Microsatellites with Macro-Influence in Ewing Sarcoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction


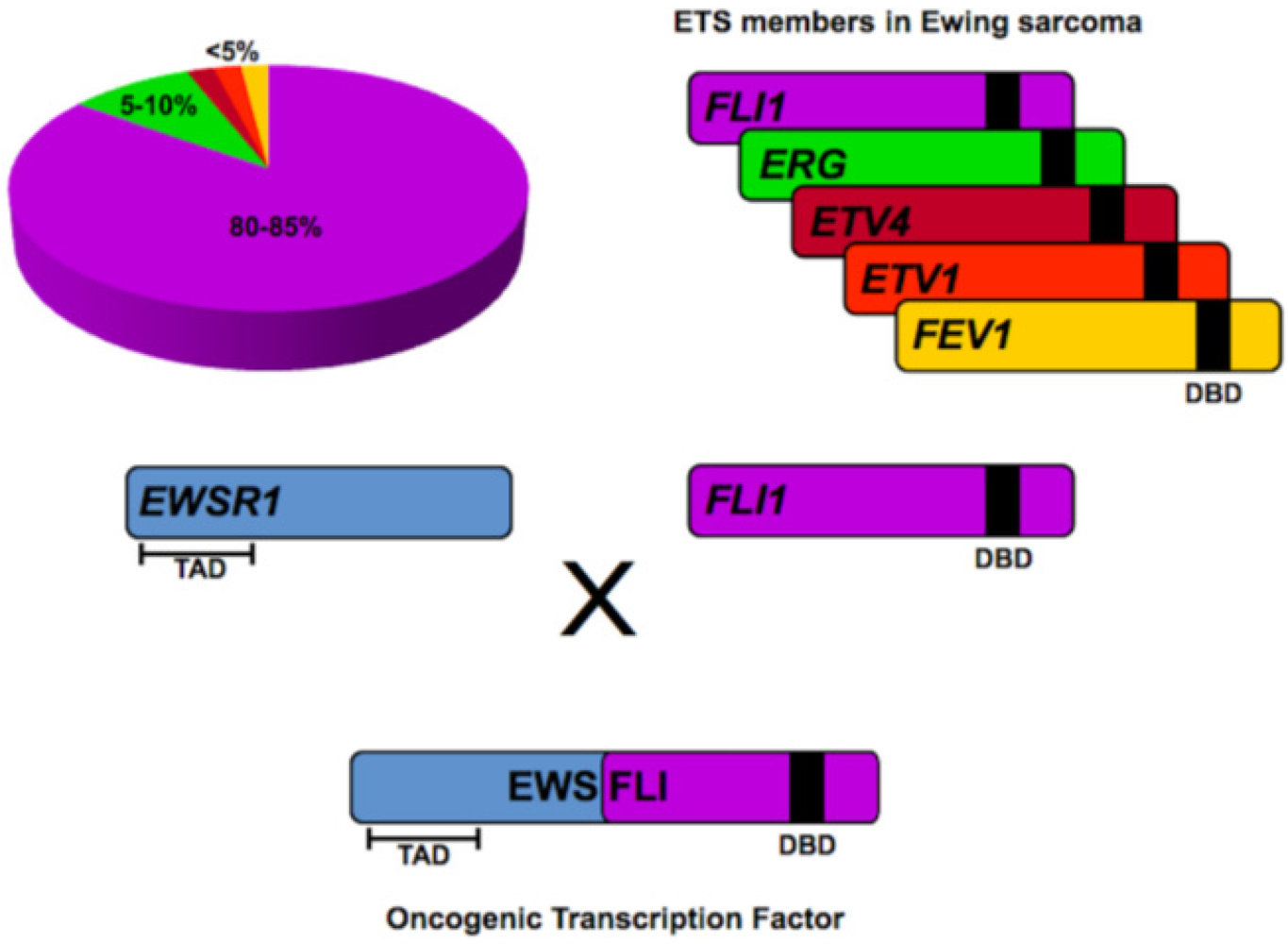
2. ETS Family of Transcription Factors
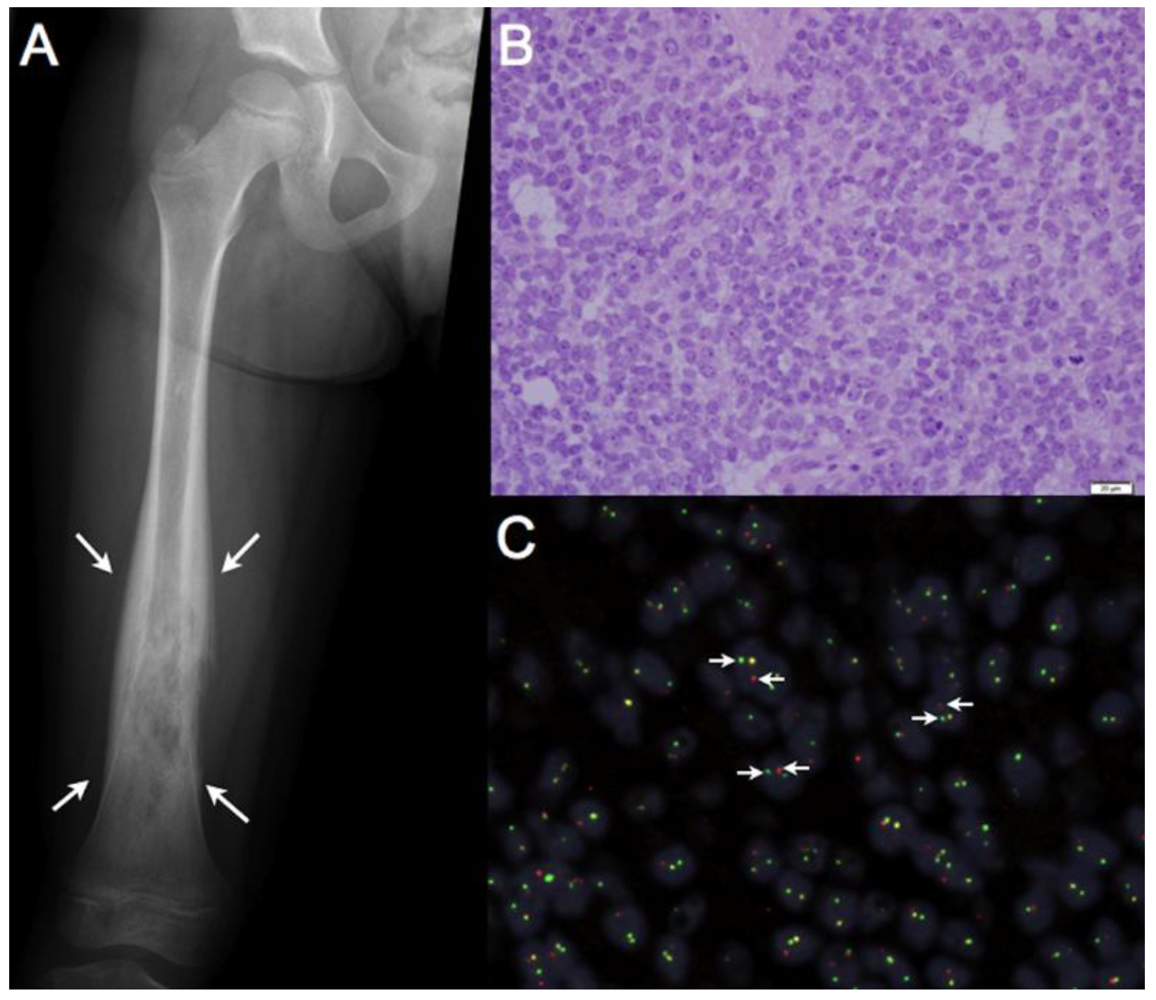
3. EWS/FLI in Ewing Sarcoma
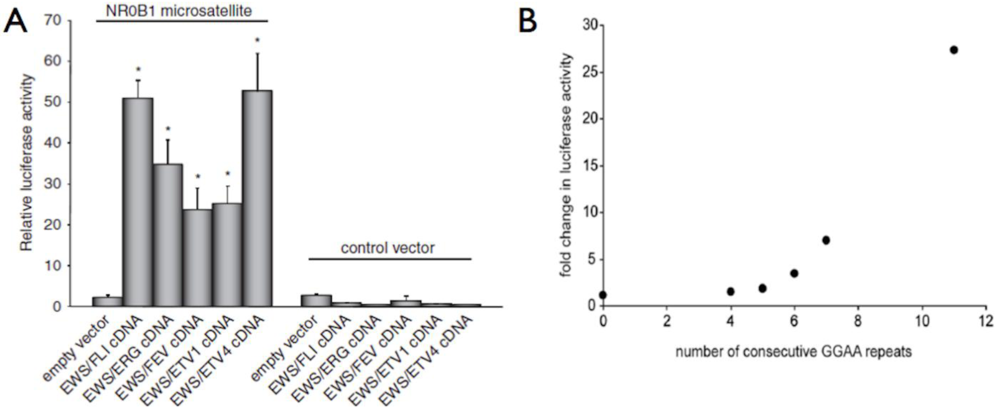
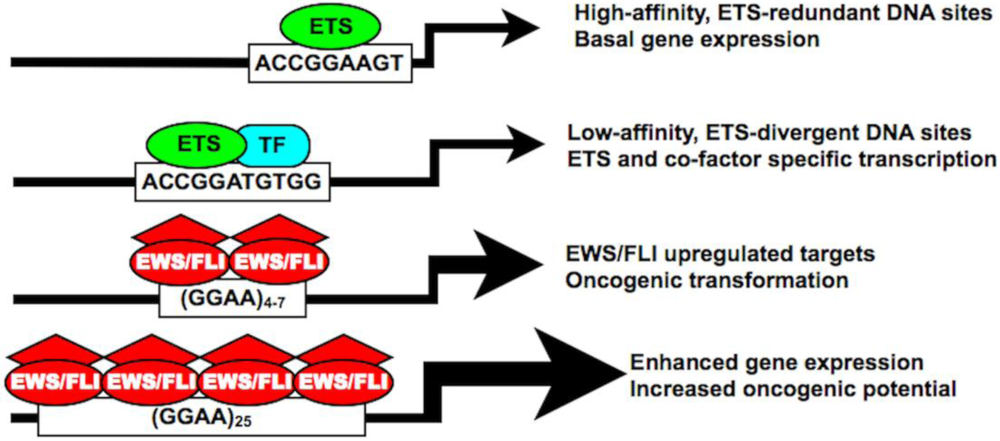
4. EWS/FLI Fusions Mediate Gene Dysregulation via a GGAA Microsatellite Response Element
5. Microsatellite Constitution Influences EWS/FLI Binding and Gene Activation


6. GGAA Microsatellites Identify Other Potential EWS/FLI Targets and Epigenetically Regulated Enhancer Loci
7. The NR0B1 Microsatellite: A Functional Assessment Tool in Ewing Sarcoma Research
8. Microsatellite DNA in Cancer Pathogenesis
9. Polymorphic EWS/FLI GGAA Microsatellites: A Novel Approach to Ethnic Patterns of Ewing Sarcoma Susceptibility and Prognosis
10. Conclusions
Conflict of Interest
Acknowledgments
References
- Helman, L.J.; Meltzer, P. Mechanisms of sarcoma development. Nat. Rev. Cancer 2003, 3, 685–694. [Google Scholar] [CrossRef]
- Martens, J.H.; Stunnenberg, H.G. The molecular signature of oncofusion proteins in acute myeloid leukemia. FEBS Lett. 2010, 584, 2662–2669. [Google Scholar]
- Maheshwari, A.V.; Cheng, E.Y. Ewing sarcoma family of tumors. J. Am. Acad. Orthop. Surg. 2010, 18, 94–107. [Google Scholar]
- Balamuth, N.J.; Womer, R.B. Ewing’s sarcoma. Lancet Oncol. 2010, 11, 184–192. [Google Scholar] [CrossRef]
- Grier, H.E.; Krailo, M.D.; Tarbell, N.J.; Link, M.P.; Fryer, C.J.; Pritchard, D.J.; Gebhardt, M.C.; Dickman, P.S.; Perlman, E.J.; Meyers, P.A.; et al. Addition of ifosfamide and etoposide to standard chemotherapy for ewing’s sarcoma and primitive neuroectodermal tumor of bone. N. Engl. J. Med. 2003, 348, 694–701. [Google Scholar]
- Sankar, S.; Lessnick, S.L. Promiscuous partnerships in ewing’s sarcoma. Cancer Genet. 2011, 204, 351–365. [Google Scholar] [CrossRef]
- May, W.A.; Gishizky, M.L.; Lessnick, S.L.; Lunsford, L.B.; Lewis, B.C.; Delattre, O.; Zucman, J.; Thomas, G.; Denny, C.T. Ewing sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by fli1 for transformation. Proc. Natl. Acad. Sci. USA 1993, 90, 5752–5756. [Google Scholar]
