Polymorphisms in Fatty Acid Desaturase (FADS) Gene Cluster: Effects on Glycemic Controls Following an Omega-3 Polyunsaturated Fatty Acids (PUFA) Supplementation


Abstract
:1. Introduction
2. Experimental Section
2.1. Study Population
2.2. Study Design and Diets
2.3. Anthropometric Measurements
2.4. Biochemical Parameters
 [7]. Plasma C-reactive protein (CRP) was measured by nephelometry (Prospec equipment Behring) using a sensitive assay, as described previously [20]. Plasma total cholesterol (TC) and TG concentrations were measured using enzymatic assays [21]. The high-density lipoprotein cholesterol (HDL-C) fraction was obtained after precipitation of very low-density lipoprotein and low-density lipoprotein particles in the infranatant with heparin manganese chloride [22]. Low-density lipoprotein cholesterol (LDL-C) was calculated with the Friedewald formula [23]. Apolipoprotein B-100 (ApoB100) concentrations were measured in plasma by the rocket immunoelectrophoretic method of Laurell, as previously described [24].
[7]. Plasma C-reactive protein (CRP) was measured by nephelometry (Prospec equipment Behring) using a sensitive assay, as described previously [20]. Plasma total cholesterol (TC) and TG concentrations were measured using enzymatic assays [21]. The high-density lipoprotein cholesterol (HDL-C) fraction was obtained after precipitation of very low-density lipoprotein and low-density lipoprotein particles in the infranatant with heparin manganese chloride [22]. Low-density lipoprotein cholesterol (LDL-C) was calculated with the Friedewald formula [23]. Apolipoprotein B-100 (ApoB100) concentrations were measured in plasma by the rocket immunoelectrophoretic method of Laurell, as previously described [24]. 2.5. SNP Selection and Genotyping
2.6. Statistical Analyses
3. Results and Discussion

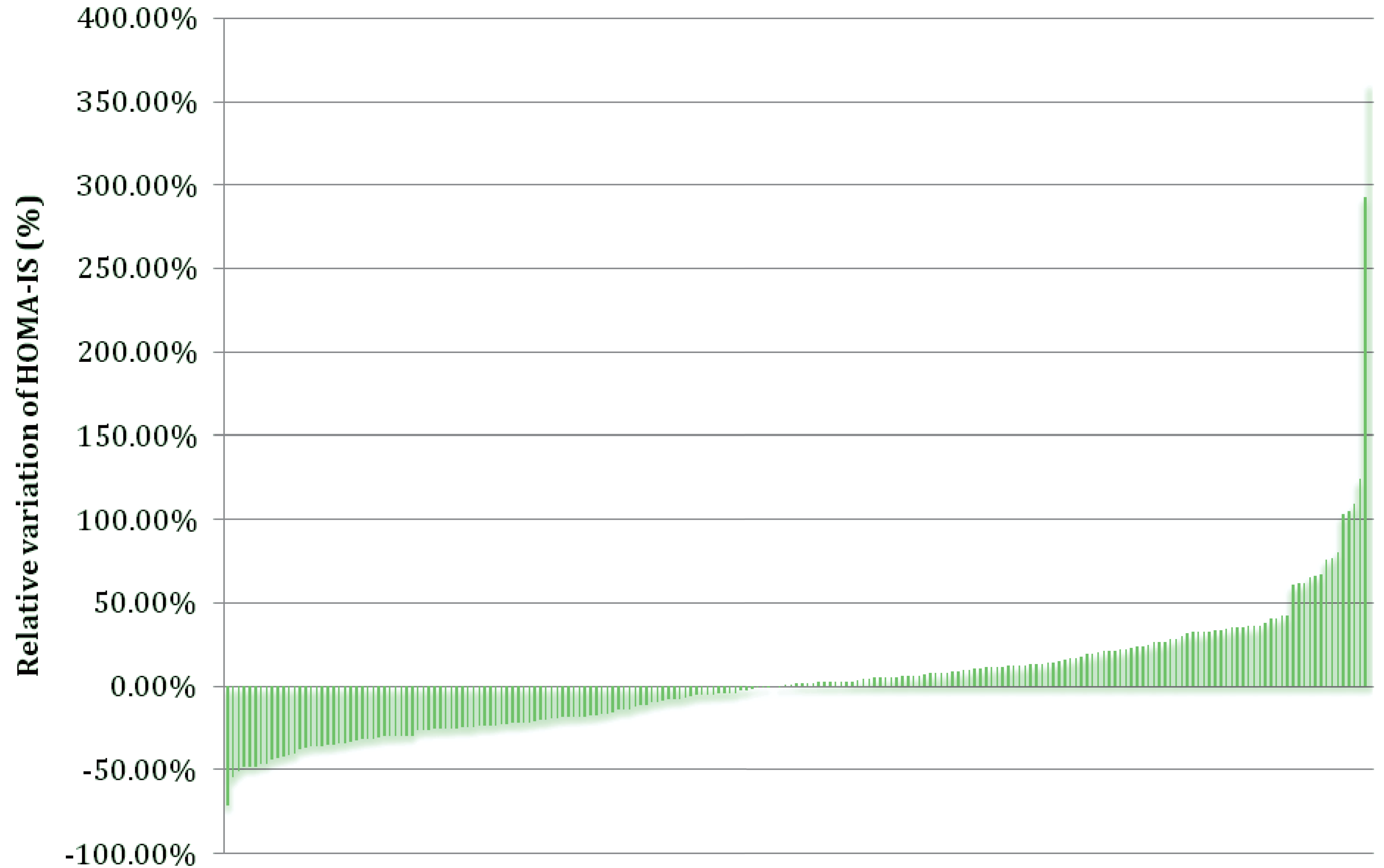{kind=link}
| All 1 | Men 1 | Women 1 | |
|---|---|---|---|
| Population. Men/Women | 208 | 96 (46.2%) | 112 (53.8%) |
