.K.; Wilson, S.J.; Tlsty, T.D.; Lees, E.; Harper, J.W.; Elledge, S.J.; Reed, S.I. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell 1994, 76, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Waga, S.; Hannon, G.J.; Beach, D.; Stillman, B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature 1994, 369, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.; Herskowitz, I. Joining the complex: Cyclin-dependent kinase inhibitory proteins, the cell cycle. Cell 1994, 79, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Datto, M.B.; Yu, Y.; Wang, X.F. Functional Analysis of the Transforming Growth Factor beta Responsive Elements in the WAF1/Cip1/p21 Promoter. J. Biol. Chem. 1995, 270, 28623–28628. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Bae, G.U.; Kang, J.K.; Park, J.W.; Lee, E.K.; Lee, H.Y.; Choi, W.S.; Lee, H.W.; Han, J.W. Cooperation of H2O2-mediated ERK activation with Smad pathway in TGF-beta1 induction of p21WAF1/Cip1. Cell. Signalling 2006, 18, 236–243. [Google Scholar] [CrossRef]
- Tu, Y.; Wu, W.; Wu, T.; Cao, Z.; Wilkins, R.; Toh, B.H.; Cooper, M.; Chai, Z. Antiproliferative autoantigen CDA1 transcriptionally upregulates p21Wafl/Cip1 by activating p53, MEK/ERK1/2 MAPK pathways. J. Biol. Chem. 2007, 282, 11722–11731. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Liao, Y.; Xia, W.; Zou, Y.; Spohn, B.; Hung, M.C. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat. Cell Biol. 2001, 3, 973–982. [Google Scholar] [CrossRef] [PubMed]
- el-Deiry, W.S.; Harper, J.W.; O’Connor, P.M.; Velculescu, V.E.; Canman, C.E.; Jackman, J.; Pietenpol, J.A.; Burrell, M.; Hill, D.E.; Wang, Y.; et al. WAF1/CIP1 is induced in p53-mediated G1 arrest, apoptosis . Cancer Res. 1994, 54, 1169–1174. [Google Scholar] [PubMed]
- Wei, C.L.; Wu, Q.; Vega, V.B.; Chiu, K.P.; Ng, P.; Zhang, T.; Shahab, A.; Yong, H.C.; Fu, Y.; Weng, Z.; Liu, J.; Zhao, X.D.; Chew, J.L.; Lee, Y.L.; Kuznetsov, V.A.; Sung, W.K.; Miller, L.D.; Lim, B.; Liu, E.T.; Yu, Q.; Ng, H.H.; Ruan, Y. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006, 124, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Schmitt, E.M.; Smith, S.W.; Osborne, B.A.; Jacks, T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 1993, 362, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Lozano, G.; Elledge, S.J. p53 sends nucleotides to repair DNA. Nature 2000, 404, 24–25. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Arakawa, H.; Yamaguchi, T.; Shiraishi, K.; Fukuda, S.; Matsui, K.; Takei, Y.; Nakamura, Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 2000, 404, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Caspari, T. How to activate p53 . Curr. Biol. 2000, 10, R315–R317. [Google Scholar] [CrossRef] [PubMed]
- Colman, M.S.; Afshari, C.A.; Barrett, J.C. Regulation of p53 stability, activity in response to genotoxic stress. Mutat. Res. 2000, 462, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.R.; Purdie, C.A.; Harrison, D.J.; Morris, R.G.; Bird, C.C.; Hooper, M.L.; Wyllie, A.H. Thymocyte apoptosis induced by p53-dependent, independent pathways. Nature 1993, 362, 849–852. [Google Scholar] [CrossRef] [PubMed]
- Hertzberg, R.P.; Busby, R.W.; Caranfa, M.J.; Holden, K.G.; Johnson, R.K.; Hecht, S.M.; Kingsbury, W.D. Irreversible trapping of the DNA-topoisomerase I covalent complex. Affinity labeling of the camptothecin binding site. J. Biol. Chem. 1990, 265, 19287–19295. [Google Scholar] [PubMed]
