A Global Expression Switch Marks Pachytene Initiation during Mouse Male Meiosis

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
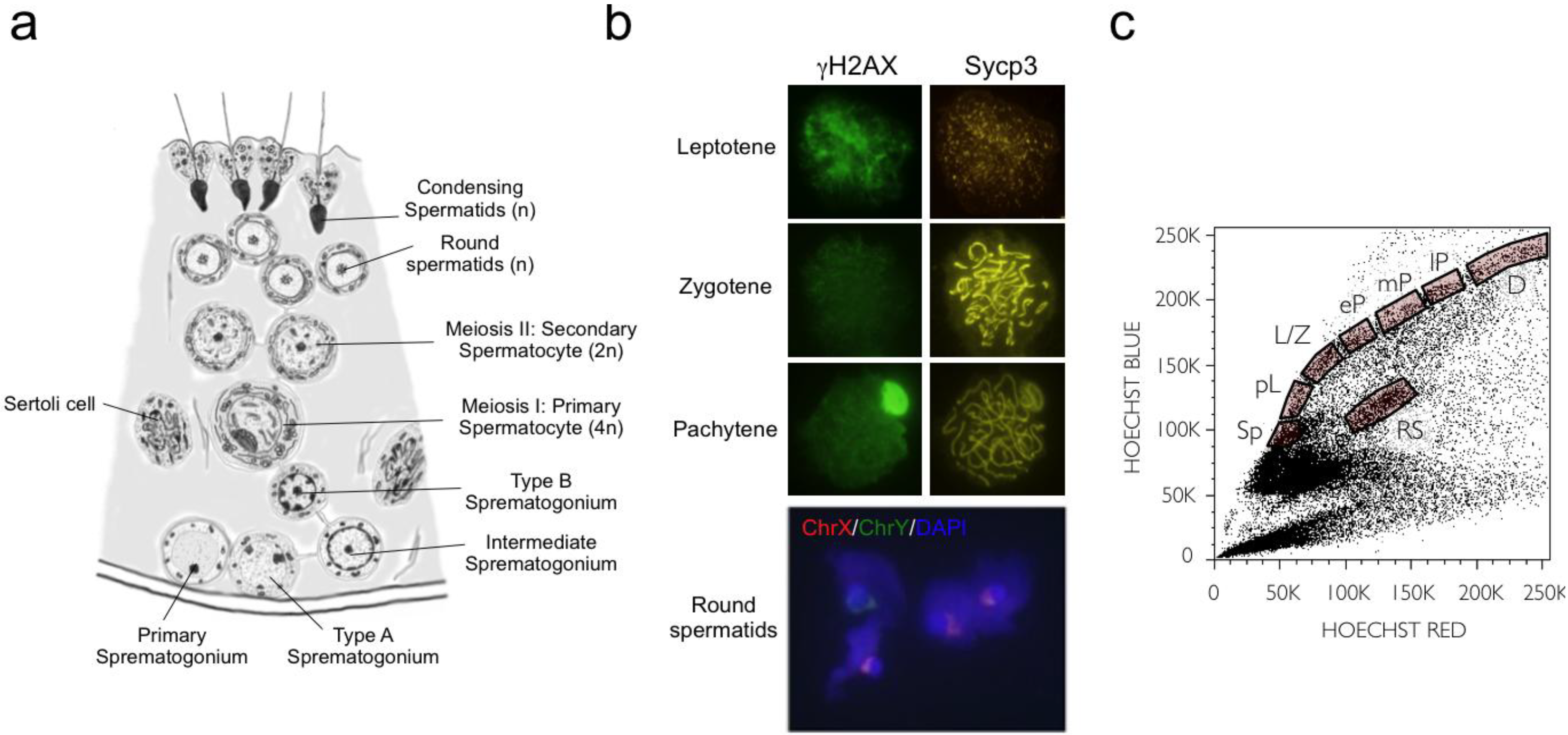
2.1. Meiotic Cell Purification

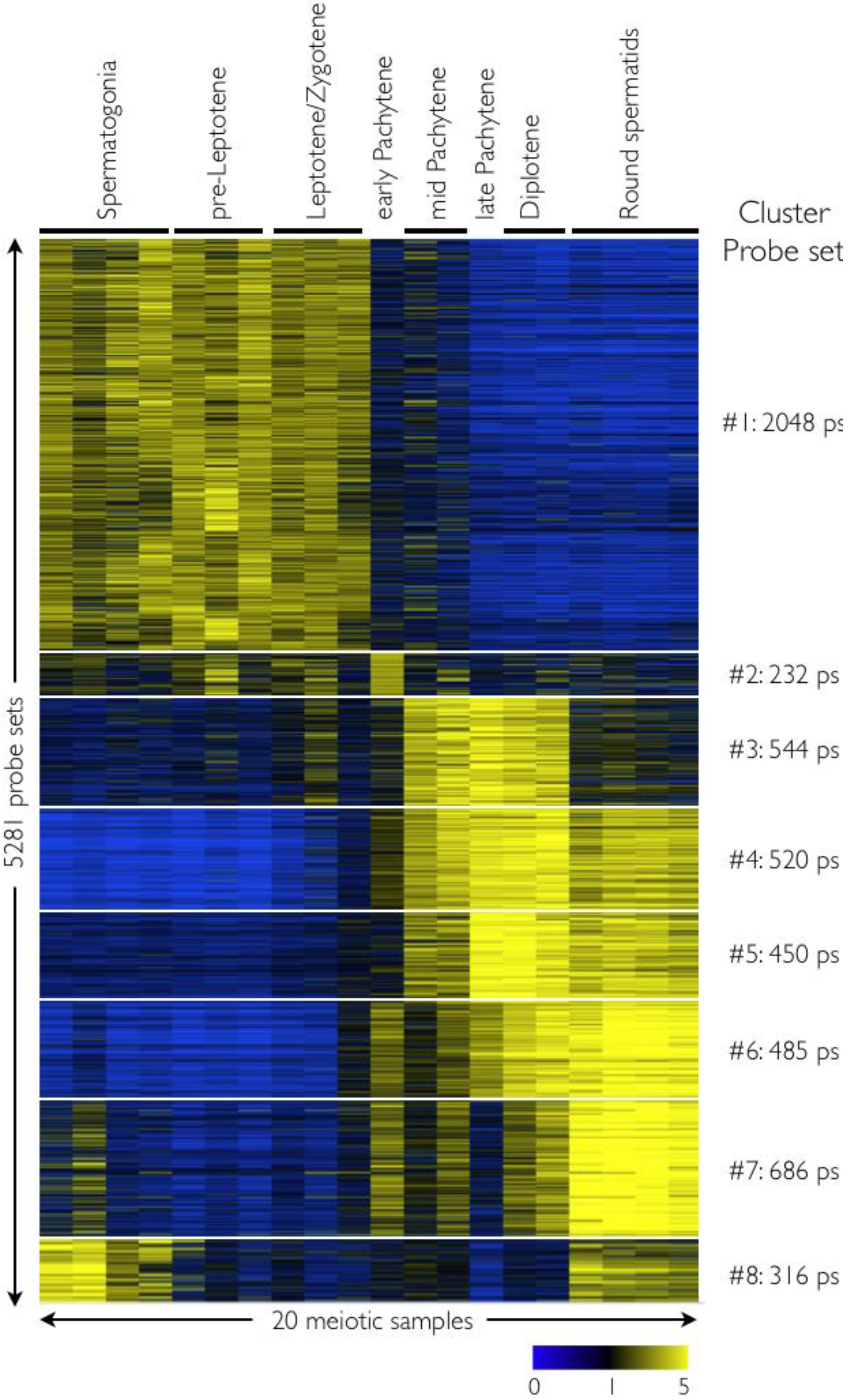
2.2. Expression Analysis Overview

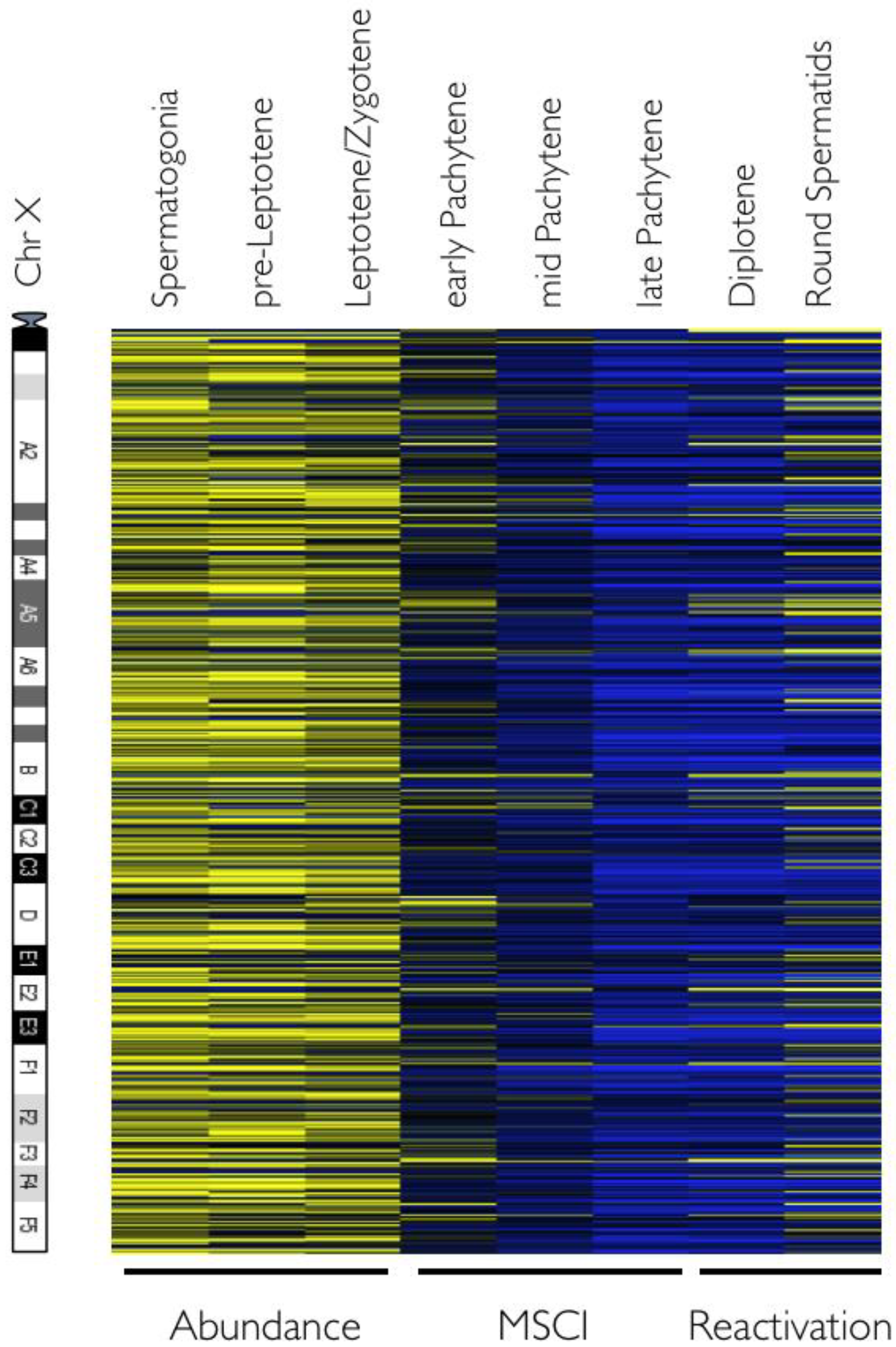
2.3. Meiotic Sex Chromosome Inactivation

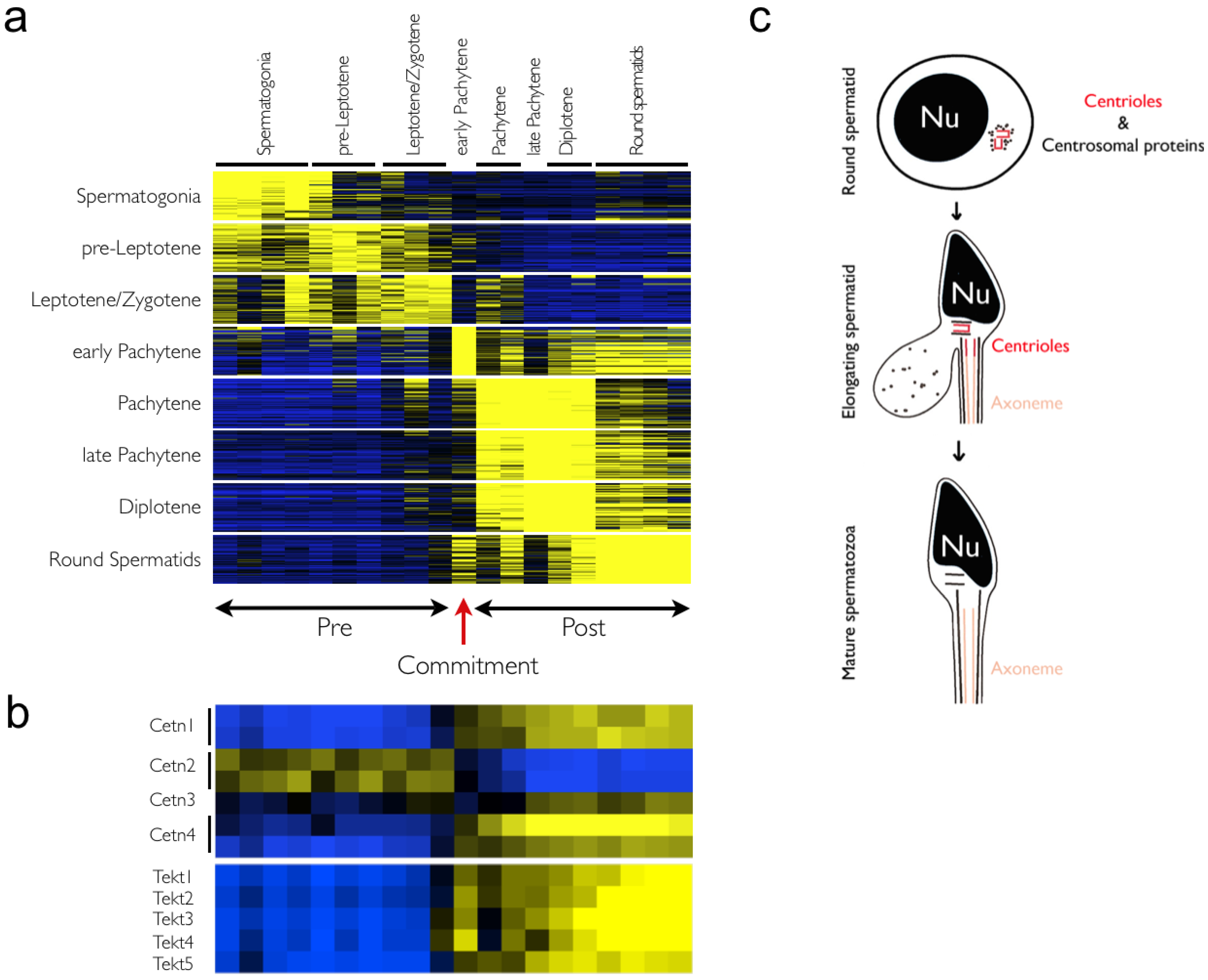
2.4. Commitment to Meiosis

2.5. Discussion
3. Experimental Section
3.1. Mice
3.2. Sample Collection and RNA Extraction
3.3. Immunofluorescence Analyses
3.4. RNA Labeling and ARRAY Hybridization
3.5. Data Analysis
4. Conclusions
Acknowledgements
Supplementary Files
{kind=link}
{kind=link}
{kind=link}
References
- McLaren, A. Primordial germ cells in the mouse. Dev. Biol. 2003, 262, 1–15. [Google Scholar]
- Sasaki, H.; Matsui, Y. Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nat. Rev. Genet. 2008, 9, 129–140. [Google Scholar] [CrossRef]
- Schultz, N.; Hamra, F.K.; Garbers, D.L. A multitude of genes expressed solely in meiotic or postmeiotic spermatogenic cells offers a myriad of contraceptive targets. Proc. Natl. Acad. Sci. USA 2003, 100, 12201–12206. [Google Scholar] [CrossRef]
