Regulation of Fibroblast Growth Factor-2 Expression and Cell Cycle Progression by an Endogenous Antisense RNA

Abstract
:
1. Introduction
2. Results and Discussion
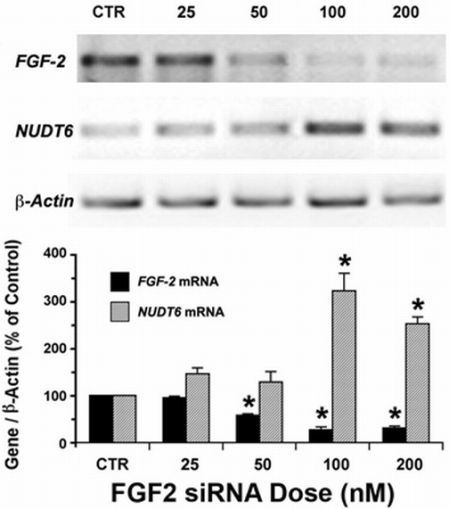
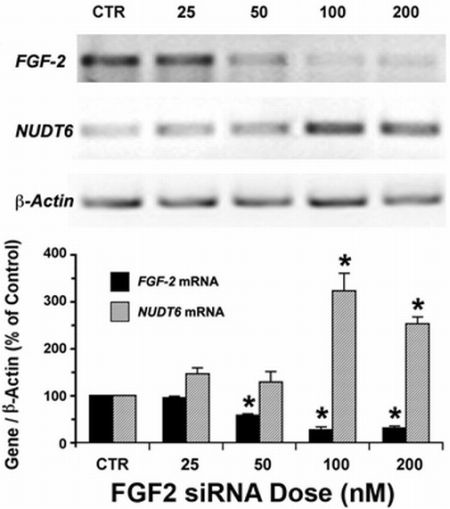
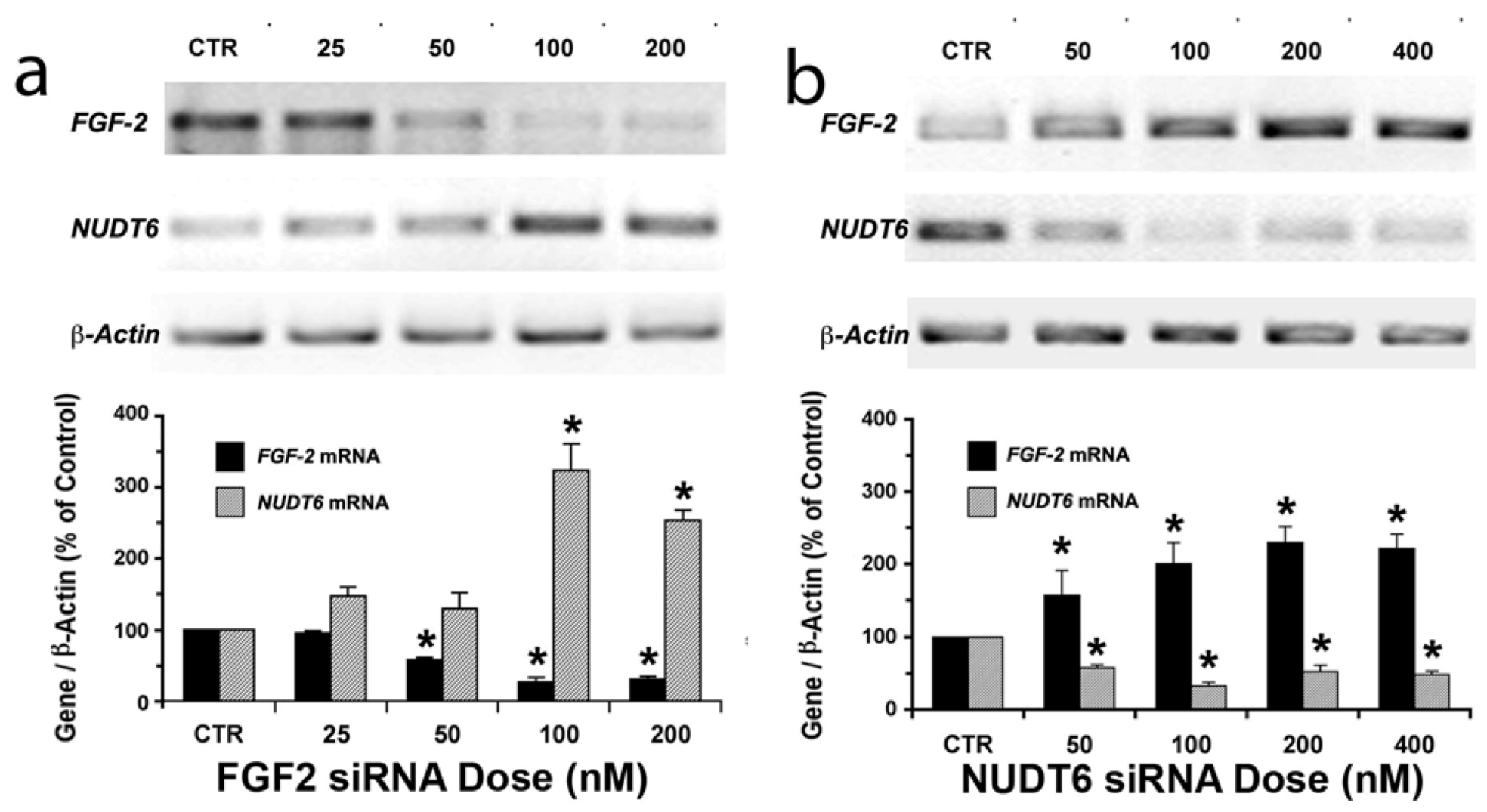
2.1. Reciprocal Effects of FGF2 and NUDT6 Knockdown on mRNA Levels
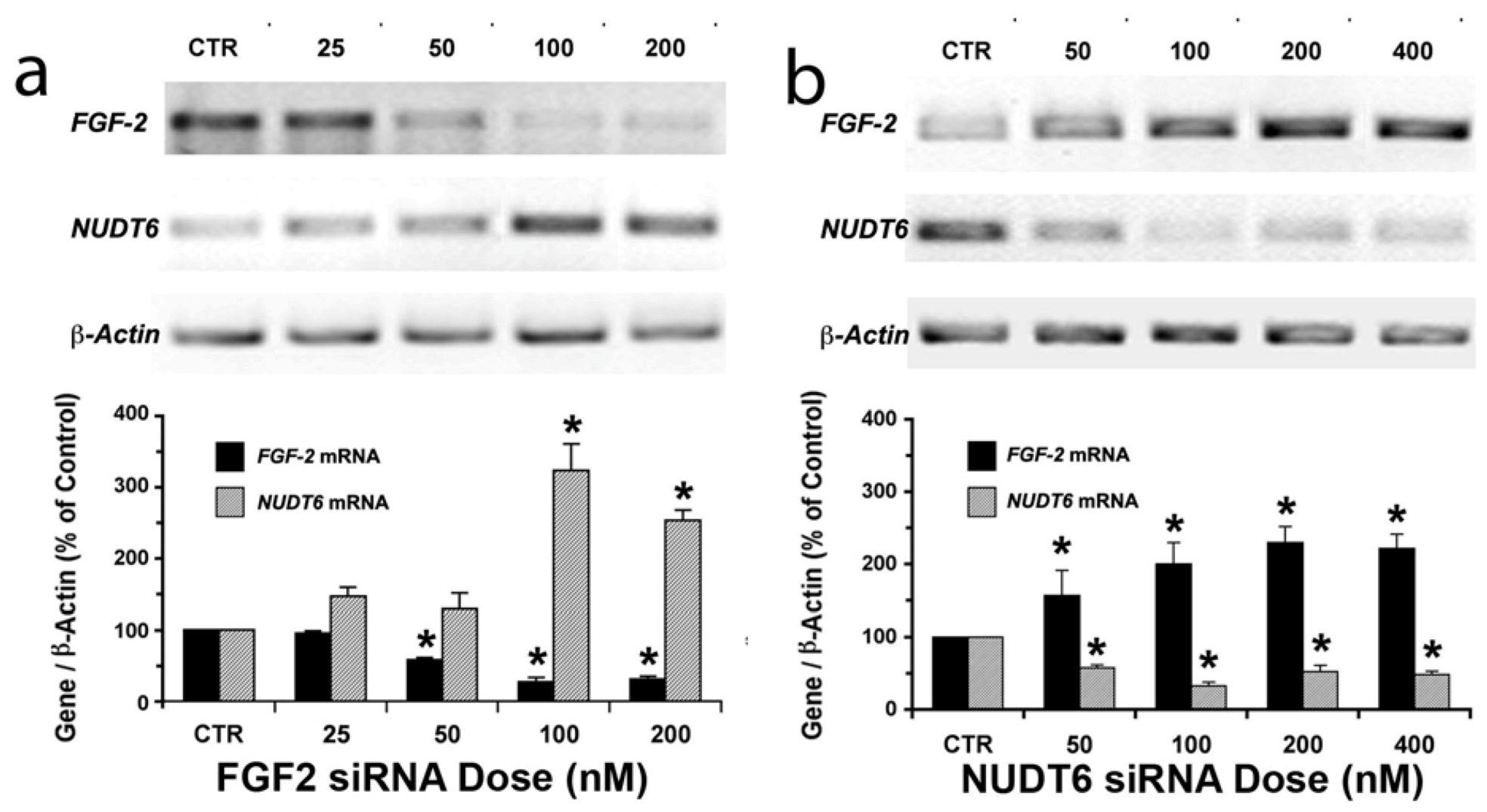
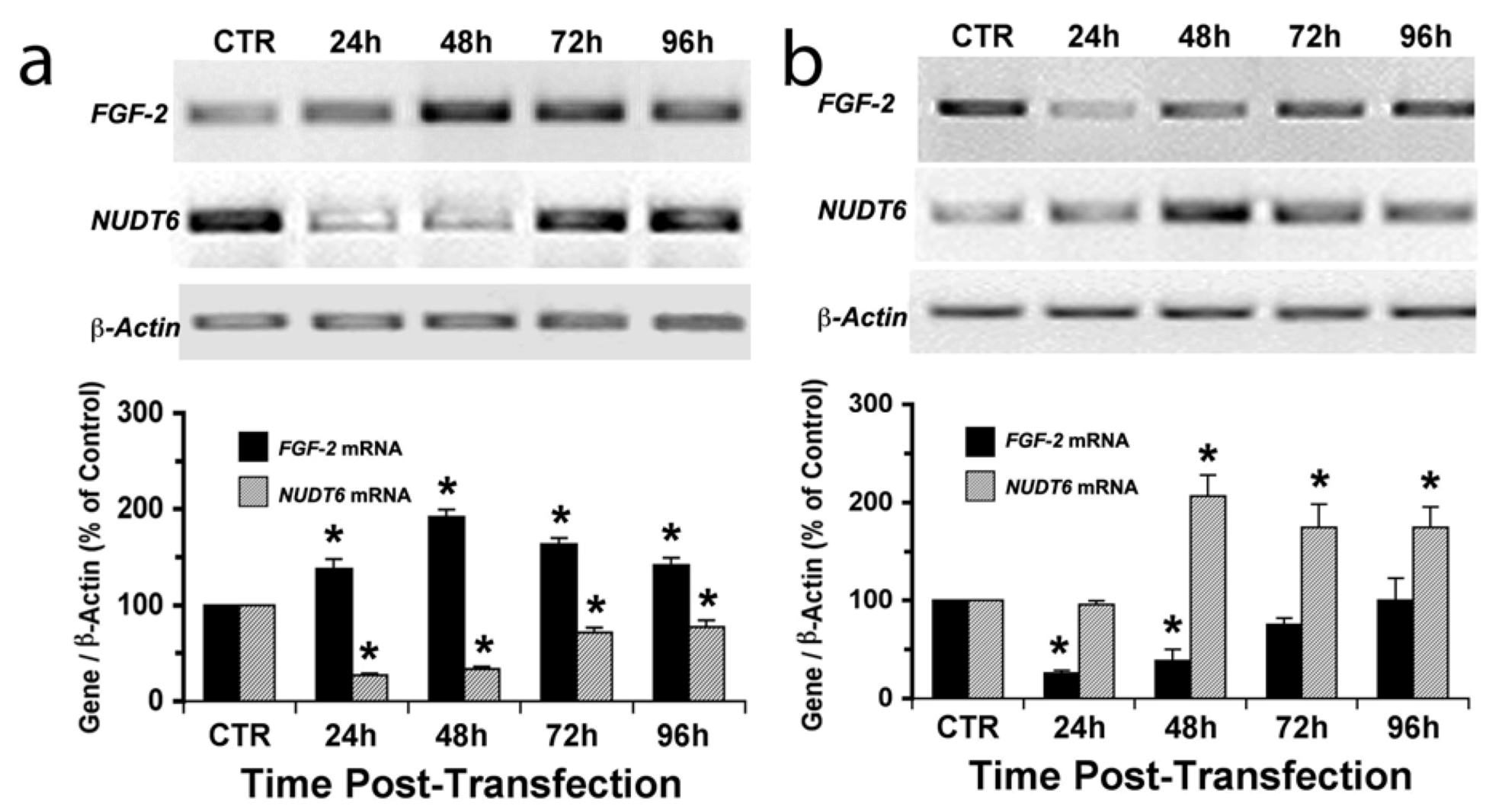
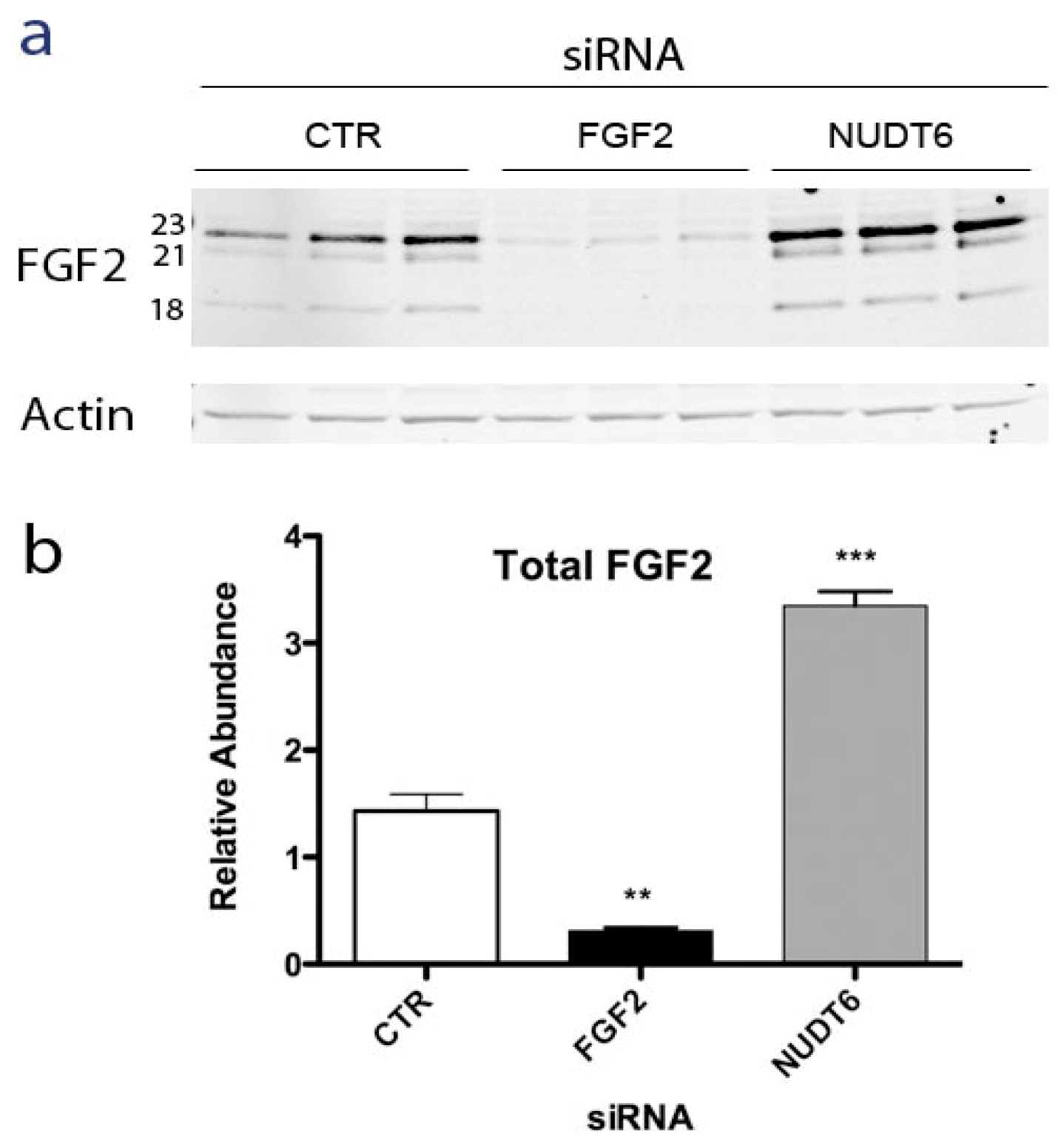
2.2. Effect of NUDT6 Knockdown on FGF2 Protein Isoform Expression
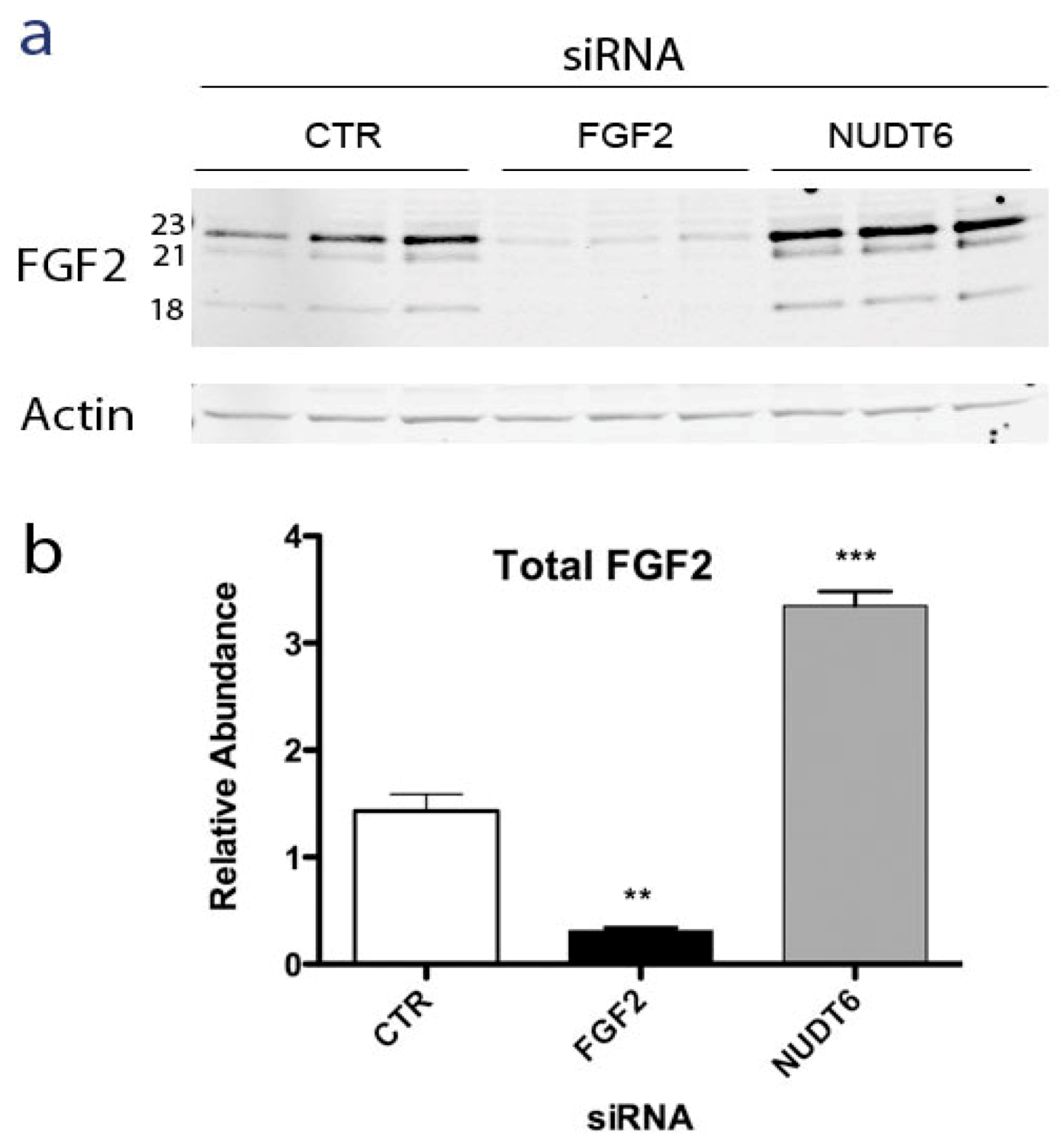
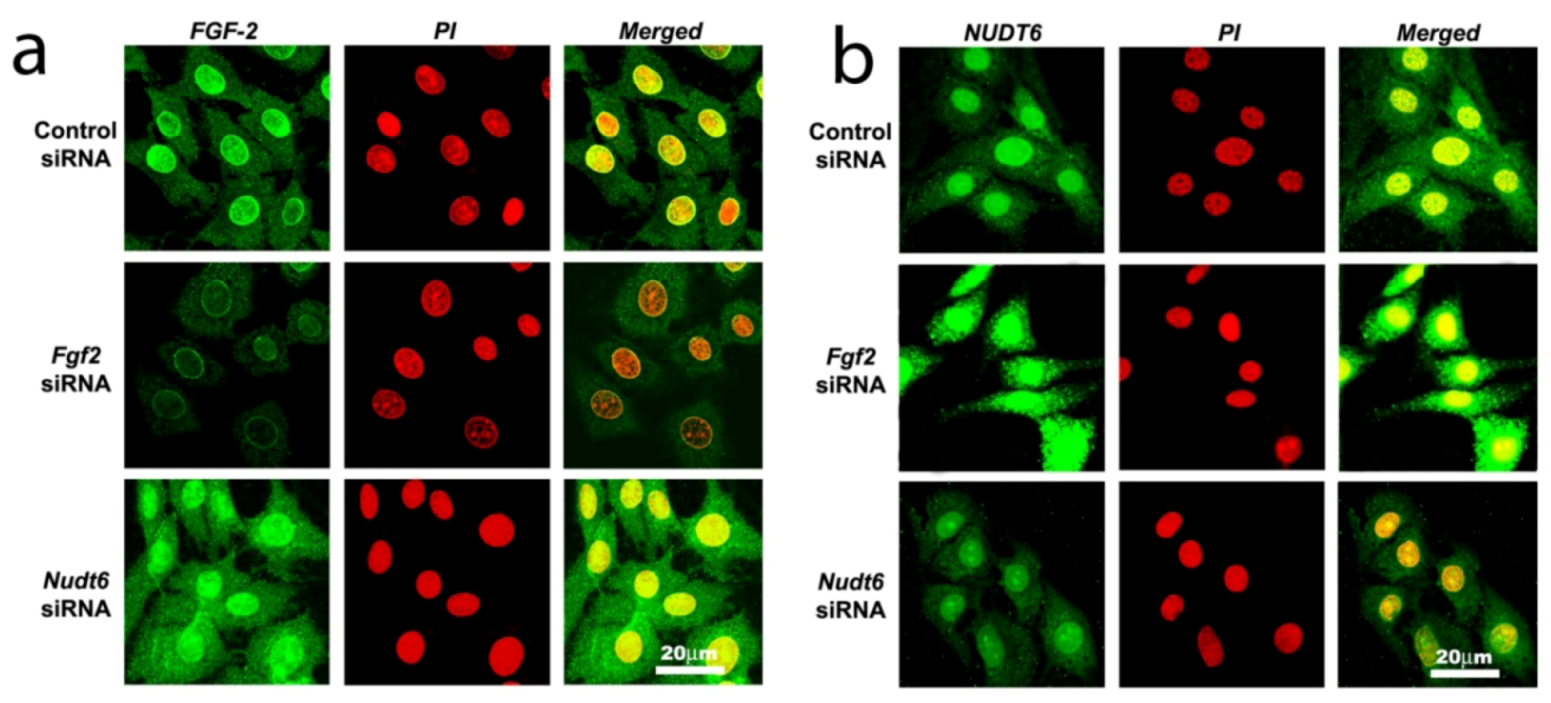
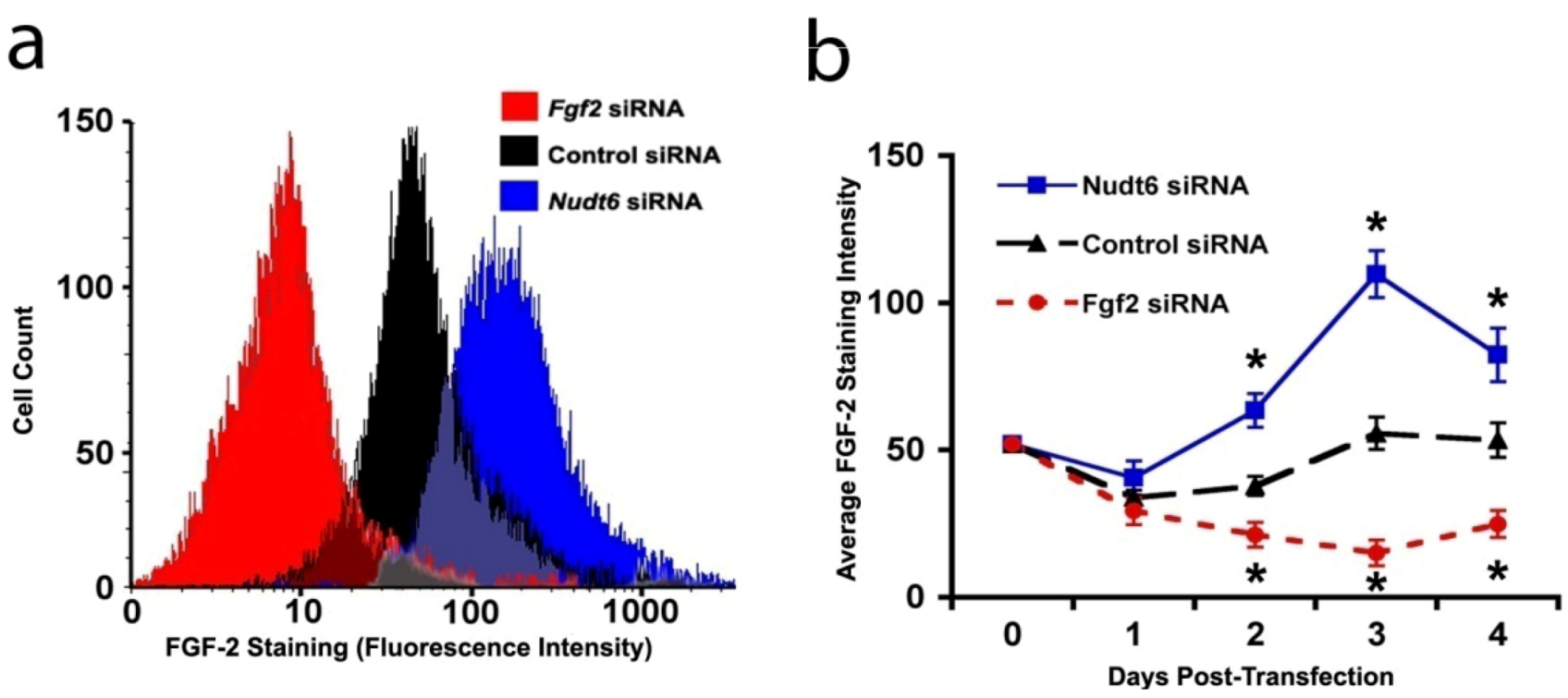
2.3. Effect of RNAi-Mediated Knockdown of FGF2 and NUDT6 mRNA on Their Cognate Proteins
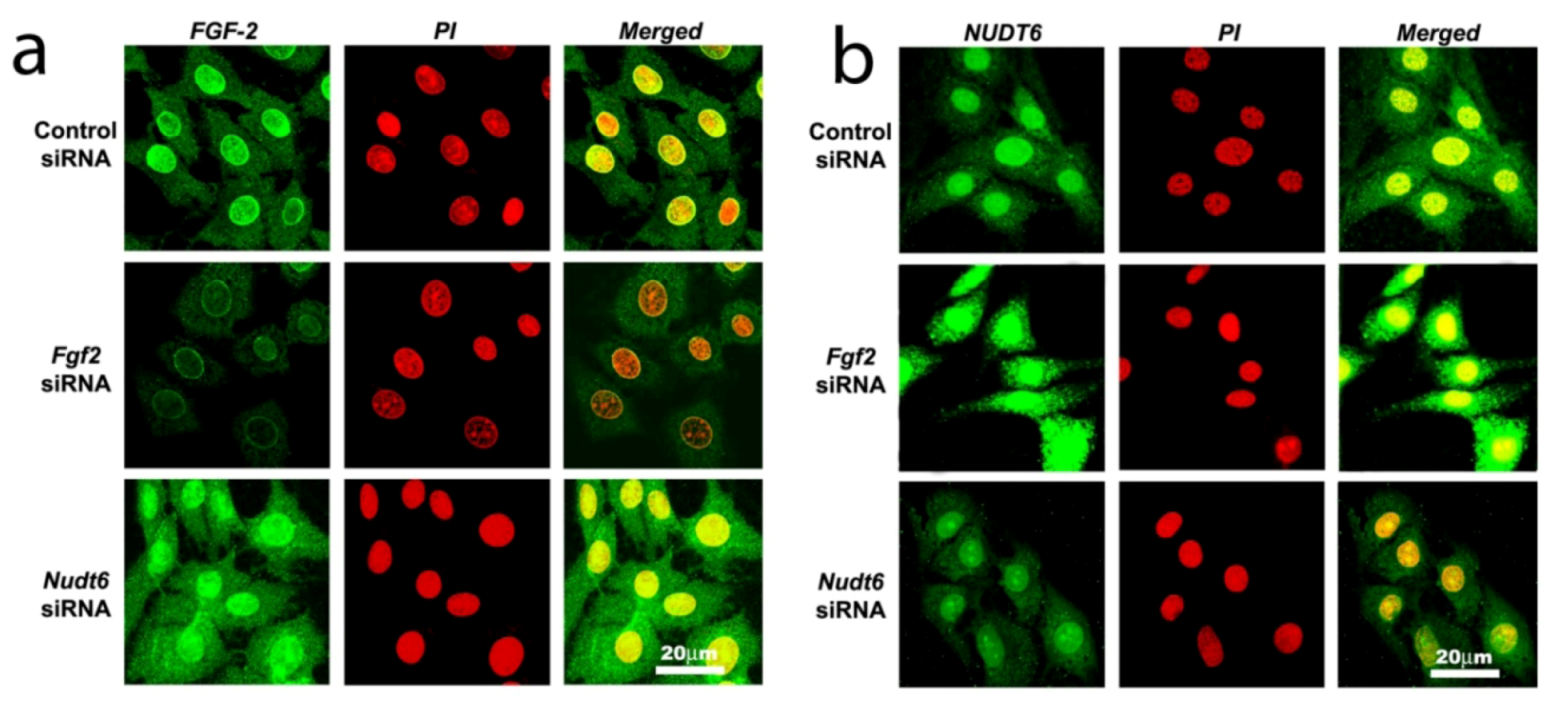
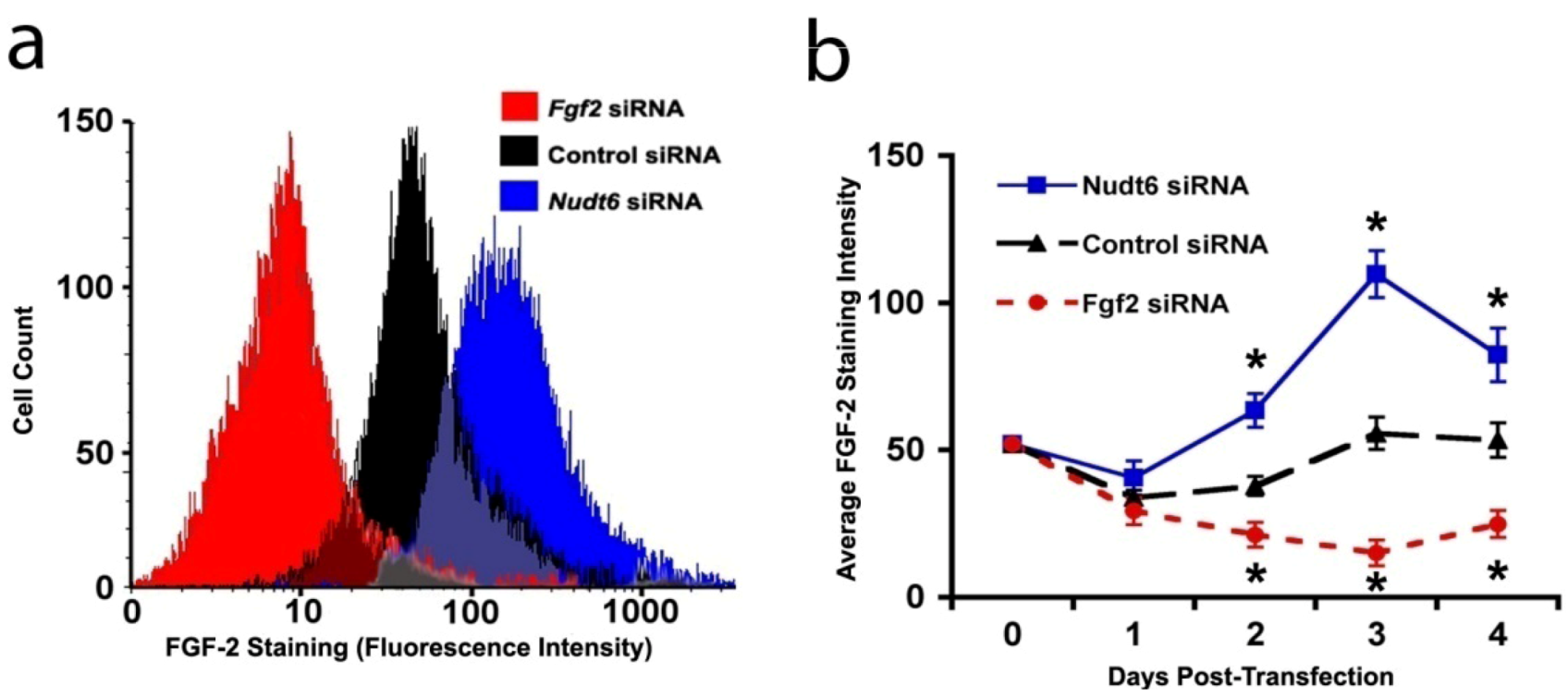
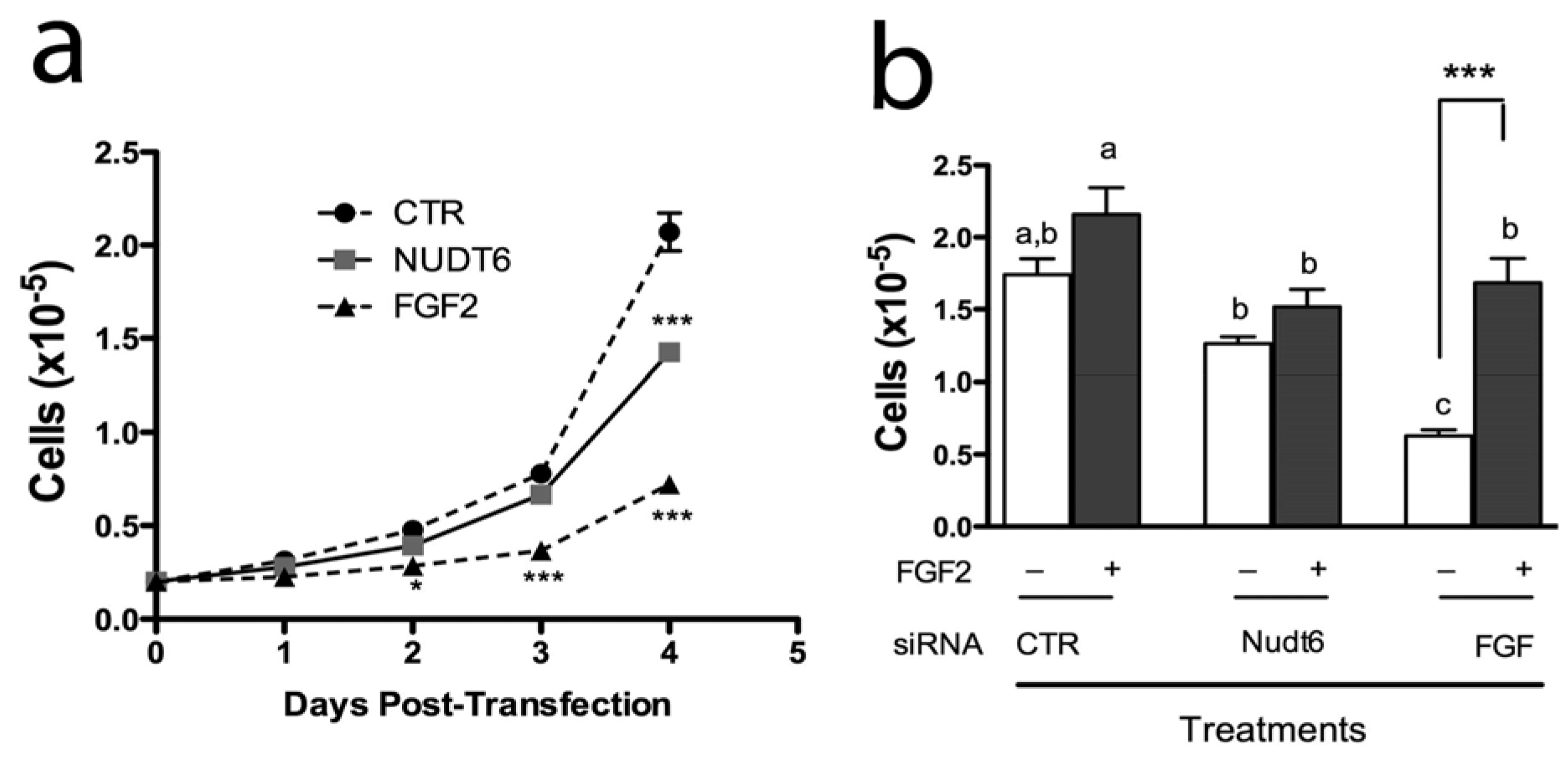
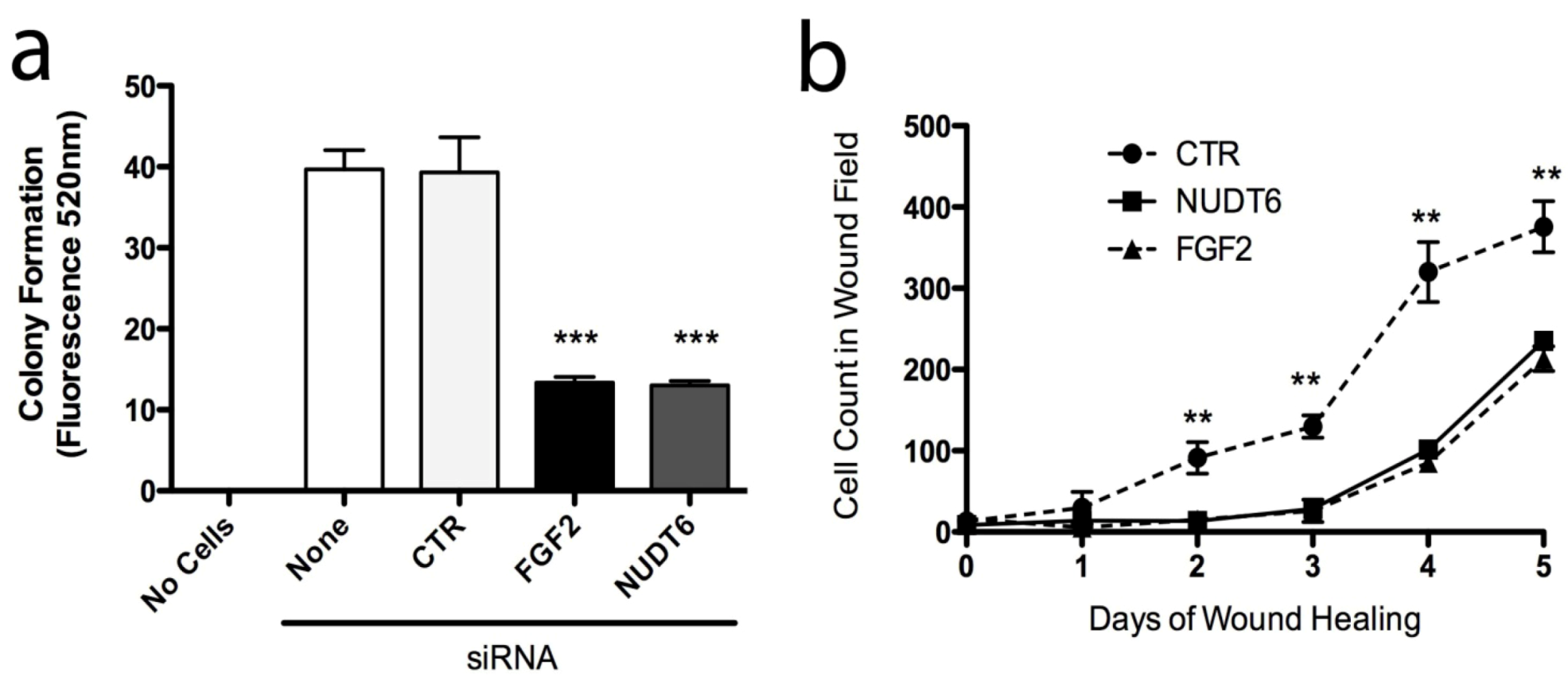
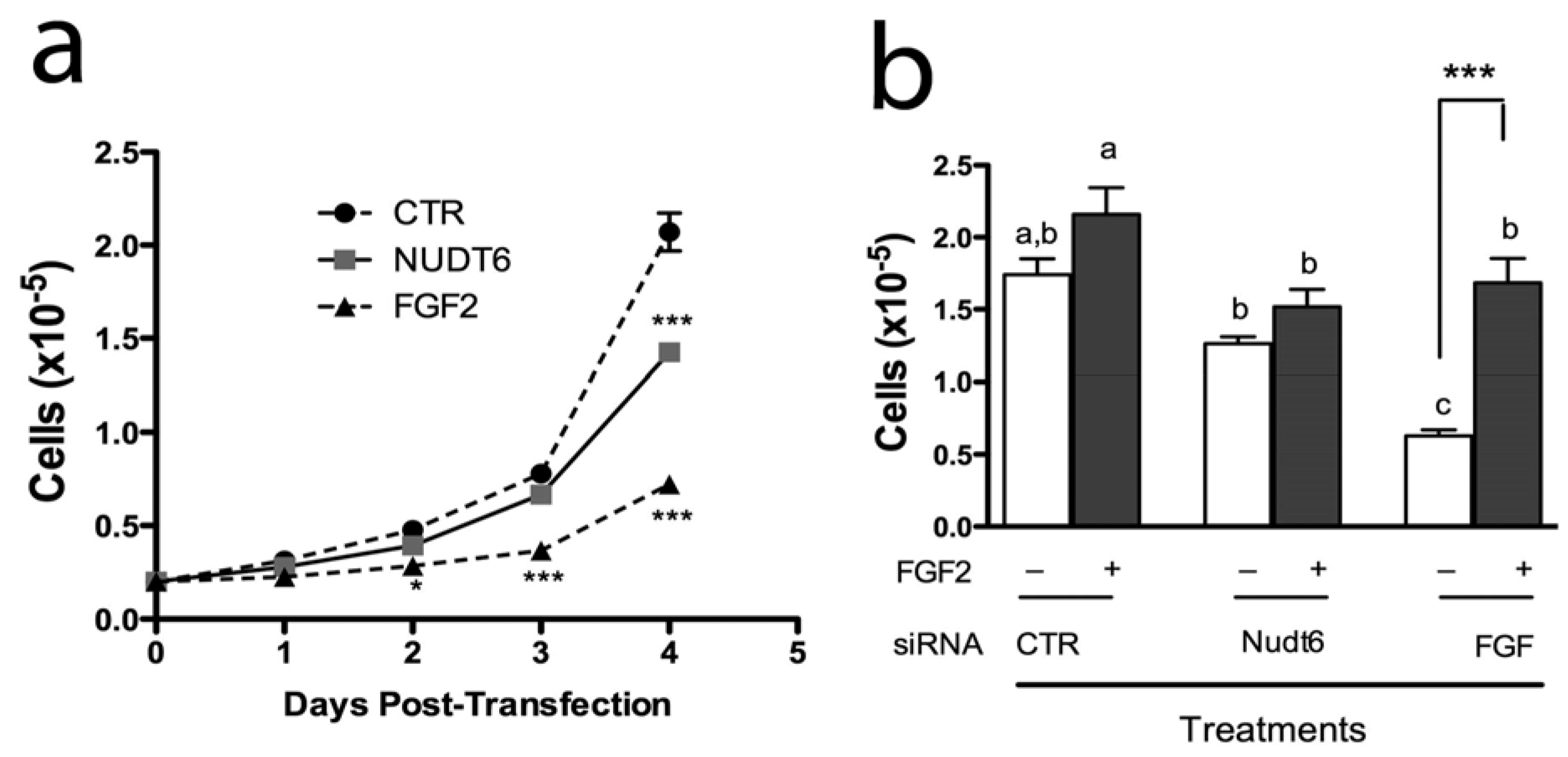
2.4. Effect of FGF2 and NUDT6 Knockdown on Cell Proliferation
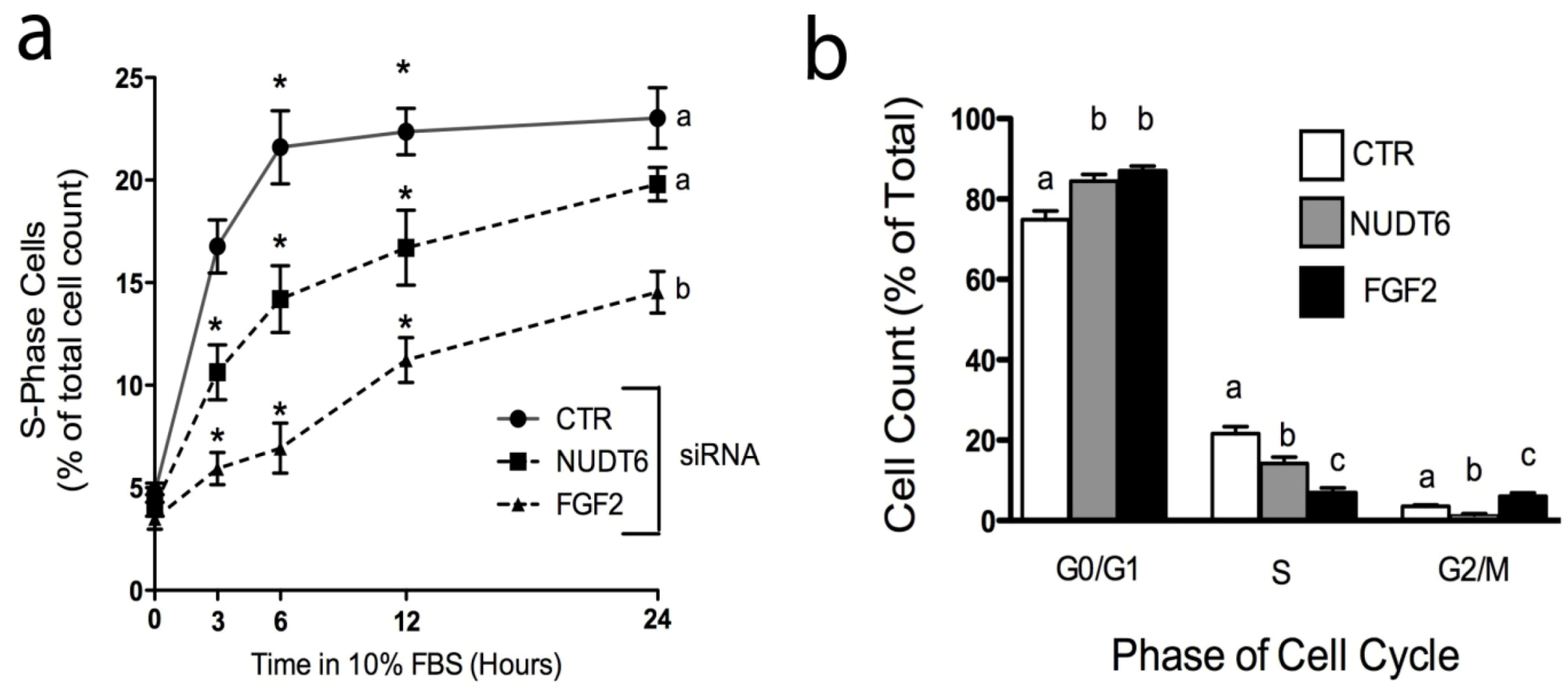
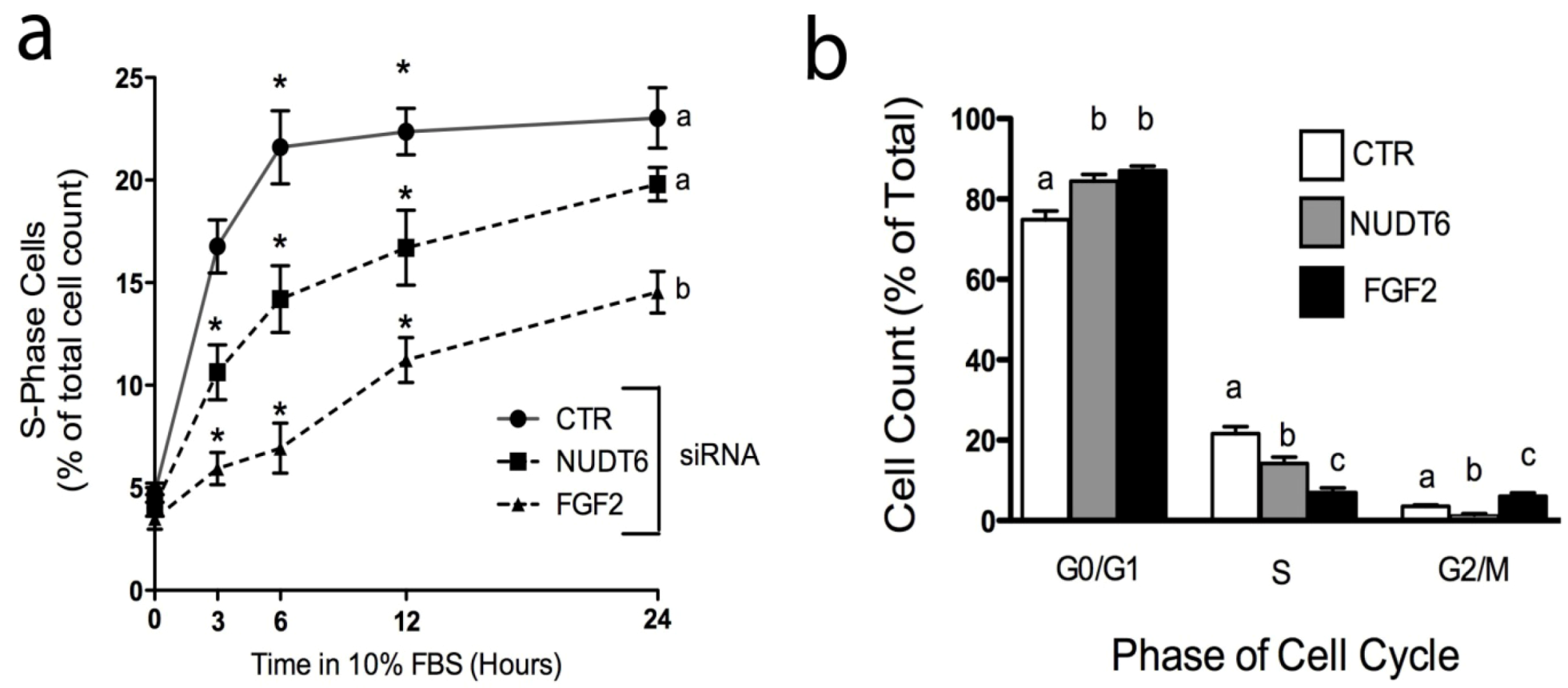
2.5. Effect of Knockdown on S-phase Entry and Cell Cycle Progression
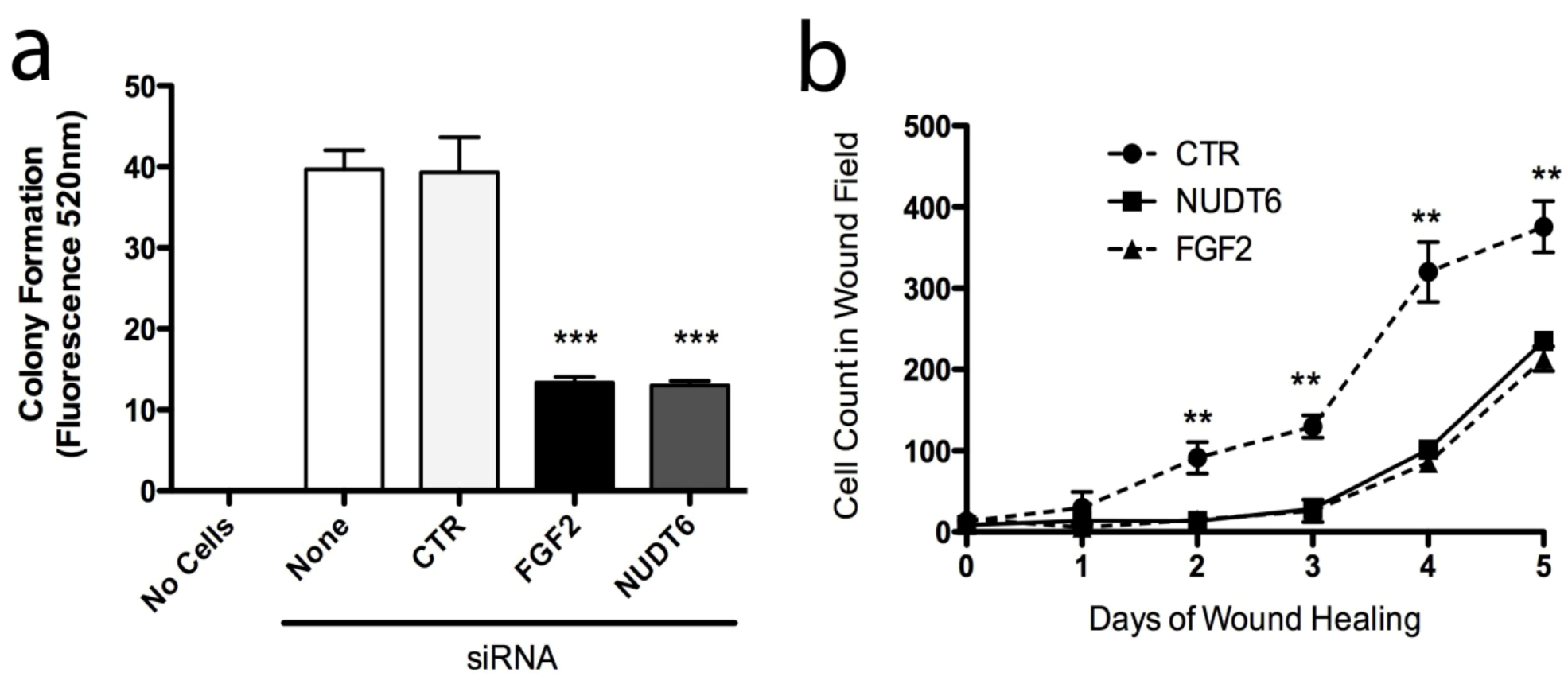
2.6. Effect of Knockdown on Cell Transformation
3. Experimental Section
