
EMAST is a Form of Microsatellite Instability That is Initiated by Inflammation and Modulates Colorectal Cancer Progression

Abstract
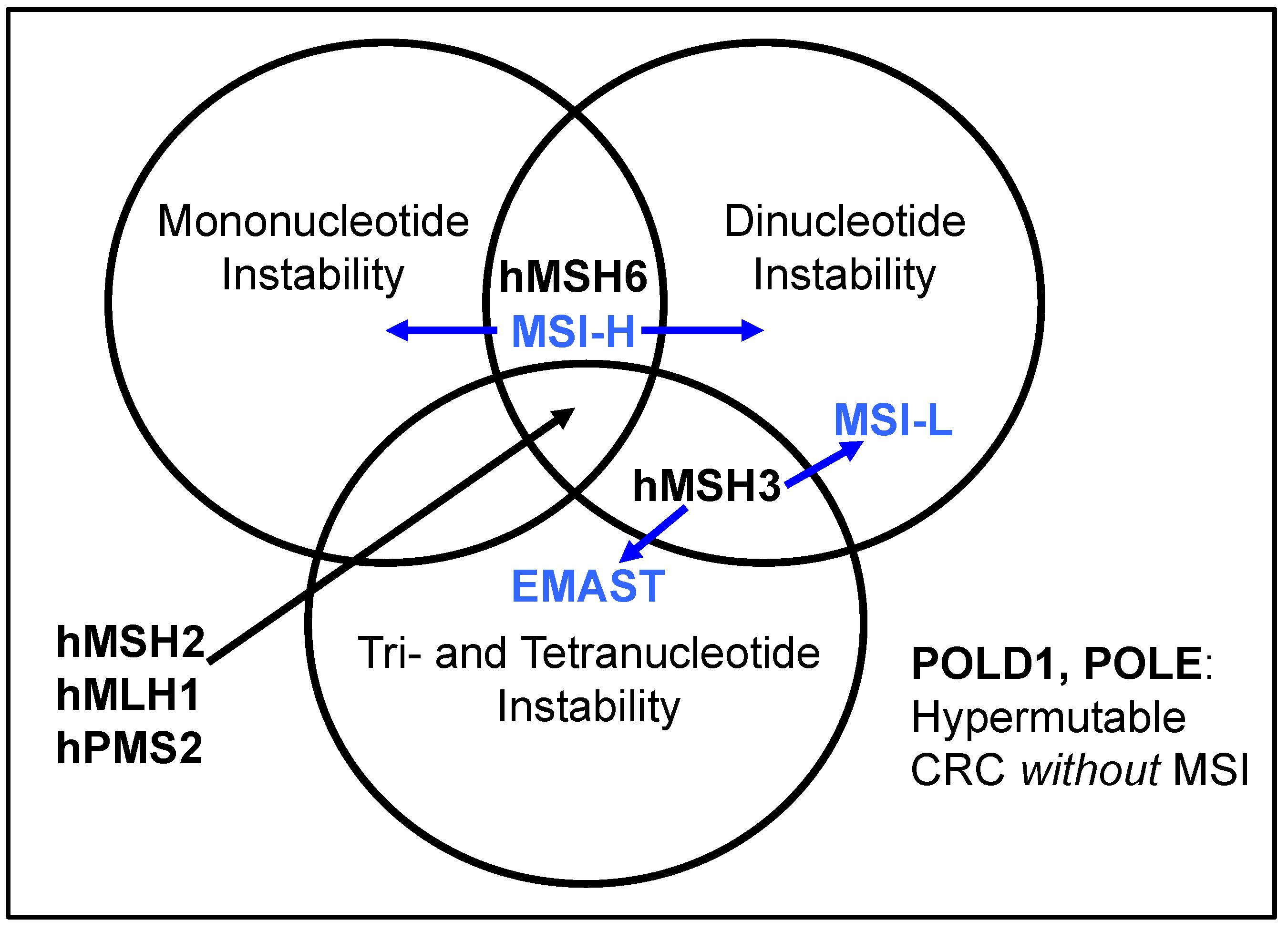
:1. Introduction: DNA Mismatch Repair, Microsatellite Instability, and EMAST

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MutSα (MSH2-MSH6) | MutSβ (MSH2-MSH3) | |
|---|---|---|
| Single mispaired nucleotides | Yes | No |
| Insertion-Deletion Loops | ||
| 1 | Yes | No |
| 2 | Yes | Yes |
| 3 | No | Yes |
| 4 | No | Yes |
| 5-fluorodeoxyuracil | Yes | Yes |
| O6-methylguanine adduct | Yes | No |
| 6-thioguanine adduct | Yes | No |
| Cisplatin, carboplatin | Yes | No data, but triggers DSBs |
| Oxaliplatin, teraplatin, transplatin, JM335, JM216 | No | No, but triggers DSBs |
| Irinotecan (CPT-11) | No | No |
| I/D = 1 | I/D = 2 | I/D = 3 | I/D = 4 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| CELL LINE | MMR-Status | BAT 25 | BAT 26 | D5S346 | D17S250 | TBP | RB | REN | HPRTII |
| SW480 | Proficient | 0/58 (0%) | 0/58 (0%) | 0/58 (0%) | 0/58 (0%) | 0/58 (0%) | 0/58 (0%) | 0/58 (0%) | 0/58 (0%) |
| HCT 116 | MLH1−/− and MSH3−/− | 26/46 (57%) | 17/53 (32%) | 13/47 (28%) | 18/38 (47%) | 13/83 (16%) | 7/51 (14%) | 23/91 (25%) | 14/50 (28%) |
| HCT 116 + 3 | MSH3−/− | 0/111 (0%) | 0/107 (0%) | 3/102 (2.94%) | 50/117 (43%) | 6/53 (11%) | 11/100 (11%) | 29/52 (56%) | 11/111 (10%) |
| DLD1 | MSH6−/− | 21/59 (36%) | 15/58 (26%) | 12/60 (20%) | 26/71 (37%) | 0/60 (0%) | 0/80 (0%) | 0/67 (0%) | 0/59 (0%) |

2. Defining EMAST, and Its Overlap with MSI-L

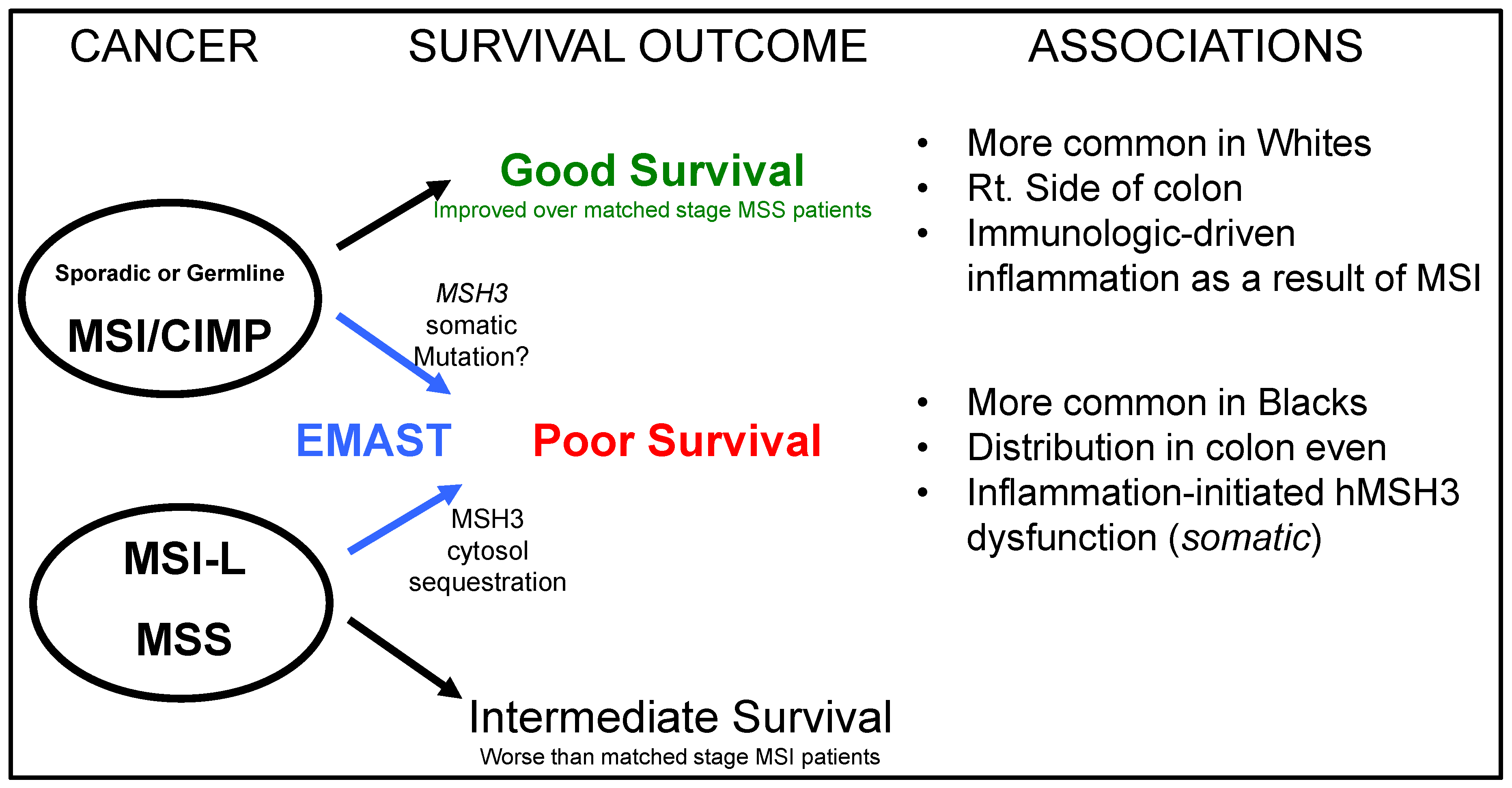
3. EMAST is a Biomarker Observed in Several Cancers and in Inflamed Non-Cancer Tissue
4. EMAST Occurs in Colorectal Cancer and Modifies Patient Outcome
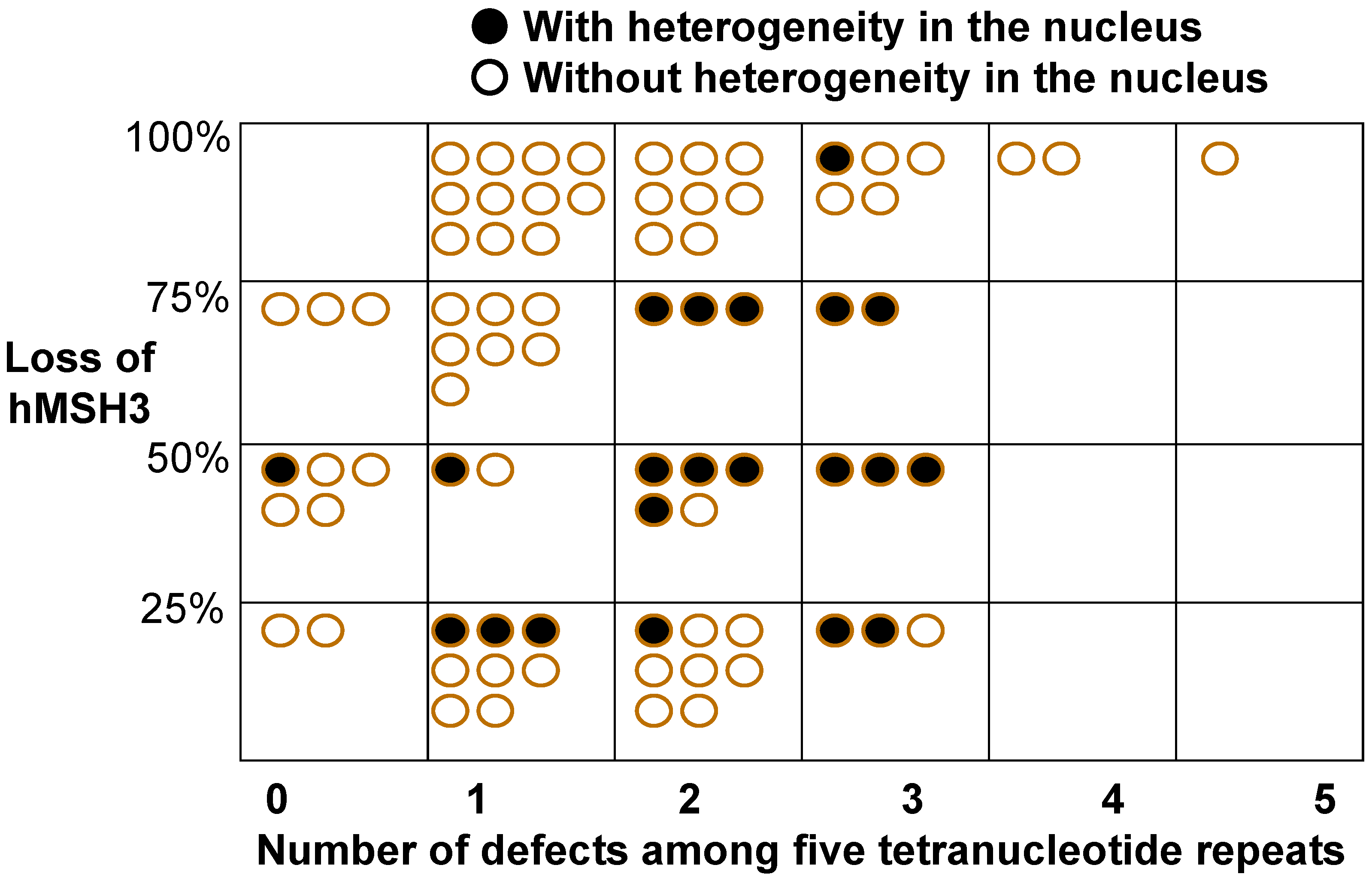
5. A Defect in the DNA Mismatch Repair Protein MSH3 is a Cause of EMAST
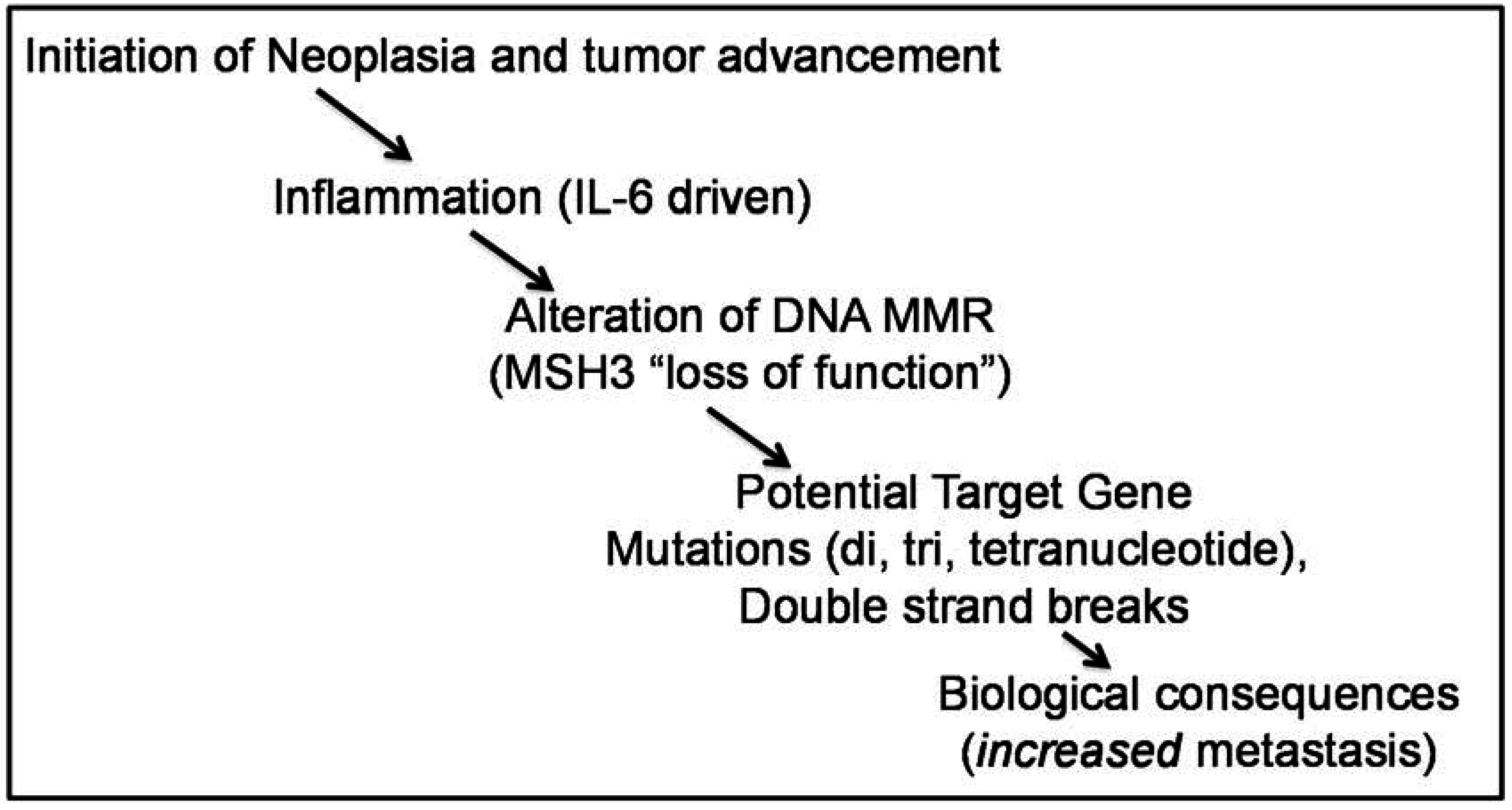
6. A Driver for MSH3 Dysfunction and EMAST Appears to Be Oxidative Stress and Inflammation-Induced Cytokines, and Potentially Hypoxia

7. Additional Considerations for Pathogenesis of Colorectal Cancer by MSH3 Dysfunction
8. MSH3 is Involved in Double Strand Break (DSB) Repair
9. Summary
| MSI-H | EMAST | References | |
|---|---|---|---|
| Genomic Instability | Microsatellite instability (MSI) | Mostly MSS and MSI-L, includes MSI-H | [16,17,18,19,49] |
| Germline cause | Mutation of DNA MMR gene | None known | [4,5] |
| Sporadic cause | MLH1 hypermethylation | Inflammation and alteration of MSH3 | [21,22,23,26,27,30,31] |
| Prevalence in sporadic CRC | ~15% | Up to 60% | [9,10,16,17,20,21,22,23] |
| Inflammation | Crohns-like around tumor (tumor margin) | Associated with tumor nests around epithelial components | [10,21,22,23] |
| Immune Reaction | Neo-peptide driven; unknown but favorable | Unknown; unfavorable | [10,32] |
| Prognosis | Better survival; early stage | Poorer survival; later stage | [8,19,21] |
| Pathogenesis | Target gene mutation | Unknown; target gene mutation? Chromosomal instability? | [9,49,57,58,59] |
