1. Introduction
The expansion of gene number within organisms is facilitated by the processes of gene duplication and polyploidization. The study of gene duplicates has burgeoned within the last two decades, owing largely to the availability of whole-genome sequences that enable the identification of a genome-wide population of paralogs for determining the dominant pattern(s) of duplicate origin and the evolutionary forces responsible for their diversification and retention, as well as new methods to assess copy-number variation in natural populations. Additionally, these large data sets enable exploration of additional hypotheses that hitherto remain unanswered (e.g., which evolutionary forces drive the fixation of duplicates at the species-wide level) and test key theoretical predictions for gene duplicate evolution that were established in the pre-genomic era [
1,
2,
3].
The canonical model of gene duplicate evolution as outlined by Ohno [
1] postulates that gene duplicates originate bearing complete sequence and functional identity to the progenitor copy (
i.e., ancestral gene structure and the entire repertoire of ancestral regulatory elements are preserved during the gene duplication process). Sequence divergence between the two paralogs over evolutionary time is affected by the gradual accumulation of mutations, ultimately leading to functional divergence or pseudogenization and eventual loss. However, we now know that this view of gene paralog evolution is overly simplistic. Paralogs are also capable of nonreciprocal recombination with each other via gene conversion, a form of concerted evolution wherein one donor sequence converts a homologous recipient sequence over some length of sequence, thereby enhancing sequence homogeneity between the two paralogs [
4]. Hence, the evolutionary trajectories of gene paralogs is governed by the relative frequencies of these two opposing forces; sequence divergence by new mutations and the erosion of sequence heterogeneity via gene conversion [
5,
6].
The 1980s witnessed accumulating evidence for gene conversion among paralogs representing a range of functions across a variety of species such as human α- and goat ξ-globins [
7,
8,
9],
Drosophila heat-shock genes [
10], human fetal globin genes [
11], mouse immunoglobulin α-heavy chain constant regions [
12], and chorion genes in
Bombyx [
13], among others. The earliest detailed insights regards the biological mechanics of ectopic gene conversion events are owing to genetic studies in microorganisms, particularly the yeast
Saccharomyces cerevisiae [
14]. Interlocus gene conversion events were studied by taking advantage of lines bearing naturally-occurring repetitive elements [
15] as well as experimentally-manipulated strains bearing an insertion of an extra, synthesized gene copy [
16,
17,
18,
19] to determine the influence of a homolog’s genomic location on the frequency of gene conversion. The yeast system has been further developed to determine the relative frequencies of intralocus [
20]
versus interlocus (ectopic) gene conversion [
17,
21,
22], the physical length of gene conversion tracts [
23,
24,
25], the degree of bias with respect to the identity of the donor sequence during gene conversion [
26], and the genome-wide rates of gene conversion events [
27]. However, Li [
4] has cautioned that generalizations from patterns and rates of gene conversion in yeast may have limited applicability to other eukaryotic genomes due to an extremely high rate of gene conversion in yeast and the widespread presence of paralogs as templates for gene conversion in its genome given its polyploid history [
28,
29]. The nematode
Caenorhabditis elegans may represent an appropriate multicellular eukaryotic model system to study the consequences of gene conversion for the evolutionary dynamics of multigene families in a genome dominated by small-scale duplication events (in contrast to polyploidy).
2. Detection of Gene Conversion in Caenorhabditis Paralogs
A handful of studies in the pre-genomic era had already hypothesized the occurrence of possible gene conversion events among recently identified
C. elegans paralogs with extremely high levels of sequence identity. Russnak and Candido [
30] hypothesized the formation of the
C. elegans heat-shock genes
hsp16-1 and
hsp16-48 via an ancestral duplication and inversion event yielding a head-to-head transcriptional orientation. A subsequent round of duplication of this ancestral 1.9 kb module yielded a second
hsp16-1/hsp16-48 cluster 416 bp apart from the ancestral module. The authors favored gene conversion (over an evolutionary recent duplication event) as an explanation to account for the 100% sequence homology between the two
hsp modules based on the observation that regions flanking the modules were completely diverged in sequence. Actin paralogs
act-1 and
act-3 were also proposed to have undergone concerted evolution via gene conversion and, interestingly, the authors proposed that the conversion events extended to intronic regions in addition to the exonic ones [
31]. Another study of two tandem collagen paralogs (
col-12 and
col-13) inferred an older duplication event leading to their formation based, once again, on conflicting patterns of sequence similarity at the spatial level, extremely low levels of sequence similarity in the flanking regions and 99.5% sequence identity in the open reading frame (ORF) regions including an intron [
32]. The identification of
col-12 and
col-13 orthologs in both
C. briggsae and
C. remanei and their patterns of sequence identity confirm this conjecture (
Figure 1), wherein the two paralogs within each species appear most closely related [
33].