3.1. Chemicals and Reagents
3.2. Cell Culture and Transfection
3.3. RNA Isolation and RT-PCR Amplification
3.4. Protein Extraction and Western Blotting

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Primer Sequence a, 5' to 3' | Product Size (bp) |
|---|---|---|
| FGF2 | f: GAG GAG TTG TGT CCA TCA AG | 230 |
| r: GGC CTT CTG TCC AGG CCC CG | ||
| NUDT6 | f: GCT CTT GCA GGC CGC CAT TCA G | 226 |
| r: AAA TAC GGC ACC TGC AAC CCC TA | ||
 | f: TGG CCT TAG GGT TCA GAG GGG | 244 |
| r: ATC GTG GGC CGC CCT AGG CA |
3.5. Immunofluorescent Confocal Laser Scanning Microscopy
3.6. Cell Cycle, FGF2 Staining Intensity and Proliferation Analyses
3.7. Cell Transformation Assays
3.7.1. Anchorage-Independent Cell Growth
3.7.2. Cell Adhesion
3.7.3. Cell Migration
3.7.4. Cell Invasion
3.7.5. Wound Healing
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
References
- Yelin, R.; Dahary, D.; Sorek, R.; Levanon, E.Y.; Goldstein, O.; Shoshan, A.; Diber, A.; Biton, S.; Tamir, Y.; Khosravi, R.; et al. Widespread occurrence of antisense transcription in the human genome. Nat. Biotechnol. 2003, 21, 379–386. [Google Scholar] [CrossRef]
- Morris, K.V. Long antisense non-coding rnas function to direct epigenetic complexes that regulate transcription in human cells. Epigenetics 2009, 4, 296–301. [Google Scholar] [CrossRef]