| Age (years) | 30.8 ± 8.7 | 31.2 ± 8.1 | 30.5 ± 9.1 |
| Weight (kg) 3 | 81.4 ± 13.9 | 87.2 ± 13.4 | 76.4 ± 12.3 |
| BMI (kg/m2) 2,3 | 27.8 ± 3.7 | 27.5 ± 3.6 | 28.2 ± 3.8 |
| Waist circumference (cm) 3 | 93.3 ± 10.8 | 94.8 ± 11.0 | 92.0 ± 10.4 |
| Cholesterol (mM) 4 | |||
| Total | 4.82 ± 1.00 | 4.80 ± 1.00 | 4.83 ± 1.02 |
| HDL | 1.46 ± 0.39 | 1.29 ± 0.31 | 1.61 ± 0.39 |
| LDL | 2.79 ± 0.87 | 2.91 ± 0.87 | 2.69 ± 0.86 |
| Total chol./HDL ratio 4 | 3.49 ± 1.04 | 3.91 ± 1.13 | 3.12 ± 0.80 |
| Triacylglycerols (mM) 2,4 | 1.23 ± 0.64 | 1.32 ± 0.74 | 1.15 ± 0.53 |
| ApoB100 (g/L) 4 | 0.86 ± 0.25 | 0.89 ± 0.25 | 0.84 ± 0.25 |
| CRP (mg/L) 2,4 | 3.13 ± 7.10 | 1.66 ± 2.45 | 4.39 ± 9.24 |
| Glycemic controls | |||
| Glucose (mM) 4 | 4.95 ± 0.52 | 5.09 ± 0.44 | 4.83 ± 0.56 |
| Insulin (ρ/L) 4 | 82.51 ± 35.61 | 79.50 ± 32.19 | 85.04 ± 38.20 |
| FG | FI 1 | HOMA-IS | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | Suppl. | Interaction | Genotype | Suppl. | Interaction | Genotype | Suppl. | Interaction | ||
| rs174456 | 0.28 | 0.06 | 0.42 | 0.04 | 0.14 | 0.16 | 0.84 | 0.77 | 0.84 | |
| rs174627 | 0.81 | 0.002 | 0.93 | 0.98 | 0.97 | 0.74 | 0.89 | 0.52 | 0.90 | |
| rs482548 | 0.05 | <0.0001 | 0.008 | 0.18 | 0.59 | 0.16 | 0.11 | 0.13 | 0.07 | |
| rs2072114 | 0.07 | 0.05 | 0.11 | 0.25 | 0.36 | 0.23 | 0.65 | 0.49 | 0.71 | |
| rs12807005 | 0.66 | 0.04 | 0.38 | 0.30 | 0.41 | 0.33 | 0.90 | 0.83 | 0.99 | |
| rs174448 | 0.79 | 0.002 | 0.65 | 0.85 | 0.66 | 0.84 | 0.61 | 0.27 | 0.45 | |
| rs2845573 | 0.35 | 0.03 | 0.55 | 0.67 | 0.81 | 0.92 | 0.41 | 0.19 | 0.24 | |
| rs7394871 | 0.58 | 0.05 | 0.44 | 0.41 | 0.55 | 0.29 | 0.05 | 0.04 | 0.03 | |
| rs7942717 | 0.93 | 0.004 | 0.83 | 0.27 | 0.67 | 0.67 | 0.99 | 0.40 | 0.61 | |
| rs7482316 | 0.48 | 0.003 | 0.83 | 0.05 | 0.39 | 0.08 | 0.03 | 0.09 | 0.05 | |
| rs174602 | 0.88 | 0.0006 | 0.79 | 0.46 | 0.72 | 0.11 | 0.09 | 0.12 | 0.01 | |
| rs498793 | 0.60 | <0.0001 | 0.16 | 0.50 | 0.84 | 0.63 | 0.26 | 0.33 | 0.25 | |
| rs174546 | 0.34 | 0.03 | 0.52 | 0.44 | 0.58 | 0.74 | 0.42 | 0.28 | 0.42 | |
