From Environment to Man: Genome Evolution and Adaptation of Human Opportunistic Bacterial Pathogens

Abstract
:1. Introduction
2. Environment is a ‘Nursery’ for Emerging OBPs

{kind=link}
{kind=link}
{kind=link}
| Environmental OBP | Habitat/ natural host | Lifestyle | Relationships with cells | Pathogenicity for humans |
|---|---|---|---|---|
| Acetic Acid Bacteria | Food | Food processing | Extracellular | HAI |
| Fruits, flowers | Free living | Diverse mild infections in ID and CF | ||
| Midgut, salivary glands of flying insects | Symbiotic | Bacteremia | ||
| Chronic granulomatous disease | ||||
| Aeromonas spp. | Freshwater, chlorinated water | Free living | Extracellular | Diarrhea |
| Polluted soils | Pathogen for fish, amphibian and mollusk | Wound infections | ||
| Nematodes | Bacteremia | |||
| Mosquitoes | ||||
| Leeches, mollusks | ||||
| Fish, Amphibians, Crustacean | ||||
| Agrobacterium radiobacter / Agrobacterium tumefaciens | Rhizosphere | Free living | Extracellular | HAI |
| Plants | Phytopathogenic | Plant transformation | Diverse mild infections in ID and CF | |
| Bacteremia | ||||
| Burkholderia cepacia complex | Soil | Free living | Extracellular | Infections in CF (ET-12 epidemic clone) |
| Rhizosphere | Plant-growth promoting | Facultative intracellular (plant, macrophage) | Chronic granulomatous disease | |
| Plant | Phytopathogenic | |||
| Amoeba | ||||
| - Acanthamoeba | ||||
| Chromobacterium violaceum | Water and soil | Free living | Extracellular | Serious or fatal infections in ID and children |
| in tropical and subtropical ecosystems | ||||
| Legionella pneumophila | Fresh water, chlorinated water | Free living | Facultative | Serious or fatal Pneumonia (Legionellosis) |
| Amoeba | Amoeba-associated | Intracellular | ||
| - Acanthamoeba | ||||
| - Naegleria | ||||
| - Echinamoeba | ||||
| - Hartmannella… | ||||
| Ciliata: | ||||
| - Tetrahymena… | ||||
| Ochrobactrum | Soil and polluted soils | Free living | Extracellular | HAI |
| Rhizosphere | Dixeny with nematodes | Diverse mild infections in ID and CF | ||
| Plants | Plant-growth promoter | Bacteremia | ||
| Insects | Nodule formation in plant | |||
| Nematodes | ||||
| Pseudomonas aeruginosa | Fresh and Sea water | Free living | Extracellular | HAI |
| Chlorinated water | Amoeba-associated | Facultative intracellular in amoeba | Wound infections | |
| Water distribution systems (hospital, domestic) | Burn infections | |||
| Pharmaceutical water and antiseptics | ||||
| Wastewater | ||||
| Terrestrial wet ecosystems | ||||
| Polluted soils | ||||
| Rhizosphere | ||||
| Amoeba | ||||
| - Acanthamoeba | ||||
| Photorhabdus luminescens | Heterorhabditis indica | Symbiosis | Extracellular | No |
| Photorhabdus asymbiotica | Unknown | Non symbiotic | Facultative intracellular in macrophage | Serious soft tissue infections |
| Bacteremia | ||||
| Serratia marcescens | Plant (phloem) | Free living | Extracellular | HAI |
| Insect | Phytopathogen | Ocular infections | ||
| Squash bug (Anisa tristis) | ||||
| Stenotrophomonas maltophilia | Natural water | Free living | Extracellular | HAI |
| Water distribution systems (hospital, domestic) | Plant-growth promoter | Infection in CF | ||
| Pharmaceutical water and antiseptics | ||||
| Wastewater | ||||
| Rhizosphere | ||||
| Deep-sea invertebrates | ||||
| Food | ||||
| Reptiles, mammals |
2.1. Rhizosphere
2.2. Protozoa
2.3. Insects
2.4. Worms
3. From Environment to Man: Lessons from OBP Genomics
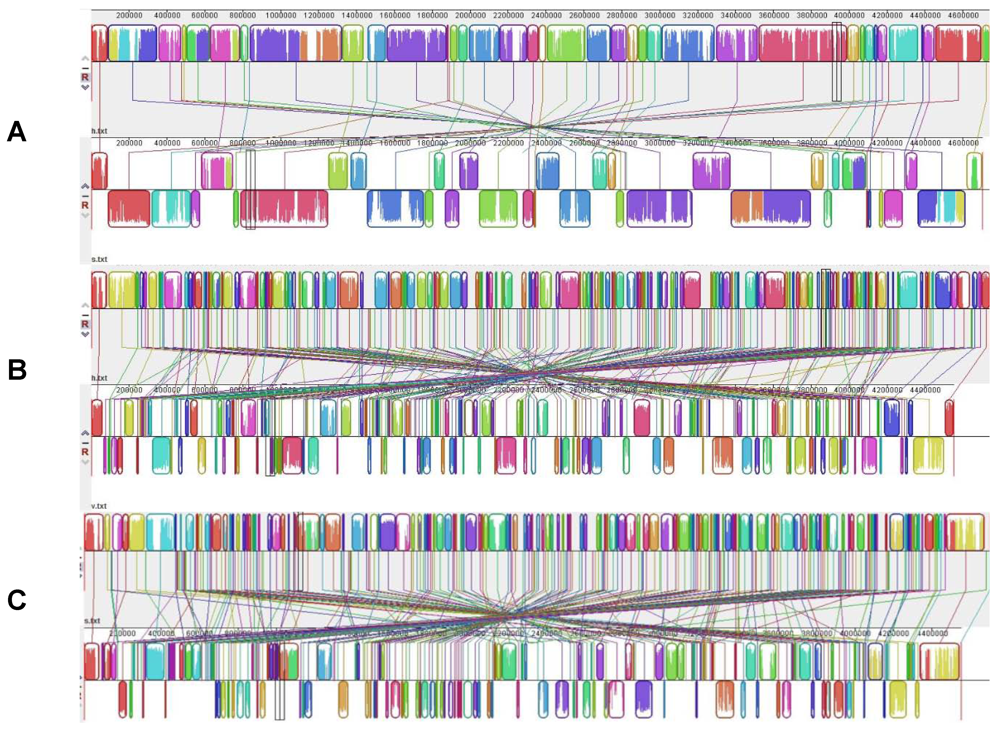
3.1. A Large and Fluid Genome Is the Key to Bacterial Versatility
3.2. Horizontal Genetic Transfers Give Evolution a Boost
3.3. Genome Reduction: No Turning Back
4. Pseudomonas aeruginosa, the Swiss Army Knife
| Strain | PAO1 | UCBPP PA14 | PA7 | LESB58 | PACS2 | PA2192 | C3719 | 39016 | PAb1 | M18 |
|---|---|---|---|---|---|---|---|---|---|---|
| Accession Number | NC_002516 | NC_008463 | NC_009656 | NC_011770 | NZ_AAQW | NZ_AAKW | NZ_AAKV | NZ_AEEX | NZ_ABKZ | CP002496 |
| Source | Wound | Clinical | Clinical | CF-patient | Clinical | CF-patient | CF-patient | Keratitis | Frost bite | Rhizophere |
| Genome size (Mbp) | 6.264 | 6.538 | 6.588 | 6.602 | 6.492 | 6.905 | 6.222 | 6.667 | 6.078 | 6,327 |
| GC-content (%) | 66.6 | 66.3 | 66.5 | 66.5 | 66 | 66.2 | 66.5 | 66 | 66 | 66.5 |
| No. of ORFs | 5570 | 5892 | 6286 | 5925 | 5676 | 6191 | 5578 | 6401 | 5943 | 5684 |
| % of coding sequences | 88.9 | 90.1 | 95.4 | 89.7 | 87.4 | 89.6 | 89.6 | 96 | 89 | 89 |