- Smith, R.; Owen, L.A.; Trem, D.J.; Wong, J.S.; Whangbo, J.S.; Golub, T.R.; Lessnick, S.L. Expression profiling of ews/fli identifies nkx2.2 as a critical target gene in ewing’s sarcoma. Cancer Cell 2006, 9, 405–416. [Google Scholar] [CrossRef]
- Kinsey, M.; Smith, R.; Iyer, A.K.; McCabe, E.R.; Lessnick, S.L. Ews/fli and its downstream target nr0b1 interact directly to modulate transcription and oncogenesis in ewing’s sarcoma. Cancer Res. 2009, 69, 9047–9055. [Google Scholar] [CrossRef]
- Hsu, T.; Trojanowska, M.; Watson, D.K. Ets proteins in biological control and cancer. J. Cell Biochem. 2004, 91, 896–903. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of tmprss2 and ets transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef]
- Clark, J.; Attard, G.; Jhavar, S.; Flohr, P.; Reid, A.; De-Bono, J.; Eeles, R.; Scardino, P.; Cuzick, J.; Fisher, G.; et al. Complex patterns of ets gene alteration arise during cancer development in the human prostate. Oncogene 2008, 27, 1993–2003. [Google Scholar]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G.; et al. Gene fusion with an ets DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar]
- May, W.A.; Lessnick, S.L.; Braun, B.S.; Klemsz, M.; Lewis, B.C.; Lunsford, L.B.; Hromas, R.; Denny, C.T. The ewing’s sarcoma ews/fli-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than fli-1. Mol. Cell Biol. 1993, 13, 7393–7398. [Google Scholar]
- Hollenhorst, P.C.; Shah, A.A.; Hopkins, C.; Graves, B.J. Genome-wide analyses reveal properties of redundant and specific promoter occupancy within the ets gene family. Genes Dev. 2007, 21, 1882–1894. [Google Scholar] [CrossRef]
- Hollenhorst, P.C.; McIntosh, L.P.; Graves, B.J. Genomic and biochemical insights into the specificity of ets transcription factors. Annu. Rev. Biochem. 2011, 80, 437–471. [Google Scholar] [CrossRef]
- Nye, J.A.; Petersen, J.M.; Gunther, C.V.; Jonsen, M.D.; Graves, B.J. Interaction of murine ets-1 with gga-binding sites establishes the ets domain as a new DNA-binding motif. Genes Dev. 1992, 6, 975–990. [Google Scholar] [CrossRef]
- Seth, A.; Watson, D.K. Ets transcription factors and their emerging roles in human cancer. Eur. J. Cancer 2005, 41, 2462–2478. [Google Scholar] [CrossRef]