Several important patterns for the gene conversion process in
C. elegans paralogs can be gleaned from these initial studies, despite the limited sample sizes. First, gene conversion was concluded to be responsible for the high sequence similarity between paralogous ORFs rather than a recent duplication event because of the low levels or absence of sequence similarity in the flanking regions. Gene conversion could certainly contribute to this observed pattern in paralogous sequences. However, we caution that the possibility of a very recent duplication event should not be ruled out completely without additional evidence. For instance, it is well-documented that gene duplication in
C. elegans is often incomplete and fails to encompass the entire ORF and/or upstream and downstream flanking regions [
34,
35]. Extremely high or complete sequence identity between paralogs in their coding regions with diminished sequence homology in flanking regions is concordant with a scenario wherein the gene duplication event was restricted to the ORFs (or an incomplete segment of the ORF) and failed to encompass ancestral flanking region sequences. Second, assuming each of these three studies truly represent interparalogous gene conversion events, it then appears that gene conversion can occur when paralogs are in direct or opposing (head-to-head or tail-to-tail) transcriptional orientation. However, whether
C. elegans paralogs with the same or opposing transcriptional orientation have equals rates of gene conversion remains to be determined. Third, two of these initial studies reported the extension of gene conversion tracts to introns [
31,
32]. Fourth, the first estimate of the length of gene conversion tracts in
C. elegans (>191 bp) was well within the range estimated for other species [
25], although with the caveat that gene conversion is characterized by exhibiting a fairly wide range in the length of conversion tracts (several base pairs to >12,000 bp in yeast) and with a high degree of locus-specificity [
24].
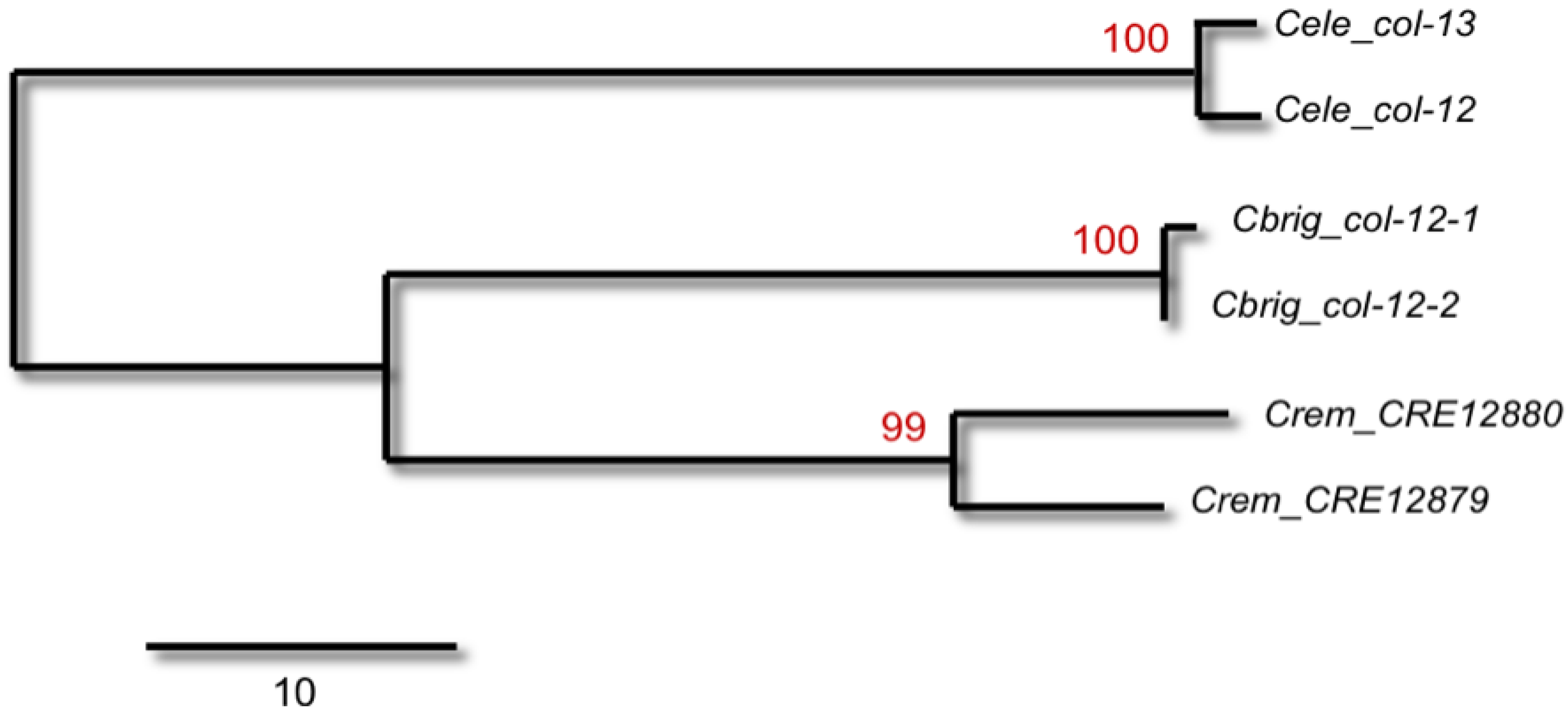
Figure 1.
