Lessons and Implications from Genome-Wide Association Studies (GWAS) Findings of Blood Cell Phenotypes

Abstract
:1. Genetics of Red Blood Cells, White Blood Cells and Platelets

{kind=link}
| Trait | Description | Unit |
|---|---|---|
| Red blood cell (RBC) count | Count of RBC per microliter | Million cells per microliter (×106/µL) |
| Hemoglobin (HGB) | Hemoglobin concentration | Gram per deciliter (g/dL) |
| Hematocrit (HCT) | Fraction of blood that contains hemoglobin | Percentage (%) |
| Mean corpuscular hemoglobin (MCH) | Amount of hemoglobin per RBC | Picogram (pg) |
| Mean corpuscular volume (MCV) | Average volume of RBC | Femtoliter (fL) |
| MCH concentration (MCHC) | Hemoglobin divided by hematocrit | Gram per deciliter (g/dL) |
| RBC distribution width (RDW) | Distribution of RBC volume | Percentage (%) |
| White blood cell (WBC) count | Number of WBC per liter (include all main subtypes) | Billion cells per liter (×109/L) |
| Platelet (PLT) count | Number of PLT per liter | Billion cells per liter (×109/L) |
| Mean platelet volume (MPV) | Average platelet volume | Femtoliter (fL) |
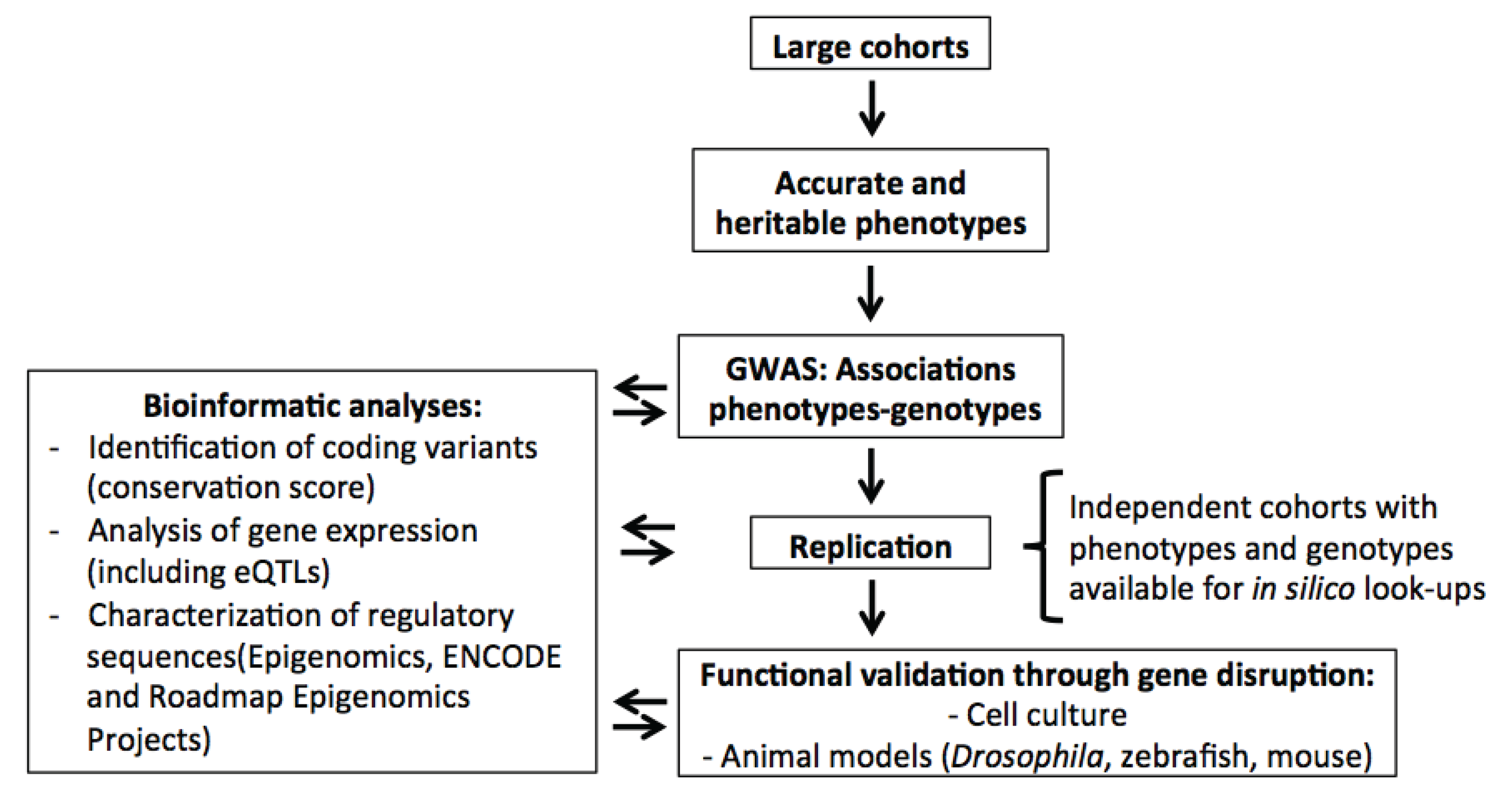
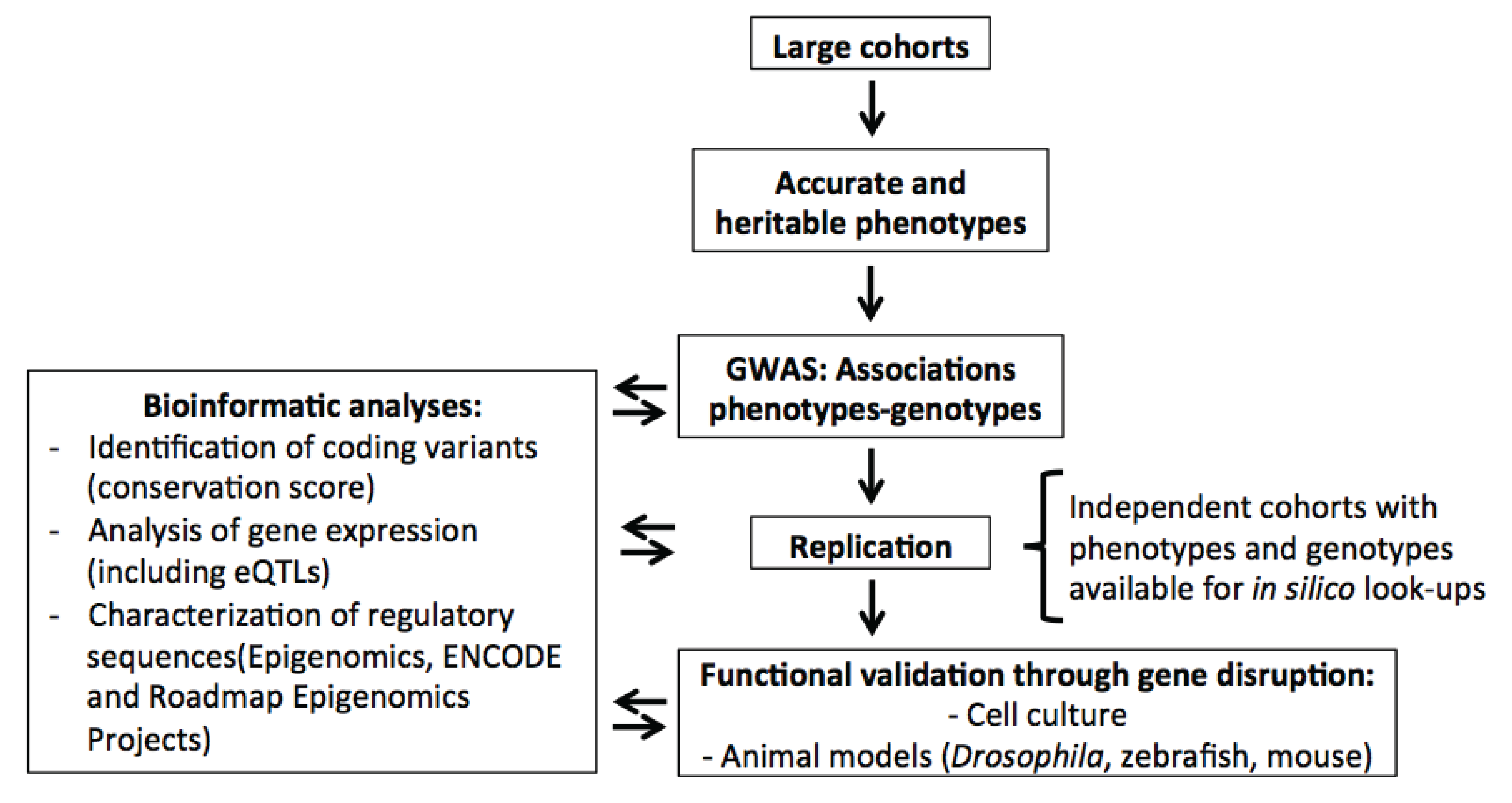
2. Genome-Wide Association Studies (GWAS) for Blood Cell Phenotypes
| Locus | Location | RBC | WBC | Platelet | References |
|---|---|---|---|---|---|
| TMCC2 | 1q32.1 | Caucasian | Caucasian | [17,18] | |
| ARHGEF3 | 3p14.3 | African American | Caucasian | [17,30,36,38] | |
| LRRC16A | 6p22.2 | African American | African American | [31,37] | |
| HBS1L-MYB | 6q22-q23.3 | African American/Caucasian/Japanese | Caucasian | African American/Caucasian | [17,18,31,32,34,35,37] |
| IL-6 | 7p21 | Japanese | Japanese | [47] | |
| RCL1 | 9p24.1-p23 | Caucasian | Caucasian/Japanese | [17,18,32,34] | |
| SH2B3 | 12q24 | Caucasian | Caucasian | Caucasian/Japanese | [17,32,33,34,35,38] |
Some Loci Associated with Blood Cell Traits Are Population-Specific
3. Genetic Modifiers of Disease Severity
4. Orphan Blood Cell Diseases
| Mendelian genetics: orphan syndromes | Genome-wide association studies | |||||||
|---|---|---|---|---|---|---|---|---|
| Locus | Disease | OMIM# | Description | SNP | Position | Phenotype | Candidate-gene(s) | Ref. |
| 5q31 | Familial eosinophilia | 131400 | Characterized by peripheral hypereosinophilia with or without other organ involvement | rs4143832 | chr5: 131,862,977 | Eosinophil count | IL5 | [33] |
| 6p21 | Macroblobulinemia, susceptibility to Waldenstrom | 153600 | Malignant B-cell neoplasm characterized by lymphoplasmacytic infiltration of the bone marrow and hypersecretion of monoclonal immunoglobulin M (IgM) protein | rs2517524 | chr6: 31,025,713 | White blood cell | HLA region | [45] |