- Svoboda, P. Long dsrna and silent genes strike back:Rnai in mouse oocytes and early embryos. Cytogenet. Genome Res. 2004, 105, 422–434. [Google Scholar] [CrossRef]
- McLennan, A.G. The nudix hydrolase superfamily. Cell. Mol. Life Sci. 2006, 63, 123–143. [Google Scholar] [CrossRef]
- Asa, S.L.; Ramyar, L.; Murphy, P.R.; Li, A.W.; Ezzat, S. The endogenous fibroblast growth factor-2 antisense gene product regulates pituitary cell growth and hormone production. Mol. Endocrinol. 2001, 15, 589–599. [Google Scholar] [CrossRef]
- Macfarlane, L.A.; Murphy, P. Nudt6 (nudix (nucleoside diphosphate linked moiety x)-type motif 6). Atlas Genet. Cytogenet. Oncol. Haematol. 2011, 15, 29–33. [Google Scholar]
- Sukhthankar, M.; Choi, C.K.; English, A.; Kim, J.S.; Baek, S.J. A potential proliferative gene, nudt6, is down-regulated by green tea catechins at the posttranscriptional level. J. Nutr. Biochem. 2009. [Google Scholar]
- Gagnon, M.L.; Moy, G.K.; Klagsbrun, M. Characterization of the promoter for the human antisense fibroblast growth factor-2 gene; regulation by ets in jurkat t cells. J. Cell. Biochem. 1999, 72, 492–506. [Google Scholar] [CrossRef]