| Race | ½ frequent in American Blacks | Twice frequent in American Blacks | [21,32] |
| Response to 5FU | Completely muted | Reduced?; not known | [7,9,10,64] |

Abbreviations
| MSI | Microsatellite instability |
| MSS | microsatellite stable |
| EMAST | elevated microsatellite alterations at selected tetranucleotide repeats |
| CRC | colorectal cancer |
| MMR | DNA mismatch repair |
| MSH3 | human MutS homolog 3 |
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Ionov, Y.; Peinado, M.A.; Malkhosyan, S.; Shibata, D.; Perucho, M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993, 363, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Thibodeau, S.N.; Bren, G.; Schaid, D. Microsatellite instability in cancer of the proximal colon. Science 1993, 260, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, L.A.; Peltomaki, P.; Leach, F.S.; Sistonen, P.; Pylkkanen, L.; Mecklin, J.P.; Jarvinen, H.; Powell, S.M.; Jen, J.; Hamilton, S.R.; et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993, 260, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Koi, M.; Chang, D.K.; Carethers, J.M. The biochemical basis of microsatellite instability and abnormal immunohistochemistry and clinical behavior in Lynch Syndrome: From bench to bedside. Fam. Cancer 2008, 7, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M. Differentiating Lynch-like from Lynch syndrome. Gastroenterology 2014, 146, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar] [PubMed]
- Carethers, J.M.; Smith, E.J.; Behling, C.A.; Nguyen, L.; Tajima, A.; Doctolero, R.T.; Cabrera, B.L.; Goel, A.; Arnold, C.A.; Miyai, K.; et al. Use of 5-fluorouracil and survival in patients with microsatellite unstable colorectal cancer. Gastroenterology 2004, 126, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic review of microsatellite instability and colorectal cancer prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Grady, W.M.; Carethers, J.M. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 2008, 135, 1079–1099. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.K.; Ricciardiello, L.; Goel, A.; Chang, C.L.; Boland, C.R. Steady-state regulation of the human DNA mismatch repair system. J. Biol. Chem. 2000, 275, 18424–18431. [Google Scholar] [CrossRef] [PubMed]
- Kolodner, R.D.; Tytell, J.D.; Schmeits, J.L.; Kane, M.F.; Gupta, R.D.; Weger, J.; Wahlberg, S.; Fox, E.A.; Peel, D.; Ziogas, A.; et al. Germ-line MSH6 mutations in colorectal cancer families. Cancer Res. 1999, 59, 5068–5074. [Google Scholar]
- Mao, L.; Schoenberg, M.P.; Scicchitano, M.; Erozan, Y.S.; Merlo, A.; Schwab, D.; Sidransky, D. Molecular detection of primary bladder cancer by microsatellite analysis. Science 1996, 271, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Kuismanen, S.A.; Liu, T.; Chadwick, R.B.; Johnson, C.K.; Stevens, M.W.; Richards, S.K.; Meek, J.E.; Gao, X.; Wright, F.A.; et al. MSH6 and MSH3 are rarely involved in genetic predisposition to nonpolypotic colon cancer. Cancer Res. 2001, 61, 1619–1623. [Google Scholar]