| 15q21 | Dyserythropoietic anemia, congenital type III | 105600 | Characterized by nonprogressive mild to moderate hemolytic anemia, macrocytosis in the peripheral blood, and giant multinucleated erythroblasts in the bone marrow | rs1532085 | chr15: 58,683,366 | Hemoglobin | LIPC | [18] |
| 19q13 | Transient erythroblastopenia of childhood | 227050 | Red blood cell aplasia | rs3892630 | chr19: 33,181484 | Mean corpuscular volume | NUDT19 | [18] |
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An evolving paradigm for stem cell biology. Cell 2008, 132, 631–644. [Google Scholar] [CrossRef]
- Pilia, G.; Chen, W.M.; Scuteri, A.; Orru, M.; Albai, G.; Dei, M.; Lai, S.; Usala, G.; Lai, M.; Loi, P.; et al. Heritability of cardiovascular and personality traits in 6,148 sardinians. PLoS Genet. 2006, 2, e132. [Google Scholar] [CrossRef]
- Garner, C.; Tatu, T.; Reittie, J.E.; Littlewood, T.; Darley, J.; Cervino, S.; Farrall, M.; Kelly, P.; Spector, T.D.; Thein, S.L. Genetic influences on F cells and other hematologic variables: A twin heritability study. Blood 2000, 95, 342–346. [Google Scholar]
- Hoffman, M.; Blum, A.; Baruch, R.; Kaplan, E.; Benjamin, M. Leukocytes and coronary heart disease. Atherosclerosis 2004, 172, 1–6. [Google Scholar] [CrossRef]
- Boos, C.J.; Lip, G.Y. Assessment of mean platelet volume in coronary artery disease—What does it mean? Thromb. Res. 2007, 120, 11–13. [Google Scholar] [CrossRef]
- Nieswandt, B.; Kleinschnitz, C.; Stoll, G. Ischaemic stroke: A thrombo-inflammatory disease? J. Physiol. 2011, 589, 4115–4123. [Google Scholar]
- Ebrahim, S.; Davey Smith, G. Mendelian randomization: Can genetic epidemiology help redress the failures of observational epidemiology? Hum. Genet. 2008, 123, 15–33. [Google Scholar] [CrossRef]
- Voight, B.F.; Peloso, G.M.; Orho-Melander, M.; Frikke-Schmidt, R.; Barbalic, M.; Jensen, M.K.; Hindy, G.; Holm, H.; Ding, E.L.; Johnson, T.; et al. Plasma HDL cholesterol and risk of myocardial infarction: A mendelian randomisation study. Lancet 2012, 380, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Do, R.; Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Gao, C.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat. Genet. 2013, 45, 1345–1352. [Google Scholar] [CrossRef]
- Zeng, S.M.; Yankowitz, J.; Widness, J.A.; Strauss, R.G. Etiology of differences in hematocrit between males and females: Sequence-based polymorphisms in erythropoietin and its receptor. J. Gend. Specif. Med.: JGSM: 2001, 4, 35–40. [Google Scholar]