- Szymczyna, B.R.; Arrowsmith, C.H. DNA binding specificity studies of four ets proteins support an indirect read-out mechanism of protein-DNA recognition. J. Biol. Chem. 2000, 275, 28363–28370. [Google Scholar] [CrossRef]
- Wei, G.H.; Badis, G.; Berger, M.F.; Kivioja, T.; Palin, K.; Enge, M.; Bonke, M.; Jolma, A.; Varjosalo, M.; Gehrke, A.R.; et al. Genome-wide analysis of ets-family DNA-binding in vitro and in vivo. EMBO J. 2010, 29, 2147–2160. [Google Scholar] [CrossRef]
- Mao, X.; Miesfeldt, S.; Yang, H.; Leiden, J.M.; Thompson, C.B. The fli-1 and chimeric ews-fli-1 oncoproteins display similar DNA binding specificities. J. Biol. Chem. 1994, 269, 18216–18222. [Google Scholar]
- Zhang, X.K.; Moussa, O.; LaRue, A.; Bradshaw, S.; Molano, I.; Spyropoulos, D.D.; Gilkeson, G.S.; Watson, D.K. The transcription factor fli-1 modulates marginal zone and follicular b cell development in mice. J. Immunol. 2008, 181, 1644–1654. [Google Scholar]
- Hart, A.; Melet, F.; Grossfeld, P.; Chien, K.; Jones, C.; Tunnacliffe, A.; Favier, R.; Bernstein, A. Fli-1 is required for murine vascular and megakaryocytic development and is hemizygously deleted in patients with thrombocytopenia. Immunity 2000, 13, 167–177. [Google Scholar] [CrossRef]
- Loughran, S.J.; Kruse, E.A.; Hacking, D.F.; de Graaf, C.A.; Hyland, C.D.; Willson, T.A.; Henley, K.J.; Ellis, S.; Voss, A.K.; Metcalf, D.; et al. The transcription factor erg is essential for definitive hematopoiesis and the function of adult hematopoietic stem cells. Nat. Immunol. 2008, 9, 810–819. [Google Scholar]
- Ohno, T.; Ouchida, M.; Lee, L.; Gatalica, Z.; Rao, V.N.; Reddy, E.S. The ews gene, involved in ewing family of tumors, malignant melanoma of soft parts and desmoplastic small round cell tumors, codes for an rna binding protein with novel regulatory domains. Oncogene 1994, 9, 3087–3097. [Google Scholar]
- Bertolotti, A.; Lutz, Y.; Heard, D.J.; Chambon, P.; Tora, L. Htaf(ii)68, a novel rna/ssdna-binding protein with homology to the pro-oncoproteins tls/fus and ews is associated with both tfiid and rna polymerase ii. EMBO J. 1996, 15, 5022–5031. [Google Scholar]
- Paronetto, M.P.; Minana, B.; Valcarcel, J. The ewing sarcoma protein regulates DNA damage-induced alternative splicing. Mol. Cell 2011, 43, 353–368. [Google Scholar]