- Namekawa, S.H.; Park, P.J.; Zhang, L.F.; Shima, J.E.; McCarrey, J.R.; Griswold, M.D.; Lee, J.T. Postmeiotic sex chromatin in the male germline of mice. Curr. Biol. 2006, 16, 660–667. [Google Scholar]
- Chalmel, F.; Rolland, A.D.; Niederhauser-Wiederkehr, C.; Chung, S.S.; Demougin, P.; Gattiker, A.; Moore, J.; Patard, J.J.; Wolgemuth, D.J.; Jegou, B.; et al. The conserved transcriptome in human and rodent male gametogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 8346–8351. [Google Scholar]
- Oakberg, E.F. Duration of spermatogenesis in the mouse and timing of stages of the cycle of the seminiferous epithelium. Am. J. Anat. 1956, 99, 507–516. [Google Scholar]
- Scherthan, H.; Weich, S.; Schwegler, H.; Heyting, C.; Harle, M.; Cremer, T. Centromere and telomere movements during early meiotic prophase of mouse and man are associated with the onset of chromosome pairing. J. Cell Biol. 1996, 134, 1109–1125. [Google Scholar]
- McKee, B.D.; Handel, M.A. Sex chromosomes, recombination, and chromatin conformation. Chromosoma 1993, 102, 71–80. [Google Scholar]
- Hoyer-Fender, S. Molecular aspects of XY body formation. Cytogenet. Genome Res. 2003, 103, 245–255. [Google Scholar]
- Lassalle, B.; Bastos, H.; Louis, J.P.; Riou, L.; Testart, J.; Dutrillaux, B.; Fouchet, P.; Allemand, I. ‘Side Population’ cells in adult mouse testis express Bcrp1 gene and are enriched in spermatogonia and germinal stem cells. Development 2004, 131, 479–487. [Google Scholar]