- McLaren, C.E.; Barton, J.C.; Gordeuk, V.R.; Wu, L.; Adams, P.C.; Reboussin, D.M.; Speechley, M.; Chang, H.; Acton, R.T.; Harris, E.L.; et al. Determinants and characteristics of mean corpuscular volume and hemoglobin concentration in white HFE C282Y homozygotes in the hemochromatosis and iron overload screening study. Am. J. Hematol. 2007, 82, 898–905. [Google Scholar] [CrossRef]
- Lin, J.P.; O’Donnell, C.J.; Jin, L.; Fox, C.; Yang, Q.; Cupples, L.A. Evidence for linkage of red blood cell size and count: Genome-wide scans in the framingham heart study. Am. J. Hematol. 2007, 82, 605–610. [Google Scholar] [CrossRef]
- Menzel, S.; Jiang, J.; Silver, N.; Gallagher, J.; Cunningham, J.; Surdulescu, G.; Lathrop, M.; Farrall, M.; Spector, T.D.; Thein, S.L. The HBS1L-MYB intergenic region on chromosome 6q23.3 influences erythrocyte, platelet, and monocyte counts in humans. Blood 2007, 110, 3624–3626. [Google Scholar] [CrossRef]
- Lohmueller, K.E.; Pearce, C.L.; Pike, M.; Lander, E.S.; Hirschhorn, J.N. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat. Genet. 2003, 33, 177–182. [Google Scholar]
- Lettre, G. The search for genetic modifiers of disease severity in the beta-hemoglobinopathies. Cold Spring Harbor Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Sankaran, V.G.; Ludwig, L.S.; Sicinska, E.; Xu, J.; Bauer, D.E.; Eng, J.C.; Patterson, H.C.; Metcalf, R.A.; Natkunam, Y.; Orkin, S.H.; et al. Cyclin D3 coordinates the cell cycle during differentiation to regulate erythrocyte size and number. Genes Dev. 2012, 26, 2075–2087. [Google Scholar] [CrossRef]
- Gieger, C.; Radhakrishnan, A.; Cvejic, A.; Tang, W.; Porcu, E.; Pistis, G.; Serbanovic-Canic, J.; Elling, U.; Goodall, A.H.; Labrune, Y.; et al. New gene functions in megakaryopoiesis and platelet formation. Nature 2011, 480, 201–208. [Google Scholar] [CrossRef]
- Van der Harst, P.; Zhang, W.; Mateo Leach, I.; Rendon, A.; Verweij, N.; Sehmi, J.; Paul, D.S.; Elling, U.; Allayee, H.; Li, X.; et al. Seventy-five genetic loci influencing the human red blood cell. Nature 2012, 492, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Lindblad-Toh, K.; Garber, M.; Zuk, O.; Lin, M.F.; Parker, B.J.; Washietl, S.; Kheradpour, P.; Ernst, J.; Jordan, G.; Mauceli, E.; et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 2011, 478, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the sift algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Wu, C.; Orozco, C.; Boyer, J.; Leglise, M.; Goodale, J.; Batalov, S.; Hodge, C.L.; Haase, J.; Janes, J.; Huss, J.W. BioGPS: An extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009, 10, R130. [Google Scholar] [CrossRef]