- Parc, Y.R.; Halling, K.C.; Wang, L.; Christensen, E.R.; Cunningham, J.M.; French, A.J.; Burgart, L.J.; Price-Troska, T.L.; Roche, P.C.; Thibodeau, S.N. MSH6 alterations in patients with microsatellite instability-low colorectal cancer. Cancer Res. 2000, 60, 2225–2231. [Google Scholar] [PubMed]
- Haugen, A.C.; Goel, A.; Yamada, K.; Marra, G.; Nguyen, T.P.; Nagasaka, T.; Kanazawa, S.; Koike, J.; Kikuchi, Y.; Zhong, X.; et al. Genetic instability caused by loss of MutS homologue 3 in human colorectal cancer. Cancer Res. 2008, 68, 8465–8472. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Kanazawa, S.; Koike, J.; Sugiyama, H.; Xu, C.; Funahashi, K.; Boland, C.R.; Koi, M.; Hemmi, H. Microsatellite instability at tetranucleotide repeats in sporadic colorectal cancer in Japan. Oncol. Rep. 2010, 23, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Hile, S.E.; Shabashev, S.; Eckert, K.A. Tumor-specific microsatellite instability: Do distinct mechanisms underlie the MSI-L and EMAST phenotypes? Mutat. Res. 2013, 743–744, 67–77. [Google Scholar] [CrossRef]
- Garcia, M.; Choi, C.; Kim, H.R.; Daoud, Y.; Toiyama, Y.; Takahashi, M.; Goel, A.; Boland, C.R.; Koi, M. Association between recurrent metastasis from stage II and III primary colorectal tumors and moderate microsatellite instability. Gastroenterology 2012, 143, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.M.; Berg, M.; Søreide, K. Prevalence and implications of elevated microsatellite alterations at selected tetranucleotides in cancer. Br. J. Cancer. 2014, 111, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, B.; Lee, A.; Cabrera, B.L.; Miyai, K.; Luo, L.; Ramamoorthy, S.; Keku, T.; Sandler, R.S.; McGuire, K.L.; Carethers, J.M. Relationship of EMAST and microsatellite instability among patients with rectal cancer. J. Gastrointest. Surg. 2010, 14, 1521–1528. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Chung, H.; Devaraj, B.; Iwaizumi, M.; Han, H.S.; Hwang, D.Y.; Seong, M.K.; Jung, B.H.; Carethers, J.M. Elevated microsatellite alterations at selected tetranucleotide repeats are associated with morphologies of colorectal neoplasia. Gastroenterology 2010, 139, 1519–1525. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Miyai, K.; Han, H.S.; Hwang, D.-Y.; Seong, M.K.; Chung, H.; Jung, B.H.; Devaraj, B.; McGuire, K.L.; Carethers, J.M. Microsatellite instability, EMAST, and morphology associations with T cell infiltration in colorectal neoplasia. Dig. Dis. Sci. 2012, 57, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Danaee, H.; Nelson, H.H.; Karagas, M.R.; Schned, A.R.; Ashok, T.D.; Hirao, T.; Perry, A.E.; Kelsey, K.T. Microsatellite instability at tetranucleotide repeats in skin and bladder cancer. Oncogene 2002, 21, 4894–4899. [Google Scholar] [CrossRef] [PubMed]
- Ahrendt, S.A.; Decker, P.A.; Doffek, K.; Wang, B.; Xu, L.; Demeure, M.J.; Jen, J.; Sidransky, D. Microsatellite instability at selected tetranucleotide repeats is associated with p53 mutations in non-small cell lung cancer. Cancer Res. 2000, 60, 2488–2491. [Google Scholar] [PubMed]
