Draft Genome Sequences of Xanthomonas sacchari and Two Banana-Associated Xanthomonads Reveal Insights into the Xanthomonas Group 1 Clade

 ,
,
Abstract
: We present draft genome sequences for three strains of Xanthomonas species, each of which was associated with banana plants (Musa species) but is not closely related to the previously sequenced banana-pathogen Xanthomonas campestris pathovar musacearum. Strain NCPPB4393 had been deposited as Xanthomonas campestris pathovar musacearum but in fact falls within the species Xanthomonas sacchari. Strain NCPPB1132 is more distantly related to Xanthomonas sacchari whilst strain NCPPB 1131 grouped in a distinct species-level clade related to X. sacchari, along with strains from ginger, rice, cotton and sugarcane. These three newly sequenced strains share many genomic features with the previously sequenced Xanthomonas albilineans, for example possessing an unsual metE allele and lacking the Hrp type III secretion system. However, they are distinct from Xanthomonas albilineans in many respects, for example showing little evidence of genome reduction. They also lack the SPI-1 type III secretion system found in Xanthomonas albilineans. Unlike X. albilineans, all three strains possess a gum gene cluster. The data reported here provide the first genome-wide survey of non-Hrp Xanthomonas species other than Xanthomonas albilineans, which is an atypical member of this group. We hope that the availability of complete sequence data for this group of organisms is the first step towards understanding their interactions with plants and identifying potential virulence factors.1. Introduction
The genus Xanthomonas includes some 27 species of plant-associated Gram-negative bacteria. Collectively these species, and their constituent pathovars, cause disease on several hundred plant species, including many economically important crops. Phylogenetic analyses of the genus Xanthomonas consistently reveal that it comprises two distinct groups [1-6]. Young and colleagues recently proposed that Group 1 and Group 2 might represent two distinct genera [6].
Complete genome sequences are available for several members of Group 2, including X. campestris pv. campestris, X. campestris pv. raphani, X. campestris pv. vesicatoria, X. citri, X. fuscans subsp. aurantifolii, X. oryzae pv. oryzae and X. oryzae pv. oryzicola [7]. Draft genome sequences are available for several further members of the Group 2 Xanthomonas species [7]. The genomes of all of the members of Group 2 investigated so far encode a type III secretion system (T3SS), known as the Hrp T3SS, which is required for delivery of effector proteins into the plant host in order to overcome the host's defences. The name “Hrp” is derived from “hypersensitive response and pathogenicity”.
Of the members of Xanthomonas Group 1, the only published genome sequence [8] is that of X. albilineans, a xylem-limited pathogen that causes leaf scorch in sugarcane (Saccharum species). Analysis of the X. albilineans genome sequence revealed that this species displays several interesting features that are quite distinct from those of the Group 2 species. For example, X. albilineans lacks the Hrp T3SS that is universally conserved and central to pathogenicity in Group 2, but it encodes an alternative non-Hrp T3SS that shares sequence similarity to the Salmonella SPI-1 T3SS [9]. This raises the question of whether these features of the X. albilineans genome are also shared with the genomes of other Group 1 Xanthomonas species. Furthermore, X. albilineans appears to have undergone significant genome reduction, perhaps as a consequence of, or adaptation to, its xylem-limited lifestyle [8]. Therefore, it would be interesting to compare its genome with those of other members of Group 2 that do not share this highly specialized lifestyle.
Until recently, the only known X. albilineans virulence factor was the toxin albicidin. The complete genome sequence of X. albilineans enabled the identification of several new candidate virulence factors via screening of a transposon mutagenesis library [10]. Many of these were not shared with the Group 2 Xanthomonas species and may reflect the distinctiveness of the pathogenic strategies adopted by Groups 1 and 2. Therefore, it raises the question of whether these newly discovered virulence factors are also found in Group 1 species other than X. albilineans.
We recently sequenced the genome of an isolate of X. campestris pv. musacearum, which is the causative agent of banana Xanthomonas wilt, a disease currently causing devastation to the banana crop in East Africa [11] and is a member of Xanthomonas Group 2. Subsequently, we performed follow-up genome-sequencing studies on additional isolates that had been deposited in the National Collection of Plant Pathogenic Bacteria (NCPPB) as X. campestris pv. musacearum. We discovered that NCPPB4393 shared little sequence similarity with the previously sequenced isolate; rather, it showed very close sequence similarity with X. sacchari, a member of Xanthomonas Group 1. On further investigation we learned that NCPPB4393 had in fact been isolated from an insect on a diseased banana plant but that there was no evidence that the strain is actually pathogenic on banana [12]. We also sequenced two additional Xanthomonas strains (NCPPB1131 and NCPPB1132) that had been isolated from banana plants in Eastern and Western Samoa.
2. Results and Discussion
2.1. Bacterial Strains
Bacterial strains (Table 1) were obtained from the National Collection of Plant Pathogenic Bacteria (NCPPB) in the United Kingdom. NCPPB1131 and NCPPB1132 had been deposited in 1961 by Hayward A.C. after isolation from banana plants. NCPPB4393 was one of several strains isolates deposited by one of the authors of the present study (V.A.) in 2007. It was deposited in the strain collection as X. campestris pv. musacearum. However, the results of this study suggest that it is actually a member of the species X. sacchari. It was originally isolated by Mgenzi Byabachwezi from an insect on a diseased banana plant (Muleba district, Kagera region, North Western Tanzania, on the shores of Lake Victoria). Although the insect was collected from diseased banana, sugarcane is commonly grown in that district of Tanzania and so it is possible that the insect acquired the bacterium from sugarcane. Pathogenicity of strain NCPPB4393 has not been tested.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Host plant | Country | Year |
|---|---|---|---|
| NCPPB1131 | Musa paradisiaca | American (Eastern) Samoa | 1961 |
| NCPPB1132 | Musa canksii var. samoensis | Western Samoa | 1961 |
| NCPPB4393 | Musa species Isolated from insect on diseased plant | Tanzania | 2007 |
2.2. Genome-Wide Sequence Data
We generated genome-wide sequence data for three strains listed in Table 1 using the Illumina GA2. After removing bar-code adaptors, the sequence reads were 70 nt long. We generated 1.7 million non-paired reads for NCPPB1131. For NCPPB1132 and NCPPB4393 we generated 1.9 million and 2.1 million paired reads respectively. These Whole Genome Shotgun project data have been deposited at DDBJ/EMBL/GenBank under the accession numbers AGHY00000000, AGHZ00000000 and AGDB00000000 respectively.