- Bastos, H.; Lassalle, B.; Chicheportiche, A.; Riou, L.; Testart, J.; Allemand, I.; Fouchet, P. Flow cytometric characterization of viable meiotic and postmeiotic cells by Hoechst 33342 in mouse spermatogenesis. Cytometry A 2005, 65, 40–49. [Google Scholar]
- Simchen, G. Commitment to meiosis: What determines the mode of division in budding yeast? BioEssays 2009, 31, 169–177. [Google Scholar] [CrossRef]
- Romrell, L.J.; Bellve, A.R.; Fawcett, D.W. Separation of mouse spermatogenic cells by sedimentation velocity. A morphological characterization. Dev. Biol. 1976, 49, 119–131. [Google Scholar] [CrossRef]
- Getun, I.V.; Wu, Z.K.; Khalil, A.M.; Bois, P.R. Nucleosome occupancy landscape and dynamics at mouse recombination hotspots. EMBO Rep. 2010, 11, 555–560. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar]
- Turner, J.M. Meiotic sex chromosome inactivation. Development 2007, 134, 1823–1831. [Google Scholar]
- Wang, P.J.; McCarrey, J.R.; Yang, F.; Page, D.C. An abundance of X-linked genes expressed in spermatogonia. Nat. Genet. 2001, 27, 422–426. [Google Scholar]