- Kinsey, M.; Smith, R.; Lessnick, S.L. Nr0b1 is required for the oncogenic phenotype mediated by ews/fli in ewing’s sarcoma. Mol. Cancer Res. 2006, 4, 851–859. [Google Scholar] [CrossRef]
- Patel, M.; Simon, J.M.; Iglesia, M.D.; Wu, S.B.; McFadden, A.W.; Lieb, J.D.; Davis, I.J. Tumor-specific retargeting of an oncogenic transcription factor chimera results in dysregulation of chromatin and transcription. Genome Res. 2011, 22, 259–270. [Google Scholar]
- Gangwal, K.; Close, D.; Enriquez, C.A.; Hill, C.P.; Lessnick, S.L. Emergent properties of ews/fli regulation via ggaa microsatellites in ewing’s sarcoma. Genes Cancer 2010, 1, 177–187. [Google Scholar] [CrossRef]
- Prieur, A.; Tirode, F.; Cohen, P.; Delattre, O. Ews/fli-1 silencing and gene profiling of ewing cells reveal downstream oncogenic pathways and a crucial role for repression of insulin-like growth factor binding protein 3. Mol. Cell Biol. 2004, 24, 7275–7283. [Google Scholar] [CrossRef]
- Gangwal, K.; Sankar, S.; Hollenhorst, P.C.; Kinsey, M.; Haroldsen, S.C.; Shah, A.A.; Boucher, K.M.; Watkins, W.S.; Jorde, L.B.; Graves, B.J.; et al. Microsatellites as ews/fli response elements in ewing’s sarcoma. Proc. Natl. Acad. Sci. USA 2008, 105, 10149–10154. [Google Scholar]
- Guillon, N.; Tirode, F.; Boeva, V.; Zynovyev, A.; Barillot, E.; Delattre, O. The oncogenic ews-fli1 protein binds in vivo ggaa microsatellite sequences with potential transcriptional activation function. PLoS One 2009, 4, e4932. [Google Scholar]
- Niakan, K.K.; McCabe, E.R. Dax1 origin, function, and novel role. Mol. Genet. Metab. 2005, 86, 70–83. [Google Scholar] [CrossRef]
- McCabe, E.R. Dax1: Increasing complexity in the roles of this novel nuclear receptor. Mol. Cell Endocrinol. 2007, 179-182, 265–266. [Google Scholar]
- Mendiola, M.; Carrillo, J.; Garcia, E.; Lalli, E.; Hernandez, T.; de Alava, E.; Tirode, F.; Delattre, O.; Garcia-Miguel, P.; Lopez-Barea, F.; et al. The orphan nuclear receptor dax1 is up-regulated by the ews/fli1 oncoprotein and is highly expressed in ewing tumors. Int. J. Cancer 2006, 118, 1381–1389. [Google Scholar]
- Garcia-Aragoncillo, E.; Carrillo, J.; Lalli, E.; Agra, N.; Gomez-Lopez, G.; Pestana, A.; Alonso, J. Dax1, a direct target of ews/fli1 oncoprotein, is a principal regulator of cell-cycle progression in ewing’s tumor cells. Oncogene 2008, 27, 6034–6043. [Google Scholar] [CrossRef]