| rs174570 | 0.67 | 0.004 | 0.80 | 0.33 | 0.61 | 0.18 | 0.07 | 0.08 | 0.03 | |
| rs174579 | 0.58 | 0.0004 | 0.77 | 0.97 | 0.88 | 0.71 | 0.38 | 0.39 | 0.35 | |
| rs174611 | 0.88 | 0.004 | 0.73 | 0.75 | 0.86 | 0.82 | 0.54 | 0.44 | 0.65 | |
| rs174616 | 0.49 | 0.0003 | 0.27 | 0.44 | 0.45 | 0.35 | 0.21 | 0.61 | 0.11 | |
| rs968567 | 0.74 | 0.0007 | 0.84 | 0.51 | 0.71 | 0.68 | 0.68 | 0.45 | 0.71 | |
| Pre-n-3 PUFA | Post-n-3 PUFA | P 1 | β-values ± SE | ||||
|---|---|---|---|---|---|---|---|
| 11 | 12 + 22 | 11 | 12 + 22 | ||||
| HOMA-IS | 0.067 ± 0.025 | 0.063 ± 0.027 | 0.068 ± 0.030 | 0.056 ± 0.022 | 0.01 | 11 | 0.4169 ± 0.0375 |
| rs174602 | 12 + 22 | 0.4391 ± 0.0379 | |||||
| HOMA-IS | 0.066 ± 0.025 | 0.065 ± 0.029 | 0.066 ± 0.029 | 0.057 ± 0.025 | 0.03 | 11 | 0.4106 ± 0.0378 |
| rs174570 | 12 + 22 | 0.4248 ± 0.0384 | |||||
| HOMA-IS | 0.065 ± 0.025 | 0.068 ± 0.033 | 0.065 ± 0.028 | 0.059 ± 0.025 | 0.03 | 11 | 0.4150 ± 0.0378 |
| rs7394871 | 12 + 22 | 0.4319 ± 0.0391 | |||||
| FG | 4.96 ± 0.43 | 4.92 ± 0.54 | 5.02 ± 0.48 | 5.15 ± 0.52 | 0.008 | 11 | 1.5753 ± 0.7327 |
| rs482548 | 12 + 22 | 1.2278 ± 0.7443 | |||||

| Gene | dbSNP No. 1 | Sequence 2 | Position | MAF | Genotype/Frequency | ||
|---|---|---|---|---|---|---|---|
| 11 | 12 | 22 | |||||
| FADS3 | rs174456 | CTAC[A/C]TGGC | Intron | 0.299 | A/A (n = 102) | A/C (n = 89) | C/C (n = 18) |
| 0.488 | 0.426 | 0.086 | |||||
| Intergenic FADS2-FADS3 | rs174627 | TCTG[C/T]GTAG | Intergenic | 0.124 | G/G (n = 159) | A/G (n = 48) | A/A (n = 2) |
| 0.761 | 0.230 | 0.010 | |||||
| FADS2 | rs482548 | ACAC[C/T]GTGG | 3' UTR | 0.126 | C/C (n = 161) | C/T (n = 40) | T/T (n = 6) |
| 0.778 | 0.193 | 0.029 | |||||
| FADS2 | rs2072114 | GTTC[A/G]GGTC | Intron | 0.110 | A/A (n = 167) | A/G (n = 38) | G/G (n = 4) |
| 0.799 | 0.182 | 0.019 | |||||
| Intergenic FADS1-FADS2 | rs12807005 | CATG[A/G]ATCA | Intergenic | 0.012 | G/G (n = 204) | A/G (n = 5) | A/A (n = 0) |
| 0.976 | 0.024 | 0.000 | |||||