A maximum-parsimony tree of the relationship between
C. elegans collagen genes
col-12 and
col-13 and their orthologs in
C. remanei and
C. briggsae using protein-coding DNA sequences downloaded from Wormbase. The numbers above the branches represent bootstrap support (1000 replications). The analysis was performed in MEGA v4.0 [
36]. The
col-12 gene has a
cis-spliced leader sequence whereas
col-13 has a
trans-spliced leader. The
cis-spliced leader sequence is well conserved in one homolog in both
C. remanei and
C. briggsae. The near-complete sequence identity between paralogs within genomes is evidence of frequent gene conversion of the coding sequence since the duplication of these genes prior to the divergence of
C. elegans and the
C. remanei/
C. briggsae clade.
Celeg,
Caenorhabditis elegans;
Cbrig,
C. briggsae;
Crem,
C. remanei.
Figure 1.
A maximum-parsimony tree of the relationship between
C. elegans collagen genes
col-12 and
col-13 and their orthologs in
C. remanei and
C. briggsae using protein-coding DNA sequences downloaded from Wormbase. The numbers above the branches represent bootstrap support (1000 replications). The analysis was performed in MEGA v4.0 [
36]. The
col-12 gene has a
cis-spliced leader sequence whereas
col-13 has a
trans-spliced leader. The
cis-spliced leader sequence is well conserved in one homolog in both
C. remanei and
C. briggsae. The near-complete sequence identity between paralogs within genomes is evidence of frequent gene conversion of the coding sequence since the duplication of these genes prior to the divergence of
C. elegans and the
C. remanei/
C. briggsae clade.
Celeg,
Caenorhabditis elegans;
Cbrig,
C. briggsae;
Crem,
C. remanei.
The first comprehensive, genome-wide study of interparalog gene conversion events in nematodes was conducted in 1999 [
37]. This landmark study comprised 7829 putative gene duplicates pairs with sequence identity between paralogs ranging from 35–99%. The analysis used the gene conversion detection software GENECONV which tests whether tracts of complete sequence identity between paralogs are larger than expected relative to sequence divergence of DNA sequences flanking these identical segments [
38]. Paralogs can display several regions of complete sequence identity either due to (i) multiple gene conversion events, or (ii) in instances where a lengthier conversion tract has incurred mutations resulting in smaller segments of complete sequence identity. Under the latter scenario, each segment of complete sequence identity is considered a gene conversion event. The relatively high sequence identity among these paralogs suggests that these gene duplicates are evolutionarily young and/or have been subjected to sequence homogenization by gene conversion. 526 gene conversion events were detected across the 7829 duplicate pairs surveyed. If locus-bias effects were absent, this would translate into gene conversion affecting 6.7% of the surveyed duplicate pairs. However, only 143 pairs (2% of the sampled duplicates) were affected, with an average of 3.75 conversion events per gene pair. This was the first indication that some duplicate pairs in the
C. elegans genome are more prone to gene conversion than others. Other relevant findings of the Semple and Wolfe study and more recent studies of interlocus gene conversion in
Caenorhabditis are discussed in subsequent sections of this review.
3. Rate of Gene Conversion in Caenorhabditis Paralogs
The rate of gene conversion is an important parameter that determines multiple facets of the evolution of a multigene family. For instance, if the rate of gene conversion is high, significant selective pressure is required for the adaptive diversification of paralogs [
6]. Interlocus gene conversion also contributes to allelic variation. Furthermore, gene conversion events with a pseudogene locus as the donor sequence can conceivably impose a significant mutational load on the remaining functional members of the multigene family.
No direct experimental measures of the spontaneous rate of interlocus gene conversion currently exist for
C. elegans. In experimental populations of
fog-2 mutants, gene conversion where the donor was an upstream paralog of
fog-2 of unknown function was found to be responsible for the reversion of null mutants to wild-type [
39]. In the same set of experiments, an exact reversion of the mutation was not detected, suggesting that the rate of gene conversion is at least as high as the base substitution rate. An early study estimated the intralocus/allelic gene conversion at the 38 kb long
unc-22 locus, a gene involved in worm muscle structure and function on Chromosome IV. Intralocus gene conversion events occurred at a frequency of 10
−5 per locus per generation [
40], at a scale similar to the rosy locus in
Drosophila [
41], although it should be cautioned that single-locus estimates of the conversion rate can be a poor proxy for predicting the genome-wide rate given the wide variation (0.8–30% in yeast) in the conversion frequency of different loci [
4,
42,
43]. Moreover, studies in yeast have found that the intralocus/allelic rate of gene conversion exceeds that of interlocus conversion. For example,
his3 paralogs in yeast exhibit an interlocus gene conversion rate of 0.5% relative to an intralocus conversion rate of 1.5% [
17].