- Cookson, W.; Liang, L.; Abecasis, G.; Moffatt, M.; Lathrop, M. Mapping complex disease traits with global gene expression. Nat. Rev. Genet. 2009, 10, 184–194. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Birney, E.; Dunham, I.; Green, E.D.; Gunter, C.; Snyder, M. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Stamatoyannopoulos, J.A.; Costello, J.F.; Ren, B.; Milosavljevic, A.; Meissner, A.; Kellis, M.; Marra, M.A.; Beaudet, A.L.; Ecker, J.R.; et al. The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol. 2010, 28, 1045–1048. [Google Scholar] [CrossRef]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef]
- Paul, D.S.; Albers, C.A.; Rendon, A.; Voss, K.; Stephens, J.; van der Harst, P.; Chambers, J.C.; Soranzo, N.; Ouwehand, W.H.; Deloukas, P. Maps of open chromatin highlight cell type-restricted patterns of regulatory sequence variation at hematological trait loci. Genome Res. 2013, 23, 1130–1141. [Google Scholar] [CrossRef] [Green Version]
- Ward, L.D.; Kellis, M. Haploreg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012, 40, D930–D934. [Google Scholar] [CrossRef]
- Schaub, M.A.; Boyle, A.P.; Kundaje, A.; Batzoglou, S.; Snyder, M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012, 22, 1748–1759. [Google Scholar] [CrossRef]
- Auer, P.L.; Johnsen, J.M.; Johnson, A.D.; Logsdon, B.A.; Lange, L.A.; Nalls, M.A.; Zhang, G.; Franceschini, N.; Fox, K.; Lange, E.M.; et al. Imputation of exome sequence variants into population- based samples and blood-cell-trait-associated loci in African Americans: NHLBI go exome sequencing project. Am. J. Hum. Genet. 2012, 91, 794–808. [Google Scholar] [CrossRef]
- Chen, Z.; Tang, H.; Qayyum, R.; Schick, U.M.; Nalls, M.A.; Handsaker, R.; Li, J.; Lu, Y.; Yanek, L.R.; Keating, B.; et al. Genome-wide association analysis of red blood cell traits in African Americans: The cogent network. Hum. Mol. Genet. 2013, 22, 2529–2538. [Google Scholar] [CrossRef]
- Ganesh, S.K.; Zakai, N.A.; van Rooij, F.J.; Soranzo, N.; Smith, A.V.; Nalls, M.A.; Chen, M.H.; Kottgen, A.; Glazer, N.L.; Dehghan, A.; et al. Multiple loci influence erythrocyte phenotypes in the charge consortium. Nat. Genet. 2009, 41, 1191–1198. [Google Scholar] [CrossRef]
- Gudbjartsson, D.F.; Bjornsdottir, U.S.; Halapi, E.; Helgadottir, A.; Sulem, P.; Jonsdottir, G.M.; Thorleifsson, G.; Helgadottir, H.; Steinthorsdottir, V.; Stefansson, H.; et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat. Genet. 2009, 41, 342–347. [Google Scholar] [CrossRef]
- Kamatani, Y.; Matsuda, K.; Okada, Y.; Kubo, M.; Hosono, N.; Daigo, Y.; Nakamura, Y.; Kamatani, N. Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat. Genet. 2010, 42, 210–215. [Google Scholar]