| Intergenic FADS2-FADS3 | rs174448 | CTGA[C/T]TTCT | Intergenic | 0.363 | A/A (n = 78) | A/G (n = 109) | G/G (n = 21) |
| 0.375 | 0.524 | 0.101 | |||||
| FADS2 | rs2845573 | CTCA[C/T]GTTA | Intron | 0.081 | A/A (n = 177) | A/G (n = 30) | G/G (n = 2) |
| 0.847 | 0.144 | 0.010 | |||||
| FADS3 | rs7394871 | GGAC[A/C]CCTG | Intron | 0.072 | C/C (n = 181) | A/C (n = 26) | A/A (n = 2) |
| 0.866 | 0.124 | 0.010 | |||||
| FADS3 | rs7942717 | AACG[A/G]GTGC | Intron | 0.117 | A/A (n = 161) | A/G (n = 47) | G/G (n = 1) |
| 0.770 | 0.225 | 0.005 | |||||
| Intergenic FADS2-FADS3 | rs7482316 | TCAA[A/G]CTGC | Intergenic | 0.103 | A/A (n = 168) | A/G (n = 39) | G/G (n = 2) |
| 0.804 | 0.187 | 0.010 | |||||
| FADS2 | rs174602 | ACCC[A/G]TCCT | Intron | 0.184 | T/T (n = 141) | C/T (n = 59) | C/C (n = 9) |
| 0.675 | 0.282 | 0.043 | |||||
| FADS2 | rs498793 | TAAC[A/G]CAGG | Intron | 0.456 | C/C (n = 62) | C/T (n = 99) | T/T (n = 43) |
| 0.098 | 0.717 | 0.186 | |||||
| FADS2 | rs7935946 | GTTC[C/T]GGGA | Intron | 0.041 | C/C (n = 195) | C/T (n = 11) | T/T (n = 3) |
| 0.933 | 0.053 | 0.014 | |||||
| FADS1 | rs174546 | CTGC[C/T]TTGG | 3' UTR | 0.297 | C/C (n = 103) | C/T (n = 86) | T/T (n = 19) |
| 0.498 | 0.412 | 0.091 | |||||
| FADS2 | rs174570 | TTGA[C/T]GTAG | Intron | 0.125 | C/C (n = 159) | C/T (n = 46) | T/T (n = 3) |
| 0.764 | 0.221 | 0.014 | |||||
| FADS2 | rs174579 | CTTT[C/T]CAGG | Intron | 0.202 | C/C (n = 127) | C/T (n = 78) | T/T (n = 3) |
| 0.611 | 0.375 | 0.014 | |||||
| FADS2 | rs174611 | TGGA[C/T]CCTG | Intron | 0.258 | T/T (n = 113) | C/T (n = 84) | C/C (n = 12) |
| 0.541 | 0.402 | 0.057 | |||||
| FADS2 | rs174616 | CTCA[C/T]GTTC | Intron | 0.498 | A/A (n = 51) | A/G (n = 108) | G/G (n = 50) |
| 0.244 | 0.517 | 0.239 | |||||
| FADS2 | rs968567 | CCGG[A/G]AGCT | 5' UTR | 0.160 | G/G (n = 144) | A/G (n = 63) | A/A (n = 2) |
| 0.689 | 0.301 | 0.010 | |||||
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- National Diabetes Fact Sheet. Available online: http://www.cdc.gov/diabetes/pubs/factsheet11.htm/ (accessed on 7 May 2013).
