
Novel PIGT Variant in Two Brothers: Expansion of the Multiple Congenital Anomalies-Hypotonia Seizures Syndrome 3 Phenotype

 , , and
, , and
Abstract
:1. Introduction
2. Experimental Section
2.1. Clinical Description
2.2. Materials and Methods
2.2.1. Whole Exome Sequencing
2.2.2. Flow Cytometry
3. Results
3.1. Genetic Analysis
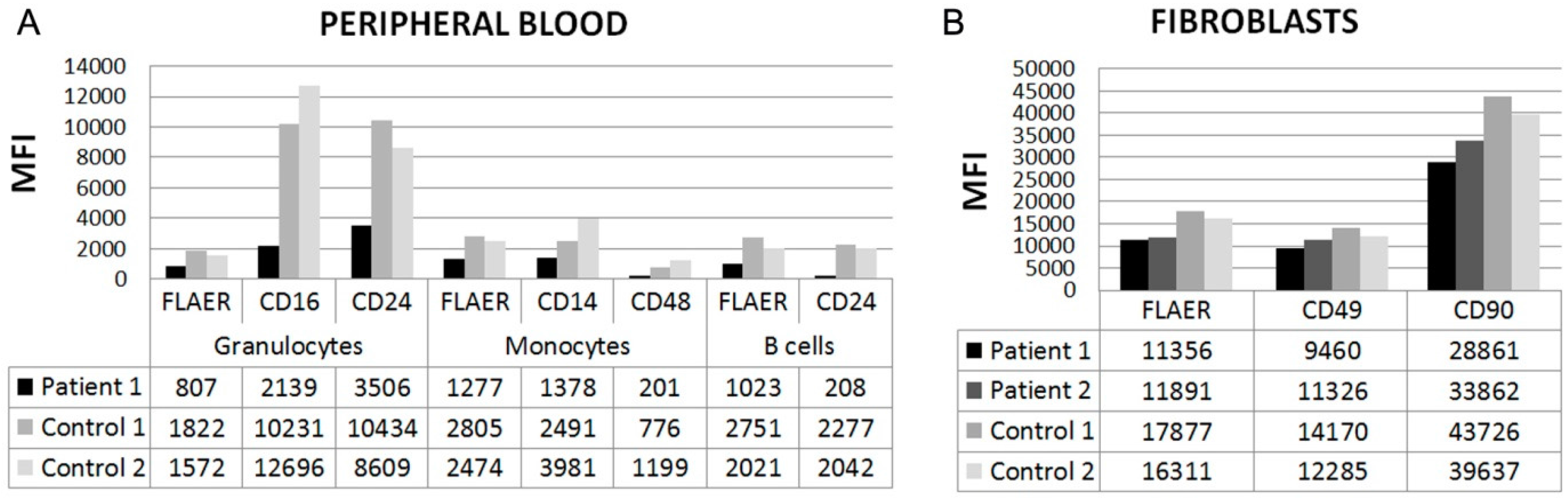
3.2. Flow Cytometry
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kinoshita, T.; Fujita, M. Biosynthesis of GPI-anchored proteins: Special emphasis on GPI lipid remodeling. J. Lipid Res. 2016, 57, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, K.; Inoue, N.; Kinoshita, T. PIG-S and PIG-T, essential for GPI anchor attachment to proteins, form a complex with GAA1 and GPI8. EMBO J. 2001, 20, 4088–4098. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, K.; Nagamune, K.; Maeda, Y.; Kinoshita, T. Two subunits of glycosylphosphatidylinositol transamidase, GPI8 and PIG-T, form a functionally important intermolecular disulfide bridge. J. Biol. Chem. 2003, 278, 13959–13967. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.M.; Murakami, Y.; Layton, D.M.; Hillmen, P.; Sellick, G.S.; Maeda, Y.; Richards, S.; Patterson, S.; Kotsianidis, I.; Mollica, L.; et al. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat. Med. 2006, 12, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Chiyonobu, T.; Inoue, N.; Morimoto, M.; Kinoshita, T.; Murakami, Y. Glycosylphosphatidylinositol (GPI) anchor deficiency caused by mutations in PIGW is associated with west syndrome and hyperphosphatasia with mental retardation syndrome. J. Med. Genet. 2014, 51, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.; Tawamie, H.; Murakami, Y.; Mang, Y.; ur Rehman, S.; Buchert, R.; Schaffer, S.; Muhammad, S.; Bak, M.; Nothen, M.M.; et al. Hypomorphic mutations in PGAP2, encoding a GPI-anchor-remodeling protein, cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2013, 92, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.F.; Murakami, Y.; Pagnamenta, A.T.; Daumer-Haas, C.; Fischer, B.; Hecht, J.; Keays, D.A.; Knight, S.J.; Kolsch, U.; Kruger, U.; et al. Mutations in PGAP3 impair GPI-anchor maturation, causing a subtype of hyperphosphatasia with mental retardation. Am. J. Hum. Genet. 