- Lo, K.S.; Wilson, J.G.; Lange, L.A.; Folsom, A.R.; Galarneau, G.; Ganesh, S.K.; Grant, S.F.; Keating, B.J.; McCarroll, S.A.; Mohler, E.R., 3rd; et al. Genetic association analysis highlights new loci that modulate hematological trait variation in caucasians and African Americans. Hum. Genet. 2011, 129, 307–317. [Google Scholar]
- Meisinger, C.; Prokisch, H.; Gieger, C.; Soranzo, N.; Mehta, D.; Rosskopf, D.; Lichtner, P.; Klopp, N.; Stephens, J.; Watkins, N.A.; et al. A genome-wide association study identifies three loci associated with mean platelet volume. Am. J. Hum. Genet. 2009, 84, 66–71. [Google Scholar] [CrossRef]
- Qayyum, R.; Snively, B.M.; Ziv, E.; Nalls, M.A.; Liu, Y.; Tang, W.; Yanek, L.R.; Lange, L.; Evans, M.K.; Ganesh, S.; et al. A meta-analysis and genome-wide association study of platelet count and mean platelet volume in African Americans. PLoS Genet. 2012, 8, e1002491. [Google Scholar] [CrossRef]
- Soranzo, N.; Spector, T.D.; Mangino, M.; Kuhnel, B.; Rendon, A.; Teumer, A.; Willenborg, C.; Wright, B.; Chen, L.; Li, M.; et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the haemgen consortium. Nat. Genet. 2009, 41, 1182–1190. [Google Scholar] [CrossRef]
- Soranzo, N.; Rendon, A.; Gieger, C.; Jones, C.I.; Watkins, N.A.; Menzel, S.; Doring, A.; Stephens, J.; Prokisch, H.; Erber, W.; et al. A novel variant on chromosome 7q22.3 associated with mean platelet volume, counts, and function. Blood 2009, 113, 3831–3837. [Google Scholar] [CrossRef]
- Chambers, J.C.; Zhang, W.; Li, Y.; Sehmi, J.; Wass, M.N.; Zabaneh, D.; Hoggart, C.; Bayele, H.; McCarthy, M.I.; Peltonen, L.; et al. Genome-wide association study identifies variants in TMPRSS6 associated with hemoglobin levels. Nat. Genet. 2009, 41, 1170–1172. [Google Scholar] [CrossRef]
- Reiner, A.P.; Lettre, G.; Nalls, M.A.; Ganesh, S.K.; Mathias, R.; Austin, M.A.; Dean, E.; Arepalli, S.; Britton, A.; Chen, Z.; et al. Genome-wide association study of white blood cell count in 16,388 African Americans: The continental origins and genetic epidemiology network (cogent). PLoS Genet. 2011, 7, e1002108. [Google Scholar] [CrossRef]
- Crosslin, D.R.; McDavid, A.; Weston, N.; Nelson, S.C.; Zheng, X.; Hart, E.; de Andrade, M.; Kullo, I.J.; McCarty, C.A.; Doheny, K.F.; et al. Genetic variants associated with the white blood cell count in 13,923 subjects in the emerge network. Hum. Genet. 2012, 131, 639–652. [Google Scholar] [CrossRef]
- Okada, Y.; Hirota, T.; Kamatani, Y.; Takahashi, A.; Ohmiya, H.; Kumasaka, N.; Higasa, K.; Yamaguchi-Kabata, Y.; Hosono, N.; Nalls, M.A.; et al. Identification of nine novel loci associated with white blood cell subtypes in a Japanese population. PLoS Genet. 2011, 7, e1002067. [Google Scholar] [CrossRef]