| Pseudogenes | 5 | 0 | 8 | 34 | 0 | 2 | 3 | 9 | 0 | 6 |
4.1. A Mosaic of Core and Accessory Genes
4.2. The Eclectic Specialist
5. Aeromonas hydrophila, Jack-of-all-trades, and Its Relatives
5.1. Water and Other ‘Nurseries’
5.2. What Does Aeromonas hydrophila ATCC 7966T Genome Teach Us?
5.3. Aeromonas salmonicida subsp salmonicida: Evolution towards Specialization

5.4. Environment as a Training Ground: The Model of A. veronii with Leeches
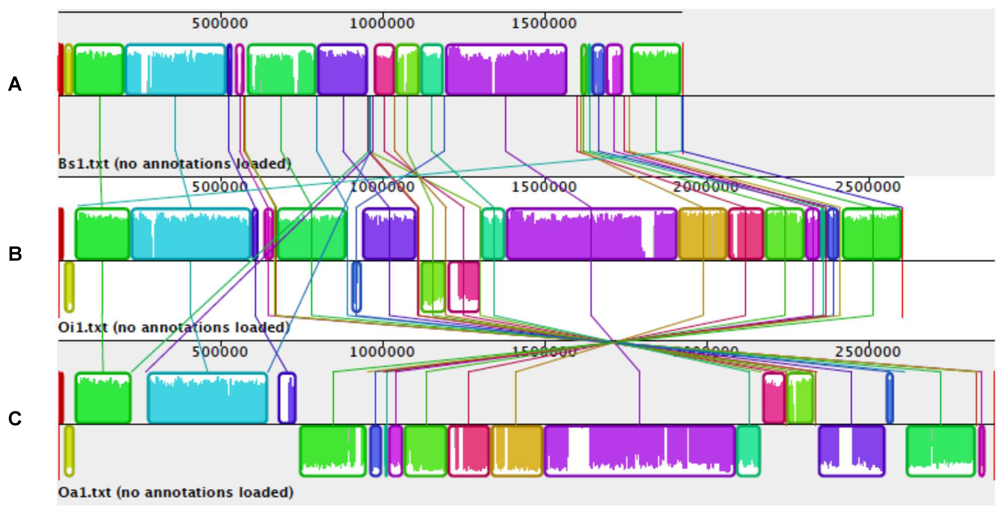
6. Is Brucella an Ochrobactrum with Reduced Genome?
6.1. Genomics of Brucellaceae
6.2. Real-Time Genomic Reduction in an Ochrobacterium intermedium Clone
7. Conclusive Remarks


| Species Strain RefSeq or INSDC | B. ovis ATCC 25840T NC_009505–009504 | B. suis 1330 CP002997–002998 | B. melitensis 16M NZ_ACJL | O. anthropi ATCC 49188T NC_009667–09668 | O. intermedium * LMG 3301T NZ_ACQA | |||||
| Chr I | Chr II | Chr I | Chr II | Chr I | Chr II | Chr I | Chr II | Chr I | Chr II | |
| Size (bp) | 2,111,370 | 1,164,20 | 2,107,792 | 1,207,381 | 2,117,144 | 1,177,787 | 2,887,297 | 1,895,911 | NA | NA |
| GC content (%) | 57.2 | 57.2 | 57.2 | 57.3 | 56 | 56 | 57.2 | 57.3 | 57 | 57 |
| Protein coding | 2,890 | 3,272 | 3,165 | 4,424 | 4,363 | |||||
| rRNA operons | 2 | 1 | 2 | 1 | 2 | 2 | 2 | 1 | 2 | 2 |
| N° of tRNAs | 53 | 55 | 54 | 73 | NA | |||||
| N° pseudogenes | 244 | 62 | 172 | 31 | 77 | |||||
| % pseudogenes | 7.8 | 1.8 | 5.1 | 0.7 | 1.7 | |||||
References
- Curtis, L.T. Prevention of hospital-acquired infections: Review of non-pharmacological interventions. J. Hosp. Infect. 2008, 69, 204–219. [Google Scholar]
- Guss, A.M.; Roeselers, G.; Newton, I.L.; Young, C.R.; Klepac-Ceraj, V.; Lory, S.; Cavanaugh, C.M. Phylogenetic and metabolic diversity of bacteria associated with cystic fibrosis. ISME J. 2011, 5, 20–29. [Google Scholar]
- Klevens, R.M.; Edwards, J.R.; Richards, C.L., Jr.; Horan, T.C.; Gaynes, R.P.; Pollock, D.A.; Cardo, D.M. Estimating health care-associated infections and deaths in U.S. hospitals, 2002. Public Health Rep. 2007, 122, 160–166. [Google Scholar]
- Lynch, P.; Pittet, D.; Borg, M.A.; Mehtar, S. Infection control in countries with limited resources. J. Hosp. Infect. 2007, 65, S148–S150. [Google Scholar]
- Bleves, S.; Viarre, V.; Salacha, R.; Michel, G.P.; Filloux, A.; Voulhoux, R. Protein secretion systems in Pseudomonas aeruginosa: A wealth of pathogenic weapons. Int. J. Med. Microbiol. 2010, 300, 534–543. [Google Scholar] [CrossRef]
- Bengis, R.G.; Leighton, F.A.; Fischer, J.R.; Artois, M.; Morner, T.; Tate, C.M. The role of wildlife in emerging and re-emerging zoonoses. Rev. Sci. Tech. 2004, 23, 497–511. [Google Scholar]
- Cutler, S.J.; Fooks, A.R.; van der Poel, W.H. Public health threat of new, reemerging, and neglected zoonoses in the industrialized world. Emerg Infect. Dis. 2010, 16, 1–7. [Google Scholar]
- Pearce-Duvet, J.M. The origin of human pathogens: Evaluating the role of agriculture and domestic animals in the evolution of human disease. Biol. Rev. Camb Philos. Soc. 2006, 81, 369–382. [Google Scholar]
- Mitchell, J. Streptococcus mitis: Walking the line between commensalism and pathogenesis. Mol. Oral Microbiol. 2011, 26, 89–98. [Google Scholar] [CrossRef]
- Theron, J.; Cloete, T.E. Emerging waterborne infections: Contributing factors, agents, and detection tools. Crit. Rev. Microbiol. 2002, 28, 1–26. [Google Scholar] [CrossRef]
- Sobsey, M.D.; Pillai, S.D. Where future emerging pathogens will come from and what approaches can be used to find them, besides VFARs. J. Water Health 2009, 7, S75–S93. [Google Scholar]
- Jackson, R.W.; Johnson, L.J.; Clarke, S.R.; Arnold, D.L. Bacterial pathogen evolution: Breaking news. Trends Genet. 2011, 27, 32–40. [Google Scholar]
- Moliner, C.; Raoult, D.; Fournier, P.E. Evidence of horizontal gene transfer between amoeba and bacteria. Clin. Microbiol. Infect. 2009, 15, S178–S180. [Google Scholar]
- Waterfield, N.R.; Wren, B.W.; Ffrench-Constant, R.H. Invertebrates as a source of emerging human pathogens. Nat. Rev. Microbiol. 2004, 2, 833–841. [Google Scholar]
- Greub, G.; Raoult, D. Microorganisms resistant to free-living amoebae. Clin. Microbiol. Rev. 2004, 17, 413–433. [Google Scholar]
- Scully, L.R.; Bidochka, M.J. Developing insect models for the study of current and emerging human pathogens. FEMS Microbiol. Lett. 2006, 263, 1–9. [Google Scholar]
- Berg, G.; Eberl, L.; Hartmann, A. The rhizosphere as a reservoir for opportunistic human pathogenic bacteria. Environ. Microbiol. 2005, 7, 1673–1685. [Google Scholar]
- Aujoulat, F.; Jumas-Bilak, E.; Masnou, A.; Salle, F.; Faure, D.; Segonds, C.; Marchandin, H.; Teyssier, C. Multilocus sequence-based analysis delineates a clonal population of Agrobacterium (Rhizobium) radiobacter (Agrobacterium tumefaciens) of human origin. J. Bacteriol. 2011, 193, 2608–2618. [Google Scholar]
- Vial, L.; Chapalain, A.; Groleau, M.C; Déziel, E. The various lifestyles of the Burkholderia cepacia complex species: A tribute to adaptation. Env. Microbiol. 2011, 13, 1–12. [Google Scholar] [CrossRef]
- Rezzonico, F.; Smits, T.H.; Montesinos, E.; Frey, J.E.; Duffy, B. Genotypic comparison of Pantoea agglomerans plant and clinical strains. BMC Microbiol. 2009, 9, 204. [Google Scholar] [CrossRef]