Reliable measures of the rate of spontaneous interlocus gene conversion events and their genomic characteristics are best answered by analyzing long-term mutation accumulation lines maintained under relaxed selective conditions with periodic assays of a subset of paralogs at different generations.
5. Population-Genetic and Phenotypic Effects of Interlocus Gene Conversion in C. elegans
Despite a plethora of studies that have found evidence of gene conversion in DNA sequences across a diverse set of organisms, the phenotypic consequences of such gene conversion events are largely obscure. In experimental populations of
C. elegans, a gene conversion event led to the repair of a loss-of-function mutation with an extreme phenotypic consequence of sex reversion [
39].
C. elegans is one of two species in its genus exhibiting an androdioecious mode of reproduction with populations composed largely of self-fertile hermaphrodites and males in low-frequency [
57].
C. elegans hermaphrodites phenotypically resemble the females of other congeneric, obligately-outcrossing gonochoristic species but have evolved the ability to produce limited amounts of sperm for self-fertilization. The evolution of hermaphroditism in
C. elegans may have been specifically promoted by the appearance of a new gene,
fog-2, via a gene duplication of
ftr-1 that enabled spermatogenesis in
C. elegans hermaphrodites [
39,
58,
59].
ftr-1, a gene of unknown function, comprises four exons encoding 314 amino acids (aa). The exon-intron structure of
fog-2, comprising five exons (327 aa) exhibits both similarities and dissimilarities relative to
ftr-1 (
Figure 2). Homology between
ftr-1 and
fog-2 commences ~170 bp upstream of the start codon, completely encompassing the first three exons and introns and partially spanning the terminal exon of
ftr-1 (first 91 of 186 bp). Synonymous sequence divergence between the two paralogs over their region of homology is approximately 28%. The novel terminal region of
fog-2 spans the latter 66 bp of exon 4, the entire intron 4 (45 bp) and entire exon 5 (157 bp) [
39]. The creation of
fog-2 likely resulted from a
partial duplication with recruitment [
35] of
ftr-1 with the incorporation of novel sequence into its reading frame at the C-terminus from it new genomic neighborhood, likely conferring the novel function of hermaphrodite spermatogenesis in this species [
47,
59]. Both paralogs reside in tandem on Chromosome V, separated by 763 bp of unique sequence, with
ftr-1 placed directly upstream of
fog-2 [
39].
A long-term mutation-accumulation study by us employed replicate lines of
C. elegans rendered obligately-outcrossing (male-female) by a loss-of-function (
lf) mutation in the
fog-2 coding region due to the presence of a premature stop codon. During the course of this experiment, several of these male-female lines reverted spontaneously to hermaphroditism during the population bottleneck phase. Hermaphroditic revertants also originated and reached fixation in a handful of lines during a subsequent fitness-recovery phase of the experiment. Two of the sequenced revertants to hermaphroditism were established to have resulted from a gene conversion event whereby a short segment of a paralogous gene,
ftr-1, recombined with the
fog-2 (lf) mutant allele, replacing the premature stop codon with a tryptophan codon, thereby restoring
fog-2 function and leading to the subsequent appearance of functional selfing hermaphrodites (
Figure 3) [
39]. This type of repair of a deleterious mutation by gene conversion has been suggested to be important in the evolutionary conservation of the human Y chromosome, where rampant gene conversion between paralogs may have stalled Y chromosome degeneration [
60,
61] and deleterious mutation accumulation in plastid genomes [
62]. The overall nucleotide sequence identity between
ftr-1 and
fog-2 over their homologous coding region is 82%; however, the converted region in
fog-2 in the experimental populations is downstream of a stretch of 70 bp of complete identity that probably facilitated these events. Moreover, the sequence tract that was converted in the experimental lines had an overall nucleotide sequence identity of 77% between
fog-2 and
ftr-1. Considering how easily these spontaneous gene conversions were obtained in experimental populations, the fact that this region has remained impervious to gene conversion over evolutionary time suggests functional divergence between the two genes in this region.