- Li, J.; Glessner, J.T.; Zhang, H.; Hou, C.; Wei, Z.; Bradfield, J.P.; Mentch, F.D.; Guo, Y.; Kim, C.; Xia, Q.; et al. GWAS of blood cell traits identifies novel associated loci and epistatic interactions in Caucasian and African-American children. Hum. Mole. Genet. 2013, 22, 1457–1464. [Google Scholar] [CrossRef]
- Nalls, M.A.; Couper, D.J.; Tanaka, T.; van Rooij, F.J.; Chen, M.H.; Smith, A.V.; Toniolo, D.; Zakai, N.A.; Yang, Q.; Greinacher, A.; et al. Multiple loci are associated with white blood cell phenotypes. PLoS Genet. 2011, 7, e1002113. [Google Scholar] [CrossRef] [Green Version]
- Shameer, K.; Denny, J.C.; Ding, K.; Jouni, H.; Crosslin, D.R.; de Andrade, M.; Chute, C.G.; Peissig, P.; Pacheco, J.A.; Li, R.; et al. A genome- and phenome-wide association study to identify genetic variants influencing platelet count and volume and their pleiotropic effects. Hum. Genet. 2014, 133, 95–109. [Google Scholar] [CrossRef]
- Okada, Y.; Takahashi, A.; Ohmiya, H.; Kumasaka, N.; Kamatani, Y.; Hosono, N.; Tsunoda, T.; Matsuda, K.; Tanaka, T.; Kubo, M.; et al. Genome-wide association study for C-reactive protein levels identified pleiotropic associations in the IL6 locus. Hum. Mol. Genet. 2011, 20, 1224–1231. [Google Scholar] [CrossRef]
- Monda, K.L.; Chen, G.K.; Taylor, K.C.; Palmer, C.; Edwards, T.L.; Lange, L.A.; Ng, M.C.; Adeyemo, A.A.; Allison, M.A.; Bielak, L.F.; et al. A meta-analysis identifies new loci associated with body mass index in individuals of African ancestry. Nat. Genet. 2013, 45, 690–696. [Google Scholar] [CrossRef]
- N’Diaye, A.; Chen, G.K.; Palmer, C.D.; Ge, B.; Tayo, B.; Mathias, R.A.; Ding, J.; Nalls, M.A.; Adeyemo, A.; Adoue, V.; et al. Identification, replication, and fine-mapping of loci associated with adult height in individuals of African ancestry. PLoS Genet. 2011, 7, e1002298. [Google Scholar] [CrossRef] [Green Version]
- Reich, D.; Nalls, M.A.; Kao, W.H.; Akylbekova, E.L.; Tandon, A.; Patterson, N.; Mullikin, J.; Hsueh, W.C.; Cheng, C.Y.; Coresh, J.; et al. Reduced neutrophil count in people of African descent is due to a regulatory variant in the duffy antigen receptor for chemokines gene. PLoS Genet. 2009, 5, e1000360. [Google Scholar] [CrossRef] [Green Version]
- Greenburg, A.G. Pathophysiology of anemia. Am. J. Med. 1996, 101, 7S–11S. [Google Scholar] [CrossRef]
- Worldwide Prevalence of Anaemia 1993–2005 (WHO Global Database on Anaemia). Available online: http://whqlibdoc.Who.Int/publications/2008/9789241596657_eng.pdf (accessed on 19 November 2013).