- Graves, B.J.; Gillespie, M.E.; McIntosh, L.P. DNA binding by the ets domain. Nature 1996, 384, 322. [Google Scholar]
- Luo, W.; Gangwal, K.; Sankar, S.; Boucher, K.M.; Thomas, D.; Lessnick, S.L. Gstm4 is a microsatellite-containing ews/fli target involved in ewing’s sarcoma oncogenesis and therapeutic resistance. Oncogene 2009, 28, 4126–4132. [Google Scholar] [CrossRef]
- Martins, A.S.; Ordonez, J.L.; Amaral, A.T.; Prins, F.; Floris, G.; Debiec-Rychter, M.; Hogendoorn, P.C.; de Alava, E. Igf1r signaling in ewing sarcoma is shaped by clathrin-/caveolin-dependent endocytosis. PLoS One 2011, 6, e19846. [Google Scholar]
- Williams, T.M.; Lisanti, M.P. Caveolin-1 in oncogenic transformation, cancer, and metastasis. Am. J. Physiol. Cell Physiol. 2005, 288, C494–C506. [Google Scholar] [CrossRef]
- Tirado, O.M.; Mateo-Lozano, S.; Villar, J.; Dettin, L.E.; Llort, A.; Gallego, S.; Ban, J.; Kovar, H.; Notario, V. Caveolin-1 (cav1) is a target of ews/fli-1 and a key determinant of the oncogenic phenotype and tumorigenicity of ewing’s sarcoma cells. Cancer Res. 2006, 66, 9937–9947. [Google Scholar] [CrossRef]
- Grohar, P.J.; Woldemichael, G.M.; Griffin, L.B.; Mendoza, A.; Chen, Q.R.; Yeung, C.; Currier, D.G.; Davis, S.; Khanna, C.; Khan, J.; et al. Identification of an inhibitor of the ews-fli1 oncogenic transcription factor by high-throughput screening. J. Natl. Cancer Inst. 2011, 103, 962–978. [Google Scholar] [CrossRef]
- Grohar, P.J.; Griffin, L.B.; Yeung, C.; Chen, Q.R.; Pommier, Y.; Khanna, C.; Khan, J.; Helman, L.J. Ecteinascidin 743 interferes with the activity of ews-fli1 in ewing sarcoma cells. Neoplasia 2011, 13, 145–153. [Google Scholar]
- Erkizan, H.V.; Scher, L.J.; Gamble, S.E.; Barber-Rotenberg, J.S.; Sajwan, K.P.; Uren, A.; Toretsky, J.A. Novel peptide binds ews-fli1 and reduces the oncogenic potential in ewing tumors. Cell Cycle 2011, 10, 3397–3408. [Google Scholar]
- Kovar, H. Context matters: The hen or egg problem in ewing’s sarcoma. Semin. Cancer Biol. 2005, 15, 189–196. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Aaltonen, L.A.; Peltomaki, P.; Leach, F.S.; Sistonen, P.; Pylkkanen, L.; Mecklin, J.P.; Jarvinen, H.; Powell, S.M.; Jen, J.; Hamilton, S.R.; et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993, 260, 812–816. [Google Scholar]