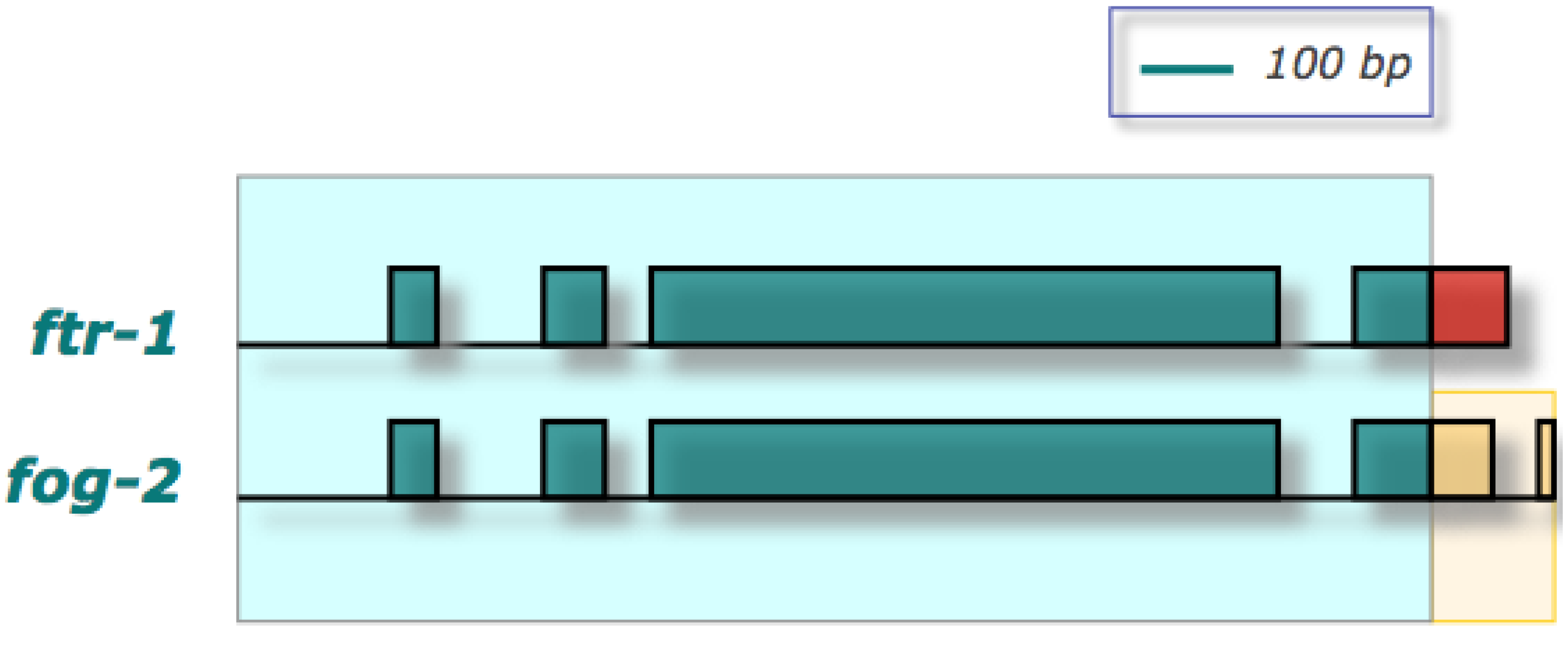
Figure 2.
Schematic depicting the regions of homology between the paralogs
fog-2 and
ftr-1 [
39,
47]. Shaded rectangles denote exons; horizontal lines represent introns and duplicated flanking regions where applicable. The duplicated region is shaded in light blue. Duplicated segments as determined by sequence homology between the two paralogs are also depicted by the correspondence of regions with identical color and pattern. Nonhomologous segments of the two paralogs are depicted in different colors. The novel portion of
fog-2 highlighted in yellow likely confers the novel function of hermaphrodite spermatogenesis.
Figure 2.
Schematic depicting the regions of homology between the paralogs
fog-2 and
ftr-1 [
39,
47]. Shaded rectangles denote exons; horizontal lines represent introns and duplicated flanking regions where applicable. The duplicated region is shaded in light blue. Duplicated segments as determined by sequence homology between the two paralogs are also depicted by the correspondence of regions with identical color and pattern. Nonhomologous segments of the two paralogs are depicted in different colors. The novel portion of
fog-2 highlighted in yellow likely confers the novel function of hermaphrodite spermatogenesis.