- Ziga, E.D.; Druley, T.; Burnham, C.A. Herbaspirillum species bacteremiabacteremia in a pediatric oncology patient. J. Clin. Microbiol. 2010, 48, 4320–4321. [Google Scholar] [CrossRef]
- Spilker, T.; Uluer, A.Z.; Marty, F.M.; Yeh, W.W.; Levison, J.H.; Vandamme, P.; Lipuma, J.J. Recovery of Herbaspirillum species from persons with cystic fibrosis. J. Clin. Microbiol. 2008, 46, 2774–2777. [Google Scholar]
- Pedrosa, F.O.; Monteiro, R.A.; Wassem, R.; Cruz, L.M.; Ayub, R.A.; Colauto, N.B.; Fernandez, M.A.; Fungaro, M.H.; Grisard, E.C.; Hungria, M.; et al. Genome of Herbaspirillum seropedicae strain SmR1, a specialized diazotrophic endophyte of tropical grasses. PLoS Genet. 2011, 7, e1002064. [Google Scholar]
- Romano, S.; Aujoulat, F.; Jumas-Bilak, E.; Masnou, A.; Jeannot, J.L.; Falsen, E.; Marchandin, H.; Teyssier, C. Multilocus sequence typing supports the hypothesis that Ochrobactrum anthropi displays a human-associated subpopulation. BMC Microbiol. 2009, 9, 267. [Google Scholar] [CrossRef]
- Wu, D.Q.; Ye, J.; Ou, H.Y.; Wei, X.; Huang, X.; He, Y.W.; Xu, Y. Genomic analysis and temperature-dependent transcriptome profiles of the rhizosphere originating strain Pseudomonas aeruginosa M18. BMC Genomics 2011, 12, 438. [Google Scholar]
- Brooke, J.S. Stenotrophomonas maltophilia: An emerging global opportunistic pathogen. Clin. Microbiol. Rev. 2012, 25, 2–41. [Google Scholar] [CrossRef]
- Ramos, J.L.; Gonzalez-Perez, M.M.; Caballero, A.; van Dillewijn, P. Bioremediation of polynitrated aromatic compounds: Plants and microbes put up a fight. Curr. Opin. Biotechnol. 2005, 16, 275–281. [Google Scholar]
- Barabote, R.D.; Thekkiniath, J.; Strauss, R.E.; Vediyappan, G.; Fralick, J.A.; San Francisco, M.J. Xenobiotic efflux in bacteria and fungi: A genomics update. Adv. Enzymol. Relat. Areas Mol. Biol. 2011, 77, 237–306. [Google Scholar]
- Brito, C.F.; Carvalho, C.B.; Santos, F.; Gazzinelli, R.T.; Oliveira, S.C.; Azevedo, V.; Teixeira, S.M. Chromobacterium violaceum genome: Molecular mechanisms associated with pathogenicity. Genet. Mol. Res. 2004, 3, 148–161. [Google Scholar]
- Allocati, N.; Federici, L.; Masulli, M.; Di Ilio, C. Glutathione transferases in bacteria. FEBS J. 2009, 276, 58–75. [Google Scholar]
- Nishino, K.; Yamaguchi, A. Role of xenobiotic transporters in bacterial drug resistance and virulence. IUBMB Life 2008, 60, 569–574. [Google Scholar]
- Piddock, L.J. Multidrug-resistance efflux pumps - not just for resistance. Nat. Rev. Microbiol. 2006, 4, 629–636. [Google Scholar]
- Nishino, K.; Latifi, T.; Groisman, E.A. Virulence and drug resistance roles of multidrug efflux systems of Salmonella enterica serovar Typhimurium. Mol. Microbiol. 2006, 59, 126–141. [Google Scholar] [CrossRef]
- Rowbotham, T.J. Preliminary report on the pathogenicity of Legionella pneumophila for freshwater and soil amoebae. J. Clin. Pathol. 1980, 33, 1179–1183. [Google Scholar]
- Barker, J.; Brown, M.R. Trojan horses of the microbial world: Protozoa and the survival of bacterial pathogens in the environment. Microbiology 1994, 140, 1253–1259. [Google Scholar]
- Singer, M. Pathogen-pathogen interaction: A syndemic model of complex biosocial processes in disease. Virulence 2010, 1, 10–18. [Google Scholar]
- Schmitz-Esser, S.; Tischler, P.; Arnold, R.; Montanaro, J.; Wagner, M.; Rattei, T.; Horn, M. The genome of the amoeba symbiont “Candidatus Amoebophilus asiaticus” reveals common mechanisms for host cell interaction among amoeba-associated bacteria. J. Bacteriol. 2010, 192, 1045–1057. [Google Scholar] [CrossRef]
- Cirillo, J.D.; Falkow, S.; Tompkins, L.S.; Bermudez, L.E. Interaction of Mycobacterium avium with environmental amoebae enhances virulence. Infect. Immun. 1997, 65, 3759–3767. [Google Scholar]
- Laskowski-Arce, M.A.; Orth, K. Acanthamoeba castellanii promotes the survival of Vibrio parahaemolyticus. Appl. Environ. Microbiol. 2008, 74, 7183–7188. [Google Scholar] [CrossRef]
- Yamada, Y.; Yukphan, P. Genera and species in acetic acid bacteria. Int. J. Food Microbiol. 2008, 125, 15–24. [Google Scholar]
- Crotti, E.; Rizzi, A.; Chouaia, B.; Ricci, I.; Favia, G.; Alma, A.; Sacchi, L.; Bourtzis, K.; Mandrioli, M.; Cherif, A.; et al. Acetic acid bacteria, newly emerging symbionts of insects. Appl. Environ. Microbiol. 2010, 76, 6963–6970. [Google Scholar]
- Snyder, R.W.; Ruhe, J.; Kobrin, S.; Wasserstein, A.; Doline, C.; Nachamkin, I.; Lipschutz, J.H. Asaia bogorensis peritonitis identified by 16S ribosomal RNA sequence analysis in a patient receiving peritoneal dialysis. Am. J. Kidney Dis. 2004, 44, e15–e17. [Google Scholar] [CrossRef]
- Alauzet, C.; Teyssier, C.; Jumas-Bilak, E.; Gouby, A.; Chiron, R.; Rabaud, C.; Counil, F.; Lozniewski, A.; Marchandin, H. Gluconobacter as well as Asaia species, newly emerging opportunistic human pathogens among acetic acid bacteria. J. Clin. Microbiol. 2010, 48, 3935–3942. [Google Scholar]
- Nadarasah, G.; Stavrinides, J. Insects as alternative hosts for phytopathogenic bacteria. FEMS Microbiol. Rev. 2011, 35, 555–575. [Google Scholar]
- Avila, F.J.; Bruton, B.D.; Fletcher, J.; Sherwood, J.L.; Pair, S.D.; Melcher, U. Polymerase chain reaction detection and phylogenetic characterization of an agent associated with yellow vine disease of cucurbits. Phytopathology 1998, 88, 428–436. [Google Scholar]