- Knee, R.; Li, A.W.; Murphy, P.R. Characterization and tissue-specific expression of the rat basic fibroblast growth factor antisense mrna and protein. Proc. Natl. Acad. Sci. USA 1997, 94, 4943–4947. [Google Scholar]
- Kimelman, D.; Kirschner, M.W. An antisense messenger rna directs the covalent modification of the transcript encoding fibroblast growth factor in xenopus oocytes. Cell 1989, 59, 687–696. [Google Scholar] [CrossRef]
- Zuniga Mejia Borja, A.; Meijers, C.; Zeller, L. Expression of alternatively spliced bfgf first coding exons and antisense mrnas during chicken embryogenesis. Dev. Biol. 1993, 157, 110–118. [Google Scholar] [CrossRef]
- Knee, R.S.; Pitcher, S.E.; Murphy, P.R. Basic fibroblast growth factor sense (fgf) and antisense (gfg) rna transcripts are expressed in unfertilized human oocytes and in differentiated adult tissues. Biochem. Biophys. Res. Comm. 1994, 205, 577–583. [Google Scholar] [CrossRef]
- Li, A.; Seyoum, G.; Shiu, R.; Murphy, P. Expression of the rat bfgf antisense rna transcript is tissue-specific and developmentally regulated. Mol. Cell. Endocrinol. 1996, 118, 113–123. [Google Scholar] [CrossRef]
- Baguma-Nibasheka, M.; Li, A.W.; Osman, M.S.; Geldenhuys, L.; Casson, A.G.; Too, C.K.; Murphy, P.R. Coexpression and regulation of the fgf-2 and fgf antisense genes in leukemic cells. Leuk. Res. 2005, 29, 423–433. [Google Scholar] [CrossRef]