2014, 94, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Ilkovski, B.; Pagnamenta, A.T.; O’Grady, G.L.; Kinoshita, T.; Howard, M.F.; Lek, M.; Thomas, B.; Turner, A.; Christodoulou, J.; Sillence, D.; et al. Mutations in PIGY: Expanding the phenotype of inherited glycosylphosphatidylinositol deficiencies. Hum. Mol. Genet. 2015, 24, 6146–6159. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.J.; Gropman, A.L.; Sapp, J.C.; Teer, J.K.; Martin, J.M.; Liu, C.F.; Yuan, X.; Ye, Z.; Cheng, L.; Brodsky, R.A.; et al. The phenotype of a germline mutation in PIGA: The gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am. J. Hum. Genet. 2012, 90, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Krawitz, P.M.; Murakami, Y.; Hecht, J.; Kruger, U.; Holder, S.E.; Mortier, G.R.; Delle Chiaie, B.; De Baere, E.; Thompson, M.D.; Roscioli, T.; et al. Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am. J. Hum. Genet. 2012, 91, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Kvarnung, M.; Nilsson, D.; Lindstrand, A.; Korenke, G.C.; Chiang, S.C.; Blennow, E.; Bergmann, M.; Stodberg, T.; Makitie, O.; Anderlid, B.M.; et al. A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J. Med. Genet. 2013, 50, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Maydan, G.; Noyman, I.; Har-Zahav, A.; Neriah, Z.B.; Pasmanik-Chor, M.; Yeheskel, A.; Albin-Kaplanski, A.; Maya, I.; Magal, N.; Birk, E.; et al. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J. Med. Genet. 2011, 48, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Tawamie, H.; Maeda, Y.; Buttner, C.; Buchert, R.; Radwan, F.; Schaffer, S.; Sticht, H.; Aigner, M.; Reis, A.; et al. Null mutation in PGAP1 impairing GPI-anchor maturation in patients with intellectual disability and encephalopathy. PLoS Genet. 2014, 10, e1004320. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.G.; Hackmann, K.; Jones, M.A.; Eroshkin, A.M.; He, P.; Wiliams, R.; Bhide, S.; Cantagrel, V.; Gleeson, J.G.; Paller, A.S.; et al. Mutations in the glycosylphosphatidylinositol gene PIGL cause chime syndrome. Am. J. Hum. Genet. 2012, 90, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Krawitz, P.M.; Schweiger, M.R.; Rodelsperger, C.; Marcelis, C.; Kolsch, U.; Meisel, C.; Stephani, F.; Kinoshita, T.; Murakami, Y.; Bauer, S.; et al. Identity-by-descent filtering of exome sequence data identifies pigv mutations in hyperphosphatasia mental retardation syndrome. Nat. Genet. 2010, 42, 827–829. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.; Golas, G.A.; Davids, M.; Huizing, M.; Kane, M.S.; Krasnewich, D.M.; Malicdan, M.C.; Adams, D.R.; Markello, T.C.; Zein, W.M.; et al. Expanding the clinical and molecular characteristics of PIGT-CDG, a disorder of glycosylphosphatidylinositol anchors. Mol. Genet. Metab. 2015, 115, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Kashii, H.; Murakami, Y.; Kato, M.; Tsurusaki, Y.; Miyake, N.; Kubota, M.; Kinoshita, T.; Saitsu, H.; Matsumoto, N. Novel compound heterozygous PIGT mutations caused multiple congenital anomalies-hypotonia-seizures syndrome 3. Neurogenetics 2014, 15, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Michalec, D. Bayley Scales of Infant Development: Third Edition. In Encyclopedia of Child Behavior and Development; Goldstein, S., Naglieri, J.A., Eds.; Springer: Boston, MA, USA, 2011; p. 215. [Google Scholar]
- Reynell, J.; Gruber, C. Reynell Developmental Language Scales; Western Psychological Services: Los Angeles, CA, USA, 1990. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SNPeff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Pritchard, B.; Rios, D.; Chen, Y.; Flicek, P.; Cunningham, F. Deriving the consequences of genomic variants with the ensembl API and SNP effect predictor. Bioinformatics 2010, 26, 2069–2070. [Google Scholar] [CrossRef] [PubMed]