- Dessi, A.; Puddu, M.; Testa, M.; Marcialis, M.A.; Pintus, M.C.; Fanos, V. Serratia marcescens infections and outbreaks in neonatal intensive care units. J. Chemother 2009, 21, 493–499. [Google Scholar]
- Levy, B. Infectious keratitis: What have we learned? Eye Contact Lens 2007, 33, 418–420, disussion 424-415.. [Google Scholar] [CrossRef]
- Labbate, M.; Zhu, H.; Thung, L.; Bandara, R.; Larsen, M.R.; Willcox, M.D.; Givskov, M.; Rice, S.A.; Kjelleberg, S. Quorum-sensing regulation of adhesion in Serratia marcescens MG1 is surface dependent. J. Bacteriol. 2007, 189, 2702–2711. [Google Scholar]
- Mellies, J.L.; Lawrence-Pine, E.R. Interkingdom signaling between pathogenic bacteria and Caenorhabditis elegans. Trends Microbiol. 2010, 18, 448–454. [Google Scholar] [CrossRef]
- Abby, S.; Daubin, V. Comparative genomics and the evolution of prokaryotes. Trends Microbiol. 2007, 15, 135–141. [Google Scholar]
- Shapiro, J.A. Letting Escherichia coli teach me about genome engineering. Genetics 2009, 183, 1205–1214. [Google Scholar] [CrossRef]
- Dobrindt, U.; Hacker, J. Whole genome plasticity in pathogenic bacteria. Curr. Opin. Microbiol. 2001, 4, 550–557. [Google Scholar]
- Konstantinidis, K.T.; Tiedje, J.M. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc. Natl. Acad. Sci. USA 2004, 101, 3160–3165. [Google Scholar]
- Georgiades, K.; Raoult, D. Defining pathogenic bacterial species in the genomic era. Front. Microbiol. 2010, 1, 151. [Google Scholar]
- Klappenbach, J.A.; Dunbar, J.M.; Schmidt, T.M. rRNA operon copy number reflects ecological strategies of bacteria. Appl. Environ. Microbiol. 2000, 66, 1328–1333. [Google Scholar]
- Brazilian National Genome Project Consortium. The complete genome sequence of Chromobacterium violaceum reveals remarkable and exploitable bacterial adaptability. Proc. Natl. Acad. Sci. USA 2003 2003, 100, 11660–11665. [CrossRef]
- Teyssier, C.; Marchandin, H.; Masnou, A.; Jeannot, J.L.; de Buochberg, M.S.; Jumas-Bilak, E. Pulsed-field gel electrophoresis to study the diversity of whole-genome organization in the genus Ochrobactrum. Electrophoresis 2005, 26, 2898–2907. [Google Scholar] [CrossRef]
- Ogier, J.C.; Calteau, A.; Forst, S.; Goodrich-Blair, H.; Roche, D.; Rouy, Z.; Suen, G.; Zumbihl, R.; Givaudan, A.; Tailliez, P.; et al. Units of plasticity in bacterial genomes: New insight from the comparative genomics of two bacteria interacting with invertebrates, Photorhabdus and Xenorhabdus. BMC Genomics 2010, 11, 568. [Google Scholar]
- Morales, G.; Wiehlmann, L.; Gudowius, P.; van Delden, C.; Tummler, B.; Martinez, J.L.; Rojo, F. Structure of Pseudomonas aeruginosa populations analysed by single nucleotide polymorphism and pulsed-field gel electrophoresis genotyping. J. Bacteriol. 2004, 186, 4228–4237. [Google Scholar]
- Johnson, J.K.; Arduino, S.M.; Stine, O.C.; Johnson, J.A.; Harris, A.D. Multilocus sequence typing compared to pulsed-field gel electrophoresis for molecular typing of Pseudomonas aeruginosa. J. Clin. Microbiol. 2007, 45, 3707–3712. [Google Scholar]
- Boussau, B.; Karlberg, E.O.; Frank, A.C.; Legault, B.A.; Andersson, S.G. Computational inference of scenarios for alpha-proteobacterial genome evolution. Proc. Natl. Acad. Sci. USA 2004, 101, 9722–9727. [Google Scholar]
- Tamas, I.; Klasson, L.; Canback, B.; Naslund, A.K.; Eriksson, A.S.; Wernegreen, J.J.; Sandstrom, J.P.; Moran, N.A.; Andersson, S.G. 50 million years of genomic stasis in endosymbiotic bacteria. Science 2002, 296, 2376–2379. [Google Scholar]
- Wilkinson, P.; Waterfield, N.R.; Crossman, L.; Corton, C.; Sanchez-Contreras, M.; Vlisidou, I.; Barron, A.; Bignell, A.; Clark, L.; Ormond, D.; et al. Comparative genomics of the emerging human pathogen Photorhabdus asymbiotica with the insect pathogen Photorhabdus luminescens. BMC Genomics 2009, 10, 302. [Google Scholar]
- Gaudriault, S.; Pages, S.; Lanois, A.; Laroui, C.; Teyssier, C.; Jumas-Bilak, E.; Givaudan, A. Plastic architecture of bacterial genome revealed by comparative genomics of Photorhabdus variants. Genome Biol. 2008, 9, R117. [Google Scholar] [CrossRef]
- Matz, C.; Moreno, A.M.; Alhede, M.; Manefield, M.; Hauser, A.R.; Givskov, M.; Kjelleberg, S. Pseudomonas aeruginosa uses type III secretion system to kill biofilm-associated amoebae. ISME J. 2008, 2, 843–852. [Google Scholar] [CrossRef]
- Tounsi, S.; Blight, M.; Jaoua, S.; de Lima Pimenta, A. From insects to human hosts: Identification of major genomic differences between entomopathogenic strains of Photorhabdus and the emerging human pathogen Photorhabdus asymbiotica. Int. J. Med. Microbiol. 2006, 296, 521–530. [Google Scholar] [CrossRef]
- Heermann, R.; Fuchs, T.M. Comparative analysis of the Photorhabdus luminescens and the Yersinia enterocolitica genomes: Uncovering candidate genes involved in insect pathogenicity. BMC Genomics 2008, 9, 40. [Google Scholar] [CrossRef]
- Holden, M.T.; Seth-Smith, H.M.; Crossman, L.C.; Sebaihia, M.; Bentley, S.D.; Cerdeno-Tarraga, A.M.; Thomson, N.R.; Bason, N.; Quail, M.A.; Sharp, S.; et al. The genome of Burkholderia cenocepacia J2315, an epidemic pathogen of cystic fibrosis patients. J. Bacteriol. 2009, 191, 261–277. [Google Scholar]
- Mil-Homens, D.; Rocha, E.P.; Fialho, A.M. Genome-wide analysis of DNA repeats in Burkholderia cenocepacia J2315 identifies a novel adhesin-like gene unique to epidemic-associated strains of the ET-12 lineage. Microbiology 2010, 156, 1084–1096. [Google Scholar] [CrossRef]