- Thibodeau, S.N.; Bren, G.; Schaid, D. Microsatellite instability in cancer of the proximal colon. Science 1993, 260, 816–819. [Google Scholar]
- Ionov, Y.; Peinado, M.A.; Malkhosyan, S.; Shibata, D.; Perucho, M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993, 363, 558–561. [Google Scholar]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic review of microsatellite instability and colorectal cancer prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar]
- Pinol, V.; Castells, A.; Andreu, M.; Castellvi-Bel, S.; Alenda, C.; Llor, X.; Xicola, R.M.; Rodriguez-Moranta, F.; Paya, A.; Jover, R.; et al. Accuracy of revised bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA 2005, 293, 1986–1994. [Google Scholar]
- Guastadisegni, C.; Colafranceschi, M.; Ottini, L.; Dogliotti, E. Microsatellite instability as a marker of prognosis and response to therapy: A meta-analysis of colorectal cancer survival data. Eur. J. Cancer 2010, 46, 2788–2798. [Google Scholar] [CrossRef]
- Alldinger, I.; Schaefer, K.L.; Goedde, D.; Ottaviano, L.; Dirksen, U.; Ranft, A.; Juergens, H.; Gabbert, H.E.; Knoefel, W.T.; Poremba, C. Microsatellite instability in ewing tumor is not associated with loss of mismatch repair protein expression. J. Cancer Res. Clin. Oncol. 2007, 133, 749–759. [Google Scholar] [CrossRef]
- Ebinger, M.; Bock, T.; Kandolf, R.; Sotlar, K.; Bultmann, B.D.; Greil, J. Standard mono- and dinucleotide repeats do not appear to be sensitive markers of microsatellite instability in the ewing family of tumors. Cancer Genet. Cytogenet. 2005, 157, 189–190. [Google Scholar] [CrossRef]
- Ohali, A.; Avigad, S.; Cohen, I.J.; Meller, I.; Kollender, Y.; Issakov, J.; Goshen, Y.; Yaniv, I.; Zaizov, R. High frequency of genomic instability in ewing family of tumors. Cancer Genet. Cytogenet. 2004, 150, 50–56. [Google Scholar] [CrossRef]
- Tzaida, O.; Gogas, H.; Dafni, U.; Kyroudi, A.; Papaspyrou, I.; Kyriakou, V.; Malamou-Mitsi, V.; Alamani, M.; Skopa, C.; Kostopoulos, I.; et al. Evaluation of the prognostic and predictive value of her-1/egfr in breast cancer patients participating in a randomized study with dose-dense sequential adjuvant chemotherapy. Oncology (Williston Park) 2007, 72, 388–396. [Google Scholar]