- Barclay, C.; Li, A.W.; Geldenhuys, L.; Baguma-Nibasheka, M.; Porter, G.A.; Veugelers, P.J.; Murphy, P.R.; Casson, A.G. Basic fibroblast growth factor (fgf-2) overexpression is a risk factor for esophageal cancer recurrence and reduced survival, which is ameliorated by coexpression of the fgf-2 antisense gene. Clin. Cancer Res. 2005, 11, 7683–7691. [Google Scholar] [CrossRef]
- Zhang, S.C.; Barclay, C.; Alexander, L.A.; Geldenhuys, L.; Porter, G.A.; Casson, A.G.; Murphy, P.R. Alternative splicing of the fgf antisense gene: Differential subcellular localization in human tissues and esophageal adenocarcinoma. J. Mol. Med. (Berlin, Germany) 2007, 85, 1215–1228. [Google Scholar] [CrossRef]
- MacFarlane, L.A.; Murphy, P.R. Regulation of fgf-2 by an endogenous antisense rna: Effects on cell adhesion and cell-cycle progression. Mol. Carcinog. 2010, 49, 1031–1044. [Google Scholar] [CrossRef]
- Mihalich, A.; Reina, M.; Mangioni, S.; Ponti, E.; Alberti, L.; Vigano, P.; Vignali, M.; di Blasio, A.M. Different basic fibroblast growth factor and fibroblast growth factor-antisense expression in eutopic endometrial stromal cells derived from women with and without endometriosis. J. Clin. Endocrinol. Metab. 2003, 88, 2853–2859. [Google Scholar] [CrossRef]