- Wilson, J.F. In the clinic. Type 2 diabetes. Ann. Intern. Med. 2007, 146, ITC1-1. [Google Scholar] [CrossRef]
- Harris, W.S.; Bulchandani, D. Why do omega-3 fatty acids lower serum triglycerides? Curr. Opin. Lipidol. 2006, 17, 387–393. [Google Scholar] [CrossRef]
- McEwen, B.; Morel-Kopp, M.C.; Tofler, G.; Ward, C. Effect of omega-3 fish oil on cardiovascular risk in diabetes. Diabetes Educ. 2010, 36, 565–584. [Google Scholar] [CrossRef]
- Thifault, E.; Cormier, H.; Bouchard-Mercier, A.; Rudkowska, I.; Paradis, A.M.; Garneau, V.; Ouellette, C.; Lemieux, S.; Couture, P.; Vohl, M.C. Effects of age, sex, body mass index and APOE genotype on cardiovascular biomarker response to an n-3 polyunsaturated fatty acid supplementation. J. Nutrigenet. Nutrigenomics 2013, 6, 73–82. [Google Scholar]
- Montori, V.M.; Farmer, A.; Wollan, P.C.; Dinneen, S.F. Fish oil supplementation in type 2 diabetes: A quantitative systematic review. Diabetes Care 2000, 23, 1407–1415. [Google Scholar] [CrossRef]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef]
- Kim, O.Y.; Lim, H.H.; Yang, L.I.; Chae, J.S.; Lee, J.H. Fatty acid desaturase (fads) gene polymorphisms and insulin resistance in association with serum phospholipid polyunsaturated fatty acid composition in healthy korean men: Cross-sectional study. Nutr. Metab. (Lond.) 2011, 8, 24. [Google Scholar] [CrossRef]
- Martinelli, N.; Girelli, D.; Malerba, G.; Guarini, P.; Illig, T.; Trabetti, E.; Sandri, M.; Friso, S.; Pizzolo, F.; Schaeffer, L.; et al. Fads genotypes and desaturase activity estimated by the ratio of arachidonic acid to linoleic acid are associated with inflammation and coronary artery disease. Am. J. Clin. Nutr. 2008, 88, 941–949. [Google Scholar]
- Warensjo, E.; Rosell, M.; Hellenius, M.L.; Vessby, B.; de Faire, U.; Riserus, U. Associations between estimated fatty acid desaturase activities in serum lipids and adipose tissue in humans: Links to obesity and insulin resistance. Lipids Health Dis. 2009, 8, 37. [Google Scholar] [CrossRef]
- Kroger, J.; Zietemann, V.; Enzenbach, C.; Weikert, C.; Jansen, E.H.; Doring, F.; Joost, H.G.; Boeing, H.; Schulze, M.B. Erythrocyte membrane phospholipid fatty acids, desaturase activity, and dietary fatty acids in relation to risk of type 2 diabetes in the european prospective investigation into cancer and nutrition (epic)-potsdam study. Am. J. Clin. Nutr. 2011, 93, 127–142. [Google Scholar] [CrossRef]
- Hodge, A.M.; English, D.R.; O’Dea, K.; Sinclair, A.J.; Makrides, M.; Gibson, R.A.; Giles, G.G. Plasma phospholipid and dietary fatty acids as predictors of type 2 diabetes: Interpreting the role of linoleic acid. Am. J. Clin. Nutr. 2007, 86, 189–197. [Google Scholar]
- Cormier, H.; Rudkowska, I.; Paradis, A.M.; Thifault, E.; Garneau, V.; Lemieux, S.; Couture, P.; Vohl, M.C. Association between polymorphisms in the fatty acid desaturase gene cluster and the plasma triacylglycerol response to an n-3 pufa supplementation. Nutrients 2012, 4, 1026–1041. [Google Scholar] [CrossRef]
- Vohl, M.C. Genes, Omega-3 Fatty Acids and Cardiovascular Disease Risk Factors (fas); Institute of Nutrition and Functional Foods (INAF), Laval University: Quebec City, QC, Canada, 2011. [Google Scholar]
- Eating Well with Canada’s Food Guide; Health Canada: Ottawa, ON, Canada, 2007.