2.3. Phylogenetic Position of the Sequenced Xanthomonas Strains
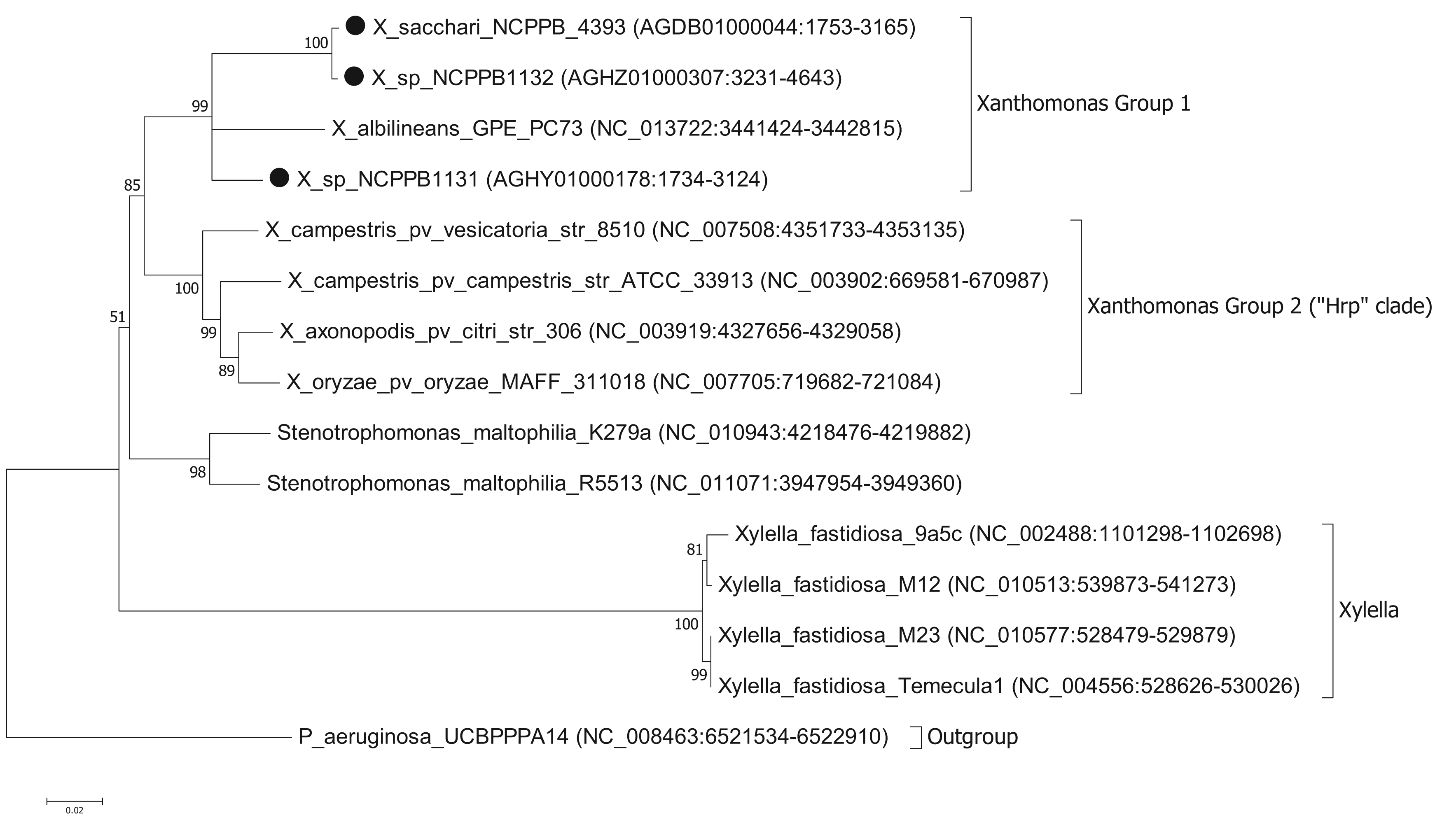
To ascertain the phylogenetic position of the three newly sequenced strains, we generated a series of phylogenetic trees based on the nucleotide sequences of house-keeping genes. We used the same set of seven genes that were used by Pieretti and colleagues [8]. For four of these genes (atpD, dnaK, groEL and recA) we were able to build trees from multiple sequence alignments using the maximum likelihood method. However, for three of the genes (efp, glnA and gyrB) we were unable to build valid multiple sequence alignments because of a lack of orthologues with detectable nucleotide sequence similarity. For example, blastn searches against the NCBI non-redundant nucleotide database, using X. albilineans gyrB (XALc_0004) as the query, yielded no significant matches in Xylella species. The phylogenetic reconstruction [13,14] based on atpD is shown in Figure 1. Phylogenetic trees based on dnaK, groEL and recA are included in the Supplementary Files. The trees for atpD, dnaK and groEL are all toplogically consistent with each other, though there is some variation in branch lengths and the precise position of X. albilineans within Group 1 is less well resolved in the atpD tree than in the others. However, analysis of the recA sequences yielded a different branching pattern in which X. albilineans falls within the Xylella fastidiosa lineage rather than within the Xanthomonas Group 1.
Our phylogenetic analyses clearly and consistently indicated that all three strains fell within the phylogenetic range of the genus Xanthomonas. Specifically, all three strains are more closely related to X. albilineans than to the Group 2 Xanthomonas species and therefore likely belong to Group 1.