- Tseng-Rogenski, S.; Chung, H.; Wilk, M.B.; Zhang, S.; Iwaizumi, M.; Carethers, J.M. Oxidative stress induces nuclear-to-cytosol shift of MSH3, a potential mechanism for EMAST in colorectal cancer cells. PLOS ONE 2012, 7, e50616. [Google Scholar] [CrossRef]
- Campregher, C.; Schmid, G.; Ferk, F.; Knasmüller, S.; Khare, V.; Kortüm, B.; Dammann, K.; Lang, M.; Scharl, T.; Spittler, A.; et al. MSH3-deficiency initiates EMAST without oncogenic transformation of human colon epithelial cells. PLOS ONE 2012, 7, e50541. [Google Scholar] [CrossRef] [PubMed]
- Brentnall, T.A.; Chen, R.; Lee, J.G.; Kimmey, M.B.; Bronner, M.P.; Haggitt, R.C.; Kowdley, K.V.; Hecker, L.M.; Byrd, D.R. Microsatellite instability and K-ras mutations associated with pancreatic adenocarcinoma and pancreatitis. Cancer Res. 1995, 55, 4264–4267. [Google Scholar] [PubMed]
- Brentnall, T.A.; Crispin, D.A.; Bronner, M.P.; Cherian, S.P.; Hueffed, M.; Rabinovitch, P.S.; Rubin, C.E.; Haggitt, R.C.; Boland, C.R. Microsatellite instability in nonneoplastic mucosa from patients with chronic ulcerative colitis. Cancer Res. 1996, 56, 1237–1240. [Google Scholar] [PubMed]
- Huang, S.C.; Lee, J.K.; Smith, E.J.; Pharm, R.T.D.; Tajima, A.; Beck, S.E.; Weidner, N.; Carethers, J.M. Evidence for an hMSH3 defect in familial hamartomatous polyps. Cancer 2011, 117, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Tseng-Rogenski, S.; Hamaya, Y.; Choi, D.Y.; Carethers, J.M. Interleukin 6 alters localization of hMSH3, leading to DNA mismatch repair defects in colorectal cancer cells. Gastroenterology 2015, 148, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M.; Murali, B.; Yang, B.; Doctolero, R.T.; Tajima, A.; Basa, R.; Smith, E.J.; Lee, M.; Janke, R.; Ngo, T.; et al. Influence of race on microsatellite instability and CD8+ T cell infiltration in colon cancer. PLOS ONE 2014, 9, e100461. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.L.; Marra, G.; Chauhan, D.P.; Ha, H.T.; Chang, D.K.; Ricciardiello, L.; Randolph, A.; Carethers, J.M.; Boland, C.R. Oxidative stress inactivates the human DNA mismatch repair system. Am. J. Physiol. Cell Physiol. 2002, 283, C148–C154. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.K.; Goel, A.; Ricciardiello, L.; Lee, D.H.; Chang, C.L.; Carethers, J.M.; Boland, C.R. Effect of H(2)O(2) on cell cycle and survival in DNA mismatch repair-deficient and -proficient cell lines. Cancer Lett. 2003, 195, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Piao, J.; Nakatsu, Y.; Ohno, M.; Taguchi, K.; Tsuzuki, T. Mismatch repair deficient mice show susceptibility to oxidative stress-induced intestinal carcinogenesis. Int. J. Biol. Sci. 2013, 10, 73–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holt, I.; Thanh Lam, L.; Tomé, S.; Wansink, D.G.; te Riele, H.; Gourdon, G.; Morris, G.E. The mouse mismatch repair protein, MSH3, is a nucleoplasmic protein that aggregates into denser nuclear bodies under conditions of stress. J. Cell. Biochem. 