- Ellis, P.J.; Clemente, E.J.; Ball, P.; Toure, A.; Ferguson, L.; Turner, J.M.; Loveland, K.L.; Affara, N.A.; Burgoyne, P.S. Deletions on mouse Yq lead to upregulation of multiple X- and Y-linked transcripts in spermatids. Hum. Mol. Genet. 2005, 14, 2705–2715. [Google Scholar] [CrossRef]
- Khalil, A.M.; Driscoll, D.J. Trimethylation of histone H3 lysine 4 is an epigenetic mark at regions escaping mammalian X inactivation. Epigenetics 2007, 2, 114–118. [Google Scholar]
- Miki, K.; Willis, W.D.; Brown, P.R.; Goulding, E.H.; Fulcher, K.D.; Eddy, E.M. Targeted disruption of the Akap4 gene causes defects in sperm flagellum and motility. Dev. Biol. 2002, 248, 331–342. [Google Scholar] [CrossRef]
- Zheng, K.; Yang, F.; Wang, P.J. Regulation of Male Fertility by X-Linked Genes. J. Androl. 2009, 31, 79–85. [Google Scholar] [CrossRef]
- Iuchi, Y.; Okada, F.; Tsunoda, S.; Kibe, N.; Shirasawa, N.; Ikawa, M.; Okabe, M.; Ikeda, Y.; Fujii, J. Peroxiredoxin 4 knockout results in elevated spermatogenic cell death via oxidative stress. Biochem. J. 2009, 419, 149–158. [Google Scholar]
- Manandhar, G.; Schatten, H.; Sutovsky, P. Centrosome reduction during gametogenesis and its significance. Biol. Reprod. 2005, 72, 2–13. [Google Scholar] [CrossRef]