- Baguma-Nibasheka, M.; Li, A.W.; Murphy, P.R. The fibroblast growth factor-2 antisense gene inhibits nuclear accumulation of fgf-2 and delays cell cycle progression in c6 glioma cells. Mol. Cell. Endocrinol. 2007, 267, 127–136. [Google Scholar] [CrossRef]
- Li, A.W.; Murphy, P.R. Expression of alternatively spliced fgf-2 antisense rna transcripts in the central nervous system: Regulation of fgf-2 mrna translation. Mol. Cell. Endocrinol. 2000, 170, 233–242. [Google Scholar]
- Arese, M.; Chen, Y.; Florkiewicz, R.Z.; Gualandris, A.; Shen, B.; Rifkin, D.B. Nuclear activities of basic fibroblast growth factor: Potentiation of low-serum growth mediated by natural or chimeric nuclear localization signals. Mol. Biol. Cell 1999, 10, 1429–1444. [Google Scholar]
- Arnaud, E.; Touriol, C.; Boutonnet, C.; Gensac, M.-C.; Vagner, S.; Prats, H.; Prats, A.-C. A new 34-kilodalton isoform of human fibroblast growth factor 2 is cap dependently synthesized by using a non-aug start codon and behaves as a survival factor. Mol. Cell. Biol. 1999, 19, 505–514. [Google Scholar]
- Vagner, S.; Gensac, M.C.; Maret, A.; Bayard, F.; Amalric, F.; Prats, H.; Prats, A.C. Alternative translation of human fibroblast growth factor 2 mrna occurs by internal entry of ribosomes. Mol. Cell. Biol. 1995, 15, 35–44. [Google Scholar]