- Vigeland, M.D.; Gjotterud, K.S.; Selmer, K.K. Filtus: A desktop GUI for fast and efficient detection of disease-causing variants, including a novel autozygosity detector. Bioinformatics 2016, 32, 1592–1594. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [PubMed]
- UniProt Consortium. Uniprot: A hub for protein information. Nucl. Acids Res. 2015, 43, D204–D212. [Google Scholar]
- Millan, J.L. The role of phosphatases in the initiation of skeletal mineralization. Calcif. Tissue Int. 2013, 93, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Di Mauro, S.; Manes, T.; Hessle, L.; Kozlenkov, A.; Pizauro, J.M.; Hoylaerts, M.F.; Millan, J.L. Kinetic characterization of hypophosphatasia mutations with physiological substrates. J. Bone Miner. Res. 2002, 17, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Waymire, K.G.; Mahuren, J.D.; Jaje, J.M.; Guilarte, T.R.; Coburn, S.P.; MacGregor, G.R. Mice lacking tissue non-specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat. Genet. 1995, 11, 45–51. [Google Scholar] [CrossRef] [PubMed]

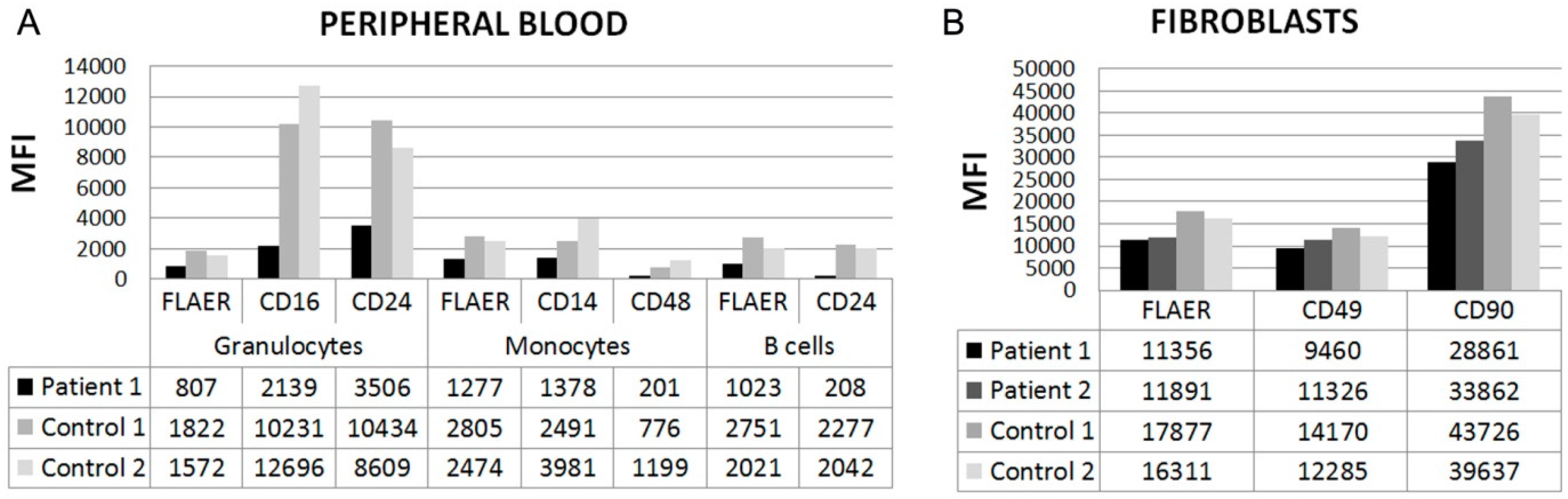

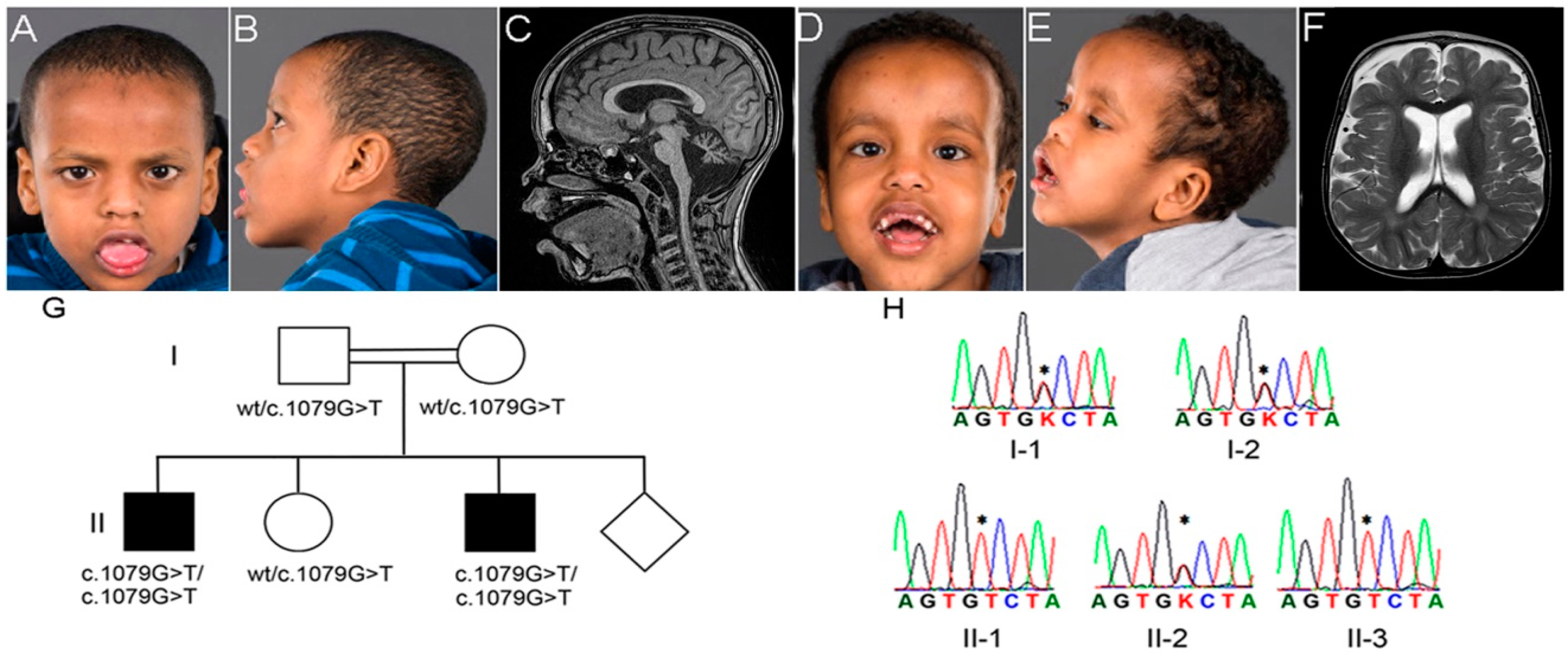{kind=link}
{kind=link}
| Reference | Skauli et al. | Lam et al. [16] | Nakashima et al. [17] | Kvarnung et al. [11] | |||||
|---|---|---|---|---|---|---|---|---|---|
| Patients | 1 | 2 | 1 | 2 | 1 | V-1 | V-2 | V-4 | V-5 |
| PIGT Variants | c.1079G>T; p.Gly360Val | c.918dupC; p.Val307Argfs*13/c.1342C>T; p.Arg488Trp | c.250G>T; p.Glu84*/c.1342C>T; p.Arg488Trp | c.547A>C; p.Thr183Pro | |||||
| Gender | M | M | F | M | F | F | F | F | F |
| Weeks GA | 40 | 40 | <32 | <32 | 40 | 40 | 39 | 37 | 37 |
| HC > 90th at birth | - | + | − | − | + | + | + | + | + |
| Progressive neurological features | + | + | + | + | + | + | + | + | + |
| ID | Se | Mo-Se | Pro | Pro | Pro | Se | Se | Se | Se |
| Hypotonia | + | + | + | + | + | + | + | + | + |
| Epileptic seizures | + | + | + | + | + | + | + | + | + |
| Cerebral and cerebellar atrophy | + | + | + | + | + | PSF | - | + | + |
| Esotropia | + | + | + | + | + | + | + | + | + |
| Nystagmus | + | + | + | + | + | + | + | + | + |
| CVI | + | + | + | + | + | + | + | + | + |
| Heart defect | − | − | + | + | + | + | - | + | + |