- Moran, N.A.; Wernegreen, J.J. Lifestyle evolution in symbiotic bacteria: Insights from genomics. Trends Ecol. Evol. 2000, 15, 321–326. [Google Scholar]
- Ogata, H.; Audic, S.; Renesto-Audiffren, P.; Fournier, P.E.; Barbe, V.; Samson, D.; Roux, V.; Cossart, P.; Weissenbach, J.; Claverie, J.M.; et al. Mechanisms of evolution in Rickettsia conorii and R. prowazekii. Science 2001, 293, 2093–2098. [Google Scholar]
- Merhej, V.; Royer-Carenzi, M.; Pontarotti, P.; Raoult, D. Massive comparative genomic analysis reveals convergent evolution of specialized bacteria. Biol. Direct 2009, 4, 13. [Google Scholar]
- Pallen, M.J.; Wren, B.W. Bacterial pathogenomics. Nature 2007, 449, 835–842. [Google Scholar]
- Kurland, C.G.; Andersson, S.G. Origin and evolution of the mitochondrial proteome. Microbiol. Mol. Biol. Rev. 2000, 64, 786–820. [Google Scholar]
- Andersson, S.G.; Dehio, C. Rickettsia prowazekii and Bartonella henselae: Differences in the intracellular life styles revisited. Int. J. Med. Microbiol. 2000, 290, 135–141. [Google Scholar] [CrossRef]
- Andersson, S.G.; Kurland, C.G. Reductive evolution of resident genomes. Trends Microbiol. 1998, 6, 263–268. [Google Scholar]
- Moreno, E. Genome evolution within the alpha Proteobacteria: Why do some bacteria not possess plasmids and others exhibit more than one different chromosome? FEMS Microbiol. Rev. 1998, 22, 255–275. [Google Scholar] [CrossRef]
- Teyssier, C.; Marchandin, H.; Simeon De Buochberg, M.; Ramuz, M.; Jumas-Bilak, E. Atypical 16S rRNA gene copies in Ochrobactrum intermedium strains reveal a large genomic rearrangement by recombination between rrn copies. J. Bacteriol. 2003, 185, 2901–2909. [Google Scholar] [CrossRef]
- Georgiades, K.; Merhej, V.; El Karkouri, K.; Raoult, D.; Pontarotti, P. Gene gain and loss events in Rickettsia and Orientia species. Biol. Direct 2011, 6, 6. [Google Scholar] [CrossRef]
- Koonin, E.V. Darwinian evolution in the light of genomics. Nucleic Acids Res. 2009, 37, 1011–1034. [Google Scholar]
- Georgiades, K.; Raoult, D. Genomes of the most dangerous epidemic bacteria have a virulence repertoire characterized by fewer genes but more toxin-antitoxin modules. PLoS One 2011, 6, e17962. [Google Scholar]
- Pellett, S.; Bigley, D.V.; Grimes, D.J. Distribution of Pseudomonas aeruginosa in a riverine ecosystem. Appl. Environ. Microbiol. 1983, 45, 328–332. [Google Scholar]
- Khan, N.H.; Ishii, Y.; Kimata-Kino, N.; Esaki, H.; Nishino, T.; Nishimura, M.; Kogure, K. Isolation of Pseudomonas aeruginosa from open ocean and comparison with freshwater, clinical, and animal isolates. Microb. Ecol. 2007, 53, 173–186. [Google Scholar] [CrossRef]
- Schwartz, T.; Volkmann, H.; Kirchen, S.; Kohnen, W.; Schon-Holz, K.; Jansen, B.; Obst, U. Real-time PCR detection of Pseudomonas aeruginosa in clinical and municipal wastewater and genotyping of the ciprofloxacin-resistant isolates. FEMS Microbiol. Ecol. 2006, 57, 158–167. [Google Scholar] [CrossRef]
- Deziel, E.; Paquette, G.; Villemur, R.; Lepine, F.; Bisaillon, J. Biosurfactant production by a soil Pseudomonas strain growing on polycyclic aromatic hydrocarbons. Appl. Environ. Microbiol. 1996, 62, 1908–1912. [Google Scholar]
- Livermore, D.M. Multiple mechanisms of antimicrobial resistance in Pseudomonas aeruginosa: Our worst nightmare? Clin. Infect. Dis. 2002, 34, 634–640. [Google Scholar] [CrossRef]
- Frimmersdorf, E.; Horatzek, S.; Pelnikevich, A.; Wiehlmann, L.; Schomburg, D. How Pseudomonas aeruginosa adapts to various environments: A metabolomic approach. Environ. Microbiol. 2010, 12, 1734–1747. [Google Scholar] [CrossRef]
- Stover, C.K.; Pham, X.Q.; Erwin, A.L.; Mizoguchi, S.D.; Warrener, P.; Hickey, M.J.; Brinkman, F.S.; Hufnagle, W.O.; Kowalik, D.J.; Lagrou, M.; et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 2000, 406, 959–964. [Google Scholar]
- Moran, N.A. Microbial minimalism: Genome reduction in bacterial pathogens. Cell 2002, 108, 583–586. [Google Scholar]
- Teyssier, C.; Marchandin, H.; Jumas-Bilak, E. The genome of alpha-proteobacteria: Complexity, reduction, diversity and fluidity. Can. J. Microbiol. 2004, 50, 383–396. [Google Scholar] [CrossRef]
- Kung, V.L.; Ozer, E.A.; Hauser, A.R. The accessory genome of Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 2010, 74, 621–641. [Google Scholar] [CrossRef]
- Schmidt, K.D.; Tummler, B.; Romling, U. Comparative genome mapping of Pseudomonas aeruginosa PAO with P. aeruginosa C, which belongs to a major clone in cystic fibrosis patients and aquatic habitats. J. Bacteriol. 1996, 178, 85–93. [Google Scholar]
- Romling, U.; Schmidt, K.D.; Tummler, B. Large genome rearrangements discovered by the detailed analysis of 21 Pseudomonas aeruginosa clone C isolates found in environment and disease habitats. J. Mol. Biol. 1997, 271, 386–404. [Google Scholar] [CrossRef]
- Silby, M.W.; Winstanley, C.; Godfrey, S.A.; Levy, S.B.; Jackson, R.W. Pseudomonas genomes: Diverse and adaptable. FEMS Microbiol. Rev. 2011, 35, 652–680. [Google Scholar] [CrossRef]
- Lee, D.G.; Urbach, J.M.; Wu, G.; Liberati, N.T.; Feinbaum, R.L.; Miyata, S.; Diggins, L.T.; He, J.; Saucier, M.; Deziel, E.; et al. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 2006, 7, R90. [Google Scholar] [CrossRef]