- Gupta, R.S.; Gupta, R.; Eng, B.; Lock, R.B.; Ross, W.E.; Hertzberg, R.P.; Caranfa, M.J.; Johnson, R.K. Camptothecin-resistant mutants of Chinese hamster ovary cells containing a resistant form of topoisomerase I. Cancer Res. 1988, 48, 6404–6410. [Google Scholar] [PubMed]
- Hu, P.P.; Shen, X.; Huang, D.; Liu, Y.; Counter, C.; Wang, X.F. The MEK pathway is required for stimulation of p21(WAF1/CIP1) by transforming growth factor-beta. J. Biol. Chem. 1999, 274, 35381–35387. [Google Scholar] [CrossRef] [PubMed]
- Kivinen, L.; Laiho, M. Ras-, mitogen-activated protein kinase kinase-dependent, -independent pathways in p21Cip1/Waf1 induction by fibroblast growth factor-2, platelet-derived growth factor,, transforming growth factor-beta1. Cell Growth Differ. 1999, 10, 621–628. [Google Scholar] [PubMed]
- Lee, B.; Moon, S.K. Ras/ERK signaling pathway mediates activation of the p21WAF1 gene promoter in vascular smooth muscle cells by platelet-derived growth factor. Arch. Biochem. Biophys. 2005, 443, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Mantel, C.; Anzai, N.; Braun, S.E.; Broxmeyer, H.E. Transcriptional, ERK1/2-dependent synergistic upregulation of p21(cip1/waf1) associated with steel factor synergy in MO7e. Biochem. Biophys. Res. Commun. 2001, 280, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Camps, M.; Nichols, A.; Arkinstall, S. Dual specificity phosphatases: A gene family for control of MAP kinase function. Faseb. J. 2000, 14, 6–16. [Google Scholar]
- Dickinson, R.J.; Keyse, S.M. Diverse physiological functions for dual-specificity MAP kinase phosphatases. J. Cell Sci. 2006, 119, 4607–4615. [Google Scholar] [CrossRef] [PubMed]
- Ozbun, L.L.; Martinez, A.; Jakowlew, S.B. Differentially expressed nucleolar TGF-beta1 target (DENTT) shows tissue-specific nuclear, cytoplasmic localization, increases TGF-beta1-responsive transcription in primates. Biochim. Biophys. Acta 2005, 1728, 163–180. [Google Scholar] [PubMed]
- Martinez, A.; Ozbun, L.L.; Angdisen, J.; Jakowlew, S.B. Expression of differentially expressed nucleolar transforming growth factor-beta1 target (DENTT) in adult mouse tissues. Dev. Dyn. 2002, 224, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Reichner, C.; Wu, X.; Levine, A.J. Analysis of wild-type, mutant p21WAF-1 gene activities. Mol. Cell Biol. 1996, 16, 1786–1793. [Google Scholar] [PubMed]
- Russo, A.A.; Jeffrey, P.D.; Patten, A.K.; Massague, J.; Pavletich, N.P. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature 1996, 382, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jackson, P.K.; Kirschner, M.W.; Dutta, A. Separate domains of p21 involved in the inhibition of Cdk kinase, PCNA. Nature 1995, 374, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Saha, P.; Kornbluth, S.; Dynlacht, B.D.; Dutta, A. Cyclin-binding motifs are essential for the function of p21CIP1. Mol. Cell Biol. 1996, 16, 4673–4682. [Google Scholar] [PubMed]
- Nakanishi, M.; Robetorye, R.S.; Adami, G.R.; Pereira-Smith, O.M.; Smith, J.R. Identification of the active region of the DNA synthesis inhibitory gene p21Sdi1/CIP1/WAF1. EMBO J. 1995, 14, 555–563. [Google Scholar] [PubMed]