- Callaway, C.W. Cwbc Standardization of Anthropometric Measurements. The Airlie (va) Consensus Conference; Human Kinetics Publishers: Champaign, IL, USA, 1988. [Google Scholar]
- Genest, J.; McPherson, R.; Frohlich, J.; Anderson, T.; Campbell, N.; Carpentier, A.; Couture, P.; Dufour, R.; Fodor, G.; Francis, G.A.; et al. 2009 canadian cardiovascular society/canadian guidelines for the diagnosis and treatment of dyslipidemia and prevention of cardiovascular disease in the adult—2009 recommendations. Can. J. Cardiol. 2009, 25, 567–579. [Google Scholar] [CrossRef]
- Desbuquois, B.; Aurbach, G.D. Use of polyethylene glycol to separate free and antibody-bound peptide hormones in radioimmunoassays. J. Clin. Endocrinol. Metab. 1971, 33, 732–738. [Google Scholar] [CrossRef]
- Richterich, R.; Kuffer, H.; Lorenz, E.; Colombo, J.P. The determination of glucose in plasma and serum (hexokinase-glucose-6-phosphate dehydrogenase method) with the greiner electronic selective analyzer gsa ii (author’s transl). Z. Klin. Chem. Klin. Biochem. 1974, 12, 5–13. [Google Scholar]
- Pirro, M.; Bergeron, J.; Dagenais, G.R.; Bernard, P.M.; Cantin, B.; Despres, J.P.; Lamarche, B. Age and duration of follow-up as modulators of the risk for ischemic heart disease associated with high plasma c-reactive protein levels in men. Arch. Intern. Med. 2001, 161, 2474–2480. [Google Scholar] [CrossRef]
- McNamara, J.R.; Schaefer, E.J. Automated enzymatic standardized lipid analyses for plasma and lipoprotein fractions. Clin. Chim. Acta Int. J. Clin. Chem. 1987, 166, 1–8. [Google Scholar] [CrossRef]
- Albers, J.J.; Warnick, G.R.; Wiebe, D.; King, P.; Steiner, P.; Smith, L.; Breckenridge, C.; Chow, A.; Kuba, K.; Weidman, S.; et al. Multi-laboratory comparison of three heparin-mn2+ precipitation procedures for estimating cholesterol in high-density lipoprotein. Clin. Chem. 1978, 24, 853–856. [Google Scholar]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin. Chem. 1972, 18, 499–502. [Google Scholar]
- Laurell, C.B. Quantitative estimation of proteins by electrophoresis in agarose gel containing antibodies. Anal. Biochem. 1966, 15, 45–52. [Google Scholar] [CrossRef]
- Cartegni, L.; Wang, J.; Zhu, Z.; Zhang, M.Q.; Krainer, A.R. Esefinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003, 31, 3568–3571. [Google Scholar] [CrossRef]
- Henkin, L.; Bergman, R.N.; Bowden, D.W.; Ellsworth, D.L.; Haffner, S.M.; Langefeld, C.D.; Mitchell, B.D.; Norris, J.M.; Rewers, M.; Saad, M.F.; et al. Genetic epidemiology of insulin resistance and visceral adiposity. The iras family study design and methods. Ann. Epidemiol. 2003, 13, 211–217. [Google Scholar]
- Connor, W.E.; Prince, M.J.; Ullmann, D.; Riddle, M.; Hatcher, L.; Smith, F.E.; Wilson, D. The hypotriglyceridemic effect of fish oil in adult-onset diabetes without adverse glucose control. Ann. N. Y. Acad. Sci. 1993, 683, 337–340. [Google Scholar] [CrossRef]
- Simopoulos, A. Fatty acid composition of the skeletal muscle membrane phospholipids, insulin resistance and obesity. Nutr. Today 1994, 2, 12–16. [Google Scholar] [CrossRef]
- Grunfeld, C.; Baird, K.L.; Kahn, C.R. Maintenance of 3t3-l1 cells in culture media containing saturated fatty acids decreases insulin binding and insulin action. Biochem. Biophys. Res. Commun. 1981, 103, 219–226. [Google Scholar] [CrossRef]