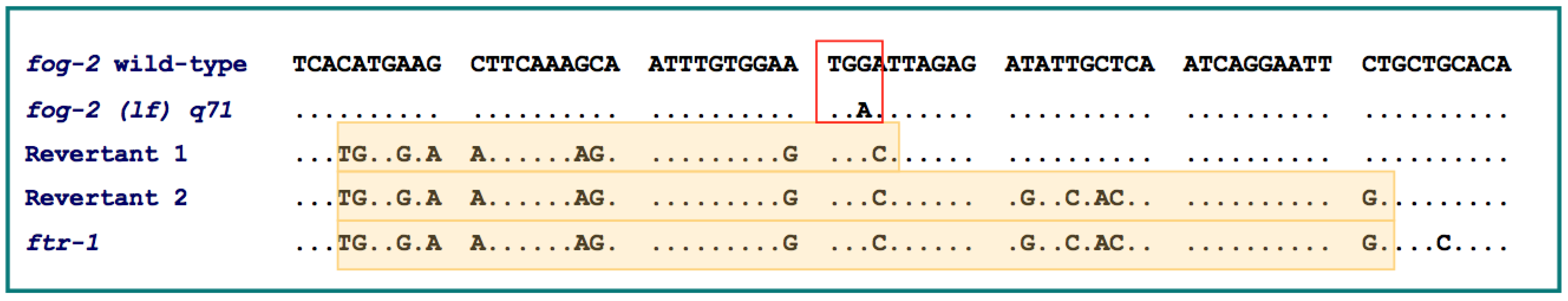
Figure 3.
Sequence alignments of in-frame nucleotide positions 200-499 of exon 3 displaying two independent gene conversion events at the
fog-2 locus by
ftr-1 resulting in a phenotypic alteration from obligate outcrossing to hermaphroditism in two
fog-2(lf) q71 mutant lines [
39]. The red box displays the nonsense mutation G → A in the
fog-2(lf)q71 allele resulting in a nonfunctional gene relative to the wild-type. Regions highlighted in yellow represent the minimum gene conversion tracts by
ftr-1 in sex-revertants 1 and 2. Dots represent identical nucleotides to the
fog-2 wild-type sequence.
Figure 3.
Sequence alignments of in-frame nucleotide positions 200-499 of exon 3 displaying two independent gene conversion events at the
fog-2 locus by
ftr-1 resulting in a phenotypic alteration from obligate outcrossing to hermaphroditism in two
fog-2(lf) q71 mutant lines [
39]. The red box displays the nonsense mutation G → A in the
fog-2(lf)q71 allele resulting in a nonfunctional gene relative to the wild-type. Regions highlighted in yellow represent the minimum gene conversion tracts by
ftr-1 in sex-revertants 1 and 2. Dots represent identical nucleotides to the
fog-2 wild-type sequence.
Interlocus gene conversion events serve to increase allelic diversity at a locus and may result in shared polymorphisms between loci [
63,
64,
65]. In a follow up study,
Rane et al. [
47] further analyzed DNA sequence variation in
fog-2 and
ftr-1 within 40 isolates of
C. elegans to investigate the population-genetic consequences of gene conversion in natural populations (
Figure 4). Gene conversion was found to contribute significantly to DNA sequence diversity at
fog-2 (22%) and
ftr-1 (34%) in these populations. This study found a region of shared polymorphism between the two paralogs that was positioned immediately upstream of a 75 bp gene conversion tract. It appears that this stretch of nucleotides, with 100% identity in the two paralogs, is facilitating gene conversion in both experimental [
39] and natural populations of
C. elegans.
In conclusion, the population-genetic consequences of gene conversion are to increase allelic diversity at paralogous loci. The standing genetic variation at synonymous sites within a species is frequently utilized to estimate the parameter Neμ as a proxy for the species effective population size (Ne). Estimates of effective population size (Ne) for a species calculated from the standing genetic variation at a converted locus will lead to inflated estimates.
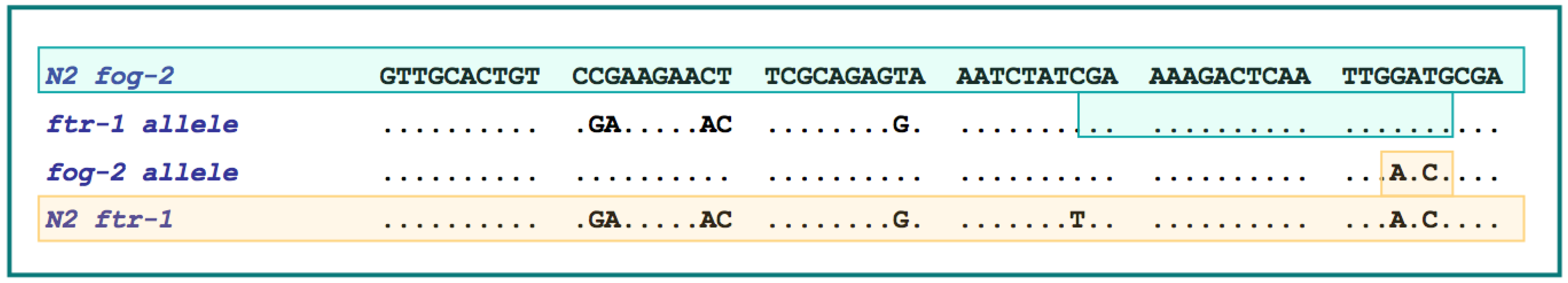
Figure 4.