- Nogi, H.; Kobayashi, T.; Suzuki, M.; Tabei, I.; Kawase, K.; Toriumi, Y.; Fukushima, H.; Uchida, K. Egfr as paradoxical predictor of chemosensitivity and outcome among triple-negative breast cancer. Oncol. Rep. 2009, 21, 413–417. [Google Scholar]
- Gebhardt, F.; Zanker, K.S.; Brandt, B. Modulation of epidermal growth factor receptor gene transcription by a polymorphic dinucleotide repeat in intron 1. J. Biol. Chem. 1999, 274, 13176–13180. [Google Scholar]
- Chamberlain, N.L.; Driver, E.D.; Miesfeld, R.L. The length and location of cag trinucleotide repeats in the androgen receptor n-terminal domain affect transactivation function. Nucleic Acids Res. 1994, 22, 3181–3186. [Google Scholar]
- Stanford, J.L.; Just, J.J.; Gibbs, M.; Wicklund, K.G.; Neal, C.L.; Blumenstein, B.A.; Ostrander, E.A. Polymorphic repeats in the androgen receptor gene: Molecular markers of prostate cancer risk. Cancer Res. 1997, 57, 1194–1198. [Google Scholar]
- Giovannucci, E.; Stampfer, M.J.; Krithivas, K.; Brown, M.; Dahl, D.; Brufsky, A.; Talcott, J.; Hennekens, C.H.; Kantoff, P.W. The cag repeat within the androgen receptor gene and its relationship to prostate cancer. Proc. Natl. Acad. Sci. USA 1997, 94, 3320–3323. [Google Scholar]
- Contente, A.; Dittmer, A.; Koch, M.C.; Roth, J.; Dobbelstein, M. A polymorphic microsatellite that mediates induction of pig3 by p53. Nat. Genet. 2002, 30, 315–320. [Google Scholar] [CrossRef]
- Kotsinas, A.; Aggarwal, V.; Tan, E.J.; Levy, B.; Gorgoulis, V.G. Pig3: A novel link between oxidative stress and DNA damage response in cancer. Cancer Lett. 2011, in press. [Google Scholar]
- Randall, R.L.; Lessnick, S.L.; Jones, K.B.; Gouw, L.G.; Cummings, J.E.; Cannon-Albright, L.; Schiffman, J.D. Is there a predisposition gene for ewing’s sarcoma? J. Oncol. 2010, 2010, 397632. [Google Scholar]
- Polednak, A.P. Primary bone cancer incidence in black and white residents of new york state. Cancer 1985, 55, 2883–2888. [Google Scholar] [CrossRef]
- Jawad, M.U.; Cheung, M.C.; Min, E.S.; Schneiderbauer, M.M.; Koniaris, L.G.; Scully, S.P. Ewing sarcoma demonstrates racial disparities in incidence-related and sex-related differences in outcome: An analysis of 1631 cases from the seer database, 1973-2005. Cancer 2009, 115, 3526–3536. [Google Scholar] [CrossRef]