- Zhang, H.; Hannon, G.J.; Beach, D. p21-containing cyclin kinases exist in both active, inactive states. Genes Dev. 1994, 8, 1750–1758. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. Living with or without cyclins, cyclin-dependent kinases. Genes Dev. 2004, 18, 2699–2711. [Google Scholar] [CrossRef] [PubMed]
- LaBaer, J.; Garrett, M.D.; Stevenson, L.F.; Slingerland, J.M.; Sandhu, C.; Chou, H.S.; Fattaey, A.; Harlow, E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997, 11, 847–862. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Olivier, P.; Diehl, J.A.; Fero, M.; Roussel, M.F.; Roberts, J.M.; Sherr, C.J. The p21(Cip1), p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999, 18, 1571–1583. [Google Scholar] [CrossRef] [PubMed]
- Basson, M. Cardiovascular disease. Nature 2008, 451, 903. [Google Scholar] [CrossRef]
- Rumble, J.R.; Cooper, M.E.; Soulis, T.; Cox, A.; Wu, L.; Youssef, S.; Jasik, M.; Jerums, G.; Gilbert, R. E. Vascular hypertrophy in experimental diabetes. Role of advanced glycation end products. J. Clin. Invest. 1997, 99, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.M.; Hulthen, U.L.; Allen, T.J.; Cooper, M.E. Angiotensin Converting Enzyme Inhibition, Calcium Antagonism Attenuate Streptozotocin-Diabetes-Associated Mesenteric Vascular Hypertrophy Independently of Their Hypotensive Action. J. Hypertens. 1998, 16, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Candido, R.; Jandeleit-Dahm, K.A.; Cao, Z.; Nesteroff, S.P.; Burns, W.C.; Twigg, S.M.; Dilley, R.J.; Cooper, M.E.; Allen, T.J. Prevention of accelerated atherosclerosis by angiotensin-converting enzyme inhibition in diabetic apolipoprotein E-deficient mice. Circulation 2002, 106, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Candido, R.; Allen, T.J.; Lassila, M.; Cao, Z.; Thallas, V.; Cooper, M.E.; Jandeleit-Dahm, K.A. Irbesartan but not amlodipine suppresses diabetes-associated atherosclerosis. Circulation 2004, 109, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, W.; Abel, E.D.; Breslow, J.L.; Maeda, N.; Davis, R.C.; Fisher, E.A.; Dansky, H.; McClain, D. A.; McIndoe, R.; Wassef, M.K.; Rabadan-Diehl, C.; Goldberg, I.J. Recipes for creating animal models of diabetic cardiovascular disease. Circ. Res. 2007, 100, 1415–1427. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Abraham, D. J. TGF-beta signaling, the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Sorescu, D. Smad3 mediates angiotensin II-, TGF-beta1-induced vascular fibrosis: Smad3 thickens the plot. Circ. Res. 2006, 98, 988–989. [Google Scholar] [CrossRef] [PubMed]
- Pham, Y.; Tu, Y.; Wu, T.; Allen, T. J.; Calkin, A.C.; Watson, A.M.; Li, J.; Jandeleit-Dahm, K.A.; Toh, B.H.; Cao, Z.; Cooper, M.E.; Chai, Z. Cell division autoantigen 1 plays a profibrotic role by modulating downstream signalling of TGF-beta in a murine diabetic model of atherosclerosis. Diabetologia 2010, 53, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Tyagi, S.C. Arteriogenesis: Angiogenesis within Unstable Atherosclerotic Plaque— Interactions with Extracellular Matrix. Curr. Interv. Cardiol. Rep. 2000, 2, 218–227. [Google Scholar] [PubMed]
- Schmidt, A.; Lorkowski, S.; Seidler, D.; Breithardt, G.; Buddecke, E. TGF-beta1 generates a specific multicomponent extracellular matrix in human coronary SMC. Eur. J. Clin. Invest. 2006, 36, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Datto, M.B.; Li, Y.; Panus, J.F.; Howe, D.J.; Xiong, Y.; Wang, X.F. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Nat. Acad. Sci. U. S. A. 1995, 92, 5545–5549. [Google Scholar] [CrossRef]