Evidence of gene conversion in both
fog-2 and
ftr-1 alleles in natural isolates of
C. elegans [
47]. The top and bottom sequences (highlighted in green and yellow throughout, respectively) represent base positions 470-589 of the
fog-2 and
ftr-1 alleles of the N2 laboratory strain. Dots represent nucleotides identical to the N2
fog-2 sequence. The second sequence labeled ‘
ftr-1 allele’ represents a gene conversion event in
ftr-1 by a
fog-2 donor sequence. The minimum gene conversion tract length of 9 bp in this converted
ftr-1 allele is highlighted in green to display its sequence identity to the N2
fog-2 allele. The region surrounding this converted tract has 100% sequence identity between N2
fog-2 and
ftr-1 alleles over a 102 bp stretch, which would represent the maximum length of the gene conversion tract. This converted
ftr-1 allele was detected in 20 of the 40 natural isolates surveyed. The third sequence labeled ‘
fog-2 allele’ represents a gene conversion event in
fog-2 by an
ftr-1 donor sequence possessed by three of 40 natural isolates. The minimum gene conversion tract length of 3 bp in this converted
fog-2 allele is highlighted in yellow to display its sequence identity to the N2
ftr-1 allele. The region surrounding the converted nucleotides is identical between
fog-2 and
ftr-1 over a span of 93 bp, representing the maximum possible length of the gene conversion tract.
Figure 4.
Evidence of gene conversion in both
fog-2 and
ftr-1 alleles in natural isolates of
C. elegans [
47]. The top and bottom sequences (highlighted in green and yellow throughout, respectively) represent base positions 470-589 of the
fog-2 and
ftr-1 alleles of the N2 laboratory strain. Dots represent nucleotides identical to the N2
fog-2 sequence. The second sequence labeled ‘
ftr-1 allele’ represents a gene conversion event in
ftr-1 by a
fog-2 donor sequence. The minimum gene conversion tract length of 9 bp in this converted
ftr-1 allele is highlighted in green to display its sequence identity to the N2
fog-2 allele. The region surrounding this converted tract has 100% sequence identity between N2
fog-2 and
ftr-1 alleles over a 102 bp stretch, which would represent the maximum length of the gene conversion tract. This converted
ftr-1 allele was detected in 20 of the 40 natural isolates surveyed. The third sequence labeled ‘
fog-2 allele’ represents a gene conversion event in
fog-2 by an
ftr-1 donor sequence possessed by three of 40 natural isolates. The minimum gene conversion tract length of 3 bp in this converted
fog-2 allele is highlighted in yellow to display its sequence identity to the N2
ftr-1 allele. The region surrounding the converted nucleotides is identical between
fog-2 and
ftr-1 over a span of 93 bp, representing the maximum possible length of the gene conversion tract.
![Genes 01 00452 g004]()
6. Evolutionary Consequences of Ectopic Gene Conversion for Caenorhabditis Multigene Families
Preceding studies have established interlocus gene conversion as an important homogenizing force in the evolution of gene duplicates in
Caenorhabditis species. Semple and Wolfe [
37] found evidence for gene conversion in only 2% of 7,829 duplicate pairs in the
C. elegans genome, and 85% of these events were restricted to members of multigene families. This may have fostered the notion that gene conversion is a less potent force in the evolution of small gene families, comprising <5 paralogs [
34]. However, statistical methods [
38,
66] currently available for the detection of gene conversion events between paralogs in the absence of an outgroup sequence fail to work under regimes of high sequence identity between the focal paralogs. Most statistical analyses of gene conversion events in
C. elegans have employed Sawyer’s GENECONV statistical software [
37,
39,
45,
47] which uses the distribution of mismatches between DNA sequences to test the null hypothesis of no gene conversion between paralogs. GENECONV performs well in simulation studies but is found to be lacking in power under regimes of frequent gene conversion [
67]. Hence, the view that gene conversion in
C. elegans is relatively infrequent may need to be reevaluated. Analysis of laboratory-based mutation accumulation lines, with the strength of selection reduced to a minimal, ought to yield the most robust estimate of the spontaneous rate of gene conversion between paralogs in
Caenorhabditis genomes, the length of gene conversion tracts, and the influence, if any, of key genomic characteristics (transcriptional orientation, chromosomal location, extent of sequence homology) on this key parameter.