- Cunha-Ferreira, I.; Bento, I.; Bettencourt-Dias, M. From zero to many: Control of centriole number in development and disease. Traffic 2009, 10, 482–498. [Google Scholar] [CrossRef]
- Salisbury, J.L. A mechanistic view on the evolutionary origin for centrin-based control of centriole duplication. J. Cell. Physiol. 2007, 213, 420–428. [Google Scholar] [CrossRef]
- Amos, L.A. The tektin family of microtubule-stabilizing proteins. Genome Biol. 2008, 9, 229. [Google Scholar]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef]
- Brunskill, E.W.; Aronow, B.J.; Georgas, K.; Rumballe, B.; Valerius, M.T.; Aronow, J.; Kaimal, V.; Jegga, A.G.; Yu, J.; Grimmond, S.; et al. Atlas of gene expression in the developing kidney at microanatomic resolution. Dev. Cell 2008, 15, 781–791. [Google Scholar]
- Bellve, A.R.; Cavicchia, J.C.; Millette, C.F.; O'Brien, D.A.; Bhatnagar, Y.M.; Dym, M. Spermatogenic cells of the prepuberal mouse. Isolation and morphological characterization. J. Cell Biol. 1977, 74, 68–85. [Google Scholar] [CrossRef]
- Guillon, H.; de Massy, B. An initiation site for meiotic crossing-over and gene conversion in the mouse. Nat. Genet. 2002, 32, 296–299. [Google Scholar] [CrossRef]
- Bellvé, A.R. Purification, culture, and fractionation of spermatogenic cells. Meth. Enzymol. 1993, 225, 84–113. [Google Scholar] [CrossRef]