- Winstanley, C.; Langille, M.G.; Fothergill, J.L.; Kukavica-Ibrulj, I.; Paradis-Bleau, C.; Sanschagrin, F.; Thomson, N.R.; Winsor, G.L.; Quail, M.A.; Lennard, N.; et al. Newly introduced genomic prophage islands are critical determinants of in vivo competitiveness in the Liverpool Epidemic Strain of Pseudomonas aeruginosa. Genome Res. 2009, 19, 12–23. [Google Scholar]
- Cheng, K.; Smyth, R.L.; Govan, J.R.; Doherty, C.; Winstanley, C.; Denning, N.; Heaf, D.P.; van Saene, H.; Hart, C.A. Spread of beta-lactam-resistant Pseudomonas aeruginosa in a cystic fibrosis clinic. Lancet 1996, 348, 639–642. [Google Scholar]
- McCallum, S.J.; Gallagher, M.J.; Corkill, J.E.; Hart, C.A.; Ledson, M.J.; Walshaw, M.J. Spread of an epidemic Pseudomonas aeruginosa strain from a patient with cystic fibrosis (CF) to non-CF relatives. Thorax 2002, 57, 559–560. [Google Scholar] [CrossRef]
- Roy, P.H.; Tetu, S.G.; Larouche, A.; Elbourne, L.; Tremblay, S.; Ren, Q.; Dodson, R.; Harkins, D.; Shay, R.; Watkins, K.; et al. Complete genome sequence of the multiresistant taxonomic outlier Pseudomonas aeruginosa PA7. PLoS One 2010, 5, e8842. [Google Scholar]
- Mathee, K.; Narasimhan, G.; Valdes, C.; Qiu, X.; Matewish, J.M.; Koehrsen, M.; Rokas, A.; Yandava, C.N.; Engels, R.; Zeng, E.; et al. Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 3100–3105. [Google Scholar]
- Jones, A.M.; Govan, J.R.; Doherty, C.J.; Dodd, M.E.; Isalska, B.J.; Stanbridge, T.N.; Webb, A.K. Spread of a multiresistant strain of Pseudomonas aeruginosa in an adult cystic fibrosis clinic. Lancet 2001, 358, 557–558. [Google Scholar]
- Stewart, R.M.; Wiehlmann, L.; Ashelford, K.E.; Preston, S.J.; Frimmersdorf, E.; Campbell, B.J.; Neal, T.J.; Hall, N.; Tuft, S.; Kaye, S.B.; et al. Genetic characterization indicates that a specific subpopulation of Pseudomonas aeruginosa is associated with keratitis infections. J. Clin. Microbiol. 2011, 49, 993–1003. [Google Scholar]
- Salzberg, S.L.; Sommer, D.D.; Puiu, D.; Lee, V.T. Gene-boosted assembly of a novel bacterial genome from very short reads. PLoS Comput. Biol. 2008, 4, e1000186. [Google Scholar]
- Klockgether, J.; Cramer, N.; Wiehlmann, L.; Davenport, C.F.; Tummler, B. Pseudomonas aeruginosa Genomic Structure and Diversity. Front. Microbiol. 2011, 2, 150. [Google Scholar]
- Wolfgang, M.C.; Kulasekara, B.R.; Liang, X.; Boyd, D.; Wu, K.; Yang, Q.; Miyada, C.G.; Lory, S. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2003, 100, 8484–8489. [Google Scholar]
- Dotsch, A.; Klawonn, F.; Jarek, M.; Scharfe, M.; Blocker, H.; Haussler, S. Evolutionary conservation of essential and highly expressed genes in Pseudomonas aeruginosa. BMC Genomics 2010, 11, 234. [Google Scholar] [CrossRef]
- Wiehlmann, L.; Wagner, G.; Cramer, N.; Siebert, B.; Gudowius, P.; Morales, G.; Kohler, T.; van Delden, C.; Weinel, C.; Slickers, P.; et al. Population structure of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2007, 104, 8101–8106. [Google Scholar]
- Aujoulat, F.; Lebreton, F.; Romano, S.; Delage, M.; Marchandin, H.; Brabet, M.; Bricard, F.; Godreuil, S.; Parer, S.; Jumas-Bilak, E. Comparative diffusion assay to assess efficacy of topical antimicrobial agents against Pseudomonas aeruginosa in burns care. Ann. Clin. Microbiol. Antimicrob. 2011, 10, 27. [Google Scholar] [CrossRef]
- Klockgether, J.; Wurdemann, D.; Reva, O.; Wiehlmann, L.; Tummler, B. Diversity of the abundant pKLC102/PAGI-2 family of genomic islands in Pseudomonas aeruginosa. J. Bacteriol. 2007, 189, 2443–2459. [Google Scholar] [CrossRef]
- Williams, K.P. Integration sites for genetic elements in prokaryotic tRNA and tmRNA genes: Sublocation preference of integrase subfamilies. Nucleic Acids Res. 2002, 30, 866–875. [Google Scholar]
- Pirnay, J.P.; Matthijs, S.; Colak, H.; Chablain, P.; Bilocq, F.; Van Eldere, J.; De Vos, D.; Zizi, M.; Triest, L.; Cornelis, P. Global Pseudomonas aeruginosa biodiversity as reflected in a Belgian river. Environ. Microbiol. 2005, 7, 969–980. [Google Scholar] [CrossRef]
- Pirnay, J.P.; De Vos, D.; Cochez, C.; Bilocq, F.; Vanderkelen, A.; Zizi, M.; Ghysels, B.; Cornelis, P. Pseudomonas aeruginosa displays an epidemic population structure. Environ. Microbiol. 2002, 4, 898–911. [Google Scholar] [CrossRef]
- Yang, L.; Jelsbak, L.; Marvig, R.L.; Damkiaer, S.; Workman, C.T.; Rau, M.H.; Hansen, S.K.; Folkesson, A.; Johansen, H.K.; Ciofu, O.; et al. Evolutionary dynamics of bacteria in a human host environment. Proc. Natl. Acad. Sci. USA 2011, 108, 7481–7486. [Google Scholar]
- Romling, U.; Fiedler, B.; Bosshammer, J.; Grothues, D.; Greipel, J.; von der Hardt, H.; Tummler, B. Epidemiology of chronic Pseudomonas aeruginosa infections in cystic fibrosis. J. Infect. Dis. 1994, 170, 1616–1621. [Google Scholar] [CrossRef]
- Hancock, R.E.; Mutharia, L.M.; Chan, L.; Darveau, R.P.; Speert, D.P.; Pier, G.B. Pseudomonas aeruginosa isolates from patients with cystic fibrosis: A class of serum-sensitive, nontypable strains deficient in lipopolysaccharide O side chains. Infect. Immun. 1983, 42, 170–177. [Google Scholar]
- Ernst, R.K.; Adams, K.N.; Moskowitz, S.M.; Kraig, G.M.; Kawasaki, K.; Stead, C.M.; Trent, M.S.; Miller, S.I. The Pseudomonas aeruginosa lipid A deacylase: Selection for expression and loss within the cystic fibrosis airway. J. Bacteriol. 2006, 188, 191–201. [Google Scholar]