- Mignatti, P.; Morimoto, T.; Rifkin, D.B. Basic fibroblast growth factor, a protein devoid of secretory signal sequence, is released by cells via a pathway independent of the endoplasmic reticulum-golgi complex. J. Cell Physiol. 1992, 151, 81–93. [Google Scholar] [CrossRef]
- Murphy, P.R.; Knee, R.S. Basic fibroblast growth factor binding and processing by human glioma cells. Mol. Cell. Endocrinol. 1995, 114, 193–203. [Google Scholar] [CrossRef]
- Amalric, F.; Bouche, G.; Bonnet, H.; Brethenou, P.; Roman, A.M.; Truchet, I.; Quarto, N. Fibroblast growth factor-2 (fgf-2) in the nucleus: Translocation process and targets. Biochem. Pharmacol. 1994, 47, 111–115. [Google Scholar]
- Delrieu, I. The high molecular weight isoforms of basic fibroblast growth factor (fgf-2): An insight into an intracrine mechanism. FEBS Lett. 2000, 468, 6–10. [Google Scholar] [CrossRef]
- Zhang, S.C.; MacDonald, K.A.; Baguma-Nibasheka, M.; Geldenhuys, L.; Casson, A.G.; Murphy, P.R. Alternative splicing and differential subcellular localization of the rat fgf antisense gene product. BMC Mol. Biol. 2008, 9. [Google Scholar]
- Murphy, P.; Sato, Y.; Knee, R. Phosphorothioate antisense oligonucleotides against basic fibroblast growth factor inhibit anchorage-dependent and anchorage-independent growth of a malignant glioblastoma cell line. Mol. Endocrinol. 1992, 6, 877–884. [Google Scholar] [CrossRef]