- Mueller, J.L.; Mahadevaiah, S.K.; Park, P.J.; Warburton, P.E.; Page, D.C.; Turner, J.M. The mouse X chromosome is enriched for multicopy testis genes showing postmeiotic expression. Nat. Genet. 2008, 40, 794–799. [Google Scholar]
- de Rooij, D.G.; de Boer, P. Specific arrests of spermatogenesis in genetically modified and mutant mice. Cytogenet. Genome Res. 2003, 103, 267–276. [Google Scholar] [CrossRef]
- Jeppesen, P.; Turner, B.M. The inactive X chromosome in female mammals is distinguished by a lack of histone H4 acetylation, a cytogenetic marker for gene expression. Cell 1993, 74, 281–289. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fallahi, M.; Getun, I.V.; Wu, Z.K.; Bois, P.R.J. A Global Expression Switch Marks Pachytene Initiation during Mouse Male Meiosis. Genes 2010, 1, 469-483. https://doi.org/10.3390/genes1030469
Fallahi M, Getun IV, Wu ZK, Bois PRJ. A Global Expression Switch Marks Pachytene Initiation during Mouse Male Meiosis. Genes. 2010; 1(3):469-483. https://doi.org/10.3390/genes1030469
Chicago/Turabian StyleFallahi, Mohammad, Irina V. Getun, Zhen K. Wu, and Philippe R.J. Bois. 2010. "A Global Expression Switch Marks Pachytene Initiation during Mouse Male Meiosis" Genes 1, no. 3: 469-483. https://doi.org/10.3390/genes1030469