We note that in our analyses based on three out of four house-keeping genes, the genus Xanthomonas comprises a single monophyletic clade, distinct from the related genera Stenotrophomonas and Xylella. This is consistent with previous studies [7,15] but contradicts recent claims that X. albilineans and Xylella fastidiosa form a monophyletic clade distinct from the Group 2 Xanthomonas species [8,10]. On the other hand, our analyses based on the recA gene were consistent with Pieretti's hypothesis that X. albilineans and Xylella fastidiosa form a monophyletic clade distinct from the Group 2. The incongruity between atpD, groEL and dnaK on the one hand and recA on the other implies that there has been recombination and that not all of these house-keeping genes truly reflect the vertical descent of the core genome. The most parsimonious explanation is that recA has undergone horizontal transfer in either the Xylella lineage or in the X. albilineans lineage. The reasons for discrepancy between our phylogenetic reconstructions for atpD, groEL and dnaK compared with that of Pieretti and colleagues [8] are two-fold. First, the analysis presented by Pieretti is a composite of genes displaying at least two distinct phylogenetic histories. Second, Pieretti's analysis [8] appears to be partly based on alignments of non-orthologous gene sequences (e.g., their gyrB sequences are not orthologous between Xylella and Xanthomonas species).
The results of our analysis are also inconsistent with those of Sharma and Patil [16]. In their study [16] they present a phylogenetic tree in which X. albilineans is the outgroup and Stenotrophomonas species fall within a monophyletic group along with the other Xanthomonas species. However, this discrepancy is explained by their misplacing the root of their tree. Sharma and Patil [16] do not explain how they chose the position of the root in their tree; they simply assume that X. albilineans is the outgroup without offering any justification for this. On the other hand, we used a phylogenetically distinct species (P. aeruginosa) as the outgroup in order to root our tree. If the tree of Sharma and Patil [16] is re-rooted with Stenotrophomonas species as the outgroup, then their tree is topologically congruent with ours. Note that Sharma and Patil do not include Xylella fastidiosa in their analysis.

To more precisely resolve the newly sequenced strains' positions within Xanthomonas Group 1, we performed phylogenetic analyses based on the gyrase B (gyrB) gene (Figure 2); partial sequences of gyrB are available from two studies [5,6] including many more Xanthomonas strains than those for which there are fully sequenced genomes. The sequences that we used are given in the Supplementary Files. We found that the gyrB sequence from NCPPB4393 was identical to those from strains of X. sacchari. Previously, X. sacchari was described as comprising strains isolated from diseased sugarcane [17]. Therefore, the description may need to be modified to include strains isolated from insects.

Strain NCPPB1132 is also closely related to X. sacchari but is more divergent than NCPPB4393 (Figure 2). It falls within a clade that included X. sacchari sensu strictu as well as X. “campestris” pv. cannae (from canna lilly, a relative of banana) and several unnamed strains isolated from sugarcane (NCPPB888 and NCPPB917), foxtail millet (NCPPB3174) and arrow leaf elephant ear (NCPPB3570).
Strain NCPPB1131 is more closely related to X. albilineans than to X. sacchari, but shows closest affinity with NCPPB2250 (Figure 2), which was originally isolated from ginger, a relative of banana. Strain NCPPB1131 is also similarly closely related to NCPPB3949, isolated from rice and erroneously deposited as X. oryzae pv. oryzicola. Parkinson and colleagues [6] describe a species-level clade (Slc 7) that included NCPPB1131, NCPPB3949 and strains isolated from ginger, cotton and sugarcane.
2.4. Comparison of the Three Genomes Versus X. albilineans
The total sizes of the genome assemblies were 3.8 Mb for NCPPB1131, 4.7 Mb for NCPPB1132 and 4.9 Mb for NCPPB4393. These can be used as estimates of genome size, but may be inaccurate since we have not closed the gaps in the draft assembly. The size for NCPPB1131 should be treated with particular caution, since the use of non-paired sequence reads yielded a very fragmented assembly. Contiguity of the assemblies can be represented by N50 scaffold lengths which were 1.1 Kb (NCPPB1131), 4.8 Kb (NCPPB1132) and 51.5 Kb (NCPPB4393). The numbers of scaffolds in each assembly were 4,158 (NCPPB1131), 1,652 (NCPPB1132) and 259 (NCPPB4393). Nevertheless, these estimates are congruent with sizes of previously sequenced Xanthomonas genomes, which range from 4.8 to 5.3 Mb for Group 2, whilst X. albilineans has a genome of less than 3.8 Mb.