- Goldberg, H.J.; Huszar, T.; Mozes, M.M.; Rosivall, L.; Mucsi, I. Overexpression of the type II transforming growth factor-beta receptor inhibits fibroblasts proliferation, activates extracellular signal regulated kinase, c-Jun N-terminal kinase. Cell Biol. Int. 2002, 26, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Ziyadeh, F.N.; Lee, E.Y.; Pyagay, P.E.; Sung, S.H.; Sheardown, S.A.; Laping, N.J.; Chen, S. Interference with TGF-beta signaling by Smad3-knockout in mice limits diabetic glomerulosclerosis without affecting albuminuria . Am. J. Physiol. Renal. Physiol. 2007, 293, F1657–F1665. [Google Scholar] [PubMed]
- Yokoyama, H.; Deckert, T. Central role of TGF-beta in the pathogenesis of diabetic nephropathy, macrovascular complications: A hypothesis. Diabet. Med. 1996, 13, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Twigg, S.M.; Cooper, M.E. The time has come to target connective tissue growth factor in diabetic complications. Diabetologia 2004, 47, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Huang, X.R.; Canlas, E.; Oka, K.; Truong, L.D.; Deng, C.; Bhowmick, N.A.; Ju, W.; Bottinger, E.P.; Lan, H.Y. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ. Res. 2006, 98, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Huang, X.R.; Zhu, H.J.; Johnson, R.; Lan, H.Y. Role of TGF-beta signaling in extracellular matrix production under high glucose conditions. Kidney Int. 2003, 63, 2010–2019. [Google Scholar] [CrossRef] [PubMed]
- Macias-Silva, M.; Abdollah, S.; Hoodless, P.A.; Pirone, R.; Attisano, L.; Wrana, J.L. MADR2 is a substrate of the TGFbeta receptor, its phosphorylation is required for nuclear accumulation, signaling. Cell 1996, 87, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, Y.; Constantinescu, S.N.; Karam, E.; Weinberg, R.A.; Lodish, H.F. Transforming growth factor beta-induced phosphorylation of Smad3 is required for growth inhibition, transcriptional induction in epithelial cells. Proc. Nat. Acad. Sci. U. S. A. 1997, 94, 10669–10674. [Google Scholar] [CrossRef]
- Li, J.H.; Wang, W.; Huang, X.R.; Oldfield, M.; Schmidt, A.M.; Cooper, M.E.; Lan, H.Y. Advanced glycation end products induce tubular epithelial-myofibroblast transition through the RAGE-ERK1/2 MAP kinase signaling pathway. Am. J. Pathol. 2004, 164, 1389–1397. [Google Scholar] [PubMed]
- Li, J.H.; Huang, X.R.; Zhu, H.J.; Oldfield, M.; Cooper, M.; Truong, L.D.; Johnson, R.J.; Lan, H.Y. Advanced glycation end products activate Smad signaling via TGF-beta-dependent, independent mechanisms: implications for diabetic renal, vascular disease. FASEB J. 2004, 18, 176–178. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Toh, B.-H.; Tu, Y.; Cao, Z.; Cooper, M.E.; Chai, Z. Role of Cell Division Autoantigen 1 (CDA1) in Cell Proliferation and Fibrosis. Genes 2010, 1, 335-348. https://doi.org/10.3390/genes1030335
Toh B-H, Tu Y, Cao Z, Cooper ME, Chai Z. Role of Cell Division Autoantigen 1 (CDA1) in Cell Proliferation and Fibrosis. Genes. 2010; 1(3):335-348. https://doi.org/10.3390/genes1030335
Chicago/Turabian StyleToh, Ban-Hock, Yugang Tu, Zemin Cao, Mark E. Cooper, and Zhonglin Chai. 2010. "Role of Cell Division Autoantigen 1 (CDA1) in Cell Proliferation and Fibrosis" Genes 1, no. 3: 335-348. https://doi.org/10.3390/genes1030335