- Katayama, S.; Tomaru, Y.; Kasukawa, T.; Waki, K.; Nakanishi, M.; Nakamura, M.; Nishida, H.; Yap, C.C.; Suzuki, M.; Kawai, J.; et al. Antisense transcription in the mammalian transcriptome. Science (New York, N.Y.) 2005, 309, 1564–1566. [Google Scholar]
- Wahlestedt, C. Natural antisense and noncoding rna transcripts as potential drug targets. Drug Discov. Today 2006, 11, 503–508. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Baguma-Nibasheka, M.; MacFarlane, L.A.; Murphy, P.R. Regulation of Fibroblast Growth Factor-2 Expression and Cell Cycle Progression by an Endogenous Antisense RNA. Genes 2012, 3, 505-520. https://doi.org/10.3390/genes3030505
Baguma-Nibasheka M, MacFarlane LA, Murphy PR. Regulation of Fibroblast Growth Factor-2 Expression and Cell Cycle Progression by an Endogenous Antisense RNA. Genes. 2012; 3(3):505-520. https://doi.org/10.3390/genes3030505
Chicago/Turabian StyleBaguma-Nibasheka, Mark, Leigh Ann MacFarlane, and Paul R. Murphy. 2012. "Regulation of Fibroblast Growth Factor-2 Expression and Cell Cycle Progression by an Endogenous Antisense RNA" Genes 3, no. 3: 505-520. https://doi.org/10.3390/genes3030505