- Skoda, R. The genetic basis of myeloproliferative disorders. Am. Soc. Hematol. Educ. Program 2007, 2007, 1–10. [Google Scholar] [CrossRef]
- Oh, S.T.; Gotlib, J. JAK2 V617F and beyond: Role of genetics and aberrant signaling in the pathogenesis of myeloproliferative neoplasms. Expert Rev. Hematol. 2010, 3, 323–337. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Pieri, L.; Guglielmelli, P. JAK2 allele burden in the myeloproliferative neoplasms: Effects on phenotype, prognosis and change with treatment. Ther. Adv. Hematol. 2011, 2, 21–32. [Google Scholar] [CrossRef]
- Hobbs, C.M.; Manning, H.; Bennett, C.; Vasquez, L.; Severin, S.; Brain, L.; Mazharian, A.; Guerrero, J.A.; Li, J.; Soranzo, N.; et al. JAK2V617F leads to intrinsic changes in platelet formation and reactivity in a knock-in mouse model of essential thrombocythemia. Blood 2013, 122, 3787–3797. [Google Scholar] [CrossRef]
- Sankaran, V.G.; Xu, J.; Orkin, S.H. Advances in the understanding of haemoglobin switching. Br. J. Haematol. 2010, 149, 181–194. [Google Scholar] [CrossRef]
- Sankaran, V.G.; Lettre, G.; Orkin, S.H.; Hirschhorn, J.N. Modifier genes in mendelian disorders: The example of hemoglobin disorders. Ann. N. Y. Acad. Sci. 2010, 1214, 47–56. [Google Scholar] [CrossRef]
- Platt, O.S.; Brambilla, D.J.; Rosse, W.F.; Milner, P.F.; Castro, O.; Steinberg, M.H.; Klug, P.P. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N. Engl. J. Med. 1994, 330, 1639–1644. [Google Scholar] [CrossRef]
- Platt, O.S.; Thorington, B.D.; Brambilla, D.J.; Milner, P.F.; Rosse, W.F.; Vichinsky, E.; Kinney, T.R. Pain in sickle cell disease. Rates and risk factors. N. Engl. J. Med. 1991, 325, 11–16. [Google Scholar] [CrossRef]
- Castro, O.; Brambilla, D.J.; Thorington, B.; Reindorf, C.A.; Scott, R.B.; Gillette, P.; Vera, J.C.; Levy, P.S. The acute chest syndrome in sickle cell disease: Incidence and risk factors. The cooperative study of sickle cell disease. Blood 1994, 84, 643–649. [Google Scholar]
- Thein, S.L.; Craig, J.E. Genetics of HB F/F cell variance in adults and heterocellular hereditary persistence of fetal hemoglobin. Hemoglobin 1998, 22, 401–414. [Google Scholar] [CrossRef]
- Menzel, S.; Garner, C.; Gut, I.; Matsuda, F.; Yamaguchi, M.; Heath, S.; Foglio, M.; Zelenika, D.; Boland, A.; Rooks, H.; et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat. Genet. 2007, 39, 1197–1199. [Google Scholar] [CrossRef]
- Thein, S.L.; Menzel, S.; Peng, X.; Best, S.; Jiang, J.; Close, J.; Silver, N.; Gerovasilli, A.; Ping, C.; Yamaguchi, M.; et al. Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proc. Natl. Acad. Sci. USA 2007, 104, 11346–11351. [Google Scholar] [CrossRef]