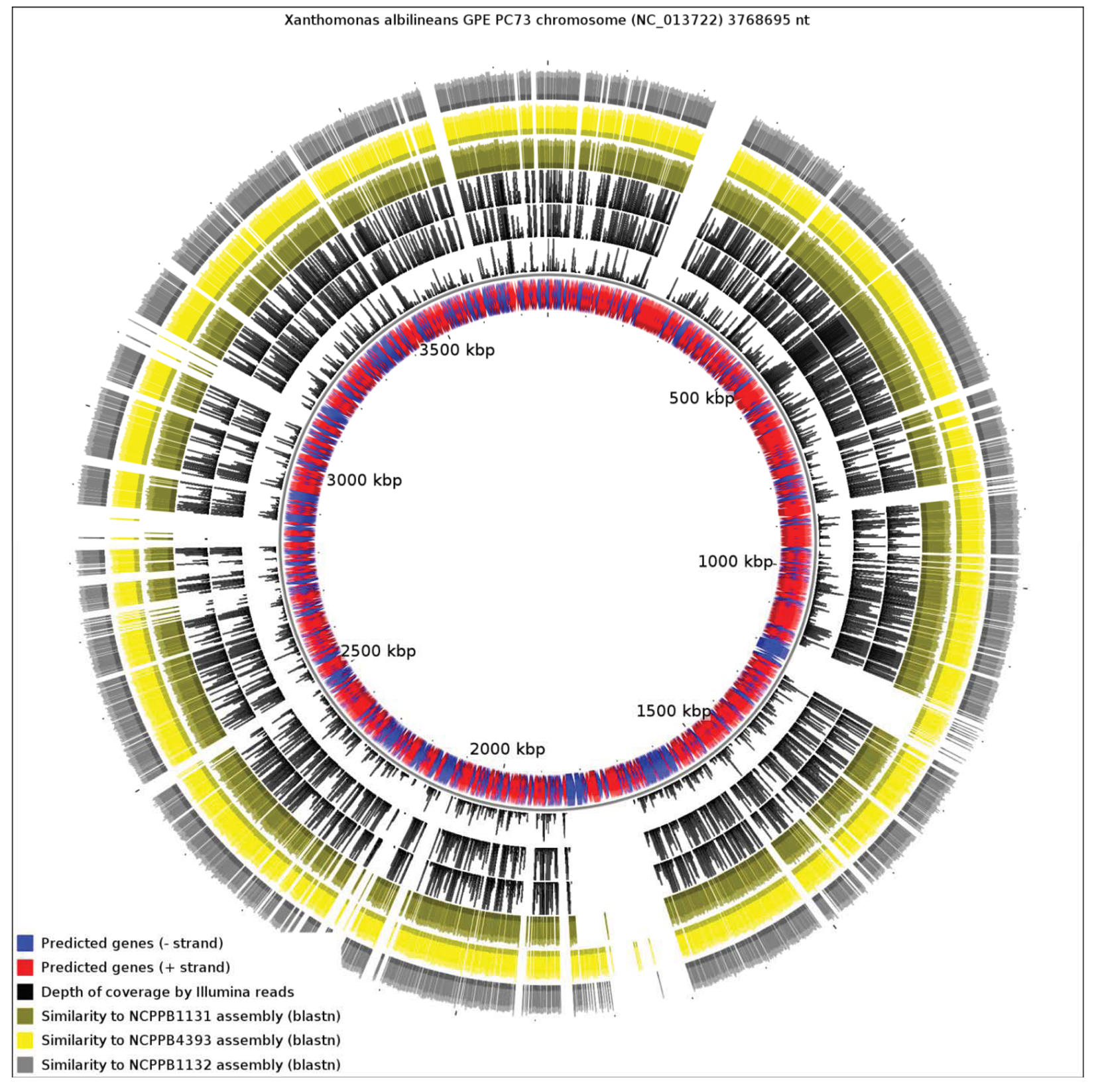
We aligned the three genome assemblies against the genome sequence of X. albilineans species. We also aligned the reads, without performing de novo assembly. Figure 3 illustrates a genome-wide overview of these alignments. It should be noted that the sequencing depths obtained for the three strains ensures complete coverage over the entire breadth of the genomes. This means that by examining alignments of sequence reads against a reference sequence, independently from any de novo assembly, we can confidently determine the presence or absence of genes. The depths of coverage of each genome, as determined by depths of alignments of raw reads against assemblies, were 20× (NCPPB1131), 67× (NCPPB1132) and 72× (NCPPB4393).
Clearly, a significant proportion of the X. albilineans genome is not conserved (at the nucleotide sequence level) in the three newly sequenced Group 1 strains (Figure 3). Prominent amongst the non-conserved regions is the gene cluster encoding the X. albilineans SPI-1 T3SS (positions 1,703,391-1,730,688). We could find no evidence for any non-flagellar T3SS in any of the three strains. All three strains also lack the albicidin biosynthesis cluster at positions 1,740,869-1,788,517 and so they likely do not produce albicidin.
Pieretti and colleagues [8] observed that the genomes of two xylem-limited pathogens, X. albilineans and Xylella fastidiosa, both share an unusual allele of the metE gene, which encodes 5-methyltetrahydropterolyl-triglutamate-homocysteine methyltransferase, an enzyme required for methionine biosynthesis. They infer that the ancestor of X. albilineans and X. fastidiosa lost metE, while the rest of the xanthomonads retained it, and then this ancestor gained a new allele of metE by horizontal transfer. We reject this interpretation, since the last common ancestor of X. albilineans and Xylella fastidiosa was also the ancestor of the other xanthomonads, including Stenotrophomonas and all Xanthomonas species (Figure 1). We found that NCPPB1131, NCPPB1132 and NCPPB4393 all contain a metE gene that most closely resembles (at least 90% amino acid sequence identity) that of X. albilineans rather than those of Group 2 Xanthomonas species. This suggests that the X. albilineans metE occurs widely in the Group 1 Xanthomonas species and is not restricted only to xylem-limited species. The incongruence between the phylogeny of metE genes and the core house-keeping genes indicates that metE has been replaced independently in at least two distinct lineages during the evolution of the xanthomonads.

2.5. Genome Reduction
Genome reduction has occurred independently in two separate lineages of xanthomonads that are specialized for a xylem-limited lifestyle. That is, Xylella fastidiosa and X. albilineans have independently converged on a xylem-limited lifestyle, each having evolved from a different ancestor with a larger genome. The only other xylem-limited bacterial species for which complete genome sequence data are available is Leifsonia xyli. Interestingly, L. xyli also appears to have a reduced genome, its chromosome being 2.6 megabases long in contrast to its non-xylem-associated relative Clavibacter michiganensis, which has a 3.3-megabase chromosome [18]. Thus, genome reduction is associated with at least three distinct lineages of xylem-limited bacteria, suggesting that a stripped-down genome may be adaptive for this relatively stable environment. Complete genome data are not yet available for the other well-known xylem-limited species, Ralstonia syzygii [19], so we cannot yet be sure that this is a universal phenomenon.