2011, 112, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wajapeyee, N.; Turker, M.S.; Glazer, P.M. Silencing of the DNA mismatch repair gene MLH1 induced by hypoxic stress in a pathway dependent on the histone demethylase LSD1. Cell Rep. 2014, 8, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Iwaizumi, M.; Tseng-Rogenski, S.; Carethers, J.M. Acidic tumor microenvironment downregulates MLH1 but does not diminish 5-fluorouracil chemosensitivity. Mutat. Res. 2013, 747–748, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, V.T.; Bindra, R.S.; Yuan, J.; Campisi, D.; Narayanan, L.; Jensen, R.; Giordano, F.; Johnson, R.S.; Rockwell, S.; Glazer, P.M. Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol. Cell. Biol. 2003, 23, 3265–3273. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Tanimoto, K.; Hiyama, K.; Yunokawa, M.; Kawamoto, T.; Kato, Y.; Yoshiga, K.; Poellinger, L.; Hiyama, E.; Nishiyama, M. Human mismatch repair gene, MLH1, is transcriptionally repressed by the hypoxia-inducible transcription factors, DEC1 and DEC2. Oncogene 2008, 27, 4200–4209. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.A.; Witherspoon, M.; Wang, K.; Afrasiabi, K.; Pham, T.; Birnbaumer, L.; Lipkin, S.M. Epigenetic repression of DNA mismatch repair by inflammation and hypoxia in inflammatory bowel disease-associated colorectal cancer. Cancer Res. 2009, 69, 6423–6429. [Google Scholar] [CrossRef] [PubMed]
- Koshiji, M.; To, K.K.; Hammer, S.; Kumamoto, K.; Harris, A.L.; Modrich, P.; Huang, L.E. HIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expression. Mol. Cell. 2005, 17, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Jiménez, F.J.; Moreno-Manzano, V.; Lucas-Dominguez, R.; Sánchez-Puelles, J.M. Hypoxia causes downregulation of mismatch repair system and genomic instability in stem cells. Stem Cells 2008, 26, 2052–2062. [Google Scholar] [CrossRef] [PubMed]
- Kondo, A.; Safaei, R.; Mishima, M.; Niedner, H.; Lin, X.; Howell, S.B. Hypoxia-induced enrichment and mutagenesis of cells that have lost DNA mismatch repair. Cancer Res. 2001, 61, 7603–7607. [Google Scholar] [PubMed]
- Li, J.; Koike, J.; Kugoh, H.; Arita, M.; Ohhira, T.; Kikuchi, Y.; Funahashi, K.; Takamatsu, K.; Boland, C.R.; Koi, M.; et al. Down-regulation of MutS homolog 3 by hypoxia in human colorectal cancer. Biochim. Biophys. Acta 2012, 1823, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Kloor, M.; Schwitalle, Y.; von Knebel Doeberitz, M.; Wentzensen, N. Tetranucleotide repeats in coding regions: No evidence for involvement in EMAST carcinogenesis. J. Mol. Med. (Berl) 2006, 84, 329–333. [Google Scholar] [CrossRef]
- Ikeda, M.; Orimo, H.; Moriyama, H.; Nakajima, E.; Matsubara, N.; Mibu, R.; Tanaka, N.; Shimada, T.; Kimura, A.; Shimizu, K. Close correlation between mutations of E2F4 and MSH3 genes in colorectal cancers with microsatellite instability. Cancer Res. 1998, 58, 594–598. [Google Scholar]