- Barth, A.L.; Pitt, T.L. Auxotrophic variants of Pseudomonas aeruginosa are selected from prototrophic wild-type strains in respiratory infections in patients with cystic fibrosis. J. Clin. Microbiol. 1995, 33, 37–40. [Google Scholar]
- Luzar, M.A.; Thomassen, M.J.; Montie, T.C. Flagella and motility alterations in Pseudomonas aeruginosa strains from patients with cystic fibrosis: Relationship to patient clinical condition. Infect. Immun. 1985, 50, 577–582. [Google Scholar]
- Oliver, A.; Canton, R.; Campo, P.; Baquero, F.; Blazquez, J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 2000, 288, 1251–1254. [Google Scholar] [CrossRef]
- Nivens, D.E.; Ohman, D.E.; Williams, J.; Franklin, M.J. Role of alginate and its O acetylation in formation of Pseudomonas aeruginosa microcolonies and biofilms. J. Bacteriol. 2001, 183, 1047–1057. [Google Scholar]
- Mena, A.; Smith, E.E.; Burns, J.L.; Speert, D.P.; Moskowitz, S.M.; Perez, J.L.; Oliver, A. Genetic adaptation of Pseudomonas aeruginosa to the airways of cystic fibrosis patients is catalyzed by hypermutation. J. Bacteriol. 2008, 190, 7910–7917. [Google Scholar] [CrossRef]
- Mathee, K.; Ciofu, O.; Sternberg, C.; Lindum, P.W.; Campbell, J.I.; Jensen, P.; Johnsen, A.H.; Givskov, M.; Ohman, D.E.; Molin, S.; et al. Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: A mechanism for virulence activation in the cystic fibrosis lung. Microbiology 1999, 145, 1349–1357. [Google Scholar] [CrossRef]
- Mahenthiralingam, E.; Campbell, M.E.; Speert, D.P. Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect. Immun. 1994, 62, 596–605. [Google Scholar]
- D’Argenio, D.A.; Wu, M.; Hoffman, L.R.; Kulasekara, H.D.; Deziel, E.; Smith, E.E.; Nguyen, H.; Ernst, R.K.; Larson Freeman, T.J.; Spencer, D.H.; et al. Growth phenotypes of Pseudomonas aeruginosa lasR mutants adapted to the airways of cystic fibrosis patients. Mol. Microbiol. 2007, 64, 512–533. [Google Scholar] [CrossRef]
- Rau, M.H.; Hansen, S.K.; Johansen, H.K.; Thomsen, L.E.; Workman, C.T.; Nielsen, K.F.; Jelsbak, L.; Hoiby, N.; Yang, L.; Molin, S. Early adaptive developments of Pseudomonas aeruginosa after the transition from life in the environment to persistent colonization in the airways of human cystic fibrosis hosts. Environ. Microbiol. 2010, 12, 1643–1658. [Google Scholar]
- Smith, E.E.; Buckley, D.G.; Wu, Z.; Saenphimmachak, C.; Hoffman, L.R.; D'Argenio, D.A.; Miller, S.I.; Ramsey, B.W.; Speert, D.P.; Moskowitz, S.M.; et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 2006, 103, 8487–8492. [Google Scholar]
- Jelsbak, L.; Johansen, H.K.; Frost, A.L.; Thogersen, R.; Thomsen, L.E.; Ciofu, O.; Yang, L.; Haagensen, J.A.; Hoiby, N.; Molin, S. Molecular epidemiology and dynamics of Pseudomonas aeruginosa populations in lungs of cystic fibrosis patients. Infect. Immun. 2007, 75, 2214–2224. [Google Scholar]
- Hoboth, C.; Hoffmann, R.; Eichner, A.; Henke, C.; Schmoldt, S.; Imhof, A.; Heesemann, J.; Hogardt, M. Dynamics of adaptive microevolution of hypermutable Pseudomonas aeruginosa during chronic pulmonary infection in patients with cystic fibrosis. J. Infect. Dis. 2009, 200, 118–130. [Google Scholar] [CrossRef]
- Bragonzi, A.; Paroni, M.; Nonis, A.; Cramer, N.; Montanari, S.; Rejman, J.; Di Serio, C.; Doring, G.; Tummler, B. Pseudomonas aeruginosa microevolution during cystic fibrosis lung infection establishes clones with adapted virulence. Am. J. Respir. Crit. Care Med. 2009, 180, 138–145. [Google Scholar] [CrossRef]
- Kresse, A.U.; Dinesh, S.D.; Larbig, K.; Romling, U. Impact of large chromosomal inversions on the adaptation and evolution of Pseudomonas aeruginosa chronically colonizing cystic fibrosis lungs. Mol. Microbiol. 2003, 47, 145–158. [Google Scholar]
- Janda, J.M.; Abbott, S.L. The genus Aeromonas: Taxonomy, pathogenicity, and infection. Clin. Microbiol. Rev. 2010, 23, 35–73. [Google Scholar] [CrossRef]
- Seshadri, R.; Joseph, S.W.; Chopra, A.K.; Sha, J.; Shaw, J.; Graf, J.; Haft, D.; Wu, M.; Ren, Q.; Rosovitz, M.J.; et al. Genome sequence of Aeromonas hydrophila ATCC 7966T: Jack of all trades. J. Bacteriol. 2006, 188, 8272–8282. [Google Scholar]
- Janda, J.M.; Abbott, S.L. Evolving concepts regarding the genus Aeromonas: An expanding Panorama of species, disease presentations, and unanswered questions. Clin. Infect. Dis. 1998, 27, 332–344. [Google Scholar]
- Monfort, P.; Baleux, B. Distribution and survival of motile Aeromonas spp. in brackish water receiving sewage treatment effluent. Appl. Environ. Microbiol. 1991, 57, 2459–2467. [Google Scholar]
- Graf, J.; Kikuchi, Y.; Rio, R.V. Leeches and their microbiota: Naturally simple symbiosis models. Trends Microbiol. 2006, 14, 365–371. [Google Scholar]
- Pidiyar, V.; Kaznowski, A.; Narayan, N.B.; Patole, M.; Shouche, Y.S. Aeromonas culicicola sp. nov., from the midgut of Culex quinquefasciatus. Int. J. Syst. Evol. Microbiol. 2002, 52, 1723–1728. [Google Scholar] [CrossRef]
- Rahman, M.; Abd, H.; Romling, U.; Sandstrom, G.; Mollby, R. Aeromonas-Acanthamoeba interaction and early shift to a viable but nonculturable state of Aeromonas by Acanthamoeba. J. Appl. Microbiol. 2008, 104, 1449–1457. [Google Scholar] [CrossRef]