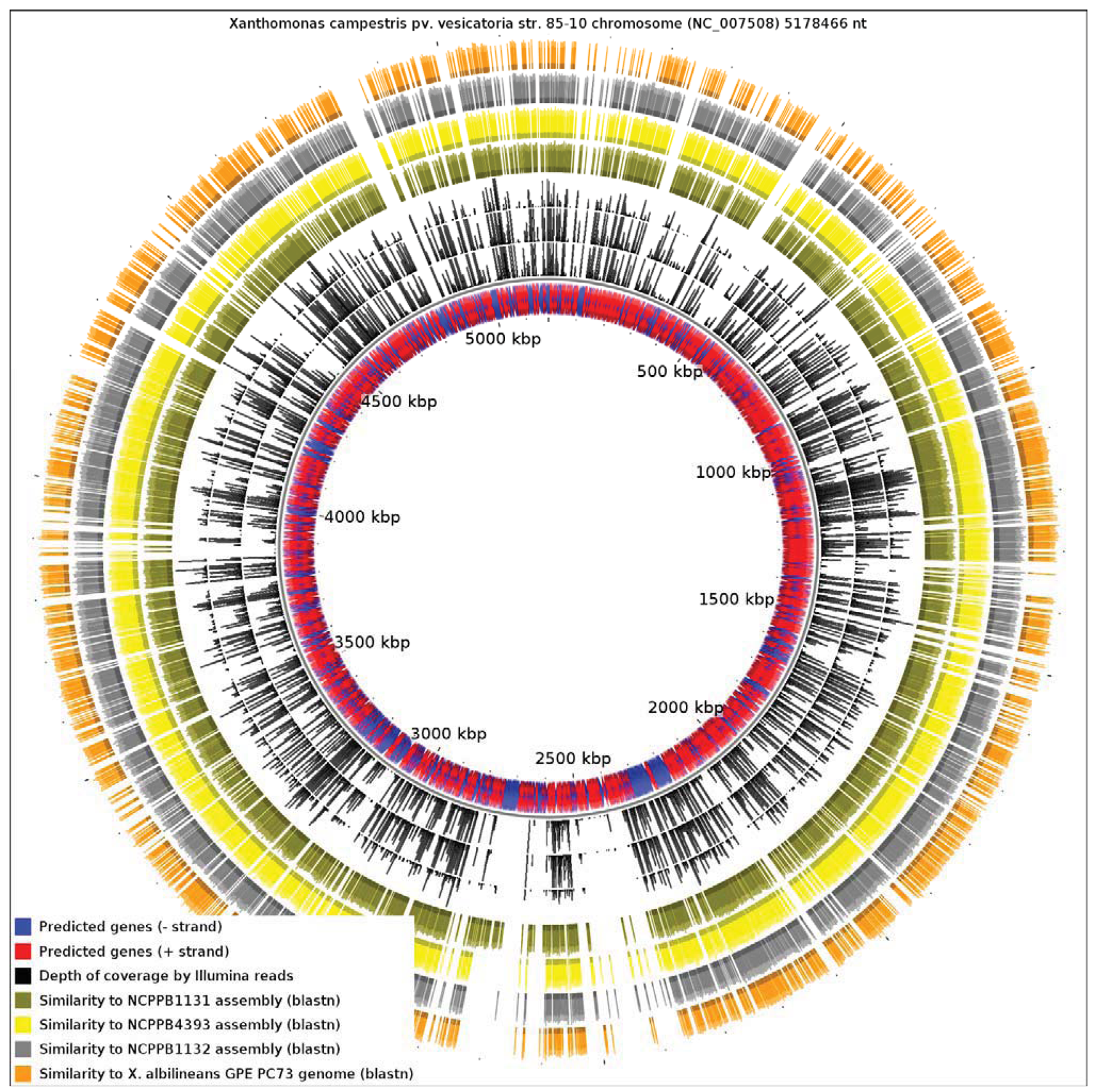
Since the only sequenced member of Xanthomonas Group 1 is a specialized xylem-limited pathogen with a reduced genome, this raises the question of whether other members of Group 1 show similar evidence of genome reduction. Our results reveal that X. albilineans has undergone significantly more reduction than NCPPB1131, NCPPB1132 and NCPPB4393. We aligned all four available genomes from Group 1 against the reference sequence of X. campestris pv. vesicatoria (Xcv) 85-10 (RefSeq: NC_007508), a member of Group 2 (Figure 4). We found that only 13.82% of the Xcv chromosome was conserved in X. albilineans. Significantly larger portions of the Xcv were conserved in NCPPB1131, NCPPB1132 and NCPPB4393 (21.69%, 26.85% and 28.44% respectively). This is consistent with X. albilineans having lost more of the Xanthomonas genome than have the other three strains.
X. albilineans produces the toxin albicidin, which is a DNA-gyrase inhibitor [20]. In addition to its action on host-plant chloroplasts, it is also deleterious to most bacteria. Pieretti and colleagues [8] propose that albicidin played a key role in the erosion of the X. albilineans genome. Specifically, they propose that exposure to sub-lethal doses of intracellular albicidin induced recombination and mutagenesis. They note that X. albilineans has, in addition to its albicidin transporter (AlbF), an unusual DNA gyrase, containing a 43-amino-acid insertion, that confers resistance to albicidin [21]. They go on to propose that “genome erosion induced by albicidin was likely arrested by the evolution of the albicidin-resistant DNA gyrase” [8]. Interesting though it is, there is no evidence to support this conjecture. Nor is there any need to invoke such a special mechanism, since genome reduction is a common phenomenon, seen in many parasitic organisms that do not produce albicidin-like antibiotics. Furthermore, sequence insertions similar to that in X. albilineans gyrase A are not uncommon among the gamma-Proteobacteria (see Supplementary Files). Therefore, it is quite plausible that albicidin-insensitivity is an ancient and/or common trait that was already present in X. albilineans before it acquired the ability to produce albicidin.
Examination of our genome-wide sequence data revealed that the Group 1 strains NCPPB1131, NCPPB1132 and NCPPB4393 lack the gene cluster required for albicidin biosynthesis, yet all three strains encode a gyrase A resembling that of X. albilineans, including the 43 amino acid insertion that is supposed to be responsible for albicidin resistance. These strains do have other non-ribosomal peptide synthesis genes, so we cannot exclude the possibility that the 43 amino acid insertion confers resistance to some other unknown toxin. Nevertheless, the presence of this insert clearly does not correlate with the incidence of genome reduction. Aside from any involvement in genome reduction, albicidin-like antibiotics probably have had profound effects on the evolution and ecology of xylem-limited parasites. For example, the genome sequence of L. xyli encodes a close homologue of AlbF that may allow it to colonise the host simultaneously with X. albilineans [22].