- Yoshitaka, T.; Matsubara, N.; Ikeda, M.; Tanino, M.; Hanafusa, H.; Tanaka, N.; Shimizu, K. Mutations of E2F-4 trinucleotide repeats in colorectal cancer with microsatellite instability. Biochem. Biophys. Res. Commun. 1996, 227, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Nework. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Calin, G.A.; Gafà, R.; Tibiletti, M.G.; Herlea, V.; Becheanu, G.; Cavazzini, L.; Barbanti-Brodano, G.; Nenci, I.; Negrini, M.; Lanza, G. Genetic progression in microsatellite instability high (MSI-H) colon cancers correlates with clinico-pathological parameters: A study of the TGRbetaRII, BAX, MSH3, MSH6, IGFIIR and BLM genes. Int. J. Cancer 2000, 89, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Planck, M.; Wenngren, E.; Borg, A.; Olsson, H.; Nilbert, M. Somatic frameshift alterations in mononucleotide repeat-containing genes in different tumor types from an HNPCC family with germline MSH2 mutation. Genes Chromosomes Cancer 2000, 29, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Ohmiya, N.; Matsumoto, S.; Yamamoto, H.; Baranovskaya, S.; Malkhosyan, S.R.; Perucho, M. Germline and somatic mutations in MSH6 and MSH3 in gastrointestinal cancers of the microsatellite mutator phenotype. Gene 2001, 272, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.R.; Lahue, E.E.; Li, G.M.; Lahue, R.S. Trinucleotide repeat expansions catalyzed by human cell-free extracts. Cell Res. 2013, 23, 565–572. [Google Scholar] [CrossRef]
- Tomé, S.; Manley, K.; Simard, J.P.; Clark, G.W.; Slean, M.M.; Swami, M.; Shelbourne, P.F.; Tillier, E.R.; Monckton, D.G.; Messer, A.; et al. MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington’s disease mice. PLOS Genet. 2013, 9, e1003280. [Google Scholar] [CrossRef] [PubMed]
- Tomé, S.; Simard, J.P.; Slean, M.M.; Holt, I.; Morris, G.E.; Wojciechowicz, K.; te Riele, H.; Pearson, C.E. Tissue-specific mismatch repair protein expression: MSH3 is higher than MSH6 in multiple mouse tissues. DNA Repair (Amst.) 2013, 12, 46–52. [Google Scholar] [CrossRef]
- Chung, H.; Lopez, C.G.; Young, D.J.; Lai, J.F.; Holmstrom, J.; Ream-Robinson, D.; Cabrera, B.L.; Carethers, J.M. Flanking sequence specificity determines coding microsatellite heteroduplex and mutation rates with defective DNA mismatch repair. Oncogene 2010, 29, 2172–2180. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Lopez, C.G.; Holmstrom, J.; Young, D.J.; Lai, J.F.; Ream-Robinson, D.; Carethers, J.M. Both microsatellite length and sequence context determine frameshift mutation rates in defective DNA mismatch repair. Hum. Mol. Genet. 2010, 19, 2638–2644. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Chaudhry, J.; Lai, J.F.; Young, D.J.; Carethers, J.M. Flanking nucleotide specificity for DNA mismatch repair-deficient frameshifts within Activin Receptor 2 (ACVR2). Mutat. Res. 2012, 729, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Lang, W.H.; Coats, J.E.; Majka, J.; Hura, G.L.; Lin, Y.; Rasnik, I.; McMurray, C.T. Conformational trapping of mismatch recognition complex MSH2/MSH3 on repair-resistant DNA loops. Proc. Natl. Acad. Sci. USA 2011, 108, E837–E844. [Google Scholar] [CrossRef]