| Nephrocalcinosis/urolithiasis | − | − | − | − | + | + | + | + | + |
| Genitourinary abnormalities | - | - | - | - | + | + | + | + | + |
| Skeletal abnormalities | − | − | + | + | + | + | + | + | + |
| Pectus excavatum | − | − | + | + | - | + | - | - | + |
| Scoliosis | − | − | + | + | + | + | + | - | - |
| Mesomelic shortening upper limbs | − | − | − | − | - | + | + | + | - |
| Slender long bones | − | − | + | + | - | - | - | - | + |
| Osteopenia | − | − | + | + | + | + | + | + | - |
| Bone age | N | N | Advanced | N | NR | Delayed | Delayed | Delayed | Delayed |
| Low serum alkaline phosphatase | − | − | − | − | + | + | + | + | + |
| High forehead, frontal bossing, bitemporal narrowing | + | + | + | + | NR | + | + | + | + |
| Short nose, anteverted nares, depressed nasal bridge | + | + | + | + | + | + | + | + | + |
| Wide, open mouth | + | + | + | + | + | + | + | + | + |
| High palate | + | + | + | + | + | NR | NR | NR | + |
| Inverted nipples | + | + | + | + | + | NR | NR | NR | + |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skauli, N.; Wallace, S.; Chiang, S.C.C.; Barøy, T.; Holmgren, A.; Stray-Pedersen, A.; Bryceson, Y.T.; Strømme, P.; Frengen, E.; Misceo, D. Novel PIGT Variant in Two Brothers: Expansion of the Multiple Congenital Anomalies-Hypotonia Seizures Syndrome 3 Phenotype. Genes 2016, 7, 108. https://doi.org/10.3390/genes7120108
Skauli N, Wallace S, Chiang SCC, Barøy T, Holmgren A, Stray-Pedersen A, Bryceson YT, Strømme P, Frengen E, Misceo D. Novel PIGT Variant in Two Brothers: Expansion of the Multiple Congenital Anomalies-Hypotonia Seizures Syndrome 3 Phenotype. Genes. 2016; 7(12):108. https://doi.org/10.3390/genes7120108
Chicago/Turabian StyleSkauli, Nadia, Sean Wallace, Samuel C. C. Chiang, Tuva Barøy, Asbjørn Holmgren, Asbjørg Stray-Pedersen, Yenan T. Bryceson, Petter Strømme, Eirik Frengen, and Doriana Misceo. 2016. "Novel PIGT Variant in Two Brothers: Expansion of the Multiple Congenital Anomalies-Hypotonia Seizures Syndrome 3 Phenotype" Genes 7, no. 12: 108. https://doi.org/10.3390/genes7120108