2.6. Novel Genes in Xanthomonas Group 1
Until the present study, X. albilineans was the only member of Group 1 for which genome-wide sequence data were available. However, X. albilineans is an atypical example of the group, since it has undergone significant genome reduction associated with adaptation to life within the xylem. Therefore, we examined the genome sequence data from NCPPB1131, NCPPB1132 and NCPPB4393 for genomic regions that are absent from X. albilineans GPE PC73. Comprehensive lists of these regions are provided in the Supplementary Files and include heavy metal resistance proteins, haemolysins, haemagglutinins, citrate synthase, persistence factor HipA, sigma54-dependent activators, type-IV pili, drug efflux pumps, vancomycin B-type resistance protein VanW, xylanase, chitinase, beta-lactamase, restriction modification systems and many others.
There are, of course, some differences among the genomes of the Group I Xanthomonas strains. The genomes of NCPPB1132 and NCPPB4393 are alignable with each other over 88.64% and 84.33% of their respective lengths. That is, 12–16% of their genomes are variable and presumably subject to relatively recent horizontal transfer. They share 96.85% nucleotide sequence identity over the alignable conserved core portion of their genomes. Similarly they share 94.47% and 94.45% identity with NCPPB1131.
The genomes of NCPPB1131, NCPPB1132 and NCPPB4393 each encode a protein with 64% amino acid sequence identity to AvrXca (also known as AvrXccA1), a protein that confers avirulence on Arabidopsis in X. campestris pv. raphani [23]. Based on this avirulence activity, it has been speculated that AvrXca might be an effector. However, all three strains lack a T3SS, so it seems unlikely that it is secreted via the T3SS. Interestingly, at least two homologues of AvrXca are effectors secreted by the type-II secretion system (T2SS) [24-26], suggesting that AvrXca might also be a T2SS-dependent effector.
Each of the three newly sequenced genomes encode two cellulose-degrading enzymes that are absent from X. albilineans. Specifically, these include an endoglucanase that shares 61% amino acid sequence identity with XCC0028 from X. campestris pv. campestris ATCC 33913 and a cellulase (glycosyl hydrolase family 5) that shares 68% identity with XCV0358 from X. campestris pv. vesicatoria 85-10. Both enzymes are widely distributed among the Group 2 Xanthomonas species as well as in the three newly sequenced Group 1 strains, but absent from X. albilineans. It is possible that they play a role in degrading plant cell walls.
The gum genes, found in Xylella fastidiosa and all studied Group 2 Xanthomonas species, play a role in producing extracellular polysaccharides and forming biofilms and are implicated in pathogenicity. However, no gum genes have been found in X. albilineans [8]. We found homologues of these (gumBGCEKDHIMJL) conserved in all three newly sequenced strains. Therefore, it appears that the gum cluster was probably present in the common ancestor of Xylella, Stenotrophomonas and Xanthomonas and was subsequently lost by X. albilineans and Stenotrophomonas but retained in X. sacchari and the other Group 1 Xanthomonas species.
3. Experimental Section
Bacterial strains were obtained from the National Collection of Plant Pathogenic Bacteria (NCPPB) at FERA. Sequence alignments were performed using MAFFT [27], BWA [28], BLAST [29] and MUMMER [30]. DNA preparation and genome sequencing using the Illumina GA2 were performed as previously described [11]. We used CGview [31] to visualize whole-genome alignments and used MEGA5 for phylogenetic analyses. Literature references for previously sequenced bacterial genomes used in this study are listed cited in [10,11]. De novo assembly of Illumina sequence reads was performed using Velvet 1.1.03 [32]. We discarded any sequence reads that contained one or more ‘N’ prior to assembly. We used the following values for the hash-length parameter: 25 for NCPPB1131, 41 for NCPPB1132 and 49 for NCPPB4493. The coverage cut-off parameter was set to 4 in all three assemblies. For NCPPB1132 and NCPPB4393 read-pairs, we used Velvet's scaffolding step. We did not perform scaffolding on the NCPPB1131 data as the reads were not paired (i.e., we used single-end sequencing). The RAST server [33] was used for automated annotation of draft assemblies.
Note that in the genome-wide alignments (Figure 3 and Figure 4), the pattern of coverage by aligned raw reads does not exactly coincide with the coverage by aligned contigs/scaffolds. This inconsistency is inevitable since the two alignment methods use different criteria for assigning a match. The BWA alignment tool tolerates mismatches so long as the edit distance does not exceed two between the raw read and the reference genome sequence. On the other hand, BLAST uses an E-value threshold (1e-10 in this case) as the criterion for whether to accept a match. Furthermore, the process of assembly leads to the correction of errors by consensus of multiple sequence reads.
4. Conclusions
The ability to survive on banana plants has evolved more than once within the genus Xanthomonas, with strains isolated from banana falling within both major phylogenetic lineages: Group 1 (NCPPB1131 and NCPPB1132) and Group 2 (X. campestris pv. musacearum). Clearly their strategies are different. Xanthomonas campestris pv. musacearum encodes an apparently intact Hrp T3SS and a suite of effectors that it presumably uses to overcome the host's defences. On the other hand, the Group 1 strains, related to X. sacchari and X. albilineans, lack the T3SS and must use some other strategy to avoid triggering host defences. In the case of X. albilineans, the strategy appears to be one of stealth, where the pathogen restricts its colonization to the dead xylem. As a result, it has undergone substantial genome reduction, shedding genes unnecessary for this restricted niche. Very little information is available about the endophytic lifestyles of X. sacchari and other related members of Xanthomonas Group 1 and there is no evidence that they are limited to the xylem. Certainly the lack of genome reduction would be consistent with having to survive in more diverse conditions. The example strain that we sequenced here, NCPPB4393, apparently spent at least part of its life cycle associated with insects. We hope that the availability of complete sequence data for this group of organisms is the first step towards understanding their interactions with plants and identifying potential virulence factors.
Acknowledgments
This study was supported by the National Agriculture Research Organisation, Uganda. The authors wish to thank Karen Moore and Alex Moorhouse for their invaluable technical assistance with sequencing. We thank Mgenzi Byabachwezi for isolation of strain NCPPB4393 and Neil Parkinson for some helpful comments on an earlier draft of the manuscript.