- Uda, M.; Galanello, R.; Sanna, S.; Lettre, G.; Sankaran, V.G.; Chen, W.; Usala, G.; Busonero, F.; Maschio, A.; Albai, G.; et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc. Natl. Acad. Sci. USA 2008, 105, 1620–1625. [Google Scholar] [CrossRef]
- Galarneau, G.; Palmer, C.D.; Sankaran, V.G.; Orkin, S.H.; Hirschhorn, J.N.; Lettre, G. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat. Genet. 2010, 42, 1049–1051. [Google Scholar] [CrossRef]
- Lettre, G.; Sankaran, V.G.; Bezerra, M.A.; Araujo, A.S.; Uda, M.; Sanna, S.; Cao, A.; Schlessinger, D.; Costa, F.F.; Hirschhorn, J.N.; et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc. Natl. Acad. Sci. USA 2008, 105, 11869–11874. [Google Scholar] [CrossRef]
- Nuinoon, M.; Makarasara, W.; Mushiroda, T.; Setianingsih, I.; Wahidiyat, P.A.; Sripichai, O.; Kumasaka, N.; Takahashi, A.; Svasti, S.; Munkongdee, T.; et al. A genome-wide association identified the common genetic variants influence disease severity in beta(0)-thalassemia/hemoglobin E. Hum. Genet. 2010, 127, 303–314. [Google Scholar]
- Sankaran, V.G.; Menne, T.F.; Xu, J.; Akie, T.E.; Lettre, G.; van Handel, B.; Mikkola, H.K.; Hirschhorn, J.N.; Cantor, A.B.; Orkin, S.H. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 2008, 322, 1839–1842. [Google Scholar] [CrossRef]
- Xu, J.; Peng, C.; Sankaran, V.G.; Shao, Z.; Esrick, E.B.; Chong, B.G.; Ippolito, G.C.; Fujiwara, Y.; Ebert, B.L.; Tucker, P.W.; et al. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science 2011, 334, 993–996. [Google Scholar] [CrossRef]
- Bauer, D.E.; Kamran, S.C.; Lessard, S.; Xu, J.; Fujiwara, Y.; Lin, C.; Shao, Z.; Canver, M.C.; Smith, E.C.; Pinello, L.; et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 2013, 342, 253–257. [Google Scholar] [CrossRef]
- Hardison, R.C.; Blobel, G.A. Genetics. Gwas to therapy by genome edits? Science 2013, 342, 206–207. [Google Scholar] [CrossRef]
- Online Mendelian Inheritance in Man. Available online: http://omim.org/ (accessed on 18 November 2013).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chami, N.; Lettre, G. Lessons and Implications from Genome-Wide Association Studies (GWAS) Findings of Blood Cell Phenotypes. Genes 2014, 5, 51-64. https://doi.org/10.3390/genes5010051
Chami N, Lettre G. Lessons and Implications from Genome-Wide Association Studies (GWAS) Findings of Blood Cell Phenotypes. Genes. 2014; 5(1):51-64. https://doi.org/10.3390/genes5010051
Chicago/Turabian StyleChami, Nathalie, and Guillaume Lettre. 2014. "Lessons and Implications from Genome-Wide Association Studies (GWAS) Findings of Blood Cell Phenotypes" Genes 5, no. 1: 51-64. https://doi.org/10.3390/genes5010051