- Sneath, P.H. Evidence from Aeromonas for genetic crossing-over in ribosomal sequences. Int. J. Syst. Bacteriol. 1993, 43, 626–629. [Google Scholar] [CrossRef]
- Morandi, A.; Zhaxybayeva, O.; Gogarten, J.P.; Graf, J. Evolutionary and diagnostic implications of intragenomic heterogeneity in the 16S rRNA gene in Aeromonas strains. J. Bacteriol. 2005, 187, 6561–6564. [Google Scholar] [CrossRef]
- Umelo, E.; Trust, T.J. Physical map of the chromosome of Aeromonas salmonicida and genomic comparisons between Areomonas strains. Microbiology 1998, 144, 2141–2149. [Google Scholar] [CrossRef]
- Reith, M.E.; Singh, R.K.; Curtis, B.; Boyd, J.M.; Bouevitch, A.; Kimball, J.; Munholland, J.; Murphy, C.; Sarty, D.; Williams, J.; et al. The genome of Aeromonas salmonicida subsp. salmonicida A449: Insights into the evolution of a fish pathogen. BMC Genomics 2008, 9, 427. [Google Scholar]
- Silver, A.C.; Kikuchi, Y.; Fadl, A.A.; Sha, J.; Chopra, A.K.; Graf, J. Interaction between innate immune cells and a bacterial type III secretion system in mutualistic and pathogenic associations. Proc. Natl. Acad. Sci. USA 2007, 104, 9481–9486. [Google Scholar]
- Silver, A.C.; Graf, J. Prevalence of genes encoding the type three secretion system and the effectors AexT and AexU in the Aeromonas veronii group. DNA Cell Biol. 2009, 28, 383–388. [Google Scholar] [CrossRef]
- Hentschel, U.; Steinert, M.; Hacker, J. Common molecular mechanisms of symbiosis and pathogenesis. Trends Microbiol. 2000, 8, 226–231. [Google Scholar]
- Sha, J.; Wang, S.F.; Suarez, G.; Sierra, J.C.; Fadl, A.A.; Erova, T.E.; Foltz, S.M.; Khajanchi, B.K.; Silver, A.; Graf, J.; et al. Further characterization of a type III secretion system (T3SS) and of a new effector protein from a clinical isolate of Aeromonas hydrophila--part I. Microb. Pathog. 2007, 43, 127–146. [Google Scholar] [CrossRef]
- Silver, A.C.; Rabinowitz, N.M.; Kuffer, S.; Graf, J. Identification of Aeromonas veronii genes required for colonization of the medicinal leech, Hirudo verbana. J. Bacteriol. 2007, 189, 6763–6772. [Google Scholar]
- Silver, A.C.; Williams, D.; Faucher, J.; Horneman, A.J.; Gogarten, J.P.; Graf, J. Complex evolutionary history of the Aeromonas veronii group revealed by host interaction and DNA sequence data. PLoS One 2011, 6, e16751. [Google Scholar]
- Li, Y.; Liu, Y.; Zhou, Z.; Huang, H.; Ren, Y.; Zhang, Y.; Li, G.; Wang, L. Complete genome sequence of Aeromonas veronii strain B565. J. Bacteriol. 2011, 193, 3389–3390. [Google Scholar]
- O'Callaghan, D.; Cazevieille, C.; Allardet-Servent, A.; Boschiroli, M.L.; Bourg, G.; Foulongne, V.; Frutos, P.; Kulakov, Y.; Ramuz, M. A homologue of the Agrobacterium tumefaciens VirB and Bordetella pertussis Ptl type IV secretion systems is essential for intracellular survival of Brucella suis. Mol. Microbiol. 1999, 33, 1210–1220. [Google Scholar]
- Chain, P.S.; Lang, D.M.; Comerci, D.J.; Malfatti, S.A.; Vergez, L.M.; Shin, M.; Ugalde, R.A.; Garcia, E.; Tolmasky, M.E. Genome of Ochrobactrum anthropi ATCC 49188 T, a versatile opportunistic pathogen and symbiont of several eukaryotic hosts. J. Bacteriol. 2011, 193, 4274–4275. [Google Scholar]
- Jumas-Bilak, E.; Michaux-Charachon, S.; Bourg, G.; Ramuz, M.; Allardet-Servent, A. Unconventional genomic organization in the alpha subgroup of the Proteobacteria. J. Bacteriol. 1998, 180, 2749–2755. [Google Scholar]
- Tsolis, R.M.; Seshadri, R.; Santos, R.L.; Sangari, F.J.; Lobo, J.M.; de Jong, M.F.; Ren, Q.; Myers, G.; Brinkac, L.M.; Nelson, W.C.; et al. Genome degradation in Brucella ovis corresponds with narrowing of its host range and tissue tropism. PLoS One 2009, 4, e5519. [Google Scholar]
- DelVecchio, V.G.; Kapatral, V.; Redkar, R.J.; Patra, G.; Mujer, C.; Los, T.; Ivanova, N.; Anderson, I.; Bhattacharyya, A.; Lykidis, A.; et al. The genome sequence of the facultative intracellular pathogen Brucella melitensis. Proc. Natl. Acad. Sci. USA 2002, 99, 443–448. [Google Scholar]
- Chain, P.S.; Comerci, D.J.; Tolmasky, M.E.; Larimer, F.W.; Malfatti, S.A.; Vergez, L.M.; Aguero, F.; Land, M.L.; Ugalde, R.A.; Garcia, E. Whole-genome analyses of speciation events in pathogenic Brucellae. Infect. Immun. 2005, 73, 8353–8361. [Google Scholar]
- Sangari, F.J.; Seoane, A.; Rodriguez, M.C.; Aguero, J.; Garcia Lobo, J.M. Characterization of the urease operon of Brucella abortus and assessment of its role in virulence of the bacterium. Infect. Immun. 2007, 75, 774–780. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Aujoulat, F.; Roger, F.; Bourdier, A.; Lotthé, A.; Lamy, B.; Marchandin, H.; Jumas-Bilak, E. From Environment to Man: Genome Evolution and Adaptation of Human Opportunistic Bacterial Pathogens. Genes 2012, 3, 191-232. https://doi.org/10.3390/genes3020191
Aujoulat F, Roger F, Bourdier A, Lotthé A, Lamy B, Marchandin H, Jumas-Bilak E. From Environment to Man: Genome Evolution and Adaptation of Human Opportunistic Bacterial Pathogens. Genes. 2012; 3(2):191-232. https://doi.org/10.3390/genes3020191
Chicago/Turabian StyleAujoulat, Fabien, Frédéric Roger, Alice Bourdier, Anne Lotthé, Brigitte Lamy, Hélène Marchandin, and Estelle Jumas-Bilak. 2012. "From Environment to Man: Genome Evolution and Adaptation of Human Opportunistic Bacterial Pathogens" Genes 3, no. 2: 191-232. https://doi.org/10.3390/genes3020191