Supplementary Material
| File Name | File Type | Description |
|---|---|---|
| GyraseA_alignment.pdf | PDF (Adobe Acrobat) | Multiple sequence alignment of partial GyrA proteins from Xanthomonas albilineans and other gamma-Proteobacteria |
| phylogenetic_trees.pdf | PDF (Adobe Acrobat) | Phylogenetic analysis of fully sequenced members of the genera Xanthomonas, Xylella and Steonotrophomonas. Maximum likelihood trees are based on nucleotide sequences of the house-keeping genes dnaK, groEL and recA |
| NCPPB1131-sequences- not-in-X_albilineans.html | HTML (web browser) | A list of regions in the genome of NCPPB1131 that share no detectable nucleotide sequence similarity with X. albilineans |
| NCPPB1132-sequences-not-in-X_albilineans.html | HTML (web browser) | A list of regions in the genome of NCPPB1132 that share no detectable nucleotide sequence similarity with X. albilineans |
| NCPPB4393-sequences-not-in-X_albilineans.html | HTML (web browser) | A list of regions in the genome of NCPPB4393 that share no detectable nucleotide sequence similarity with X. albilineans |
| NCPPB1131-RAST.xls | Microsoft Excel spreadsheet | Automated annotation results for NCPPB1131 draft genome sequence generated by the RAST server |
| NCPPB1132-RAST.xls | Microsoft Excel spreadsheet | Automated annotation results for NCPPB1132 draft genome sequence generated by the RAST server |
| NCPPB4393-RAST.xls | Microsoft Excel spreadsheet | Automated annotation results for NCPPB4393 draft genome sequence generated by the RAST server |
| NCPPB1131_genes_comparison.pdf | PDF (Adobe Acrobat) | A list of predicted genes in NCPPB1131 that are absent from NCPPB1132 and/or NCPPB4393 as judged by BWA alignments of raw Illumina sequence reads against the NCPPB1131 genome. Genomic coordinates are given on GenBank accession numbers |
| NCPPB1132_genes_comparison.pdf | PDF (Adobe Acrobat) | A list of predicted genes in NCPPB1132 that are absent from NCPPB1131 and/or NCPPB4393 as judged by BWA alignments of raw Illumina sequence reads against the NCPPB1132 genome. Genomic coordinates are given on GenBank accession numbers |
| NCPPB4393_genes_comparison.pdf | PDF (Adobe Acrobat) | A list of predicted genes in NCPPB4393 that are absent from NCPPB1131 and/or NCPPB1132 as judged by BWA alignments of raw Illumina sequence reads against the NCPPB4393 genome. Genomic coordinates are given on GenBank accession numbers |
| atpD_aligned_fasta.txt | Gapped FastA / plain text | Multiple sequence alignment of atpD used to generate the phylogenetic tree in Figure 1 in the main text |
| gyrB_aligned_fasta.txt | Gapped FastA / plain text | Multiple sequence alignment of gyrB used to generate the phylogenetic tree in Figure 1 in the main text |
References
- Hauben, L.; Vauterin, L.; Swings, J.; Moore, E.R. Comparison of 16S ribosomal DNA sequences of all Xanthomonas species. Int. J. Syst. Bacteriol. 1997, 47, 328–335. [Google Scholar]
- Gonçalves, E.R.; Rosato, Y.B. Phylogenetic analysis of Xanthomonas species based upon 16S-23S rDNA intergenic spacer sequences. Int. J. Syst. Evol. Microbiol. 2002, 52, 355–361. [Google Scholar]
- Rademaker, J.L.; Louws, F.J.; Schultz, M.H.; Rossbach, U.; Vauterin, L.; Swings, J.; de Bruijn, F.J. A comprehensive species to strain taxonomic framework for Xanthomonas. Phytopathology 2005, 95, 1098–1111. [Google Scholar]
- Parkinson, N.; Aritua, V.; Heeney, J.; Cowie, C.; Bew, J.; Stead, D. Phylogenetic analysis of Xanthomonas species by comparison of partial gyrase B gene sequences. Int. J. Syst. Evol. Microbiol. 2007, 57, 2881–2887. [Google Scholar]
- Young, J.M.; Park, D.C.; Shearman, H.M.; Fargier, E. A multilocus sequence analysis of the genus Xanthomonas. Syst. Appl. Microbiol. 2008, 31, 366–377. [Google Scholar]
- Parkinson, N.; Cowie, C.; Heeney, J.; Stead, D. Phylogenetic structure of Xanthomonas determined by comparison of gyrB sequences. Int. J. Syst. Evol. Microbiol. 2009, 59, 264–274. [Google Scholar]
- Ryan, R.P.; Vorhölter, F.J.; Potnis, N.; Jones, J.B.; van Sluys, M.A.; Bogdanove, A.J.; Dow, J.M. Pathogenomics of Xanthomonas: Understanding bacterium-plant interactions. Nat. Rev. Microbiol. 2011, 9, 344–355. [Google Scholar]
- Pieretti, I.; Royer, M.; Barbe, V.; Carrere, S.; Koebnik, R.; Cociancich, S.; Couloux, A.; Darrasse, A.; Gouzy, J.; Jacques, M.A.; et al. The complete genome sequence of Xanthomonas albilineans provides new insights into the reductive genome evolution of the xylem-limited Xanthomonadaceae. BMC Genomics 2009, 10, 616. [Google Scholar]
- Marguerettaz, M.; Pieretti, I.; Gayral, P.; Puig, J.; Brin, C.; Cociancich, S.; Poussier, S.; Rott, P.; Royer, M. Genomic and evolutionary features of the SPI-1 type III secretion system that is present in Xanthomonas albilineans but is not essential for xylem colonization and symptom development of sugarcane leaf scald. Mol. Plant Microbe Interact. 2011, 24, 246–259. [Google Scholar]
- Rott, P.; Fleites, L.; Marlow, G.; Royer, M.; Gabriel, D.W. Identification of new candidate pathogenicity factors in the xylem-invading pathogen Xanthomonas albilineans by transposon mutagenesis. Mol. Plant Microbe Interact. 2011, 24, 594–605. [Google Scholar]
- Studholme, D.J.; Kemen, E.; MacLean, D.; Schornack, S.; Aritua, V.; Thwaites, R.; Grant, M.; Smith, J.; Jones, J.D. Genome-wide sequencing data reveals virulence factors implicated in banana Xanthomonas wilt. FEMS Microbiol. Lett. 2010, 310, 182–192. [Google Scholar]
- Byachwezi, M. Belgian Development Agency, Dar Es Salaam, Tanzania. Personal communication, 2011.