We next consider the evolutionary impacts of gene conversion on the functional fate of gene duplicates. Under environmental conditions where increased gene dosage is beneficial, natural selection is expected to favor concerted evolution of paralogs via gene conversion [
68,
69,
70]. On the flip side, if sequence homogenization of paralogous genes via gene conversion is sufficiently frequent, it begs the question as to how duplicates are ever able to establish independent evolutionary trajectories and neofunctionalize. With increasing sequence divergence, gene conversion between paralogs is expected to eventually taper in frequency, enabling the probability of functional divergence [
5]. However, we have no estimate for this threshold of sequence divergence between
Caenorhabditis paralogs that would serve to decrease the frequency of gene conversion to levels that effectively render it evolutionarily impotent. Even more intriguing is the question as to how this threshold of sequence divergence is ever reached under the onslaught of frequent gene conversion. Earlier theoretical work exploring the conundrum of gene duplicate neofunctionalization in the face of gene conversion pressure have suggested that “terminator mutations” such as large insertions/deletions, obstruction of sequence homology via mobile element insertion, or translocation of one paralog to another chromosome via reverse transcription may serve to interrupt sequence homology between paralogs and retard further homogenization of the two copies [
5]. More recent theoretical studies have explored the role of diversifying natural selection in the maintenance of paralog sequence diversity under conversion pressure [
6]. The patterns of DNA variation in human antigen-coding paralogs RHCE and RHD appear consistent with a model of selection maintaining antigen diversity despite frequent gene conversion, although the strength of selection required to counterbalance homogenization by gene conversion was inferred to be extremely high [
6].
We additionally suggest that structural heterogeneity between paralogs is yet another factor that likely plays an important role in restricting complete homogenization of paralogs via gene conversion, thereby promoting neofunctionalization. The mechanisms of gene duplication often fail to respect gene boundaries, resulting in the duplication of gene fractions rather than the complete ORFs of the ancestral copy [
34,
35]. More than 50% of gene duplicates with synonymous sequence divergence less than 10% in the
C. elegans genome comprised structurally heterogeneous paralogs, wherein one or both copies had unique exonic regions to the exclusion of their sister copy [
34]. If these unique coding regions in one or both paralog(s) encode novel functional domains, neofunctionalization can be promoted and maintained despite ongoing gene conversion in homologous regions of the two paralogs. As a case and point, we revisit the
fog-2/ftr-1 scenario (
Figure 2). The putative ancestral copy,
ftr-1, is of unknown function.
fog-2, implicated in the origin of hermaphroditism in
C. elegans, likely originated from a partial duplication of
ftr-1 that prematurely terminated in the terminal exon of
ftr-1, and then recruited additional unique sequence from its new genomic neighborhood to complete its ORF [
39]. Intriguingly, the recruitment of this unique sequence in the 3' end of
fog-2 likely facilitated its neofunctionalization after duplication [
47]. Frequent gene conversions of
fog-2 by
ftr-1 in both laboratory and wild
C. elegans populations fail to diminish or compromise the function of
fog-2 in hermaphrodite spermatogenesis, given that this neofunctionalized sequence tract in
fog-2 bears no homology to
ftr-1 sequence.
7. Conclusions
Independence of mutations is an important assumption in much of evolutionary genetic analysis. Gene conversion and other mechanisms of reticulate evolution resulting in shared mutations between loci can adversely affect the results of evolutionary analyses in a number of different ways. First, gene conversions can obscure and mislead conclusions on the phylogenetic relationship between genes. Second, gene conversions can lead to gene duplicates appearing more evolutionarily recent than they really are, leading to erroneous calculations with respect to the dynamics of duplicates genes in genomes. Third, gene conversion appears to occur more frequently between paralogs residing in genomic proximity and can thereby skew our understanding of gene movement around the genome. Fourth, gene conversion can result in inferences of selection when there are none [
71]. Lastly, gene conversion serves to increase allelic diversity leading to inflated estimates of the effective population size (
Ne) in instances where base substitution parameters are used to infer
Ne.
Semple and Wolfe’s study found evidence of gene conversion in only 2% of gene duplicates in the
C. elegans genome [
37]. GENECONV software [
38], often used to statistically detect the presence of gene conversion between paralogs, may underestimate gene conversion frequencies for sequences with low divergence, representing a pool of duplicates most likely to undergo conversion. Hence, the view that gene conversion is infrequent in
C. elegans may need to be revaluated. Due to low levels of DNA sequence variation in natural populations of
C. elegans, shared polymorphisms between paralogs (a key signature of gene conversion) in this species may be more challenging to detect compared to other species such as
Drosophila. High-throughput sequencing of
C. elegans mutation accumulation lines will eventually yield data on the spontaneous gene conversion rate whereas a combination of methods will be needed to evaluate the role of gene conversion in the evolution of
Caenorhabditis genomes. These will undoubtedly take advantage of various
Caenorhabditis genome sequencing projects currently in progress by analyzing relationships of paralogs whose synteny context has been conserved since speciation.



{kind=link}
{kind=link}
{kind=link}
{kind=link}