- Ginsberg, B.H.; Jabour, J.; Spector, A.A. Effect of alterations in membrane lipid unsaturation on the properties of the insulin receptor of ehrlich ascites cells. Biochim. Biophys. Acta 1982, 690, 157–164. [Google Scholar] [CrossRef]
- Riserus, U. Trans fatty acids and insulin resistance. Atheroscler. Suppl. 2006, 7, 37–39. [Google Scholar] [CrossRef]
- Das, U.N. Essential fatty acids: Biochemistry, physiology and pathology. Biotechnol. J. 2006, 1, 420–439. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Genetic variants in the metabolism of omega-6 and omega-3 fatty acids: Their role in the determination of nutritional requirements and chronic disease risk. Exp. Biol. Med. (Maywood) 2010, 235, 785–795. [Google Scholar] [CrossRef]
- Simopoulos, A.P. The omega-6/omega-3 fatty acid ratio, genetic variation, and cardiovascular disease. Asia Pac. J. Clin. Nutr. 2008, 17, 131–134. [Google Scholar]
- Brenner, R.R. Hormonal modulation of delta6 and delta5 desaturases: Case of diabetes. Prostaglandins Leukot. Essent. Fatty Acids 2003, 68, 151–162. [Google Scholar] [CrossRef]
- Kroger, J.; Schulze, M.B. Recent insights into the relation of delta5 desaturase and delta6 desaturase activity to the development of type 2 diabetes. Curr. Opin. Lipidol. 2012, 23, 4–10. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Evolutionary aspects of the dietary omega-6:Omega-3 fatty acid ratio: Medical implications. World Rev. Nutr. Diet. 2009, 100, 1–21. [Google Scholar] [CrossRef]
- Manning, A.K.; Hivert, M.F.; Scott, R.A.; Grimsby, J.L.; Bouatia-Naji, N.; Chen, H.; Rybin, D.; Liu, C.T.; Bielak, L.F.; Prokopenko, I.; et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat. Genet. 2012, 44, 659–669. [Google Scholar] [CrossRef]
- Ding, K.; Kullo, I.J. Geographic differences in allele frequencies of susceptibility snps for cardiovascular disease. BMC Med. Genet. 2011, 12, 55. [Google Scholar] [CrossRef]
- Park, M.H.; Kim, N.; Lee, J.Y.; Park, H.Y. Genetic loci associated with lipid concentrations and cardiovascular risk factors in the korean population. J. Med. Genet. 2011, 48, 10–15. [Google Scholar] [CrossRef]
- Zietemann, V.; Kroger, J.; Enzenbach, C.; Jansen, E.; Fritsche, A.; Weikert, C.; Boeing, H.; Schulze, M.B. Genetic variation of the fads1 fads2 gene cluster and n-6 pufa composition in erythrocyte membranes in the european prospective investigation into cancer and nutrition-potsdam study. Br. J. Nutr. 2010, 104, 1748–1759. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cormier, H.; Rudkowska, I.; Thifault, E.; Lemieux, S.; Couture, P.; Vohl, M.-C. Polymorphisms in Fatty Acid Desaturase (FADS) Gene Cluster: Effects on Glycemic Controls Following an Omega-3 Polyunsaturated Fatty Acids (PUFA) Supplementation. Genes 2013, 4, 485-498. https://doi.org/10.3390/genes4030485
Cormier H, Rudkowska I, Thifault E, Lemieux S, Couture P, Vohl M-C. Polymorphisms in Fatty Acid Desaturase (FADS) Gene Cluster: Effects on Glycemic Controls Following an Omega-3 Polyunsaturated Fatty Acids (PUFA) Supplementation. Genes. 2013; 4(3):485-498. https://doi.org/10.3390/genes4030485
Chicago/Turabian StyleCormier, Hubert, Iwona Rudkowska, Elisabeth Thifault, Simone Lemieux, Patrick Couture, and Marie-Claude Vohl. 2013. "Polymorphisms in Fatty Acid Desaturase (FADS) Gene Cluster: Effects on Glycemic Controls Following an Omega-3 Polyunsaturated Fatty Acids (PUFA) Supplementation" Genes 4, no. 3: 485-498. https://doi.org/10.3390/genes4030485