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar]
- Lu, H.; Patil, P.; van Sluys, M.A.; White, F.F.; Ryan, R.P.; Dow, J.M.; Rabinowicz, P.; Salzberg, S.L.; Leach, J.E.; Sonti, R.; et al. Acquisition and evolution of plant pathogenesis-associated gene clusters and candidate determinants of tissue-specificity in Xanthomonas. PLoS ONE 2008, 3, e3828. [Google Scholar]
- Sharma, V.; Patil, P.B. Resolving the phylogenetic and taxonomic relationship of Xanthomonas and Stenotrophomonas strains using complete rpoB gene sequence. PLoS Curr. 2011, 3, RRN1239. [Google Scholar]
- Vauterin, L.; Hoste, B.; Kersters, K.; Swings, J. Reclassification of Xanthomonas. Int. J. Syst. Bacteriol. 1995, 45, 472–489. [Google Scholar]
- Gartemann, K.H.; Abt, B.; Bekel, T.; Burger, A.; Engemann, J.; Flügel, M.; Gaigalat, L.; Goesmann, A.; Gräfen, I.; Kalinowski, J.; et al. The genome sequence of the tomato-pathogenic actinomycete Clavibacter michiganensis subsp. michiganensis NCPPB382 reveals a large island involved in pathogenicity. J. Bacteriol. 2008, 190, 2138–2149. [Google Scholar]
- Purcell, A.H.; Hopkins, D.L. Fastidious xylem-limited bacterial plant pathogens. Annu. Rev. Phytopathol. 1996, 34, 131–151. [Google Scholar]
- Birch, R.G.; Patil, S.S. Preliminary characterization of an antibiotic produced by Xanthomonas albilineans which inhibits DNA synthesis in Escherichia coli. J. Gen. Microbiol. 1985, 131, 1069–1075. [Google Scholar]
- Hashimi, S.M.; Huang, G.; Maxwell, A.; Birch, R.G. DNA gyrase from the albicidin producer Xanthomonas albilineans has multiple-antibiotic-resistance and unusual enzymatic properties. Antimicrob. Agents Chemother. 2008, 52, 1382–1390. [Google Scholar]
- Monteiro-Vitorello, C.B.; Camargo, L.E.; van Sluys, M.A.; Kitajima, J.P.; Truffi, D.; do Amaral, A.M.; Harakava, R.; de Oliveira, J.C.; Wood, D.; de Oliveira, M.C.; et al. The genome sequence of the gram-positive sugarcane pathogen Leifsonia xyli subsp. xyli. Mol. Plant Microbe Interact. 2004, 17, 827–836. [Google Scholar]
- Parker, J.E.; Barber, C.E.; Fan, M.J.; Daniels, M.J. Interaction of Xanthomonas campestris with Arabidopsis thaliana: Characterization of a gene from X. c. pv. raphani that confers avirulence to most A. thaliana accessions. Mol. Plant Microbe Interact. 1993, 6, 216–224. [Google Scholar]
- Corbett, M.; Virtue, S.; Bell, K.; Birch, P.; Burr, T.; Hyman, L.; Lilley, K.; Poock, S.; Toth, I.; Salmond, G. Identification of a new quorum-sensing-controlled virulence factor in Erwinia carotovora subsp. atroseptica secreted via the type II targeting pathway. Mol. Plant Microbe Interact. 2005, 18, 334–342. [Google Scholar]
- Kazemi-Pour, N.; Condemine, G.; Hugouvieux-Cotte-Pattat, N. The secretome of the plant pathogenic bacterium Erwinia chrysanthemi. Proteomics 2004, 4, 3177–3186. [Google Scholar]
- Jha, G.; Rajeshwari, R.; Sonti, R.V. Bacterial type two secretion system secreted proteins: Double-edged swords for plant pathogens. Mol. Plant Microbe Interact. 2005, 18, 891–898. [Google Scholar]
- Katoh, K.; Asimenos, G.; Toh, H. Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 2009, 537, 39–64. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
- Delcher, A.L.; Phillippy, A.; Carlton, J.; Salzberg, S.L. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002, 30, 2478–2483. [Google Scholar]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genomics 2008, 9, 75. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Studholme, D.J.; Wasukira, A.; Paszkiewicz, K.; Aritua, V.; Thwaites, R.; Smith, J.; Grant, M. Draft Genome Sequences of Xanthomonas sacchari and Two Banana-Associated Xanthomonads Reveal Insights into the Xanthomonas Group 1 Clade. Genes 2011, 2, 1050-1065. https://doi.org/10.3390/genes2041050
Studholme DJ, Wasukira A, Paszkiewicz K, Aritua V, Thwaites R, Smith J, Grant M. Draft Genome Sequences of Xanthomonas sacchari and Two Banana-Associated Xanthomonads Reveal Insights into the Xanthomonas Group 1 Clade. Genes. 2011; 2(4):1050-1065. https://doi.org/10.3390/genes2041050
Chicago/Turabian StyleStudholme, David J., Arthur Wasukira, Konrad Paszkiewicz, Valente Aritua, Richard Thwaites, Julian Smith, and Murray Grant. 2011. "Draft Genome Sequences of Xanthomonas sacchari and Two Banana-Associated Xanthomonads Reveal Insights into the Xanthomonas Group 1 Clade" Genes 2, no. 4: 1050-1065. https://doi.org/10.3390/genes2041050