- Takahashi, M.; Koi, M.; Balaguer, F.; Boland, C.R.; Goel, A. MSH3 mediates sensitization of colorectal cancer cells to cisplatin, oxaliplatin, and a poly(ADP-ribose) polymerase inhibitor. J. Biol. Chem. 2011, 286, 12157–12165. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Huang, S.; Tougeron, D.; Sinicrope, F.A. MSH3 mismatch repair protein regulates sensitivity to cytotoxic drugs and a histone deacetylase inhibitor in human colon carcinoma cells. PLOS ONE 2013, 8, e65369. [Google Scholar] [CrossRef] [PubMed]
- Van Oers, J.M.; Edwards, Y.; Chahwan, R.; Zhang, W.; Smith, C.; Pechuan, X.; Schaetzlein, S.; Jin, B.; Wang, Y.; Bergman, A.; et al. The MutSβ complex is a modulator of p53-driven tumorigenesis through its functions in both DNA double-strand break repair and mismatch repair. Oncogene 2014, 33, 3939–3946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietlein, F.; Thelen, L.; Jokic, M.; Jachimowicz, R.D.; Ivan, L.; Knittel, G.; Leeser, U.; van Oers, J.; Edelmann, W.; Heukamp, L.C.; et al. A functional cancer genomics screen identifies a druggable synthetic lethal interaction between MSH3 and PRKDC. Cancer Discov. 2014, 4, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Tajima, A.; Iwaizumi, M.; Tseng-Rogenski, S.; Cabrera, B.L.; Carethers, J.M. Both MutSα and MutSβ complexes participate in 5-fluoruracil cytotoxicity. PLOS ONE 2011, 6, e28117. [Google Scholar] [CrossRef] [PubMed]
- Vilar, E.; Scaltriti, M.; Balmaña, J.; Saura, C.; Guzman, M.; Arribas, J.; Baselga, J.; Tabernero, J. Microsatellite instability due to MLH1 deficiency is associated with increased cytotoxicity to irinotecan in human colorectal cancer cell lines. Br. J. Cancer 2008, 99, 1607–1612. [Google Scholar] [CrossRef] [PubMed]
- Bras-Gonçalves, R.A.; Rosty, C.; Laurent-Puig, P.; Soulié, P.; Dutrillaux, B.; Poupon, M.F. Sensitivity to CPT-11 of xenografted human colorectal cancers as a function of microsatellite instability and p53 status. Br. J. Cancer 2000, 82, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Magrini, R.; Bhonde, M.R.; Hanski, M.L.; Notter, M.; Scherübl, H.; Boland, C.R.; Zeitz, M.; Hanski, C. Cellular effects of CPT-11 on colon carcinoma cells: Dependence on p53 and MLH1 status. Int. J. Cancer 2002, 101, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Hansen, L.T.; Phear, G.; Scorah, J.; Spang-Thomsen, M.; Cox, A.; Helleday, T.; Meuth, M. Thymidine selectively enhances growth suppressive effects of camptothecin/irinotecan in MSI+ cells and tumors containing a mutation of MRE11. Clin. Cancer Res. 2008, 14, 5476–5483. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, A.; Poindessous, V.; Ouaret, D.; Regairaz, M.; Bastian, G.; Guérin, E.; Escargueil, A.E.; Larsen, A.K. Acquired irinotecan resistance is accompanied by stable modifications of cell cycle dynamics independent of MSI status. Int. J. Oncol. 2013, 42, 1644–1653. [Google Scholar] [PubMed]
- Fallik, D.; Borrini, F.; Boige, V.; Viguier, J.; Jacob, S.; Miquel, C.; Sabourin, J.C.; Ducreux, M.; Praz, F. Microsatellite instability is a predictive factor of the tumor response to irinotecan in patients with advanced colorectal cancer. Cancer Res. 2003, 63, 5738–5744. [Google Scholar] [PubMed]