- Worch, J.; Matthay, K.K.; Neuhaus, J.; Goldsby, R.; DuBois, S.G. Ethnic and racial differences in patients with ewing sarcoma. Cancer 2010, 116, 983–988. [Google Scholar]
- Zucman-Rossi, J.; Batzer, M.A.; Stoneking, M.; Delattre, O.; Thomas, G. Interethnic polymorphism of ews intron 6: Genome plasticity mediated by alu retroposition and recombination. Hum. Genet. 1997, 99, 357–363. [Google Scholar] [CrossRef]
- Dubois, S.G.; Goldsby, R.; Segal, M.; Woo, J.; Copren, K.; Kane, J.P.; Pullinger, C.R.; Matthay, K.K.; Witte, J.; Lessnick, S.L.; et al. Evaluation of polymorphisms in ewsr1 and risk of ewing sarcoma: A report from the childhood cancer survivor study. Pediatr. Blood Cancer 2011, 59, 52–56. [Google Scholar]
- Eckert, K.A.; Hile, S.E. Every microsatellite is different: Intrinsic DNA features dictate mutagenesis of common microsatellites present in the human genome. Mol. Carcinog. 2009, 48, 379–388. [Google Scholar] [CrossRef]
- Jorde, L.B.; Rogers, A.R.; Bamshad, M.; Watkins, W.S.; Krakowiak, P.; Sung, S.; Kere, J.; Harpending, H.C. Microsatellite diversity and the demographic history of modern humans. Proc. Natl. Acad. Sci. USA 1997, 94, 3100–3103. [Google Scholar]
- Beck, R.; Monument, M.J.; Watkins, W.S.; Smith, R.; Boucher, K.M.; Schiffman, J.D.; Jorde, L.B.; Randall, R.L.; Lessnick, S.L. EWS/FLI-responsive GGAA microsatellites exhibit polymorphic differences between European and African populations. Cancer Genet. 2012, 205, 304–312. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Monument, M.J.; Johnson, K.M.; Grossmann, A.H.; Schiffman, J.D.; Randall, R.L.; Lessnick, S.L. Microsatellites with Macro-Influence in Ewing Sarcoma. Genes 2012, 3, 444-460. https://doi.org/10.3390/genes3030444
Monument MJ, Johnson KM, Grossmann AH, Schiffman JD, Randall RL, Lessnick SL. Microsatellites with Macro-Influence in Ewing Sarcoma. Genes. 2012; 3(3):444-460. https://doi.org/10.3390/genes3030444
Chicago/Turabian StyleMonument, Michael J., Kirsten M. Johnson, Allie H. Grossmann, Joshua D. Schiffman, R. Lor Randall, and Stephen L. Lessnick. 2012. "Microsatellites with Macro-Influence in Ewing Sarcoma" Genes 3, no. 3: 444-460. https://doi.org/10.3390/genes3030444