- Bertagnolli, M.M.; Niedzwiecki, D.; Compton, C.C.; Hahn, H.P.; Hall, M.; Damas, B.; Jewell, S.D.; Mayer, R.J.; Goldberg, R.M.; Saltz, L.B.; et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and Leukemia Group B Protocol 89803. J. Clin. Oncol. 2009, 27, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Klingbiel, D.; Saridaki, Z.; Roth, A.D.; Bosman, F.T.; Delorenzi, M.; Tejpar, S. Prognosis of stage II and III colon cancer treated with adjuvant 5-fluorouracil or FOLFIRI in relation to microsatellite status: Results of the PETACC-3 trial. Ann. Oncol. 2015, 26, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.; Tran, B.; Ensor, J.; Gibbs, P.; Wong, H.L.; Wong, S.F.; Vilar, E.; Tie, J.; Broaddus, R.; Kopetz, S.; et al. Multicenter retrospective analysis of metastatic colorectal cancer (CRC) with high-level microsatellite instability (MSI-H). Ann. Oncol. 2014, 25, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Hong, Y.S.; Ryu, M.H.; Lee, J.L.; Chang, H.M.; Lim, S.B.; Kim, J.H.; Jang, S.J.; Kim, M.J.; Yu, C.S.; et al. Association between deficient mismatch repair system and efficacy to irinotecan-containing chemotherapy in metastatic colon cancer. Cancer Sci. 2011, 102, 1706–1711. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M. One colon lumen but two organs. Gastroenterology 2011, 141, 411–412. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Morikawa, T.; Kuchiba, A.; Imamura, Y.; Qian, Z.R.; Nishihara, R.; Liao, X.; Waldron, L.; Hoshida, Y.; Huttenhower, C.; et al. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut 2012, 61, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M. Screening for colorectal cancer in African Americans: Determinants and rationale for an earlier age to commence screening. Dig. Dis. Sci. 2015, 60, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Di Caro, G.; Marchesi, F.; Laghi, L.; Grizzi, F. Immune cells: Plastic players along colorectal cancer progression. J. Cell. Mol. Med. 2013, 17, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M. Secondary prevention of colorectal cancer: is there an optimal follow-up for patients with colorectal cancer? Curr. Colorectal Cancer Rep. 2010, 6, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Chia, W.K.; Ali, R.; Toh, H.C. Aspirin as adjuvant therapy for colorectal cancer—Reinterpreting paradigms. Nat. Rev. Clin. Oncol. 2012, 9, 561–570. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carethers, J.M.; Koi, M.; Tseng-Rogenski, S.S. EMAST is a Form of Microsatellite Instability That is Initiated by Inflammation and Modulates Colorectal Cancer Progression. Genes 2015, 6, 185-205. https://doi.org/10.3390/genes6020185
Carethers JM, Koi M, Tseng-Rogenski SS. EMAST is a Form of Microsatellite Instability That is Initiated by Inflammation and Modulates Colorectal Cancer Progression. Genes. 2015; 6(2):185-205. https://doi.org/10.3390/genes6020185
Chicago/Turabian StyleCarethers, John M., Minoru Koi, and Stephanie S. Tseng-Rogenski. 2015. "EMAST is a Form of Microsatellite Instability That is Initiated by Inflammation and Modulates Colorectal Cancer Progression" Genes 6, no. 2: 185-205. https://doi.org/10.3390/genes6020185